

Abstract
Although peroxisome proliferator-activated receptor α (PPARα) is highly expressed in the heart, the effects of PPARα on cardiac remodelling and the underlying mechanisms are unclear. The present study was undertaken to test the hypothesis that PPARα activator fenofibrate plays a key role in left ventricular hypertrophic remodelling via the formation of c-fos/c-jun heterodimers in spontaneous hypertensive rats (SHRs). Twenty-four male 8-week-old SHRs were randomly divided into two groups, one group treated with oral saline (n= 10) and another treated with oral fenofibrate (60 mg.kg−1.d−1, n= 14). Ten same-aged Wistar–Kyoto (WKY) rats were selected as a normal control group. Using echocardiography, immunohistochemistry, co-immunoprecipitation, Western blot analysis and real-time RT-PCR, we showed that the left ventricular wall thickness and significantly reduced and left ventricular diastolic function improved in SHRs treated with fenofibrate compared with SHRs treated with saline. Similarly, the excessive collagen deposition and the up-regulation of collagen I, collagen III, c-fos and c-jun seen in SHRs receiving saline were significantly attenuated in SHRs receiving fenofibrate. In addition, fenofibrate markedly decreased the expression of AP-1 and c-fos/c-jun heterodimers (P < 0.01). These results demonstrated that PPARα activator fenofibrate may exert a protective effect on cardiac remodelling in SHRs by decreasing the expression of c-fos and c-jun and suppressing the formation of c-fos/c-jun heterodimers, which may further inhibit transcription of the downstream genes involved in the pathogenesis of left ventricular hypertrophy induced by hypertension.
Keywords: peroxisome proliferator-activated receptor, fenofibrate, hypertension, cardiac remodelling
Introduction
Left ventricular hypertrophy is one of the most common target organ damages in primary hypertension. The major pathological changes involved in left ventricular hypertrophy are cardiomyocyte hypertrophy and apoptosis as well as extracellular matrix remodelling of the myocardium [1]. Cardiac remodelling is an important adaptive physiological response to hemodynamic overload to the heart with excessive accumulation of collagen fibres [2]. The content of the collagen may increase substantially or the composition of the collagen may be significantly altered. Cardiac fibroblasts are the most abundant cell type present in the myocardium and are mainly responsible for the deposition of extracellular matrix [3]. The majority of the extracellular matrix proteins are collagens and two-thirds of the collagens are in type I and type III [4]. Interstitial fibrosis contributes to left ventricular wall stiffness and thus impairs left ventricular diastolic function [5, 6].
Peroxisome proliferator-activated receptors (PPARs) are a family of at least three nuclear receptors (α, β and γ) [7]. PPARα is expressed mainly in the liver, heart, kidney and muscle [8] and plays an important role in catabolism of fatty acid. PPARα is a ligand-activated transcription factor that regulates gene expression by heterodimerizing with retinoid X receptor and binding to PPAR response elements [9]. Fibric acid derivatives, including fenofibrate, are thought to act as specific activators for PPARα[10, 11]. PPAR activators negatively regulate the vascular inflammatory gene response by negative cross-talk with transcription factors, nuclear factor kappa B (NF-κB) and activator protein-1 (AP-1) [12]. Moreover, PPARα is deactivated during cardiac hypertrophic growth [13], which suggests a crucial role of PPARα in regulation of cardiac remodelling.
In the relation between collagen gene regulation and PPARα activation, AP-1 is a common molecule, because several stimuli up-regulate collagen type I gene via AP-1 activation, and PPARα is reported to interfere negatively with AP-1 in endothelial cells [2]. AP-1 plays a biological role mainly through c-fos/c-jun het-erodimers [14]. In the present study, we hypothesized that PPARα activation may reduce collagen type I production and ameliorate cardiac remodelling by inhibiting the formation of c-fos/c-jun het-erodimers. To test this hypothesis, we analysed the effects of the PPARα activator fenofibrate on cardiac remodelling and its relationship with the AP-1 exprssion and collagen density in the myocardium of spontaneous hypertensive rat (SHR).
Materials and methods
Animal models
Twenty-four male 8-week-old SHRs were purchased from Wei Tong Li Hua Corp. (Beijing, China) and 10 male 8-week-old Wistar–Kyoto (WKY) rats from the experimental animal centre of Shandong University (Jinan, China). All rats were kept on a 12-hr light/12-hr dark cycle with food and water freely available. SHRs were randomly divided into two groups for treatment every day for 8 weeks: SHR + saline group which received oral 0.9% saline (n= 10) and SHR + fenofibrate group which received oral fenofibrate (Laboratoires Fournier S.A., 60 mg.kg−1.d−1 dissolved in distilled water, n= 14). The WKY rats received no treatment and served as controls. Systolic blood pressure (SBP) was measured by the tail-cuff method. Echocardiography was performed at the beginning and the end of the experiment and hemodynamic measurement was conducted immediately before rats were killed. The heart was excised and weighed. A portion of the left ventricle was fixed in Bouin's solution for collagen evaluation and immunohistological assay, and the rest of the left ventricle was snap frozen and kept at −80°C. All experimental procedures were performed in accordance with animal protocols approved by the Shandong University Animal Care Committee.
