

Abstract
Passive smoking is an independent risk factor for cardiovascular diseases. Industrial fibrous dust, e.g. the asbestos group member, amosite, causes lung cancer and fibrosis. No data are available on renal involvement after inhalational exposure to these environmental pollutants or of their combination, or on cardiovascular and renal toxicity after exposure to amosite. Male Wistar rats were randomized into four groups (n= 6): control and amosite group received initially two intratracheal instillations of saline and amosite solution, respectively. Smoking group was subjected to standardized daily exposure to tobacco smoke for 2 hrs in a concentration resembling human passive smoking. Combined group was exposed to both amosite and cigarette smoke. All rats were killed after 6 months. Rats exposed to either amosite or passive smoking developed significant glomerulosclerosis and tubulointerstitial fibrosis. Combination of both exposures had additive effects. Histomorphological changes preceded the clinical manifestation of kidney damage. In both groups with single exposures, marked perivascular and interstitial cardiac fibrosis was detected. The additive effect in the heart was less pronounced than in the kidney, apparent particularly in changes of vascular structure. Advanced oxidation protein products, the plasma marker of the myeloperoxidase reaction in activated monocytes/macrophages, were increased in all exposed groups, whereas the inflammatory cytokines did not differ between the groups. In rats, passive smoking or amosite instillation leads to renal, vascular and cardiac fibrosis potentially mediated via increased myeloperoxidase reaction. Combination of both pollutants shows additive effects. Our data should be confirmed in subjects exposed to these environmental pollutants, in particular if combined.
Keywords: passive smoking, environmental pollutants, fibrosis, glomerulosclerosis, oxidative stress, AOPP
Introduction
Smoking unequivocally promotes carcinogenesis, atherosclerosis, as well as lung and renal diseases [1–5]. The evidence that passive smoking is a considerable cardiovascular risk factor continues to grow [6]. The relative risk of coronary heart disease (CHD) for active smokers is 1.78, but as high as 1.31 for passive smokers [7]. Passive smoking was also reported to increase the relative risk of lung and renal cell carcinoma [8, 9]. There is clear clinical evidence that smoking per se is nephrotoxic [4, 5], yet no clinical or experimental data concerning the renal effects of passive smoking are available. The question of the potential nephrotoxicity of passive smoking has been raised recently by nephrologists and the data on the results are awaited [10].
Various air pollutants, including asbestos fibres, were shown to be involved in the development and progression of lung diseases, i.e. of pulmonary fibrosis, lung cancer and pleural malignant mesothelioma [11–13]. Amosite, a naturally occurring silicate of the amphibole asbestos group, represents a typical industrial fibrous dust. It was the second most common form of asbestos fibres used in the building industry in the United States. Despite the fact that the use of asbestos fibres was banned in the 1980s in most western countries, there are huge remnant amounts of asbestos, which were used in civil engineering. Professional groups such as mine workers, workers of asbestos handling industries (nowadays mainly at destruction of old buildings and at construction material waste handling) and inhabitants of areas of natural occurrence of this mineral across the globe still suffer from high exposure to asbestos fibres.
There are no definite experimental data on the renal and cardiovascular effects of asbestos fibres inhalation, of passive smoking, or of their combination. Here we show that exposure of rats to these single environmental pollutants or their combination leads to prominent renal and cardiac fibrosis and vessel damage.
Materials and methods
All experiments were conducted according to the guidelines for studies using laboratory animals, after the approval by the local Ethics Committee (Bratislava, Slovakia).
Animals and protocol
Two rats per cage were housed under controlled humidity, temperature and 12/12 hrs light/dark cycle. Animals had free access to drinking water and a standard rat chow (Top Dovo, Prague, Czech Republic). Twenty-four young male Wistar rats (VELAZ, Prague, Czech Republic) weighing 151.5 ± 19.9 g (approx. 6 weeks of age) at the beginning of the study were randomized into 4 groups:
The healthy control group (ctrl, n= 6), which at the beginning of the study was given two (1-week interval) intratracheal instillations of 0.2 ml of 0.9% saline solution, under light ether anaesthesia.
The amosite group (amos, n= 6) received in the same manner two intratracheal instillations of 0.2 ml of 0.9% saline solution containing 2 mg of industrial fibre dust amosite.
The passive smoking group (smoke, n= 6) was exposed to a concentration of cigarette smoke attainable at human passive smoking daily (except weekends) during 6 months for 2 hrs/day (a model of human passive smoking). Exposure to diluted mainstream tobacco smoke was at the concentration of 85 mg of total particulate matter (TPM)/m3 air in a whole-body exposure chamber (THRI, Lexington, KY, USA). The exposure per day required burning 8 standard research cigarettes of the 1R1 type (THRI, Lexington, USA).
The combined group (combi, n= 6) received both the intratracheal instillation of amosite as the amosite group (amos) and the diluted mainstream tobacco smoke exposure as the passive smoking group (smoke).
The experimental design and the concentrations of amosite and tobacco smoke (approx. 10% equivalent of the smoke inhaled by a human smoker) used in this study were based on established and reliable standard experimental protocols and our previous results showing development of pulmonary fibrosis and chronic inflammation after these exposures [14–17].
Six months after beginning of the exposure, all animals were killed under thiopental anaesthesia (150 mg/kg, i.p.). Following killing, venous blood was taken, kidneys and hearts were removed, weighed and processed for further analyses. No rats were excluded from the analyses or died during the experiment.
