

Abstract
Angiogenesis, the development of new blood vessel from pre-existing vessels, is a key process in the formation of the granulation tissue during wound healing. The appropriate development of new blood vessels, along with their subsequent maturation and differentiation, establishes the foundation for functional wound neovasculature. We performed studies in vivo and used a variety of cellular and molecular approaches in vitro to show that insulin stimulates angiogenesis and to elucidate the signalling mechanisms by which this protein stimulates microvessel development. Mice skin injected with insulin shows longer vessels with more branches, along with increased numbers of associated α-smooth muscle actin-expressing cells, suggesting the appropriate differentiation and maturation of the new vessels. We also found that insulin stimulates human microvascular endothelial cell migration and tube formation, and that these effects occur independently of VEGF/VEGFR signalling, but are dependent upon the insulin receptor itself. Downstream signalling pathways involve PI3K, Akt, sterol regulatory element-binding protein 1 (SREBP-1) and Rac1; inhibition of these pathways results in elimination of endothelial cell migration and tube formation and significantly decreases the development of microvessels. Our findings strongly suggest that insulin is a good candidate for the treatment of ischaemic wounds and other conditions in which blood vessel development is impaired.
Keywords: human microvascular endothelial cell, migration, signal transduction, wound healing, microvessels
Introduction
Wound healing is a dynamic, interactive process consisting of three major phases that overlap in time: inflammation, granulation tissue development (including re-epithelialisation) and tissue remodelling [1]. Granulation tissue development involves the generation of new connective tissue and new blood vessels within the wound during the healing process. Angiogenesis, the process of generation of new blood vessels from pre-existing vessels, functions to provide oxygen and nutrients to the injured area and is a key step in the development of the healing (granulation) tissue. The proper development of this tissue establishes the foundation for the subsequent events during the healing process, including wound contraction and generation of wound tensile strength.
Angiogenesis is necessary for normal wound healing. Indeed, when angiogenesis is decreased, the result is impaired healing [2]. The development of new blood vessels requires the proliferation and migration of endothelial cells [3], which is initiated by a cross-talk between cells and the surrounding microenvironment. This includes signals stimulated by numerous factors, such as acidic and basic FGF, VEGF, TGF-β and angiogenin [4, 5]. The work presented here shows that insulin is another stimulator of angiogenesis.
The use of insulin for non-diabetic purposes began in the early part of the twentieth century [6, 7] and has been used to improve bone healing in rats [8], incision or burn wounds of the skin [9, 10] and cutaneous ulcerations in diabetic and non-diabetic mice [11]. Insulin also appears to exert some effects on the vasculature. Previous studies have shown that insulin has vasculo-protective effects on endothelial cells by stimulating the release of NO from these cells [12, 13]. NO is a potent modulator of microvascular permeability [14]; it inhibits oxidant-induced dysfunction of the endothelial cell barrier [15] and reduces reperfusion-induced cardiac injury [16]. Furthermore, it has been shown in an experimental mouse model system for retinopathy developed during premature birth that mice lacking expression of the insulin receptor in vascular endothelial cells exhibit reduced retinal neovascularisation when compared with controls. A similar, though weaker, effect was seen in mice lacking insulin-like growth factor (IGF) receptor in endothelial cells [17], suggesting the important role of insulin and, to a lesser extent, of IGF in angiogenesis. However, little is known about the mechanisms by which insulin stimulates angiogenesis. The question still remains: Is insulin an angiogenic molecule?
To determine whether insulin is angiogenic in vivo, we performed subcutaneous injections of this protein in the dermis of mice, and to determine the mechanism of action, we used human microvascular endothelial cells (HMEC) in culture. Our results show for the first time that insulin stimulates the development of new microvessels, and that these microvessels are associated with α-smooth muscle actin (α-SMA)-expressing cells and that they are more branched than those in vehicle-injected animals. The studies in culture suggest that this occurs by stimulation of endothelial cell migration and tube formation through a PI3K-Akt-sterol regulatory element-binding protein-1 (SREBP-1) signalling pathway that leads to the activation of Rac1. Our findings strongly suggest that insulin is a useful tool for the improvement of wounds impaired in vascu-larisation and also for situations in which ischaemia is present.
Materials and Methods
Reagents
A human microvascular endothelial cell line, HMEC-1, was a gift from the Center for Disease Control (Atlanta, GA, USA). Plasmids expressing the constitutively active form of Rac1 (V12, Rac1-CA), the dominant-negative mutant of Rac1 (N17, Rac1-DN) and the wild-type Rac1 (Rac1-WT) were the gifts from Miguel Del Pozo (Centro National de Investigaciones Cardiovasculares, Madrid, Spain). DMEM was purchased from Mediatech (Manassas, VA, USA) and FBS from Atlanta Biologicals (Lawrenceville, GA, USA). Anti-SREBP-1(2A4), anti-Akt, anti-RhoA (sc-179) and anti-VEGF (A-20) antibodies (Ab) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-Akt Ser (473) and anti-insulin receptor (29B4) Ab as well as the PI3K inhibitor LY294002 were purchased from Cell Signaling (Danvers, MA, USA). The TRITC-antimouse Rac1 and rat antimouse CD31 Ab (PECAM-1, IHC) were purchased from Becton Dickinson and Company (Franklin Lakes, NJ, USA). Anti-GAPDH Ab was obtained from RDI Research Diagnostics (Concord, MA, USA). The IGF-1 receptor inhibitor picropodophyllin and the VEGFR2 inhibitor SU1498 came from EMD Biosciences (San Diego, CA, USA). Recombinant human insulin, 25-hydroxycholesterol (25-HC) and anti-α-SMA Ab were purchased from Sigma (St. Louis, MO, USA). FITC-conjugated goat antimouse immunoglobulin came from Dako (DK, Glostrup, Denmark), antimouse Texas Red Ab from Amersham, and FITC-conjugated goat antirat immunoglobulin from Zymed Laboratories, Inc. (South Francisco, CA, USA). Humulin Ultralene human insulin (rDNA origin) extended zinc suspension was purchased from Eli Lilly and Company (Indianapolis, IN, USA). Human VEGF was purchased from Peprotech (Rocky Hill, NJ, USA), and Lipofectin® transfection reagent was purchased from Invitrogen (Carlsbad, CA, USA). The Rac activation assay kit came from Cell Biolabs, Inc. (San Diego, CA, USA).