Echocardiographic examination
Echocardiographic examination was undertaken by use of a 12-MHz linear array transducer (Sonos 7500, Philips, MA, USA) at the beginning of the study and 24 hrs before rats were killed under anaesthesia with 10% chloral hydrate (0.3 ml/100 g). Echocardiographic images were obtained in the parasternal long-axis and short-axis views and left ventricular end-diastolic and end-systolic diameters, posterior wall and septum thickness, fractional shortening and ejection fraction were measured according to the American Society of Echocardiography guidelines [15]. The relative wall thickness (RWT) was calculated by dividing the sum of interventricular septal wall thickness (IVSTd) and left ventricular posterior wall thickness (LVPWTd) at end-diastole to left ventricular end-diastolic diameter (LVEDD). Doppler measurements included the peak early (E) and late (A) mitral inflow velocities, E/A ratio and deceleration time of the E wave velocities (DT). To derive quantitative analysis of integrated backscatter (IBS) of the left ventricle, a commercially available software package (acoustic densitometry; Phillips Medical Systems, the Netherlands) was applied [16]. Two-dimensional echocardiographic images including left ventricular short-axis view at the papillary muscle level and apical four- and two-chamber views were recorded and stored digitally on an optical magnetic disk for later IBS analysis. For all echocardiographic measurements, values of three consecutive cardiac cycles were averaged.
Hemodynamic measurement
Hemodynamic measurement was carried out in all rats under deep anaesthesia. A fluid-filled catheter was advanced from the right carotid artery into the left ventricle and the left ventricular end-diastolic pressure (LVEDP) and the maximal rate of left ventricular pressure increase (+dP/dtmax) and decrease (−dP/dtmax) were measured by use of a pressure transducer interfaced with a physiological recorder (Digi-Med, Japan) [17].
Evaluation of collagen deposition
Paraffin-embedded tissue sections (5 μm) of the median part of the left ventricle were hydrated and stained with Masson Accustain Trichrome stain (Sigma, St. Louis, MO, USA). The proportion of blue staining, indicative of interstitial collagen, was measured in 10 fields randomly selected on each of the three non-consecutive serial sections from each heart, and the values were averaged.
Ultrastructural observation
Left ventricular myocardial tissues, about 0.5 × 1 × 5 mm in each group, were fixed with 2% glutaraldehyde overnight and washed three times with 0.2 mol/l phosphate buffer solution (PBS), then fixed with 1% osmium tetraoxide, washed with 0.2 mol/l PBS again and dehydrated by ethanol at different concentrations. The samples were immersed in Epon812 resin/acetone (1:1) for 30 min., immersed in fresh Epon812 resin for 30 min. and then embedded for convergence overnight at 70°C. The tissue was cut into pieces, and slices 50-nm thick were made by use of an ultramicrotomy (LKB 8800, LKB Produkter AB, Sweden). The ultrastructure of cardiac muscle cells in media was observed on transmission electron microscopy (TEM, H-7000FA, Hitachi, Tokyo, Japan).
Quantitative real-time RT-PCR
Total RNA was isolated from frozen myocardial tissues with use of TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) [18]. The RNA was treated with DNase (DNAfreeTM, Ambion Inc., Austin, TX, USA) to remove contaminating genomic DNA. The quality of RNA was checked on spectrophotometry (DU®800, Beckman, Palo Alto, CA, USA). Total RNA was processed in a two-step procedure as described by the SYBR RT-PCR Kit (Perfect Real Time; TaKaRa, Kyoto, Japan). Briefly, in the first step, cDNA was prepared from 500 ngRNA by reverse transcription in a final volume of 20 μl in a thermal cycler (Tgradient 96, Whatman Biometra, Niedersachsen, Germany). The samples were incubated at 37°C for 60 min. and 95°C for 5 min. The cDNA was stored at −20°C. The rat-specific primers for the genes were designed by use of Primer Premier 5 (Premier Biosoft, Palo Alto, CA, USA). The sequences of oligonucleotide primers were as follows: C-FOS: sense 5′-TGT AGT GAC ACC TGA GAG C-3′, antisense 5′-TAA TTC CAA TAA TGA ACC C-3′; C-JUN: sense 5′-CCT CCC GTC TGG TTG TAG-3′, antisense 5′-AGT GGG CTG TCC CTC TCC-3′; Collagen type I: sense 5′-TTC ACC TAC AGC ACG CTT GT-3′, antisense 5′-TTG GGA TGG AGG GAG TTT AC-3′; Collagen type III: sense 5′-GGT CAC TTT CAC TGG TTG ACG A-3′, antisense 5′-TTG AAT ATC AAA CAC GCA AGG C-3′; PPARα: sense 5′-GAC GCT GGG TCC TCT GGT-3′, antisense 5′-GTC TTG GCT CGC CTC TAA -3′; β-actin: sense 5′-CGT TGA CAT CCG TAA AGA CC-3′, antisense 5′-TAGAGCCACCAATCCACACA-3′. All primers were synthesized by BioAsia Corp. (Shanghai, China). In the second step, quantitative real-time PCR involved LightCycler (Roche Diagnostics, Mannheim, Germany) with SYBR Green I in the SYBR RT-PCR Kit (Perfect Real Time). The reaction involved an initial denaturing at 95°C for 10 sec., then 40 cycles of 55°C for 5 sec. and 72°C for 10 sec., and was terminated by a cooling step at 40°C for 30 sec. A melting-curve analysis was performed to confirm the absence of primer dimmers in specific PCR products. The efficiency of PCR was assessed with serial dilutions of a sample of cDNA from the normal control group. Each experiment was performed in duplicate, and the data were analysed with the use of LightCycler Software 4.0 (Roche Diagnostic). The ‘2 −□□CT method’ for comparing relative expression results between treatments in real-time PCR was applied as described [19].