Three days before killing, systolic blood pressure (SBP) was measured by tail plethysmography. Two days before killing, all rats were placed in metabolic cages for stool-free 24 hrs urine collection.
Biochemical analyses
Standard blood (plasma glucose, urea, creatinine, albumin, sodium, potassium, cholesterol and triacylglycerol concentration) and urine parameters (creatinine and protein concentration) were determined (Vitros 250 analyzer, J&J, Rochester, NY, USA). Creatinine clearance was calculated. Plasma advanced oxidation protein products (AOPPs) were determined as described in Ref. [18]. Plasma advanced glycation end-products (AGE) were measured as AGE-associated fluorescence (λexcitation=350nm, λemision=450nm; data expressed as arbitrary units) according to [19]. Urinary tumour necrosis factor α (TNF-α), interleukin 6 (IL-6); both rat-specific ELISA kits (Bender MedSystems, Vienna, Austria) and 8-oxo-2′-deoxyguanosine (Oxis Research, Foster City, CA, USA) were analysed according to manufacturers’ instructions.
Kidney tissue preparation
The kidneys were removed, weighed and dissected in a plane perpendicular to the interpolar axis, yielding slices of 1-mm width. Ten small specimens of one kidney were selected by area weighted sampling for embedding in Epon-Araldite. Semithin (1 μm) slices were prepared and stained with methylene blue and basic fuchsin. The remaining tissue slices were fixed with formaldehyde and embedded in paraffin; 4 μm sections were prepared and stained with haematoxylin/eosin (HE), periodic acid Schiff’s and Sirius red. All histomorphological evaluations were performed in a blinded manner.
Morphological investigations of the kidney
The degree of sclerosis within the glomerular tuft (glomerulosclerosis, GSI) was determined on PAS-stained paraffin sections (magnification ×400) as described previously [20, 21]. In brief, in each animal the mean score of 100 glomeruli was obtained. Each glomerulus received a score from 0 to 4: grade 0: normal glomerulus, grade 1: presence of mesangial expansion/thickening of the basement membrane, grade 2: mild to moderate segmental hyalinosis/sclerosis involving less than 50% of the glomerular tuft, grade 3: diffuse glomerular hyalinosis/sclerosis > 50% of the tuft, and grade 4: diffuse glomerulosclerosis with total tuft obliteration and/or collapse.
Tubulointerstitial fibrosis score and renal vessel wall thickness were assessed on Sirius red and PAS-stained paraffin sections, respectively, at a magnification of ×100 using a scoring system as described previously [20, 21].
Morphological investigations of the heart and the aorta
Wall thickness and wall to lumen ratio of intramyocardial arteries and of the aorta were analysed using a semiautomatic image analysing system at a magnification of ×400 on HE sections [22, 23]. In addition, on Sirius red stained sections of the heart and of the aorta tissue collagen content and architecture were semiquantitatively assessed using a scoring system (score 0–4) at magnifications of ×200 and ×400, respectively [22, 23]. In the heart, fibrosis was scored 0 when absolutely no increase in fibrous tissue was present. Score 1 was given for minor increased in fibrous tissue content, score 2 for moderate, score 3 for strong and score 4 for extremely strong fibrosis. In the aorta, fibrous tissue content as well as architectural alterations of the wall were scored from 0 to 4 taking into account also the extent of the alterations, i.e. score 1: <25% of wall area, score 2: 25–50%, score 3: 50–75% and score 4: 75–100%.
Analysis of renal gene expression
Glomeruli and cortical tubulointerstitium were isolated by differential sieving as described previously [24]. Total RNA was isolated using the standard Trizol protocol (MRC, Cincinnati, OH, USA) and the concentration and purity of RNA were measured spectrophotometrically using Nanodrop (NanoDrop Technologies, Wilmington, DE, USA). For real-time PCR, Qiagen QuantiFast Sybr Green RT-PCR kit (Qiagen, Hilden, Germany) was used according to the manufacturers’ instructions. For the normalization of the data, the standard delta Ct method was used with peptidylprolyl isomerase A (cyclophilin A) as the housekeeping gene. The expression of genes of interest was calculated as relative expression units in comparison to the ctrl group (arbitrary set as 1). Primers used for real time PCR are shown in Table 1.
Table 1.
Primers used in this study for gene expression analysis using real time PCR
| Gene / Gene ID | Forward primer | Reverse primer |
|---|---|---|
| ppia / 25518 | gtctcttttcgccgcttgct | tctgctgtctttggaactttgtctg |
| col1a1 / 29393 | caacctcaagaagtccctgc | acaagcgtgctgtaggtgaa |
| fn1 / 25661 | caaggtccgagaagaggttg | ccgtgtaagggtcaaagcat |
| il6 / 24498 | accaggaacgaaagtcaactcc | ttgtgaagtagggaaggcagt |
| nephrin / 64563 | ctaaggatgggctgcttctg | gtggaactcgcctttagcac |
| sod2 / 24787 | ccaaaggagagttgctggag | gaaccttggactcccacaga |
| tgfb1 / 59086 | gaaggacctgggttggaagt | tactgtgtgtccaggctcca |
| tnfa / 24835 | atccgagatgtggaactggc | tcagtagacagaagagcgtggtg |
| mpo / 303413 | ggaagtggcgaactcagtgg | aggagttccatagggctggc |
ppia, peptidylprolyl isomerase A (cyclophilin A)
col1a1, collagen type I alpha 1 chain
fn1, fibronectin
il6, interleukin 6
sod2, superoxide dismutase 2
tgfb1, transforming growth factor beta 1
tnfa, tumour necrosis factor alpha
mpo, myeloperoxidase.
