

Abstract
Heat shock protein 70 (HSP70) is frequently overexpressed in a variety of human malignancies and protects cancer cells against apoptosis in response to various stresses. The bioflavonoid quercetin inhibits HSP70 expression and induces cancer cells apoptosis. In the present study, we have investigated the effects of HSP70 down-regulation on the unfolded protein response (UPR) and addressed a novel strategy to enhance the proapoptotic effect of quercetin by suppressing GRP78 induction simultaneously. Treatment of human breast cancer cells with quercetin down-regulates HSP70 expression, but up-regulates GRP78 expression in a dose-dependent manner. Down-regulation of HSP70 by small interfering RNA also leads to induction of GRP78. Moreover, our studies reveal that HSP70 knockdown or quercetin induces other typical components of the UPR, including CHOP expression, eIF2α and JNK phosphorylation, caspases activation and XBP-1 splicing. Abrogating the induction of pro-survival chaperone GRP78 by small interfering RNA sensitizes breast cancer cells to quercetin. Colony survival assays demonstrate that treatment of breast cancer cells with green tea (−)-epigallocatechin gallate (EGCG), which binds to the ATP-binding domain of GRP78 and blocks its protective function, synergistically promoted quercetin-induced cell death. These studies reveal that HSP70 down-regulation leads to the induction of UPR. The pro-survival GRP78 induction contributes to quercetin resistance. Abrogation of GRP78 induction or inhibition of GRP78 activity increases the effectiveness of quercetin. These findings indicate that combinational administration of flavonoids capable of suppressing HSP70 and GRP78 such as quercetin and EGCG might represent a novel approach for cancer therapy or chemoprevention.
Keywords: GRP78, heat shock protein, molecular chaperone, breast cancer
Introduction
Heat shock proteins (HSPs) are highly conserved and functionally interactive chaperone proteins that maintain protein homeostasis in all living organisms [1]. Heat shock cognate (HSC) proteins are constitutively synthesized, whereas others are expressed following stress. A variety of environmental or physiological stresses, such as heat shock, hypoxia, reactive oxygen species and virus transformation, can induce HSPs expression [2, 3]. HSPs act in many fundamental cellular processes, including the catalysation of substrate refolding, the prevention of irreversible aggregation of unfolded proteins, the maturation of nuclear hormone receptors and other signalling molecules, vesicle formation and protein trafficking [4–6]. A number of functionally important HSPs have been identified. Based on the molecular mass, HSPs can be generally classified into large and small HSPs. Small HSPs include HSP27, HSP 26, HSP22 and HSP30 [7]. According to sequence homology, HSPs can be classified into different subfamilies. Two well-characterized subgroups of HSPs are the HSP70 family and HSP90 family. The HSP70 family includes at least 11 distinct proteins in human beings, such as HSP70, HSP70A, HSP70B, GRP78, GRP75 and HSP75 [8]. The HSP90 family consists of at least nine proteins in human, such as HSP90, HSP90α, HSP90β, HSP86, HSP84 and HSP83 [9]. In addition, another two large HSP families, HSP100 and HSP60, are relatively less characterized.
Previous studies have established a strong connection of HSPs to cancer. In normal cells under unstressed conditions, the inducible members of HSP family are poorly expressed. In contrast, HSP70 and HSP90 are elevated in various kinds of malignant tumours, such as breast, uterine cervix and renal cell cancinoma [10–12]. High levels of HSP70 and HSP90 expression confer enhanced survival and drug resistance [13, 14]. Many client proteins of HSP70 and HSP90 are critical components of diverse signalling pathways. For example, the direct interaction among HSP70, HSP90 and glucocorticoid receptor is critical for efficient hormone binding and subsequent transcriptional activation [15]. Recent studies from in vivo yeast system and in vitro mammalian cell-free system also indicate that progesterone receptor requires the molecular chaperone HSP90 for efficient ligand binding [16]. In addition to hormone receptors, another important set of client proteins of both HSP70 and HSP90 is protein kinases, such as c-raf, c-src and the receptor tyrosine kinase ErbB2 [17, 18]. GRP78 shares 60% amino acid homology with HSP70, but it is distinct from HSP70 in that it is generally non-inducible or only weakly inducible by heat [19, 20]. As an endoplasmic reticulum chaperone, GRP78 can facilitate the folding of newly synthesized proteins, target terminally misfolded proteins for proteasomal degradation, regulate calcium homeostasis and control the activation of ER stress sensors [21]. Elevated expression of GRP78 has been observed in a variety of human cancer, including breast cancer, lung cancer, gastric cancer and malignant gliomas [21–23]. During tumour onset and progression, GRP78 is capable of enhancing tumour cell proliferation, protecting tumour cells against apoptosis and promoting tumour angiogenesis [24].