Nuclear proteins preparation and Western blot analysis of AP-1
Isolation of myocardial nuclei was prepared by a commercially available Nuclear and Cytoplasmic Extraction Reagents (Bipec Biopharma Corporation, Cambridge, MA, USA) according to the manufacturer’s instructions. Protein contents of the nuclear fraction were measured according to Bradford [20]. An equal amount of protein (30 μg) or nuclear protein (30 μg) was fractionated on 10% SDS-polyacrylamide gels in running buffer [25 mM Tris, 192 mM glycine, 0.1% (wt/vol) SDS, pH 8.3] at 90 V and then electroblotted to nitrocellulose membranes. Membranes were blocked at 4°C with 5% non-fat milk in Tris-buffered saline (25 mM Tris, 137 mM NaCl, and 2.7 mM KCl) containing 0.05% Tween-20 and then incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-rat AP-1 antibody (Abcam, USA; dilution: 1:500; molecular weight of AP-1: 36 kD). Then the membranes were washed three times in TBS-T and incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody at room temperature for 2 hrs. Immunoreactive bands were visualized using enhanced chemoluminescence and quantified by image analyzer (AlphaImager 2200, Alpha Innotech, San Leandro, CA, USA). Protein levels of AP-1 were normalized to β-actin or histone levels.
Co-immunoprecipitation and Western blot analysis of c-fos/c-jun heterodimers
Frozen myocardial tissues were homogenized in 1 ml of ice-cold lysis buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 5 mM MgCl2, 0.5% Triton X-100 and complete protease inhibitor mixture). The solution was incubated on ice for 1 hr and centrifuged for 15 min. (4°C) at 15,000 rpm, and then supernatant was collected. The concentration of total protein was determined by a modified Bradford method (Bio-Rad, Hercules, CA, USA). Co-immunoprecipitation assays involved use of the ProFound Mammalian Co-Immunoprecipitation kit (Pierce, Rockford, IL, USA). Briefly, lysates were precleared by incubation with the control gel component at 4°C for 4 hrs, and then added to anti-C-Fos antibody. After incubation at 4°C for 16 hrs, immunoprecipitates were washed four times with the immunoprecipitation buffer and once with reduced-salt immunoprecipitation buffer (125 mM NaCl) before elution. For the detection of RyR2, protein samples were separated on a 3∼12% gradient SDS-PAGE, and then transferred to a PVDF membrane (Millipore, Bedford, MA, USA). Western blot analysis of c-fos/c-jun heterodimers was performed as described previously.
Immunohistochemical staining for c-fos and c-jun
Paraffin-embedded 5-μm sections were rehydrated, and antigen retrieval involved 0.01 mol/l citrate buffer (pH 6.0) at 95°C for 20 min. Endogenous peroxidase activity was quenched by incubation in PBS containing 3% H2O2. Sections were blocked with goat serum for 30 min., incubated with respective primary antibodies (Abcam for c-fos and c-jun), then secondary-conjugated IgG antibody for 1 hr at 37°C and then DAB chromogen was applied. The sections were counterstained with haematoxylin and eosin. Negative controls were incubated with PBS instead of primary antibody. The results were viewed on a confocal FV 1000 SPD Laser Scanning microscope (Olympus, Japan).
Statistical analysis
Values were presented as mean ± S.E.M. Results were compared by one-way ANOVA, followed by a Tukey–Kramer post hoc test and independent samples t-test. A P-value <0.05 was considered statistically significant.
Results
Animal characteristics
The characteristics of three groups of rats at week 1 and week 8 were given in Table 1. At week 1, both groups of SHRs showed higher SBP than the WKY rats. Two rats in the SHR + fenofibrate group died of infection and excessive anaesthesia during the experiment. After treatment for 8 weeks, the ratio of heart weight to body weight (HW/BW) was reduced in SHRs treated with fenofibrate compared with those receiving saline. On the other hand, there was no significant difference in heart rate, SBP and body weight between the two SHR groups.
Table 1.
Characteristics of three groups of rats at week 1 and week 8
| Week 1 | Week 8 | |||||
|---|---|---|---|---|---|---|
| WKY | SHR + saline | SHR + fenofibrate | WKY | SHR + saline | SHR + fenofibrate | |
| (n= 10) | (n= 10) | (n= 14) | (n= 10) | (n= 10) | (n= 12) | |
| HR (bpm) | 459.50 ± 23.45 | 433.38 ± 36.80 | 449.20 ± 43.51 | 437.25 ± 33.98□ | 417.00 ± 38.56 | 424.40 ± 35.63 |
| BW (g) | 241.60 ± 13.40 | 198.38 ± 12.64** | 204.61 ± 9.03** | 360.98 ± 18.27□□ | 300.95 ± 14.02**□□ | 305.47 ± 17.61**□□ |
| SBP (mmHg) | 93.13 ± 1.81 | 165.80 ± 5.06** | 164.03 ± 4.51** | 114.69 ± 2.70□□ | 211.06 ± 5.65**□□ | 212.05 ± 7.85**□□ |
| HW/BW (mg/g) | 2.19 ± 0.13 | 2.95 ± 0.15** | 2.36 ± 0.10## | |||
BW: body weight;
HW: heart weight;
HR: heart rate;
bmp: beats per minute;
SBP: systolic blood pressure.