Statistics
Results are given as means ± S.D. Means between the groups were compared using one-way ANOVA with a post hoc Scheffe’s or Dunett test (RT-PCR data). P < 0.05 was considered significant.
Results
In line with clinical observations [4], rats in both groups subjected to passive smoking gained less weight during the experiment, resulting in lower body weight compared with controls (–11% at killing; not significant) (Table 2). Plasma glucose, albumin, cholesterol, triacylglycerol and electrolyte concentrations were within the normal range and did not differ between the groups (data not shown). SBP was within the normal range and comparable between the groups (Table 2). However, after adjustment for body weight, a correction often used in clinical studies with smoking patients [4], both groups exposed to cigarette smoke had higher blood pressure by 10%; (overall ANOVA P= 0.048, post hoc test closely failed the level of statistical significance).
Table 2.
Functional parameter, blood pressure and biochemical measurements
| Control (n= 6) | Amosite (n= 6) | Smoke (n= 6) | Combination (n= 6) | |
|---|---|---|---|---|
| Body weight [g] | 658 ± 92 | 698 ± 63 | 593 ± 44 | 595 ± 54 |
| Kidney weight [g] | 3.7 ± 0.6 | 4.0 ± 0.7 | 3.5 ± 0.4 | 3.3 ± 0.3 |
| SBP [mmHg] | 102 ± 5 | 101 ± 10 | 101 ± 4 | 103 ± 7 |
| SBP/BW [mmHg/100 g] | 15.7 ± 2.3 | 14.5 ± 1.4 | 17.1 ± 1.5 | 17.4 ± 2.1 |
| P-Crea [μmol/l] | 56 ± 8 | 62 ± 8 | 63 ± 9 | 63 ± 11 |
| Clcrea[μ/min/100 g] | 293 ± 95 | 272 ± 140 | 351 ± 69 | 319 ± 71 |
| P-Urea [mmol/l] | 4.6±0.7 | 5.2±1.0 | 5.0±0.7 | 5.1±0.6 |
| Diuresis [ml/24 hrs] | 15 ± 5 | 22 ± 9 | 16 ± 6 | 15 ± 8 |
| Proteinuria [mg/24 hrs] | 6.9 ± 5.6 | 7.9 ± 4.9 | 4.4 ± 0.8 | 5.4 ± 3.0 |
| AOPPs [μmol/l] | 11.5 ± 2.2 | 16.7 ± 4.4* | 22.9 ± 8.7* | 16.3 ± 3.9* |
BW, body weight
SBP, systolic blood pressure
P, plasma
Crea, creatinine
Cl, clearance
AOPPs, advanced oxidation protein products
P < 0.05 versus controls.
Renal function (Table 2)
Plasma creatinine and urea concentrations were higher by 10–14% in all exposed groups, but statistical significance was not reached. Diuresis and proteinuria were comparable between the groups.
Morphology of the kidney
No significant differences in renal hypertrophy measured by kidney weight were observed (or kidney weight corrected for body weight; data not shown) (Table 2). Indices of fibrosis in both glomeruli (glomerulosclerosis) and the tubulointerstitium were significantly higher in all exposed groups if compared with the controls (Fig. 1). The combination of both exposures resulted in additive effects mainly in the tubulointerstitium. The marker of renal vessel wall damage (vessel thickness) was only insignificantly higher in the amosite and passive smoking groups. The combination of both exposures led to a significant increase of intrarenal arterial wall thickness (Fig. 1C).
Figure 1.
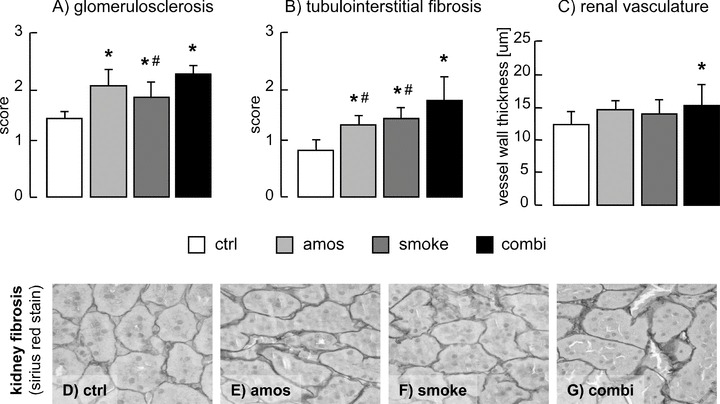
Indices of renal damage: glomerulosclerosis (A), tubulointerstitial fibrosis (B) and thickness of renal vasculature (C). Representative pictures of renal cortical tubulointerstitium stained with Sirius red of control rats (D) or rats exposed to amosite (E), passive smoking (F) or combination of both (G). ctrl, control group; amos, amosite group; smoke, passive smoking group; combi, combination of amosite and passive smoking group. *P < 0.05 versus control, #P < 0.05 versus combination.