In vivo angiogenesis model
C57BL/6J mice were purchased from The Jackson Laboratory (USA) and housed at the University of California, Riverside (UCR) vivarium. All experimental protocols were approved by the UCR Institutional Animal Care and Use Committee. Experiments were performed with 8- to 12-week-old mice. Mice were anesthetized with a single intraperitoneal injection of ketamine (80 mg/kg body weight)/xylazine (16 mg/kg body weight). One day prior to treatment, the hair was removed from the back of the mice using the Nair hair remover (Madera, CA, USA). Insulin (0.03 U/20 μl saline) or saline (20 μl) were injected at symmetric sites on the back using an insulin syringe every 24 hrs for 5 days, and skin samples from the injected areas were collected at day 6. Experiments involving the use of 25-HC were performed by injecting each mouse at four sites on the back with insulin (0.03 U/20 μl saline), saline (20 μl), 25-HC (0.5 mg/ml, 20 μl) or 25-HC (1 mg/ml, 10 μl) + insulin (0.03 U/10 μl saline). The injection sites were rotated clockwise in different mice in order to compensate for the variations from different anatomic distribution. At the sites in which 25-HC was used, this inhibitor was pre-injected 24 hrs before the first insulin treatment at the assigned injection site, then with insulin every 24 hrs for 5 days. In all cases, the injection sites were labelled using a permanent marker pen to ensure that all injections for the same site were located in the same place, and that the two injection sites on the same animal were not less than 2 cm apart to avoid possible cross-over effects. Skin samples were collected at day 6 after initiation of insulin treatment.
Preparation of tissues for histology
The animals were anesthetized, as described above, and skin samples were collected using a skin punch biopsy (7 mm in diameter) (Acuderm, Inc.). The tissues were fixed in 4% paraformaldehyde, incubated with 0.1 M glycine/PBS for 1 hr, then incubated with 15% and 30% sucrose before embedding in OCT (Tissue-Tek; Sakura Finetek, Inc., USA) and then frozen and stored at –80°C. For CD31 staining samples, the tissues were embedded directly in OCT and were frozen. After sample collection, the mice were killed using CO2.
Cell culture
HMEC-1 cells were cultured in 5% CO2 at 37°C in DMEM supplemented with 10% FBS, 10 units/ml penicillin and 10 μg/ml streptomycin sulphate (GIBCO; Invitrogen Corp.).
Cell proliferation assay
DNA synthesis was assessed by BrdU incorporation using a BrdU proliferation assay kit (EMD Biosciences). Cells seeded in the 96-well tissue culture plates (1 × 104 cells/well) were subjected to treatment with different concentrations of insulin for 24 or 48 hrs in the presence of BrdU. After fixation, the cells were processed according to the manufacturer’s protocol. Briefly, 100 μl anti-BrdU Ab was added to each well and incubated for 1 hr. After washing, 100 μl/well peroxidase goat antimouse IgG HRP was added to the cells, followed by incubation for 30 min. and addition of 100 μl of substrate solution for 15 min. and then the reaction was stopped by adding 100 μl of stop solution. The relative incorporation of BrdU was determined by reading the absorbance at 450 nm using a spectrometer.
In vitro cell migration assay
The cloning ring assay was used to monitor cell migration. In total, 2.0 × 104 cells were plated in a cloning ring with a 6-mm-diameter set within a 35-mm culture dish. Two hours after seeding, the cylinder was removed, the cell edges were marked and migration was measured at the indicated time-points by quantification of the distance from the initial cell edge to the edge of the migrating cells.
Endothelial cell tube formation assay
Twenty microlitres of Matrigel (Becton Dickinson) were applied to the centre of the 35-mm cell culture dishes to evenly coat an area with a diameter of 1.5 cm. The coated plates were incubated for 1 hr at room temperature. The endothelial cells, with or without transfection of different mutants of Rac1, were plated on the Matrigel-coated area at a density of 1.5 × 104 cells, then treated as indicated above and incubated for 20 hrs. Tube formation was observed with an inverted phase contrast microscope (Nikon, Tokyo, Japan).
Immunoblotting
The cells were treated as indicated above, washed with ice-cold 1× PBS and lysed on ice with a lysis buffer containing 10 mM Tris, 0.5% Triton X-100, 0.5% Nonidet P-40, pH 7.5, 2.5 mM KCl, 150 mM NaCl, 30 mM β-glycerophosphate, 50 mM NaF, 1 mM Na3VO4 and 0.1% SDS and with additional protease and phosphatase inhibitor cocktails (Sigma). Protein concentrations were measured using the DC protein assay kit (Bio-Rad). Equal protein concentrations of the cell extracts were mixed with the sample buffer, boiled and analysed using 10% acrylamide SDS-PAGE. Immunoblotting was performed with the indicated primary Ab and the appropriate HRP-conjugated secondary Ab, followed by incubation with West Dura extended duration substrate (Pierce Biotechnology). Blots were then re-probed for housekeeping proteins to ensure equal loading.
Rac1 pull-down assay
The cells were treated as indicated above, washed with ice-cold 1× PBS, and lysed on ice with 1× lysis buffer and the cell lysates collected and cleared by centrifugation. Thirty microlitres of cell lysates were mixed with the sample buffer for loading control. The remainder of the cell lysates were then mixed with PAK PBD agarose bead slurry and incubated at 4°C for 1 hr with gentle agitation. The beads were pelleted by centrifugation and washed three times with 0.5 ml of 1× assay buffer. The bead pellets were resuspended in 40 μl of 2× reducing SDS-PAGE sample buffer and boiled for 5 min. The pull-down supernatant was then analysed by SDS-PAGE and immunoblotting with an anti-Rac1 Ab.
Transient transfections
Plasmids expressing Rac1-CA, Rac1-DN and Rac1-WT were transfected using Lipofectin (Invitrogen, USA), according to the manufacturer’s protocol. Briefly, one day before transfection, the cells were plated in a growth medium without antibiotics. Transfections were performed at 40–60% cell confluency. DNA (2 μg) in 100 μl of DMEM without serum was mixed gently with 5 μl Lipofectin in 100 μl of DMEM without serum. This mixture was incubated for 15 min. at room temperature and added to the cells, which were then incubated at 37°C and 5% CO2 for 5 hrs. At this time, the medium was replaced with a serum-containing medium.
Immunolabelling
The cells were cultured in chamber slides (Nunc), fixed in 4% paraformaldehyde for 30 min., rinsed with PBS, incubated in 0.1 M glycine/PBS for 20 min. and blocked with 3% BSA, 0.1% Triton X-100/PBS for 30 min. Primary Ab in 1% BSA/PBS were applied to the sample for 2 hrs at room temperature, washed and incubated with Texas Red or FITC-conjugated secondary Ab for 1 hr at room temperature. After washing, the cells were mounted in Vectashield-containing DAPI (Vector Laboratories, Inc.). Immunolabelling was visualised and imaged using a Leica SP2 laser scanning confocal microscope (Allendale, NJ, USA). For frozen tissues, 8-μm cryosections were washed to remove the OCT, fixed in 2% paraformaldehyde for 30 min. and incubated in 0.1 M glycine/PBS for 20 min., followed by the primary and secondary Ab using the same procedure as indicated above. For CD31 staining samples, cryosections were fixed by incubation with ice-cold acetone for 30 min. and incubated in 0.1 M glycine/PBS for 20 min.; the remainder of the procedure was identical to that indicated above.
Statistical analysis
Data are shown as mean ± standard deviation (SD). Data analysis was performed using the unpaired Student’s t-test on raw data with GraphPad Instat software (GraphPad Software, Inc.).