P < 0.05,
P < 0.01 versus WKY group,
P < 0.05,
P < 0.01 versus SHR + saline group,
P < 0.05,
P < 0.01 versus week 1 data.
Echocardiographic measurements
After 1 week of the experiment, LVPWTd and IVSTd at end-diastole, and RWT in both SHR groups were significantly higher than those in the WKY group (Table 2). At week 8 of the experiment, LVPWTd, IVSTd, RWT and DT were significantly increased and E/A ratio was decreased in SHRs receiving saline compared with their week 1 levels and with those measured at week 8 in the WKY group. In contrast, these parameters were significantly improved at week 8 in SHRs treated with fenofibrate in comparison with SHRs treated with saline (Table 2). On the other hand, left ventricular fractional shortening and ejection fraction did not differ in SHRs treated with fenofibrate and saline (Table 2). SHRs receiving saline at week 8 showed significantly increased IBS percentage and decreased cyclic variation of IBS in the left ventricular posterior walls and interventricular septum in comparison with their week 1 levels and the WKY group, whereas fenofibrate treatment substantially improved these parameters in SHRs measured at week 8 (Table 2).
Table 2.
Echocardiographic measurements in three groups of rats at week 1 and week 8
| Week 1 | Week 8 | |||||
|---|---|---|---|---|---|---|
| WKY | SHR + saline | SHR + fenofibrate | WKY | SHR + saline | SHR + fenofibrate | |
| (n= 10) | (n= 10) | (n= 14) | (n= 10) | (n= 10) | (n= 12) | |
| IVSTd (mm) | 1.34 ± 0.02 | 1.75 ± 0.06** | 1.69 ± 0.08** | 1.82 ± 0.10□□ | 2.60 ± 0.34**□□ | 2.07 ± 0.22##□□ |
| LVPWTd (mm) | 1.33 ± 0.04 | 1.66 ± 0.05** | 1.69 ± 0.07** | 1.84 ± 0.07□□ | 2.49 ± 0.24**□□ | 1.96 ± 0.15##□□ |
| LVEDD (mm) | 4.21 ± 0.30 | 4.24 ± 0.50 | 4.44 ± 0.39 | 4.76 ± 0.10□ | 4.92 ± 0.67□ | 5.04 ± 0.30□ |
| LVESD (mm) | 1.75 ± 0.11 | 1.77 ± 0.18 | 1.79 ± 0.16 | 2.01 ± 0.20□ | 2.13 ± 0.36□ | 2.04 ± 0.24□ |
| RWT | 0.63 ± 0.05 | 0.79 ± 0.09** | 0.77 ± 0.07** | 0.77 ± 0.03 | 1.04 ± 0.29*□ | 0.79 ± 0.09# |
| LVFS | 0.59 ± 0.03 | 0.58 ± 0.02 | 0.60 ± 0.05 | 0.58 ± 0.04 | 0.58 ± 0.04 | 0.59 ± 0.07 |
| LVEF | 91.15 ± 4.02 | 90.55 ± 3.11 | 89.63 ± 4.04 | 89.78 ± 3.51 | 87.69 ± 4.45 | 87.81 ± 4.15 |
| E-wave (cm/sec.) | 81.59 ± 12.87 | 80.49 ± 13.77 | 79.48 ± 18.03 | 81.81 ± 10.10 | 82.58 ± 16.84 | 83.11 ± 15.53 |
| A-wave (cm/sec.) | 45.32 ± 7.29 | 47.09 ± 9.03 | 45.75 ± 11.51 | 45.20 ± 7.26 | 61.84 ± 13.50□ | 48.34 ± 10.69 |
| E/A | 1.82 ± 0.07 | 1.72 ± 0.07 | 1.75 ± 0.11 | 1.84 ± 0.08 | 1.34 ± 0.05**□□ | 1.74 ± 0.13## |
| DT (sec.) | 37.08 ± 2.52 | 37.29 ± 1.55 | 37.87 ± 3.23 | 38.46 ± 2.49 | 43.25 ± 3.35*□□ | 38.93 ± 3.20# |
| IBS%LVPWd | 0.27 ± 0.03 | 0.30 ± 0.06 | 0.34 ± 0.07 | 0.30 ± 0.05 | 0.50 ± 0.09**□□ | 0.37 ± 0.07 ## |
| CVIBLVPWd (dB) | 7.78 ± 1.38 | 6.58 ± 1.13 | 6.35 ± 1.49 | 6.96 ± 1.49 | 4.53 ± 0.80**□□ | 6.08 ± 0.77# |
| IBS%IVS | 0.37 ± 0.04 | 0.41 ± 0.05 | 0.44 ± 0.08 | 0.42 ± 0.06 | 0.58 ± 0.12**□□ | 0.46 ± 0.09# |
| CVIBIVS (dB) | 8.38 ± 1.57 | 7.53 ± 1.43 | 6.91 ± 1.12 | 7.68 ± 1.25 | 5.22 ± 0.95**□□ | 6.83 ± 1.14# |
IVSTd: interventricular septal wall thickness
LVPWTd: left ventricular posterior wall thickness
IVSTd: interventricular septal wall thickness
LVPWTd: left ventricular posterior wall thickness
LVEDD: left ventricular end-diastolic diameter
LVESD: left ventricular end-systolic diameter
RWT: relative wall thickness
LVFS: left ventricular fractional shortening
LVEF: left ventricular ejection fraction
E/A: ratio of E wave to A wave velocities
DT: deceleration time of E wave velocities
IBS: integrated backscatter
CVIB: cyclic variation of IBS
P < 0.05,
P < 0.01 versus WKY group,
P < 0.05,
P < 0.01 versus SHR + saline group,
P < 0.05,
P < 0.01 versus week 1 data.