Renal gene expression
Glomerular expression of the extracellular matrix protein fibronectin was increased in the combination group (Fig. 2A). Collagen type I was significantly up-regulated in all exposed groups in the tubulointerstitium (Fig. 2D), but only rats exposed to passive smoking had increased tubulointerstitial production of TGF-β1 mRNA (Fig. 2E). Glomerular nephrin expression remained similar, indicating no podocyte loss or dedifferentiation (Fig. 2C). Expression of the antioxidative enzyme superoxide dismutase 2 (SOD2) was increased in all exposed groups in the tubulointerstitum (Fig. 2F) but the increase in glomeruli was inconsistent (Fig. 2B). All groups had similar quantities of glomerular as well as of tubulointerstitial mRNA for IL-6 and TNF-α (data not shown). The relative expression of myeloperoxidase was not significantly altered, neither in the tubulointerstitum (ctrl: 1.00 ± 0.33, amos: 1.03 ± 0.20, smoke: 0.82 ± 0.13, combi: 1.23 ± 0.57 fold increase in mRNA expression) nor in the glomeruli (ctrl: 1.00 ± 0.63, amos: 1.02 ± 0.37, smoke: 3.19 ± 5.62, combi: 2.23 ± 4.03 fold increase in mRNA expression).
Figure 2.
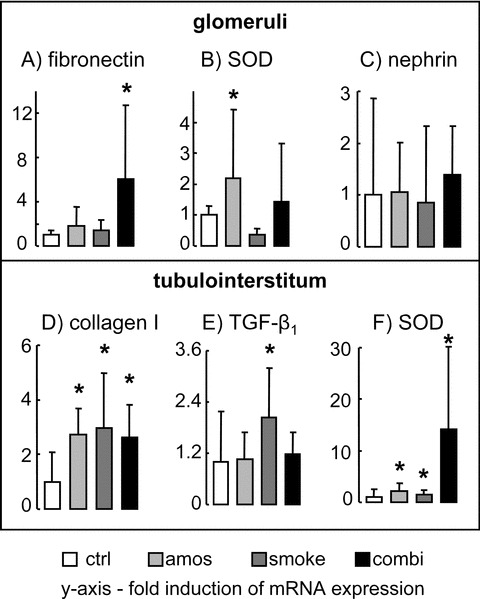
Glomerular mRNA expression of fibronectin (A), SOD (B) and nephrin (C) and cortical tubulointerstitial mRNA expression of collagen type I (D), TGF-β1 (E) and SOD (F). mRNA was measured by RT-PCR, normalized to housekeeping gene peptidylprolyl isomerase A (cyclophilin A) and expressed as fold induction compared to control group, which was set as 1. *P < 0.05 versus control.
Morphology of the heart
The wall thickness of intramyocardial arteries was significantly higher in all three exposed groups if compared with the controls (Fig. 3A). Significant cardiac fibrosis, observed as marked accumulation of connective tissue in both the interstitial and perivascular spaces, was observed in all three exposed groups (Fig. 3C–H).
Figure 3.
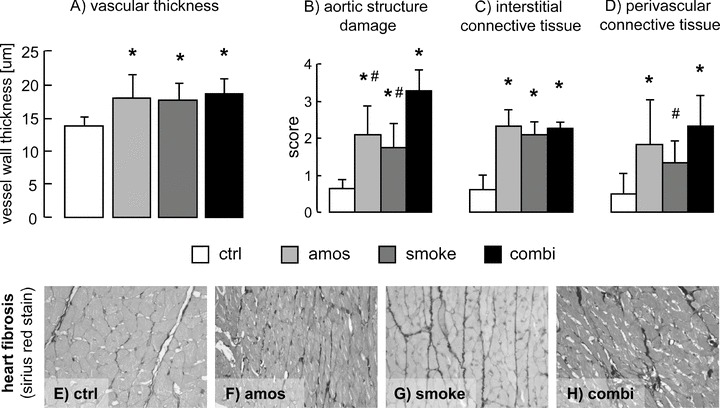
Indices of cardiovascular damage: heart vessel wall thickness (A), aortic damage score (B) and heart interstitial (C) and perivascular (D) fibrosis scores. Representative pictures of the heart stained with Sirius red of control rats (E) or rats exposed to amosite (F), passive smoking (G) or combination thereof (H). ctrl, control group; amos, amosite group; smoke, passive smoking group; combi, combination of amosite and passive smoking group. *P < 0.05 versus control, #P < 0.05 versus combination.
Morphology of the aorta
Aortic wall thickness (also if corrected for body weight) did not differ between the groups (data not shown). Exposure to amosite dust or cigarette smoke resulted in marked damage of the aortic vascular structure, assessed as altered media structure with increased fibrous tissue content. The combination of both significantly aggravated the injury (Fig. 3B).
Systemic markers of oxidative stress and inflammation
Plasma advanced oxidation protein products (AOPPs), marker of oxidative protein damage via the myeloperoxidase reaction [18], were higher in all exposed rats compared with controls (Table 1). Similar results were obtained if corrected for plasma albumin (data not shown). Urinary excretion of 8-oxo-2′-deoxyguanosine, a marker of oxidative damage to DNA, did not differ significantly between the groups (ctrl: 292 ± 133, amos: 325 ± 164, smoke: 248 ± 174, combi: 195 ± 50 ng per day or after normalization to urinary creatinine, data not shown), possibly due to very high interindividual variability. Similarly, the plasma total AGE-associated fluorescence did not differ between the groups (ctrl: 248 ± 60, amos: 244 ± 81, smoke: 249 ± 79, combi: 268 ± 37 AU). All groups had comparable levels of two markers of inflammation - TNF-α and IL-6 (data not shown).