Results
Subcutaneous injections of insulin stimulate angiogenesis in vivo
Insulin stimulates wound healing in mouse excision wounds and rat burn wounds [10]. Angiogenesis is a critical and necessary step for wound healing. Therefore, we hypothesize that insulin affects angiogenesis during wound healing, and that this is one of the ways by which it stimulates improvement of the healing process. To test this possibility, we injected 0.03 units of insulin subcutaneously in the mouse dorsum every 24 hrs. After 5 days of insulin treatment, samples were collected from the injected area, and the extent of angiogenesis was determined by quantification of CD31 staining, a marker of endothelial cells, to determine the overall blood vessel area. Insulin increases the area occupied by the CD31-positive cells (Fig. 1 A–C) and stimulates the development of longer vessels with more branches (Fig. 1A and B). We also determined the numbers of blood vessels expressing α-SMA, a marker of pericytes and smooth muscle cells, that are associated with vessels which have undergone maturation and differentiation and found that insulin stimulates a significant increase in the number of α-SMA-expressing cells associated with blood vessels (Fig. 1D).
Figure 1.
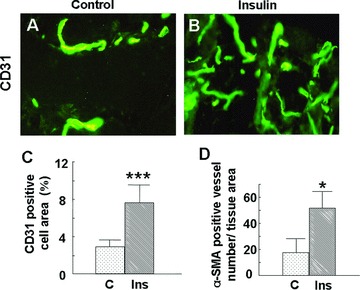
Subcutaneous injection of insulin stimulates angiogenesis in mouse skin: C57BL/6J mice (n= 6) were injected with insulin (0.03 U/20 μl saline) or saline (20 μl) subcutaneously at symmetric sites on the dorsum every 24 hrs for 5 days, and skin samples from the injected areas were collected at day 6. Tissue slides were stained with either anti-CD31 (PECAM-1) or anti-α-SMA, followed by FITC-conjugated goat antirat or goat antimouse secondary antibodies. Staining was visualised and pictures were taken using a Nikon immunofluorescence microscope (Bethesda, ML, USA). (A, B) Representative immunofluorescence images of CD31 staining: (A) control, (B) insulin; magnification 40×. Insulin injection results in the formation of longer blood vessels with more branches. (C) Percentage of CD31-positive cell area: the fraction of CD31-positive cell area relative to the total tissue area of each high-power field was measured using ImageJ software. At least eight fields per group were analysed. Insulin increases the percentage of the CD31-positive cell area. Data are shown as the mean value ± SD. Statistics indicate differences between the treatment and control. ***P < 0.001 versus control. (D) The number of blood vessels that express α-SMA were counted using fluorescence microscopy, and the vessel number was normalised by dividing by the total tissue section area. Insulin increased the number of α-SMA-positive blood vessels. Data are shown as the mean value ± SD. Statistics indicate differences between the treatment and control.*P < 0.05 versus control.
Insulin stimulates microvascular endothelial cell migration in a time- and dose-dependent manner
We used HMEC to determine the effects of insulin on endothelial proliferation and migration, processes involved in angiogenesis. BrdU incorporation assays were performed to detect DNA synthesis, and thus proliferation, in endothelial cells after treatment with multiple concentrations of insulin. Concentrations ranging from 10−8 M to 10−5 M insulin did not significantly change BrdU incorporation, showing that insulin does not stimulate endothelial cell proliferation (Fig. 2A). Cell migration was measured using the cloning ring migration assay. In this assay, the cells were treated with different concentrations of insulin to identify potential effects of insulin on migration and examine dose-dependent effects. A concentration of 10−8 M insulin slightly increased endothelial cell migration after 48 hrs of treatment. Concentrations ranging from 10−7 M to 10−5 M insulin significantly enhanced endothelial cell migration after 24 hrs of treatment (Fig. 2B and C).
Figure 2.
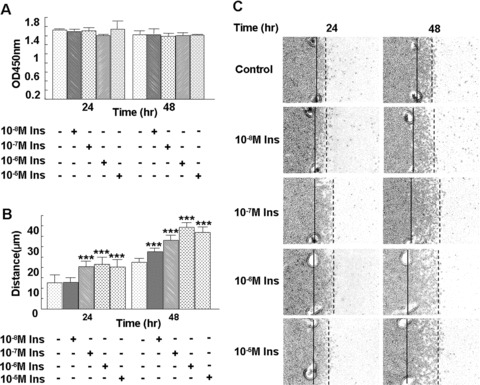
Insulin stimulates microvascular endothelial cell migration, but not proliferation, in a time- and dose-dependent manner. (A) HMEC-1 cells seeded in the 96-well tissue culture plates (1.0 × 104 cells/well) were subjected to treatment with different concentrations of insulin for 24 or 48 hrs in the presence of BrdU. After fixation, the cells were processed according to the protocol of the manufacturer. Data are shown as the mean value ± SD. There were no statistically significant differences in endothelial cell proliferation following insulin treatment. (B, C) Cells were plated within cloning rings, the edges were marked and then the cells were treated with 10−8 M, 10−7 M, 10−6 M or 10−5 M insulin, respectively. Migration distance was measured at 24 and 48 hrs after treatment. Each treatment group was performed in triplicate. Data are shown as the mean value ± SD. Statistics are shown as comparisons between the treatment and control. **P < 0.01. Insulin significantly increased HMEC-1 migration, and the effect of insulin on cell migration correlated with the dose of insulin.
Insulin stimulates microvascular endothelial cell migration via the insulin receptor
Insulin and IGF-1 receptors are tyrosine kinase receptors that share 60% homology. Insulin and IGF-1 bind, and activate, both receptors, although they bind the receptors with different affinities [18]. To determine which receptors mediate the effect of insulin on endothelial cell migration, we pre-treated the microvascular endothelial cells with either the neutralizing insulin receptor Ab, Ab 29B4, or the IGF-1 receptor tyrosine kinase inhibitor, picropodophyllin, and then treated the cells with 10−7 M or 10−6 M insulin. Pre-treatment with the insulin receptor Ab completely abolished 10−7 M insulin-induced migration but only partially blocked 10−6 M insulin-induced migration (Fig. 3A). We also found that picropodophyllin did not affect 10−7 M insulin-induced migration but did inhibit 10−6 M insulin-induced migration (Fig. 3B). These results suggest that the effects of higher concentrations (10−6 M) of insulin on endothelial cell migration are mediated by both the insulin and the IGF-1 receptors, whereas the effects of lower concentrations of insulin (10−7 M) are primarily mediated by the insulin receptor. To study the effects of insulin that are mediated only through the insulin receptor and its associated downstream signalling pathways, 10−7 M insulin was chosen for the subsequent studies, except when otherwise indicated.
Figure 3.