Hemodynamic indices
Compared with the WKY group, SHRs treated with saline demonstrated significantly elevated LVEDP and depressed ±dP/dtmax as measured at week 8. In contrast, values of LVEDP and ±dP/dtmax in SHRs treated with fenofibrate were significantly improved compared with SHRs treated with saline and showed insignificant difference from those in the WKY group (Table 3).
Table 3.
Hemodynamic indices measured at week 8 in three groups of rats
| Groups | LVEDP (mmHg) | +dp/dtmax (mmHg/sec.) | −dp/dtmax(mmHg/sec.) |
|---|---|---|---|
| WKY (n= 10) | 5.33 ± 1.51 | 3671 ± 578 | 3446 ± 190 |
| SHR + saline (n= 10) | 10.48 ± 2.69** | 2748 ± 404** | 2727 ± 399** |
| SHR + fenofibrate (n= 12) | 7.33 ± 2.17## | 3553 ± 419## | 3141 ± 313## |
LVEDP, left ventricular end-diastolic pressure;
dP/dt, change in left ventricular pressure over time;
P < 0.01 versus WKY group,
P < 0.05,
P < 0.01 versus SHR + saline group.
Ultrastructural analysis
Ultrastructural observations revealed normal quantity and distribution of collagen fibres in extracellular matrix of the WKY rats (Fig. 1). In contrast, SHRs treated with saline showed excessive deposition of collagen fibres. However, SHRs treated with fenofibrate demonstrated approximately normal configuration of collagen fibres as compared with SHRs receiving saline treatment.
Figure 1.
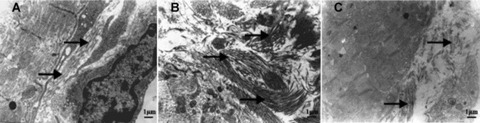
Representative myocardial ultrastructure of the left ventricle in three groups of rats. (A) A cardiac myocyte in a WKY rat showing normal quantity and distribution of extracellular collagen fibres (8000×). (B) A cardiac myocyte in an SHR treated with saline exhibited excessive deposition of collagen fibres in extracellular matrix (8000×). (C) A cardiac myocyte in an SHR treated with fenofibrate showed approximately normal configuration of the collagen fibres (8000×). Arrows indicate collagen fibres.
Collagen deposition and protein expression of c-fos and c-jun
SHRs showed a significantly higher content of left ventricular interstitial collagen than the WKY rats. However, treatment with fenofibrate in SHRs effectively prevented excessive interstitial collagen deposition (Fig. 2). The protein expression of c-fos and c-jun was increased in the myocardium of SHRs receiving saline but was inhibited in SHRs treated with fenofibrate as depicted by nuclei immunohistochemical staining, although the expression of c-fos and c-jun in the latter group was still higher than that in the WKY rats (Fig. 3A–F).
Figure 2.
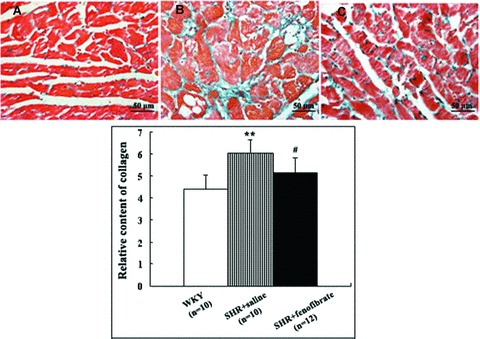
Quantitation of histology of myocardial tissues after Masson staining. Top panel: (A) Myocardial histology in a WKY rat showing normal quantity and distribution of collagen fibres (400×). (B) Myocardial histology in an SHR treated with saline showing hyperplastic and disorganized collagen fibres (400×). (C) Myocardial histology in an SHR treated with fenofibrate demonstrating almost normal appearance of collagen fibres (400×). Bottom panel: comparison of relative content of collagen in three groups of rats. Data shown were means ± S.E.M. (WKY: n= 10, SHR + saline: n= 10, SHR + fenofibrate: n= 12). **P < 0.01 versus WKY group, #P < 0.05, ##P < 0.01 versus SHR + saline group.
Figure 3.
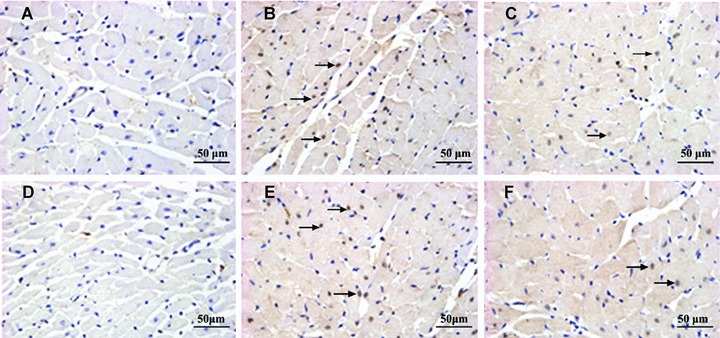
Representative immunohistochemical nuclei staining for c-fos (A, B, C) and c-jun (D, E, F) in the myocardium of three groups of rats. (A and D) c-fos and c-jun staining in WKY rats showing scanty nuclear-positive cells; (B and E) c-fos and c-jun staining in SHRs treated with saline showing a large number of nuclear-positive cells; (C and F) c-fos and c-jun staining in SHRs treated with fenofibrate depicting significantly a small number of nuclear-positive cells (magnification, 400×). Arrows indicate nuclear-positive cells.