Discussion
Cigarette smoking is a widely recognized independent risk factor for ischaemic and atherosclerotic heart and cerebrovascular disease [2, 3]. Even passive exposure to tobacco smoke is prospectively related to the risk profiles [6, 25, 26]. Cigarette smoking in human beings is associated with endothelial dysfunction, intimal hyperplasia as well as wall-thickening of myocardial and renal arterioles and arteries. It has been associated with idiopathic nodular glomerulosclerosis and with microalbuminuria or overt proteinuria in healthy individuals, whereas it appears to aggravate proteinuria in preexisting renal disease such as diabetic nephropathy [4, 5, 27–29]. In our animal model in which approximately a 10% equivalent of the daily dose of inhaled cigarette smoke by a human smoker was applied for only 1 hr a day, we show for the first time that passive smoking leads to renal fibrosis. Renal lesions are manifested histomorphologically within the glomeruli (glomerulosclerosis comparable to the findings in idiopathic nodular glomerulosclerosis in human beings) and the interstitium and by up-regulated expression of fibrosis-related genes. This goes without detectable changes in clinical parameters, such as proteinuria, suggesting that the renal injury in this model precedes the clinical manifestation of the disease. We suppose that our histopathologic findings of glomerulosclerosis and vessel thickening in rats exposed to passive smoking support the notion that the diagnosis of idiopathic nodular glomerulosclerosis might not be ‘idiopathic’ but rather ‘smoking-related’[28, 30, 31]. In their brief report, Culhaci et al. showed that only approx. 10% of rats exposed to passive smoking showed signs of glomerulosclerosis, whereas no hyalinosis or reduced number of renal vessels were observed [32]. This study was performed in a different animal strain, on 3-week-old rats of both sexes. The rats were exposed to commercial cigarette smoke for 4 months and the concentration of inhaled particulate matter was not indicated. These issues might have accounted for the different results compared to our data. Moreover, in our study, fibrotic changes were also observed in the heart and vasculature in line with previous animal studies on the cardiovascular effects of smoking [6, 25, 26]. Elevated sympathetic activity and blood pressure with an increase of glomerular filtration rate (GFR) and intraglomerular capillary pressure are candidate mechanisms of smoking-induced renal damage [4, 5]. Although our data did not reach significance, in the rats exposed to tobacco smoke blood pressure normalized to body weight was 10% higher, and GFR increased by 20%. We found no difference in proteinuria between smoke-exposed and control rats. Although the study period of 6 months corresponded to approx. 25% of a rat’s lifespan, the exposure started in early adolescence, and differences between human and rodent species may account for this lack in functional abnormality as compared to human beings. The still mild degree of fibrosis is reassuring. Alternatively, we may not exclude that a relative proteinuria in proportion to protein intake was masked by lower protein-caloric intake in our animals exposed to tobacco [33, 34].
In search of a potential mediator of the observed renal and cardiac organ fibrosis, we detected increased levels of a marker of oxidative protein damage derived from the myeloperoxidase reaction in activated monocytes/macrophages, namely AOPPs [18]. AOPPs were found to directly accelerate renal fibrosis in a model of subtotal nephrectomy [35] and to promote inflammation in the diabetic kidney [36]. Furthermore, it was shown recently that the activation state of macrophages is more important in driving renal injury than their mere numbers [37]. We did not find any differences between the glomerular or tubulointerstitial expression of myeloperoxidase, suggesting that activated monocytes/macrophages arise largely from an extrarenal source (most possibly pulmonary). These monocytes/macrophages might have been crucially involved in the renal fibrotic changes in our study, especially in combination with other insults, e.g. direct induction of oxidative stress. Chronic environmental tobacco smoke exposure induces systemic oxidative stress, which may subsequently trigger production of various profibrotic factors [38–41]. Increased renal expression of SOD2, the antioxidative defense enzyme, supports the idea of enhanced systemic oxidative stress. Increase in SOD2 mRNA as a possible compensatory mechanism to oxidative stress is however only an indirect marker. 8-oxo-2′-deoxyguanosine is a marker of DNA damage that has been shown to be strongly increased in the heart, liver and lungs of rats after 30 days of cigarette smoke inhalation [41]. In our study, we found no difference in 8-oxo-2′-deoxyguanosine between the groups, similarly to another report [33], probably due to a relatively mild exposure protocol. Although not analysed in our study, another mechanism linking smoke exposure to cardiac and renal fibrosis might be protein carbamylation as shown for atherosclerosis [42].