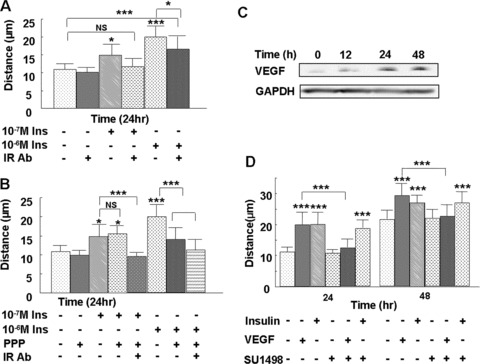
Insulin stimulates microvascular endothelial cell migration in an insulin receptor-dependent and a VEGF-independent manner. Endothelial cells were plated for the cloning ring migration assay, as described in Fig 2. (A) Cells were pre-treated with 1.5 μg of the neutralising insulin receptor Ab 29B4 for 1 hr, and then treated with 10−7 M or 10−6 M insulin for 24 hrs. The neutralising insulin receptor Ab completely inhibited 10−7 M insulin-induced cell migration and only partially inhibited 10−6 M insulin-induced cell migration, showing that the effect of 10−7 M insulin on endothelial cell migration is mediated primarily through the insulin receptor. (B) Cells were pre-treated with either 50 nM IGF-1 receptor inhibitor picropodophyllin or a combination of 1.5 μg of the insulin receptor Ab 29B4 and picropodophyllin for 1 hr, then treated with 10−7 M or 10−6 M insulin or left untreated for 24 hrs. The effects of the higher insulin concentration are mediated by both the insulin receptor and the IGF-1 receptor, whereas lower insulin concentrations are mainly mediated by the insulin receptor alone. (C) Cells were treated with 10−7 M insulin for the indicated time-points, the cell culture medium was then collected and VEGF levels were detected by Western blot. Insulin stimulates endothelial cell VEGF secretion. (D) Cells were either left untreated or pre-treated with 8 nM VEGFR inhibitor SU1498 for 1 hr, followed by treatment with 10−7 M insulin or 20 ng/ml VEGF; migration distance was measured at 24 and 48 hrs after treatment. Insulin-induced migration was not prevented with the VEGFR inhibitor, whereas VEGF-induced migration was prevented with this inhibitor. Each treatment group was performed in triplicate. Data are shown as the mean value ± SD. Statistics are shown as comparisons between the treatment and control, unless otherwise indicated. ***P < 0.001.
Insulin is known to stimulate VEGF secretion in various cell types, and this growth factor is known to stimulate endothelial cell migration and angiogenesis [3, 19, 20]. Indeed, following 24 and 48 hrs of insulin treatment, we detected elevated VEGF levels in the endothelial cell culture medium (Fig. 3C). In order to determine whether insulin-induced endothelial cell migration is mediated by VEGF, we pre-treated the cells with SU1498, a selective inhibitor of the VEGF receptor 2 (VEGFR-2), prior to treatment with insulin or VEGF and measured migration distances at 24 and 48 hrs. This VEGFR inhibitor completely abolished VEGF-induced cell migration but did not alter insulin-induced migration (Fig. 3D), suggesting that insulin-induced endothelial cell migration does not require VEGFR signalling.
PI3K and Akt mediate insulin-induced microvascular endothelial cell migration and tube formation
The activation of PI3K-Akt is important for growth factor-induced cell proliferation, migration and survival. Therefore, we investigated whether insulin promotes the phosphorylation/activation of Akt and found that the levels of phosphorylated Akt increased after 3 min. of insulin treatment and remained elevated for at least 60 min. (Fig. 4A). This effect was dose-dependent, with the higher insulin concentrations stimulating stronger Akt phosphorylation (Fig. 4B). Akt is phosphorylated and activated downstream of PI3K [21]; indeed, inhibition of PI3K with LY294002 prevented insulin-induced Akt phosphorylation (Fig. 4C). We also used this inhibitor to determine the importance of PI3K-Akt activity in insulin-induced cell migration and tube formation and found that inhibition of the PI3K-Akt pathway abolished both insulin-mediated processes (Fig. 4D and E). These results show that insulin stimulation of endothelial cell migration and tube formation is dependent upon PI3K and Akt.
Figure 4.
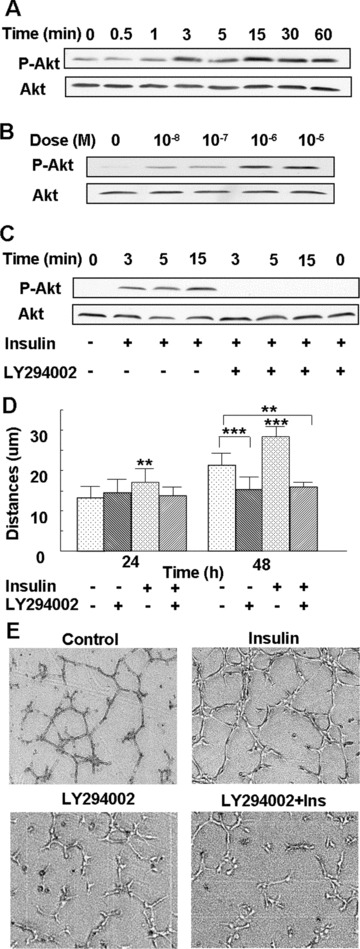
PI3K-Akt mediates insulin-induced microvascular endothelial cell migration and tube formation: (A) Endothelial cells were treated with 10−7 M insulin for the indicated time-points, followed by Western blot analysis using an antibody directed against Akt phosphorylated at Ser 473. The blot was re-probed with an anti-Akt Ab to ensure equal loading. Insulin increased Akt phosphorylation over time, with peak phosphorylation seen at 3 and 15 min. (B) Cells were either left untreated or treated with 10−8 M to10−5 M insulin for 3 min., and then analysed as in (A). Insulin stimulated phosphorylation of Akt in a dose-dependent manner, with 10−6 M and 10−5 M insulin inducing maximum phosphorylation. (C) Cells were incubated with 25 μM of the PI3K inhibitor LY294002 for 1 hr, followed by treatment with 10−7 M insulin for the indicated time-points, and Akt phosphorylation was detected by Western blot analysis, as mentioned above. Insulin-induced Akt phosphorylation was abrogated by pre-treatment with a PI3K inhibitor. (D) Cells were incubated with 25 μM of the PI3K inhibitor LY294002 for 1 hr, followed by treatment with 10−7 M insulin. The migration distances were measured at 24 and 48 hrs after treatment. Insulin-induced endothelial cell migration was abrogated by pre-treatment with a PI3K inhibitor. The results are representative of at least three independent experiments. **P < 0.01 versus control; ***P < 0.001 versus control. (E) Cells were either left untreated as a control or treated with 25 μM of LY294002 for 1 hr, then treated with or without 10−7 M insulin, and the tube formation assay was performed. Each treatment group was performed in triplicate. Insulin-induced tube formation was prevented by pre-treatment with the PI3K inhibitor.