mRNA expression of c-fos, c-jun, PPARα and collagen
The mRNA expression of c-fos, c-jun, and type I and type III collagen in the myocardium of SHRs treated with saline was significantly increased but was decreased in that of SHRs treated with fenofibrate (Fig. 4). The mRNA expression of PPARα in the myocardium of SHRs treated with saline was lower than that in WKY rats and was significantly increased in the myocardium of SHRs treated wtih fenofibrate.
Figure 4.
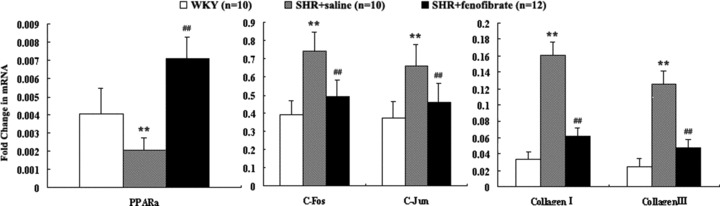
mRNA expression of PPARα, c-fos, c-jun and type I and III collagen in the myocardium of three groups of rats. Data shown were means ± S.E.M. (WKY: n= 10, SHR + saline: n= 10, SHR + fenofibrate: n= 12). *P < 0.05, **P < 0.01 versus WKY group, #P < 0.05, ##P < 0.01 versus SHR + saline group.
Protein expression of AP-1 and c-fos/c-jun heterodimers
The protein expression of AP-1 and c-fos/c-jun heterodimers was significantly higher in SHRs treated with saline than that in WKY rats (Fig. 5). On the other hand, fenofibrate-treated SHRs exhibited normal levels of protein expression of AP-1 and c-fos/c-jun heterodimers.
Figure 5.
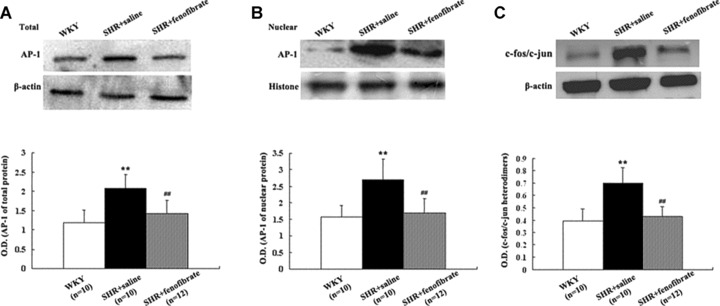
Top panel: Western blot analysis of AP-1 total (A) and nuclear (B) proteins and Western blot analysis of co-immunoprecipitation of c-jun/c-fos heterodimers (C) in the myocardium of three groups of rats (WKY group n= 10, SHR + saline group n= 10, SHR + fenofibrate group n= 12). Bottom panel: Bar graph showing mean ± S.E.M. of Western blot analysis. **P < 0.01 versus WKY group. ##P < 0.01 versus SHR + saline group.
Discussion
The present study demonstrated that PPARα activator fenofibrate had direct beneficial effects on cardiac remodelling and collagen deposition in SHRs. These findings were associated with increased PPARα level and decreased AP-1 activities through down-regulation of c-fos/c-jun heterodimers. These encouraging results indicate that the favourable cardiac effects of fenofibrate in SHRs depend, at least in part, on PPARα activation.
A common target organ damage of hypertension is left ventricular hypertrophy. In vivo studies have demonstrated that hypertension exerted a direct simulative action on cardiac hypertrophy [21, 22]. In vitro studies have also implicated cardiac hypertrophy and fibrosis associated with hypertension [23, 24]. Compared with the WKY rats, SHRs at 8–9 weeks showed clear evidence of cardiac hypertrophy caused by arterial hypertension [25]. At the first week of our study, echocardiography had already revealed a significant difference in the left ventricular wall thickness between 8-week-old SHRs and WKY rats. A recent study found activation of PPARα-inhibited hypertrophic signalling in cardiac myocytes [26], suggesting that PPARα activation may regress left ventricular hypertrophy in SHRs. Our study showed that, at week 8, the left ventricular wall thickness and the ratio of heart weight/body weight were decreased significantly and left ventricular diastolic function assessed by the E/A ratio and DT were substantially improved in SHRs treated with fenofibrate compared with SHRs treated with saline. Similarly, the IBS percentage and CVIB data, indicative of the extent of myocardial fibrosis, were also significantly improved in SHRs treated with fenofibrate when compared with SHRs treated with saline. Notably, these observed benefits of fenofibrate on cardiac remodelling were independent of blood pressure in this study, because fenofibrate has no anti-hypertensive effects and the SBP at week 8 was essentially the same in SHRs receiving fenofibrate or saline. Nonetheless, the values of left ventricular wall thickness in fenofibrate-treated SHRs were still significantly higher at week 8 than those at week 1, suggesting that fenofibrate treatment can only attenuate the progression rather than prevent the development of left ventricular hypertrophy in SHRs. Interestingly, the beneficial effects of fenofibrate on myocardial hypertrophy in SHR were associated with suppressive up-regulation of c-fos and c-jun, which supports the role of AP-1 in the anti-remodelling process. However, whether the PPARα activator directly regulates the formation of c-fos/c-jun heterodimers remains to be elucidated.