Our study provides the first evidence that rats challenged with the industrial fibre dust, amosite, develop renal and cardiovascular fibrosis and vascular changes. The administered amosite-challenge was comparable to the body of previous work where applied total dosages ranged from 0.5 mg/animal to exceeding 5 mg/animal. Intriguingly, in our study the presence and degree of vascular and fibrotic changes induced by amosite were similar to the ones induced by passive smoking. The combination of both environmental pollutants shows additive effects in the fibrosis parameters. Exposure to asbestos fibres, apart from lung and pleural malignancies, is associated with pleural and pulmonary fibrosis [13]. After dust exposure in the lungs, massive inflammatory infiltration occurs, paralleled with production of growth factors, chemokines, cytokines and reactive oxygen species [16, 17, 43]. Our study suggests that the activation state of monocytes/macrophages induced by pulmonary amosite and tobacco smoke exposure might be one of the major common mechanisms of the observed extrapulmonal fibrosis and vascular changes. Asbestos fibres have been demonstrated to translocate from the lung into the circulation [44]. The fibres have been found in all organs of exposed individuals but accumulated mostly in organs with high microvascular filtration, such as the kidneys [44]. Thus a direct profibrotic effect of asbestos fibres in the kidneys or even the heart cannot be excluded. It should be noted that the process of translocation seems to be slow (many years), at least in human beings [44].
Despite the fact that a higher mortality in white males employed at an asbestos textile plant (among others due to heart disease) was reported already in 1994 [45, 46], the data on the effects of asbestos inhalation on the cardiovascular system or other extrapulmonary organs, including the kidneys, are lacking. To the best of our knowledge, studies examining the renal and cardiovascular effects of smoking in subjects exposed to asbestos have not been performed. Thus, it remains unknown whether these combined insults go along with any histological or functional abnormalities in human beings. In silica-exposed industrial workers, renal alterations may occur prior to pulmonary involvement, and smoking exerts nephrotoxic effects synergistic to those of silica exposure [39]. Whether exposure to asbestos induces renal damage, such as microalbuminuria, proteinuria, or altered GFR in human beings, is unknown. However, when considering other industrial dust fibres, e.g. silica exposure, kidney injury emerges as a higher risk than either mortality from silicosis or lung cancer [47]. Thus, subjects exposed to asbestos, but also to analogous airborne fibres, should be carefully examined not only for occurrence of lung injury, but also for cardiac and renal diseases, particularly in the case of concurrent smoking.
In conclusion, cardiovascular and renal fibrosis developed in rats after exposure to passive smoking or the industrial fibrous dust, amosite and their combination was additive. Vascular damage might have preceded the development of organ fibrosis. Although we did not find differences in urinary or renal TNF-α and IL-6, an increased myeloperoxidase reaction – reflecting activated macrophages – might have been one of the key mechanisms. Our study urges closer monitoring not only of pulmonary but also of cardiovascular and kidney functions in patients exposed to passive smoking or industrial fibrous dust.
Acknowledgments
This study was supported in part by EU grant No. QLK4-CT-1999-01629, by the Slovak grant No. APVT-21-0111-04, LPP-0133-06 by the Ministry of Health 2006/24-UK-03, the IZKF Erlangen (project A11) and by the Verein zur Bekämpfung der Hochdruck- und Nierenkrankheiten, Würzburg e.V., Germany. The skilful technical assistance of M. Klewer, M. Reutelshöfer, S. Söllner, Erlangen and the assistance of Mr. André Klassen, Würzburg in preparation of the manuscript are gratefully acknowledged.
Conflict of interest: none declared.
References
- 1.Mallampalli A, Guntupalli KK. Smoking and systemic disease. Clin Occup Environ Med. 2006;5:173–92. doi: 10.1016/j.coem.2005.10.002. x. [DOI] [PubMed] [Google Scholar]
- 2.Freund KM, Belanger AJ, D’Agostino RB, et al. The health risks of smoking. The Framingham Study: 34 years of follow-up. Ann Epidemiol. 1993;3:417–24. doi: 10.1016/1047-2797(93)90070-k. [DOI] [PubMed] [Google Scholar]
- 3.Jee SH, Park J, Jo I, et al. Smoking and atherosclerotic cardiovascular disease in women with lower levels of serum cholesterol. Atherosclerosis. 2007;190:306–12. doi: 10.1016/j.atherosclerosis.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 4.Orth SR. Effects of smoking on systemic and intrarenal hemodynamics: influence on renal function. J Am Soc Nephrol. 2004;15:S58–63. doi: 10.1097/01.asn.0000093461.36097.d5. [DOI] [PubMed] [Google Scholar]