SREBP-1 is involved in insulin-induced microvascular endothelial cell migration and angiogenesis
SREBPs are key transcription factors that regulate cholesterol and fatty acid biosynthesis pathways and are thus important in membrane lipid biosynthesis [22]. Because of the relationship between the microviscosity of the cell membrane and cell motility, it is possible that SREBP activation is important in the effects of insulin on endothelial cell migration. Therefore, we investigated whether insulin treatment promotes SREBP-1 activation in endothelial cells, and whether this activation is involved in insulin-induced cell migration. Immunoblot analysis using Ab that recognise specifically the precursor and the cleaved/mature forms of SREBP-1 showed an increase in the mature form within 3 min. of insulin treatment (Fig. 5A). To determine the involvement of SREBP-1 in insulin-induced endothelial cell migration, we pre-treated these cells with an SREBP inhibitor, 25-HC, and found that this inhibitor blocks cell migration (Fig. 5B). In addition, the PI3K inhibitor LY294002 inhibits SREBP-1 activation (Fig. 5C), suggesting that insulin-stimulated SREBP-1 activation is mediated by PI3K-Akt, and that this activation is important in insulin-induced cell migration.
Figure 5.
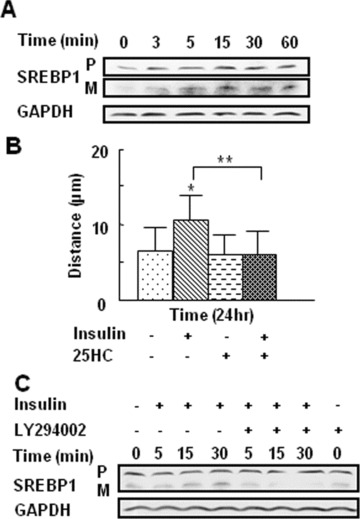
SREBP-1 is involved in insulin-induced endothelial cell migration and activation by insulin, which requires the PI3K-Akt: (A, B) Endothelial cells were treated with 10−7 M insulin for the indicated time-points. The levels of the SREBP-1 precursor (‘P’) and mature/active (‘M’) forms were analysed by Western blot. Blots were re-probed with GAPDH as a loading control. (A) Insulin increased the levels of the mature form of SREBP-1, with the active form peaking 15 min. after treatment. The levels of the mature form of SREBP-1 were quantified by determining the ratio of the integrated density of the mature form of SREBP-1 to GAPDH loading control, and further normalised to the control using ImageJ software. Data are shown as the mean value ± SD. Statistics are shown as comparisons between the treatment and control. *P < 0.05, **P < 0.01. (B) Cell migration was measured using the cloning ring assay. For 25-HC ± insulin-treated cells, cells were pre-treated with 10 μg/ml 25-HC for 24 hrs, followed by 10−7 M insulin treatment for 24 hrs. 25-HC significantly inhibited insulin-induced endothelial cell migration. Each treatment group was performed in triplicate. Data are shown as the mean value ± SD. Statistics are shown as comparisons between the treatment and control. *P < 0.05, **P < 0.01. (C) Cells were pre-treated with 25 μM of LY294002 for 1 hr, followed by treatment with 10−7 M insulin for the indicated time-points. SREBP-1 levels were detected by Western blotting. Insulin-induced activation of SREBP-1 was inhibited by LY294002, as shown by the decreased levels of mature SREBP-1.
To determine whether the PI3k-Akt-SREBP signalling pathway activated by insulin in cultured endothelial cells is involved in insulin-induced angiogenesis in vivo, we injected 25-HC to inhibit SREBP prior to insulin treatment. This pre-treatment inhibited insulin-induced angiogenesis, as shown by the decrease in the area of CD31-positive cells (Fig. 6A–E), and a decreased number of blood vessels staining with α-SMA (Fig. 6F). These data, taken together, show that the PI3k-Akt-SREBP signalling pathway is involved in insulin-induced angiogenesis in vivo.
Figure 6.
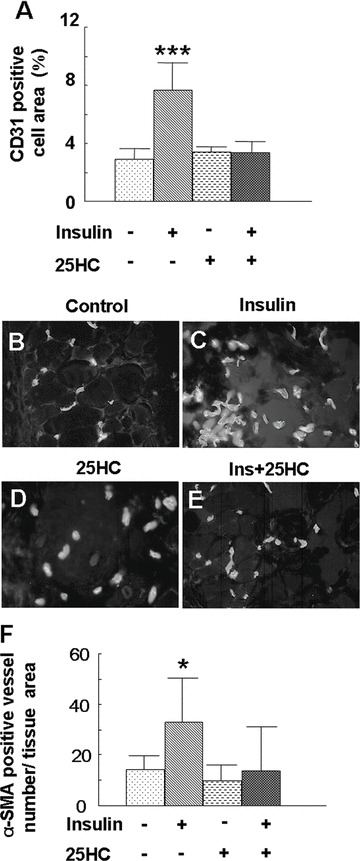
Inhibition of SREBP prevented insulin-induced angiogenesis in mice skin: (A–E) Mice (n= 4) were injected with insulin (0.03 U/20 μl saline), saline (20 μl), 25-HC (20 μl) or 25-HC (10 μl) + insulin (0.03 U/10 μl saline) at symmetric sites on the mouse dorsum every 24 hrs for 5 days, and skin samples were collected on day 6. Tissue slides were stained with anti-CD31 antibody, as mentioned above. (A) 25-HC treatment decreased the CD31-positive cell area and inhibited insulin-induced blood vessel formation. Data are shown as the mean value ± SD. Statistics indicate differences between the treatment and control. ***P < 0.001 versus control. (B–E) Representative immunofluorescence images of CD31 staining. All pictures were taken at 40× and enlarged to the same scale. (F) 25-HC treatment decreased the number of α-SMA-positive blood vessels. α-SMA-positive blood vessel number was calculated as described in Fig. 1. Data are shown as the mean value ± SD. Statistics indicate differences between the treatment and control. *P < 0.05 versus control.
Insulin stimulates Rac1 membrane translocation and this effect occurs downstream of the PI3K-Akt-SREBP-1 pathway
SREBPs are the major regulators of HMG CoA reductase (HMGR) [23, 24], which regulates the biosynthesis of farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), two components of isoprenoides that mediate the function of the RhoA family of GTPases [25–27]. As a consequence, we investigated whether RhoA, a member of the RhoA family of GTPases, is activated by insulin. No significant difference in translocation of RhoA to the plasma membrane (reflecting activation) was found after insulin treatment (data not shown). However, Rac1, another member of the RhoA family of GTPases, translocated from the cytosol to the plasma membrane 3 min. after insulin treatment (Fig. 7A and B). We also observed plasma membrane ruffling at the leading edge of migrating endothelial cells, with Rac-1 present in the membrane of ruffles (Fig. 7C). To examine whether the translocation of Rac1 is associated with its activation, we performed a Rac pull-down assay, which specifically pulls down and quantifies the active form of Rac, and found elevated levels of active Rac1 after insulin treatment (Fig. 7D).
Figure 7.