AP-1 is not a single molecule but a complex group of proteins whose components are products of the fos and jun gene families [27] and are involved in the altered collagen expression. Previous studies have demonstrated that TGF-β may up-regulate the transcriptional activity of human α1 procollagen gene, at least in part, through modulating the expression of AP-1 [28, 29]. AP-1 is a transcriptional inhibitor of the expression of the α1 procollagen gene, and alterations in this regulatory mechanism are associated with increased production of collagen [30].
Although recent in vivo studies have showed that PPARα activators prevent cardiac fibrosis in mineralocorticoid-dependent hypertension [31] and inhibit hypertrophy of neonatal rat cardiac myocytes [32], little is known about the role of PPARα activators in the process of cardiac remodelling in hypertension. In the present study, we first demonstrated that the PPARα activator fenofibrate markedly reduced collagen deposition in the myocardium, attenuated the progression of left ventricular hypertrophy and improved left ventricular diastolic function in SHRs. These novel findings revealed a direct beneficial effect of fenofibrate on cardiac remodelling in hypertension. Fenofibrate may, via activation of cardiac PPARα, lead to down-regulation of c-fos and c-jun expression and modulation of type I collagen metabolism mediated by the transcription factor AP-1. Our results are in agreement with recent reports that fenofibrate inhibited endothelin-1-induced cardiac hypertrophy via blockade of the c-jun NH2-terminal kinase pathway [33] and that activation of PPAR resulted in inhibition of the AP-1 pathways that regulated the expression of type I collagen [34]. In addition, we found that the fenofibrate had an effect on the activation of the transcription factor AP-1 by inhibiting the formation of c-jun/c-fos heterodimers.
In summary, our study demonstrated an important role of the PPARα activator fenofibrate in hypertension-induced cardiac remodelling. Fenofibrate ameliorated cardiac hypertrophy and extracellular matrix production. The possible underlying mechanism involves inhibition of formation of AP-1 and c-fos/c-jun heterodimers, which may further inhibit transcription of the downstream genes involved in the pathogenesis of left ventricular hypertrophy induced by hypertension.
Acknowledgments
This study was supported by the National 973 Basic Research Program of China (No. 2009CB521904), the National High-tech Research and Development Program of China (No. 2006AA02A406), the Program of Introducing Talents of Discipline to Universities (No. B07035) and grants from the Natural Science Foundation of Shandong Province (No.Y2004C21) and the Department of Science and Technology of Shandong Province (No. 2006GG2202006), China. We thank Rong Wang, Jin-Bo Feng, Hong Jiang, Chun-Xi Liu and Xu-Ping Wang for their technical assistance.
References
- 1.Hayden MR, Chowdhury N, Govindarajan G, et al. Myocardial myocyte remodeling and fibrosis in the cardiometabolic syndrome. J Cardiometab Syndr. 2006;1:326–33. doi: 10.1111/j.1559-4564.2006.05626.x. [DOI] [PubMed] [Google Scholar]
- 2.Ogata T, Miyauchi T, Sakai S, et al. Stimulation of peroxisome-proliferator-activated receptor alpha (PPAR alpha) attenuates cardiac fibrosis and endothelin-1 production in pressure-overloaded rat hearts. Clin Sci. 2002;103(Suppl 48):284S–8S. doi: 10.1042/CS103S284S. [DOI] [PubMed] [Google Scholar]
- 3.Grove D, Zak R, Nair KG, et al. Biochemical correlates of cardiac hypertrophy, observations on the cellular organization of growth during myocardial hypertrophy in the rat. Circ Res. 1969;25:473–85. doi: 10.1161/01.res.25.4.473. [DOI] [PubMed] [Google Scholar]
- 4.Heeneman S, Cleutjens JP, Faber BC, et al. The dynamic extracellular matrix: intervention strategies during heart failure and atherosclerosis. J Pathol. 2003;200:516–25. doi: 10.1002/path.1395. [DOI] [PubMed] [Google Scholar]
- 5.Doering CW, Jalil JE, Janicki JS, et al. Collagen network remodeling and diastolic stiffness of the rat left ventricle with pressure overload hypertrophy. Cardiovasc Res. 1988;22:686–95. doi: 10.1093/cvr/22.10.686. [DOI] [PubMed] [Google Scholar]
- 6.Jalil JE, Doering CW, Janicki JS, et al. Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res. 1989;64:1041–50. doi: 10.1161/01.res.64.6.1041. [DOI] [PubMed] [Google Scholar]