- 5.Orth SR. Smoking and the kidney. J Am Soc Nephrol. 2002;13:1663–72. doi: 10.1097/01.asn.0000018401.82863.fd. [DOI] [PubMed] [Google Scholar]
- 6.Raupach T, Schafer K, Konstantinides S, et al. Secondhand smoke as an acute threat for the cardiovascular system: a change in paradigm. Eur Heart J. 2006;27:386–92. doi: 10.1093/eurheartj/ehi601. [DOI] [PubMed] [Google Scholar]
- 7.Law MR, Morris JK, Wald NJ. Environmental tobacco smoke exposure and ischaemic heart disease: an evaluation of the evidence. BMJ. 1997;315:973–80. doi: 10.1136/bmj.315.7114.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu J, Ugnat AM. Active and passive smoking and risk of renal cell carcinoma in Canada. Eur J Cancer. 2005;41:770–8. doi: 10.1016/j.ejca.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Sasco AJ, Secretan MB, Straif K. Tobacco smoking and cancer: a brief review of recent epidemiological evidence. Lung Cancer. 2004;45:S3–9. doi: 10.1016/j.lungcan.2004.07.998. [DOI] [PubMed] [Google Scholar]
- 10.Narkiewicz K. Second-hand smoke – a license to kill due to expire. Nephrol Dial Transplant. 2007;22:1508–11. doi: 10.1093/ndt/gfm046. [DOI] [PubMed] [Google Scholar]
- 11.Reid A, De Klerk N, Ambrosini GL, et al. The effect of asbestosis on lung cancer risk beyond the dose related effect of asbestos alone. Occup Environ Med. 2005;62:885–9. doi: 10.1136/oem.2005.020834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki Y, Yuen SR. Asbestos fibers contributing to the induction of human malignant mesothelioma. Ann N Y Acad Sci. 2002;982:160–76. doi: 10.1111/j.1749-6632.2002.tb04931.x. [DOI] [PubMed] [Google Scholar]
- 13.Robledo R, Mossman B. Cellular and molecular mechanisms of asbestos-induced fibrosis. J Cell Physiol. 1999;180:158–66. doi: 10.1002/(SICI)1097-4652(199908)180:2<158::AID-JCP3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 14.Churg A, Tron V, Wright JL. Effects of cigarette smoke exposure on retention of asbestos fibers in various morphologic compartments of the guinea pig lung. Am J Pathol. 1987;129:385–93. [PMC free article] [PubMed] [Google Scholar]
- 15.Hecht SS. Carcinogenicity studies of inhaled cigarette smoke in laboratory animals: old and new. Carcinogenesis. 2005;26:1488–92. doi: 10.1093/carcin/bgi148. [DOI] [PubMed] [Google Scholar]
- 16.Beno M, Hurbankova M, Dusinska M, et al. Multinucleate cells (MNC) as sensitive semiquantitative biomarkers of the toxic effect after experimental fibrous dust and cigarette smoke inhalation by rats. Exp Toxicol Pathol. 2005;57:77–87. doi: 10.1016/j.etp.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Kovacikova Z, Hurbankova M, Cerna S, et al. Effect of exposure to ceramic fibres and cigarette smoke on antioxidant status of the lung. Neuro Endocrinol Lett. 2006;27:23–6. [PubMed] [Google Scholar]
- 18.Witko-Sarsat V, Friedlander M, Nguyen Khoa T, et al. Advanced oxidation protein products as novel mediators of inflammation and monocyte activation in chronic renal failure. J Immunol. 1998;161:2524–32. [PubMed] [Google Scholar]
- 19.Schinzel R, Munch G, Heidland A, et al. Advanced glycation end products in end-stage renal disease and their removal. Nephron. 2001;87:295–303. doi: 10.1159/000045934. [DOI] [PubMed] [Google Scholar]
- 20.Boor P, Konieczny A, Villa L, et al. PDGF-D inhibition by CR002 ameliorates tubulointerstitial fibrosis following experimental glomerulonephritis. Nephrol Dial Transplant. 2007;22:1323–31. doi: 10.1093/ndt/gfl691. [DOI] [PubMed] [Google Scholar]
- 21.Sebekova K, Ramuscak A, Boor P, et al. The selective TP receptor antagonist, S18886 (terutroban), attenuates renal damage in the double transgenic rat model of hypertension. Am J Nephrol. 2008;28:47–53. doi: 10.1159/000108760. [DOI] [PubMed] [Google Scholar]
- 22.Amann K, Wolf B, Nichols C, et al. Aortic changes in experimental renal failure: hyperplasia or hypertrophy of smooth muscle cells. Hypertension. 1997;29:770–5. doi: 10.1161/01.hyp.29.3.770. [DOI] [PubMed] [Google Scholar]
- 23.Tornig J, Amann K, Ritz E, et al. Arteriolar wall thickening, capillary rarefaction and interstitial fibrosis in the heart of rats with renal failure: the effects of ramipril, nifedipine and moxonidine. J Am Soc Nephrol. 1996;7:667–75. doi: 10.1681/ASN.V75667. [DOI] [PubMed] [Google Scholar]
- 24.Bokemeyer D, Panek D, Kramer HJ, et al. In vivo identification of the mitogen-activated protein kinase cascade as a central pathogenic pathway in experimental mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 2002;13:1473–80. doi: 10.1097/01.asn.0000017576.50319.ac. [DOI] [PubMed] [Google Scholar]
- 25.Barnoya J, Glantz SA. Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation. 2005;111:2684–98. doi: 10.1161/CIRCULATIONAHA.104.492215. [DOI] [PubMed] [Google Scholar]
- 26.Whincup PH, Gilg JA, Emberson JR, et al. Passive smoking and risk of coronary heart disease and stroke: prospective study with cotinine measurement. BMJ. 2004;329:200–5. doi: 10.1136/bmj.38146.427188.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Black HR, Zeevi GR, Silten RM, et al. Effect of heavy cigarette smoking on renal and myocardial arterioles. Nephron. 1983;34:173–9. doi: 10.1159/000183005. [DOI] [PubMed] [Google Scholar]