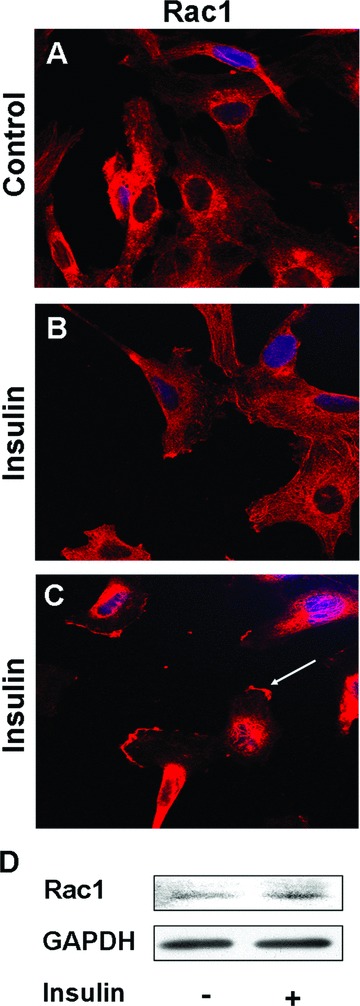
Insulin stimulates Rac1 translocation to the plasma membrane: (A–C) Representative immunofluorescence images of Rac1 staining. Cells were stained with the TRITC-anti-Rac1 Ab. Insulin induced Rac1 translocation to the plasma membrane. (C) The arrowhead indicates membrane ruffling at the leading edge of a migrating cell. (D) Rac1 pull-down assay. Endothelial cells were treated with insulin for 3 min., and the Rac1 pull-down assay was then performed to quantify Rac activation. Cell lysates were washed and mixed with PAK PBD agarose beads, and the proteins pulled down were analysed by SDS-PAGE and immunoblotting with an anti-Rac1 antibody. Insulin induced Rac1 activation.
To determine whether the Rac1 activation is essential in insulin-induced endothelial cell migration, the cells were transfected with plasmids containing mutant forms of Rac1, the constitutively active form of Rac1 (V12, Rac1-CA), dominant-negative mutant Rac1 (N17, Rac1-DN) or wild type Rac1 (Rac1-WT). Twenty-four hours after transfection, the cells were seeded for cloning ring migration assays and plated on Matrigel for tube formation assays. Cell migration distances were measured in non-transfected and transfected cells at 24 and 48 hrs after insulin treatment. Insulin-induced migration was observed in non-transfected cells as well as in cells transfected with Rac1-CA or Rac1-WT but was prevented in cells transfected with Rac1-DN (Fig. 8A). Similarly, insulin-induced tube formation was observed in non-transfected cells (Fig. 8B and C) and in cells transfected with Rac1-CA (Fig. 8D and E) or Rac1-WT (Fig. 8H and I) but not in cells transfected with Rac1-DN (Fig. 8F and G). This suggests that Rac1 is required for insulin-induced migration and tube formation. To determine the involvement of the PI3K-Akt-SREBP pathway in insulin-induced Rac activation, we pre-treated the cells with the PI3K inhibitor LY294002 or the SREBP inhibitor 25-HC and found that inhibition of these signalling molecules prevented insulin-induced Rac1 activation (Fig. 8J and K), implicating the PI3K-Akt-SREBP-1 pathway in insulin-induced Rac1 activation.
Figure 8.
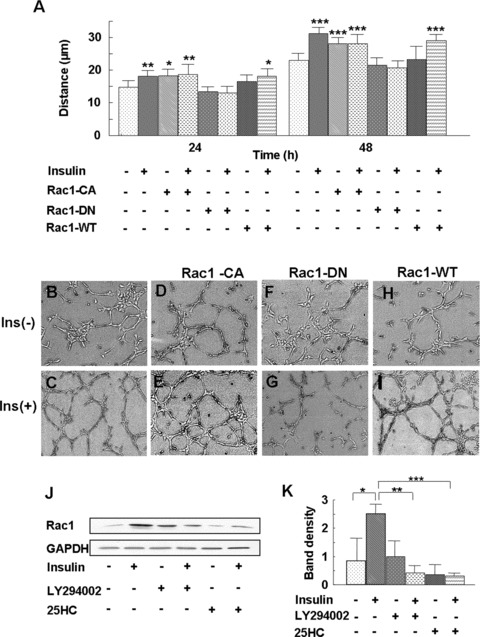
Insulin-induced translocation of Rac1 occurs downstream of PI3K-Akt- SREBP-1. (A–I) Cells were transfected with plasmids expressing the constitutively active form of Rac1 (V12, Rac1-CA), the dominant-negative mutant of Rac1 (N17, Rac1-DN) and the wild-type Rac1 (Rac1-WT) using Lipofectin. Forty-eight hours after transfection, the cells were seeded for the cloning ring migration assay and tube formation assay. Cell migration was monitored at 24 and 48 hrs after insulin treatment. Tube formation was observed after treatment for 20 hrs with insulin. (A) Insulin-induced endothelial cell migration was eliminated by Rac1-DN, whereas Rac1-CA increased cell migration even without insulin treatment. Each treatment group was performed in triplicate. Data are shown as the mean value ± SD. Statistics are shown as comparisons between the treatment and control. *P < 0.05, **P < 0.01, ***P < 0.001. (B–I) Insulin-induced endothelial cell tube formation was abolished by Rac1-DN. (J) Cells were pre-treated either with 25 μM PI3K inhibitor LY294002 for 1 hr or with 10 μg/ml SREBP inhibitor 25-HC for 24 hrs, followed by 10−7 M insulin treatment for 3 min. The Rac1 pull-down assay was then performed, as mentioned in Fig. 7. Insulin induced Rac1 activation, and this activation was prevented by inhibition of either PI3K or SREBP. (K) Rac1 expression was quantified by the ratio of band density of activated Rac1/GAPDH using ImageJ software. Statistics are shown as comparisons between the treatment and control, unless otherwise indicated, using three different gels.
Discussion
The findings that insulin has a vascular protective effect on endothelial cells and that it stimulates healing suggest that this protein may stimulate angiogenesis, which in turn is instrumental in accelerating wound healing. We show that: (1) Subcutaneous injection of insulin stimulates the development of blood vessels that are longer and have more branches than the vessels in vehicle-treated animals and are associated with α-SMA-expressing cells. (2) Insulin stimulates microvascular endothelial cell migration, but not proliferation, in a dose- and time-dependent manner. (3) Insulin stimulates endothelial tube formation in culture. (4) The activities of insulin are dependent on the insulin receptor itself and are independent of VEGF/VEGFR. (5) The PI3K-Akt-SREBP-1-Rac1 signalling pathway is involved in insulin-induced endothelial cell migration and tube formation in vitro and angiogenesis in vivo. These results show for the first time the pro-angiogenic effects of insulin and elucidate the signalling pathways stimulated by this protein that result in angiogenesis.