- 7.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–50. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 8.Braissant O, Foufelle F, Scotto C, et al. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–66. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 9.Schoonjans K, Martin G, Staels B, et al. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr Opin Lipidol. 1997;8:159–66. doi: 10.1097/00041433-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 10.Kliewer SA, Sundseth SS, Jones SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci USA. 1997;94:4318–23. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu HE, Lambert MH, Montana VG, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- 12.Delerive P, De Bosscher K, Besnard S, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J Biol Chem. 1999;274:32048–54. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- 13.Barger PM, Brandt JM, Leone TC, et al. Deactivation of peroxisome proliferator-activated receptor-α during cardiac hypertrophic growth. J Clin Invest. 2000;105:1723–30. doi: 10.1172/JCI9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 15.Sahn DJ, DeMaria A, Kisslo J, et al. Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation. 1978;58:1072–83. doi: 10.1161/01.cir.58.6.1072. [DOI] [PubMed] [Google Scholar]
- 16.Di Paola R, Di Guardo G, Lo Giudice P, et al. Preclinical thalassemic cardiopathy: a study by acoustic densitometry. Ital Heart J. 2003;4:413–8. [PubMed] [Google Scholar]
- 17.Cingolani OH, Yang XP, Cavasin MA, et al. Increased systolic performance with diastolic dysfunction in adult spontaneously hypertensive rats. Hypertension. 2003;41:249–54. doi: 10.1161/01.hyp.0000052832.96564.0b. [DOI] [PubMed] [Google Scholar]
- 18.Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15:532–4. 536–7. [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (delta delta CT–method) Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Albaladejo P, Carusi A, Apartian A, et al. Effect of chronic heart rate reduction with ivabradine on carotid and aortic structure and function in normotensive and hypertensive rats. J Vasc Res. 2003;40:320–8. doi: 10.1159/000072696. [DOI] [PubMed] [Google Scholar]
- 22.Baker KM, Booz GW, Dostal DE. Cardiac actions of angiotensin II: role of intracardiac renin–angiotensin system. Annu Rev Physiol. 1992;54:227–41. doi: 10.1146/annurev.ph.54.030192.001303. [DOI] [PubMed] [Google Scholar]
- 23.Miyata S, Haneda T. Hypertrophic growth of cultured neonatal rat heart cells mediated by type I angiotensin II receptor. Am J Physiol. 1994;266:H2443–51. doi: 10.1152/ajpheart.1994.266.6.H2443. [DOI] [PubMed] [Google Scholar]
- 24.Zhang YG, Li YG, Liu BG, et al. Urotensin II accelerates cardiac fibrosis and hypertrophy of rats induced by isoproterenol. Acta Pharmacol Sin. 2007;28:36–43. doi: 10.1111/j.1745-7254.2007.00485.x. [DOI] [PubMed] [Google Scholar]
- 25.Kokubo M, Uemura A, Matsubara T, et al. Noninvasive evaluation of the time course of change in cardiac function in spontaneously hypertensive rats by echocardiography. Hypertens Res. 2005;28:601–9. doi: 10.1291/hypres.28.601. [DOI] [PubMed] [Google Scholar]
- 26.Liang F, Wang F, Zhang S, et al. Peroxisome proliferator activated receptor (PPAR) alpha agonists inhibit hypertrophy of neonatal rat cardiac myocytes. Endocrinology. 2003;144:4187–94. doi: 10.1210/en.2002-0217. [DOI] [PubMed] [Google Scholar]
- 27.Bakiri L, Matsuo K, Wisniewska M, et al. Promoter specificity and biological activity of tethered AP-1 dimers. Mol Cell Biol. 2002;22:4952–64. doi: 10.1128/MCB.22.13.4952-4964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eghbali M, Tomek R, Sukhatme VP, et al. Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts. Regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res. 1991;69:483–90. doi: 10.1161/01.res.69.2.483. [DOI] [PubMed] [Google Scholar]
- 29.Armendariz-Borunda J, Simkevich CP, Roy N, et al. Activation of Ito cells involves regulation of AP-1 binding proteins and induction of type I collagen gene expression. Biochem J. 1994;304:817–24. doi: 10.1042/bj3040817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philips N, Bashey RI, Jiménez SA. Increased alpha 1(I) procollagen gene expression in tight skin (TSK) mice myocardial fibroblasts is due to a reduced interaction of a negative regulatory sequence with AP-1 transcription factor. J Biol Chem. 1995;270:9313–21. doi: 10.1074/jbc.270.16.9313. [DOI] [PubMed] [Google Scholar]
- 31.Iglarz M, Touyz RM, Viel EC, et al. Peroxisome proliferator-activated receptor-alpha and receptor-gamma activators prevent cardiac fibrosis in mineralocorticoid-dependent hypertension. Hypertension. 2003;42:737–43. doi: 10.1161/01.HYP.0000083511.91817.B1. [DOI] [PubMed] [Google Scholar]
- 32.Liang F, Wang F, Zhang S, et al. Peroxisome proliferator activated receptor (PPAR) alpha agonists inhibit hypertrophy of neonatal rat cardiac myocytes. Endocrinology. 2003;144:4187–94. doi: 10.1210/en.2002-0217. [DOI] [PubMed] [Google Scholar]
- 33.Irukayama-Tomobe Y, Miyauchi T, Sakai S, et al. Endothelin-1-induced cardiac hypertrophy is inhibited by activation of peroxisome proliferator-activated receptor-alpha partly via blockade of c-Jun NH2-terminal kinase pathway. Circulation. 2004;109:904–10. doi: 10.1161/01.CIR.0000112596.06954.00. [DOI] [PubMed] [Google Scholar]
- 34.Chen J, Mehta JL. Angiotensin II-mediated oxidative stress and procollagen-1 expression in cardiac fibroblasts: blockade by pravastatin and pioglitazone. Am J Physiol Heart Circ Physiol. 2006;291:H1738–45. doi: 10.1152/ajpheart.00341.2006. [DOI] [PubMed] [Google Scholar]