- 28.Markowitz GS, Lin J, Valeri AM, et al. Idiopathic nodular glomerulosclerosis is a distinct clinicopathologic entity linked to hypertension and smoking. Hum Pathol. 2002;33:826–35. doi: 10.1053/hupa.2002.126189. [DOI] [PubMed] [Google Scholar]
- 29.Oberai B, Adams CW, High OB. Myocardial and renal arteriolar thickening in cigarette smokers. Atherosclerosis. 1984;52:185–90. doi: 10.1016/0021-9150(84)90116-3. [DOI] [PubMed] [Google Scholar]
- 30.Nasr SH, D’Agati VD. Nodular glomerulosclerosis in the nondiabetic smoker. J Am Soc Nephrol. 2007;18:2032–6. doi: 10.1681/ASN.2006121328. [DOI] [PubMed] [Google Scholar]
- 31.Liang KV, Greene EL, Oei LS, et al. Nodular glomerulosclerosis: renal lesions in chronic smokers mimic chronic thrombotic microangiopathy and hypertensive lesions. Am J Kidney Dis. 2007;49:552–9. doi: 10.1053/j.ajkd.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 32.Culhaci N, Meteoglu I, Dundar M, et al. Histopathological evaluation of renal vascular changes in rats exposed to passive smoking. Pathol Oncol Res. 2005;11:121–4. doi: 10.1007/BF02893379. [DOI] [PubMed] [Google Scholar]
- 33.Arif JM, Vadhanam MV, De Groot AJ, et al. Effect of cigarette smoke exposure on the modulation of 8-oxo-2’-deoxyguanosine in rat lungs as analyzed by 32P-postlabeling and HPLC-ECD. Int J Oncol. 2001;19:763–6. doi: 10.3892/ijo.19.4.763. [DOI] [PubMed] [Google Scholar]
- 34.Gentry-Nielsen MJ, Top EV, Snitily MU, et al. A rat model to determine the biomedical consequences of concurrent ethanol ingestion and cigarette smoke exposure. Alcohol Clin Exp Res. 2004;28:1120–8. doi: 10.1097/01.alc.0000136383.45378.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li HY, Hou FF, Zhang X, et al. Advanced oxidation protein products accelerate renal fibrosis in a remnant kidney model. J Am Soc Nephrol. 2007;18:528–38. doi: 10.1681/ASN.2006070781. [DOI] [PubMed] [Google Scholar]
- 36.Shi XY, Hou FF, Niu HX, et al. Advanced oxidation protein products promote inflammation in diabetic kidney through activation of renal nicotinamide adenine dinucleotide phosphate oxidase. Endocrinology. 2008;149:1829–39. doi: 10.1210/en.2007-1544. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Wang Y, Cai Q, et al. By homing to the kidney, activated macrophages potently exacerbate renal injury. Am J Pathol. 2008;172:1491–9. doi: 10.2353/ajpath.2008.070825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bachi A, Zuccato E, Baraldi M, et al. Measurement of urinary 8-Epi-prostaglandin F2alpha, a novel index of lipid peroxidation in vivo, by immuno-affinity extraction/gas chromatography-mass spectrometry. Basal levels in smokers and nonsmokers. Free Radic Biol Med. 1996;20:619–24. doi: 10.1016/0891-5849(95)02087-x. [DOI] [PubMed] [Google Scholar]
- 39.Visioli F, Galli C, Plasmati E, et al. Olive phenol hydroxytyrosol prevents passive smoking-induced oxidative stress. Circulation. 2000;102:2169–71. doi: 10.1161/01.cir.102.18.2169. [DOI] [PubMed] [Google Scholar]
- 40.Flores L, Vidal M, Abian J, et al. The effects of smoking and its cessation on 8-epi-PGF2alpha and transforming growth factor-beta 1 in type 1 diabetes mellitus. Diabet Med. 2004;21:285–9. doi: 10.1111/j.1464-5491.2004.01133.x. [DOI] [PubMed] [Google Scholar]
- 41.Park EM, Park YM, Gwak YS. Oxidative damage in tissues of rats exposed to cigarette smoke. Free Radic Biol Med. 1998;25:79–86. doi: 10.1016/s0891-5849(98)00041-0. [DOI] [PubMed] [Google Scholar]
- 42.Wang Z, Nicholls SJ, Rodriguez ER, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007;13:1176–84. doi: 10.1038/nm1637. [DOI] [PubMed] [Google Scholar]
- 43.Cerna S, Beno M, Hurbankova M, et al. Evaluation of bronchoalveolar lavage fluid cytotoxic parameters after inhalation exposure to amosite and wollastonite fibrous dusts combined with cigarette smoke. Cent Eur J Public Health. 2004;12:S20–3. [PubMed] [Google Scholar]
- 44.Miserocchi G, Sancini G, Mantegazza F, et al. Translocation pathways for inhaled asbestos fibers. Environ Health. 2008;7:4. doi: 10.1186/1476-069X-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dement JM, Brown DP, Okun A. Follow-up study of chrysotile asbestos textile workers: cohort mortality and case-control analyses. Am J Ind Med. 1994;26:431–47. doi: 10.1002/ajim.4700260402. [DOI] [PubMed] [Google Scholar]
- 46.Hein MJ, Stayner LT, Lehman E, et al. Follow-up study of chrysotile textile workers: cohort mortality and exposure-response. Occup Environ Med. 2007;64:616–25. doi: 10.1136/oem.2006.031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.IA EL-S, Gadallah M, Shouman AE, et al. Subclinical nephrotoxicity caused by smoking and occupational silica exposure among Egyptian industrial workers. Arch Med Res. 2003;34:415–21. doi: 10.1016/S0188-4409(03)00077-8. [DOI] [PubMed] [Google Scholar]