The increase in microvessel branching stimulated by insulin in vivo is intriguing because it suggests stimulation of endothelial cell proliferation. However, we were unable to detect insulin-induced endothelial cell proliferation in the cultured endothelial cells. This indicates that in vivo insulin cooperates with other factors that stimulate endothelial cell proliferation such as VEGF. One possible explanation for this cooperation is that insulin stimulation of glucose transport provides the energy that the cell needs to then respond to proliferation stimuli. Our observation that more α-SMA-expressing cells are associated with the microvessels in vivo indicates that insulin also promotes blood vessel maturation. This might be another process in which insulin could cooperate with VEGF because it has been shown that VEGF-induced angiogenesis results in leaky vessels [28, 29]. Indeed, following new microvessel development, the nascent vessels must undergo a maturation process, both within the vascular network and within the vessel wall, in order to function properly. Maturation of the vascular network involves vascular branching, expansion and pruning [31], whereas maturation of the vascular wall involves the recruitment of mural cells, such as pericytes and smooth muscle cells that express α-SMA [32]. These cells are important support structures for the blood vessels [33–35]. Increases in both microvessel branching and association with α-SMA-expressing cells are important for the transport of nutrients and proper O2/CO2 exchanges in the tissue and may contribute to the development of a more regenerative tissue. Considering the importance of maturation on the function of new blood vessel, the potential effects of insulin on promoting the formation of more mature, functional vessels puts insulin in the category of a valuable pro-angiogenic molecule. Interestingly, despite our characterisation of insulin as a pro-angiogenic factor, the insulin injection sites of diabetic patients do not appear to exhibit pronounced angiogenesis. These seemingly contradictory findings may result from a variety of factors, including the altered responsiveness of many diabetic patients to insulin due to their reduced insulin sensitivity, dysfunctional insulin-induced signalling pathways or reduced effectiveness of insulin due to its non-enzymatic glycation or proteolysis following injection. In addition, frequent sites for insulin injection include the abdomen or buttocks, tissues with large amounts of fat and relatively few blood vessels; the low numbers of pre-existing endothelial cells in these tissues might result in their lack of obvious angiogenesis compared with the dermis in our animal models. Furthermore, the insulin injection sites have not been fully characterised; local insulin injection frequently results in a problem usually described as ‘hard lumps or extra fat deposits’, which could contain granulation tissue and associated blood vessels that could only be detected through a more thorough analysis; indeed, our own analysis of blood vessels in insulin-treated mouse skin involved tissue removal and specific staining for endothelial cells.
The activation of Rac1 downstream of a signalling pathway involving PI3K, Akt and SREBP-1 is essential for insulin-induced angiogenesis. SREBPs are potent regulators of lipogenic enzymes and regulate the biosynthesis of cholesterol and fatty acids [36–38], important components of the plasma membrane. Increasing evidence suggests that SREBPs are the primary regulators of HMGR [39, 24]. This enzyme regulates the biosynthesis of FPP and GGPP [40], two components of isoprenoids. Isoprenoids mediate the function of the RhoA family of GTPases to which Rac1 belongs. These GTPases are geranylgeranylated proteins, and inhibition of HMGR blocks the translocation or redistribution of Rho family of proteins from the cytoplasm to the membrane [41]. Also, Rac1 is known to regulate actin assembly [42] and stimulate formation of lamellipodia [43], thereby promoting cell movement in response to external signals from cytokines, growth factors and/or the ECM. Therefore, inhibition of SREBP-1 or the upstream signalling molecules PI3K/Akt prevents insulin-induced Rac1 translocation and activation, thus inhibiting cell migration.
Although insulin activates both its own receptor and/or IGF-1 receptor, our work shows that at lower doses insulin activates only its own receptor to stimulate endothelial cell migration in culture and angiogenesis in vivo. At higher concentrations, insulin has additional growth factor-like activities through the IGF receptor, which is known to be a potent regulator of cell proliferation [44], differentiation [45] and survival [46]. Therefore, it is possible to take advantage of these differences to treat various types of wounds. This may be particularly useful in the treatment of chronic or impaired wounds in which tissue insulin or its receptor might be deficient or dysfunctional. However, it is important to realise that dysregulated IGF signalling is associated with many human cancers [47–51], suggesting that insulin is a safer therapeutic option for the stimulation of wound healing when compared with IGF-1 or other growth factors. Indeed, insulin has a proven safety record, as it has been used in human beings for a century, without serious side effects.
In conclusion, because of the pro-angiogenic effects of insulin and its safety for human applications, insulin may prove to be a good therapy for the treatment of impaired wounds deficient in revascularisation and other conditions in which re-vascularisation is needed. However, problems could occur with insulin treatment of impaired wounds in diabetic patients, which are generally associated with peripheral vascular disease, endothelial dysfunction and localised abnormalities in cellular and cytokine activity [52]. Insulin dysfunction could occur through the non-enzymatic glycation of this protein because of either the excessive levels of glucose in the injured tissue or the deficiencies in the signalling downstream from the insulin receptor. Furthermore, in other types of chronic wounds, proper debridement might be a key to the success of insulin therapy. As a consequence, more studies are needed to examine the effects of insulin on angiogenesis in chronic wounds, in particular in wounds of diabetic patients, as well as to determine the most effective means of insulin delivery, including considerations of the specific conditions found in the wound environment.
Acknowledgments
We thank Dr. Miguel Del Pozo for the Rac1 wild-type and mutant plasmids and Hongwei Yuan and Chongze Ma for helpful discussions.
References
- 1.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–46. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 2.Li YJ, Guan H, Hazarika S, et al. Impaired angiogenesis following hind-limb ischemia in diabetes mellitus mice. Chin Med Sci J. 2007;22:232–7. [PubMed] [Google Scholar]
- 3.Hoeben A, Landuyt B, Highley MS, et al. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549–80. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J, D’Amore PA. Blood vessel formation: what is its molecular basis. Cell. 1996;87:1153–5. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- 5.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 6.King L, Kennaway EL, Piney A. A note on the action of insulin in normal persons. J Physiol. 1928;66:400–2. doi: 10.1113/jphysiol.1928.sp002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joseph B. Insulin in the treatment of non-diabetic bed sores. Ann Surg. 1930;92:318–9. doi: 10.1097/00000658-193008000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gregory WB., Jr Effect of insulin on the healing of bone wounds in albino rats. J Dent Res. 1965;44:487–92. doi: 10.1177/00220345650440030801. [DOI] [PubMed] [Google Scholar]
- 9.Rosenthal SP. Acceleration of primary wound healing by insulin. Arch Surg. 1968;96:53–5. doi: 10.1001/archsurg.1968.01330190055012. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Zhang X, Zhang Z, et al. Effects of topical application of insulin on the wound healing in scalded rats. Zhonghua Shao Shang Za Zhi. 2004;20:98–101. [PubMed] [Google Scholar]
- 11.Hanam SR, Singleton CE, Rudek W. The effect of topical insulin on infected cutaneous ulcerations in diabetic and nondiabetic mice. J Foot Surg. 1983;22:298–301. [PubMed] [Google Scholar]
- 12.Montagnani M, Chen H, Barr VA, et al. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179) J Biol Chem. 2001;276:30392–8. doi: 10.1074/jbc.M103702200. [DOI] [PubMed] [Google Scholar]
- 13.Fulton D, Gratton JP, McCabe TJ, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Persson J, Ekelund U, Grande PO. Nitric oxide and prostacyclin play a role in the regulation of microvascular protein and hydraulic permeability in cat skeletal muscle. Microcirculation. 2003;10:233–43. doi: 10.1038/sj.mn.7800187. [DOI] [PubMed] [Google Scholar]
- 15.Rath S, Kalogeris T, Mai N, et al. Insulin prevents oxidant-induced endothelial cell barrier dysfunction via nitric oxide-dependent pathway. Surgery. 2006;139:82–91. doi: 10.1016/j.surg.2005.06.056. [DOI] [PubMed] [Google Scholar]
- 16.Wang G, Liem DA, Vondriska TM, et al. Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2005;288:H1290–5. doi: 10.1152/ajpheart.00796.2004. [DOI] [PubMed] [Google Scholar]
- 17.Kondo T, Vicent D, Suzuma K, et al. Knockout of insulin and IGF-1 receptors on vascular endothelial cells protects against retinal neovascularization. J Clin Invest. 2003;111:1835–42. doi: 10.1172/JCI17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garza-Garcia A, Patel DS, Gems D, et al. RILM: a web-based resource to aid comparative and functional analysis of the insulin and IGF-1 receptor family. Hum Mutat. 2007;28:660–8. doi: 10.1002/humu.20491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carnesecchi S, Carpentier JL, Foti M, et al. Insulin-induced vascular endothelial growth factor expression is mediated by the NADPH oxidase NOX3. Exp Cell Res. 2006;312:3413–24. doi: 10.1016/j.yexcr.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Q, Liu LZ, Fu B, et al. Reactive oxygen species regulate insulin-induced VEGF and HIF-1alpha expression through the activation of p70S6K1 in human prostate cancer cells. Carcinogenesis. 2007;28:28–37. doi: 10.1093/carcin/bgl085. [DOI] [PubMed] [Google Scholar]
- 21.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 22.Eberlé D, Hegarty B, Bossard P, et al. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86:839–48. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 23.Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol. 2007;19:215–22. doi: 10.1016/j.ceb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 25.Rando RR. Chemical biology of protein isoprenylation/methylation. Biochim Biophys Acta. 1996;1300:5–16. doi: 10.1016/0005-2760(95)00233-2. [DOI] [PubMed] [Google Scholar]
- 26.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–69. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 27.Yao M, Zhou RH, Petreaca M, et al. Activation of sterol regulatory element-binding proteins (SREBPs) is critical in IL-8-induced angiogenesis. J Leukoc Biol. 2006;80:608–20. doi: 10.1189/jlb.0304175. [DOI] [PubMed] [Google Scholar]
- 28.Zacchigna S, Tasciotti E, Kusmic C, et al. In vivo imaging shows abnormal function of vascular endothelial growth factor-induced vasculature. Hum Gene Ther. 2007;18:515–24. doi: 10.1089/hum.2006.162. [DOI] [PubMed] [Google Scholar]
- 29.Thurston G. Complementary actions of VEGF and angiopoietin-1 on blood vessel growth and leakage. J Anat. 2002;200:575–80. doi: 10.1046/j.1469-7580.2002.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laschke MW, Elitzsch A, Vollmar B, et al. Combined inhibition of vascular endothelial growth factor (VEGF), fibroblast growth factor and platelet-derived growth factor, but not inhibition of VEGF alone, effectively suppresses angiogenesis and vessel maturation in endometriotic lesions. Hum Reprod. 2006;21:262–8. doi: 10.1093/humrep/dei308. [DOI] [PubMed] [Google Scholar]
- 31.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 32.Verbeek MM, Otte-Holler I, Wesseling P, et al. Induction of alpha-smooth muscle actin expression in cultured human brain pericytes by transforming growth factor-beta 1. Am J Pathol. 1994;144:372–82. [PMC free article] [PubMed] [Google Scholar]
- 33.Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–64. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–23. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 35.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 36.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horton JD, Shimomura I. Sterol regulatory element-binding proteins: activators of cholesterol and fatty acid biosynthesis. Curr Opin Lipidol. 1999;10:143–50. doi: 10.1097/00041433-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Sakakura Y, Shimano H, Sone H, et al. Sterol regulatory element-binding proteins induce an entire pathway of cholesterol synthesis. Biochem Biophys Res Commun. 2001;286:176–83. doi: 10.1006/bbrc.2001.5375. [DOI] [PubMed] [Google Scholar]
- 39.Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol. 2007;19:215–22. doi: 10.1016/j.ceb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Brown MS, Goldstein JL. Multivalent feedback regulation of HMG-CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–17. [PubMed] [Google Scholar]
- 41.Lee J, Lee I, Park C, et al. Lovastatin-induced RhoA modulation and its effect on senescence in prostate cancer cells. Biochem Biophys Res Commun. 2006;339:748–54. doi: 10.1016/j.bbrc.2005.11.075. [DOI] [PubMed] [Google Scholar]
- 42.Aspenstrom P. Effectors for the Rho GTPases. Curr Opin Cell Biol. 1999;11:95–102. doi: 10.1016/s0955-0674(99)80011-8. [DOI] [PubMed] [Google Scholar]
- 43.Ridley AJ, Paterson HF, Johnston CL, et al. The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–10. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 44.Machida S, Booth FW. Insulin-like growth factor 1 and muscle growth: implication for satellite cell proliferation. Proc Nutr Soc. 2004;63:337–40. doi: 10.1079/PNS2004354. [DOI] [PubMed] [Google Scholar]
- 45.Sanchez-Calderon H, Milo M, Leon Y, et al. A network of growth and transcription factors controls neuronal differentiation and survival in the developing ear. Int J Dev Biol. 2007;51:557–70. doi: 10.1387/ijdb.072373hs. [DOI] [PubMed] [Google Scholar]
- 46.Kurmasheva RT, Houghton PJ. IGF-I mediated survival pathways in normal and malignant cells. Biochim Biophys Acta. 2006;1766:1–22. doi: 10.1016/j.bbcan.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 47.Durai R, Yang W, Gupta S, et al. The role of the insulin-like growth factor system in colorectal cancer: review of current knowledge. Int J Colorectal Dis. 2005;20:203–20. doi: 10.1007/s00384-004-0675-4. [DOI] [PubMed] [Google Scholar]
- 48.Cocca C, Nuñnez M, Gutiérrez A, et al. IGF-I in mammary tumorigenesis and diabetes. Anticancer Res. 2004;24:2953–65. [PubMed] [Google Scholar]
- 49.Jenkins PJ, Bustin SA. Evidence for a link between IGF-I and cancer. Eur J Endocrinol. 2004;151:S17–22. doi: 10.1530/eje.0.151s017. [DOI] [PubMed] [Google Scholar]
- 50.Jerome L, Shiry L, Leyland-Jones B. Deregulation of the IGF axis in cancer: epidemiological evidence and potential therapeutic interventions. Endocr Relat Cancer. 2003;10:561–78. doi: 10.1677/erc.0.0100561. [DOI] [PubMed] [Google Scholar]
- 51.LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–37. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 52.Laing T, Hanson R, Chan F, et al. The role of endothelial dysfunction in the pathogenesis of impaired diabetic wound healing: a novel therapeutic target. Med Hypotheses. 2007;69:1029–31. doi: 10.1016/j.mehy.2007.02.040. [DOI] [PubMed] [Google Scholar]