Down-regulation of HSP70 and HSP90 results in apoptosis in cancer cells but not in untransformed cells, which makes HSP70 and HSP90 attractive targets for molecular cancer therapeutics and chemoprevention [25, 26]. Preclinical studies have demonstrated that the HSP90 inhibitor 17-allylamino-17-demethoxygel-danamycin possesses potent anti-tumour activity [27]. In addition, the bioflavonoid quercetin could inhibit HSP70 expression by blocking heat shock transcrition factor (HSF) 1 and HSF2. Treatment of cancer cells with quercetin leads to cell death [28], which indicates that quercetin may be a potential anti-tumour compound. One of the major problems in cancer chemotherapy or molecular cancer therapeutics is drug resistance. Compensatory pathways are frequently activated in response to the inhibition of one target molecule, which may lead to poor response to the targeted therapy. Like the response to other agents, cancer cells may respond to HSP70 down-regulation or quercetin to a different extent. Also, resistance to these treatments may be an obstacle to their use in clinical setting. So far, little is known about the mechanisms that are involved in quercetin sensitivity or resistance. In the present study, we have studied whether the unfolded protein response (UPR) is involved in HSP70 down-regulation- or quercetin-induced breast cancer cells apoptosis. Our approach is to assess GRP78 and CHOP expression, XBP-1 splicing, eIF2α and JNK phosphorylation induced by quercetin. We demonstrate that both HSP70 knockdown and quercetin can induce multiple arms of the UPR, including the pro-survival GRP78 induction, the pro-apoptotic JNK activation and caspase cleavage. Because GRP78 can confer resistance to some chemotherapeutic agents [29–31], we speculate that GRP78 induction may be a drawback for the anti-tumour activity of quercetin. Here, we show that abrogation of GRP78 induction by small interfering RNA or inhibition of GRP78 by the green tea (-)-epigallocatechin gallate (EGCG) synergistically promotes quercetin-induced cancer cells death. These findings identify a novel connection between HSP70 and ER homeostasis and reveal a critical role of GRP78 in the resistance to quercetin.
Materials and methods
Reagents
Quercetin and EGCG were purchased from Sigma-Aldrich, Inc (St. Louis, MO, USA). The PI3K inhibitors LY294002 and wortmanin, JNK inhibitor SP600125, caspase-3 and −7 inhibitor z-DEVD-fmk were provided by Beyotime Institute of Biotechnology (Jiangsu, China). GRP78, CHOP, XBP1, HSP70 and phosphorylated eIF2α antibodies were purchased from Santa Cruz Biotech (Santa Cruz, CA, USA). The anti-JNK, caspase-4, -3, -7 and PARP antibody was provided by Cell Signaling Technology (Beverly, MA, USA).
Cell culture
Cancer cells were grown in tissue culture flasks at 37°C in a humidified atmosphere of 5% CO2 and were maintained as monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI 1640 supplemented with 5% foetal bovine serum (FBS) and 1% penicillin-streptomycin.
Transfection of siRNA
The target sequence used for knockdown of GRP78 and HSP70 were 5′-GGAGCGCAUUGAUACUAGA-3′ and 5′-GGACAUCAGCCAGAACAAG-3′, respectively. The negative control siRNA was purchased from Ribobio Co., Ltd. (Guangzhou, China). The double-stranded siRNA duplex was dissolved in DEPC-treated water. For transfection, 1 × 105 cells were plated into 24-well plates and incubated overnight. Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) was diluted in 250 μl of Opti-MEM I Reduced Serum Medium and incubated at room temperature for 5 min. In addition, siRNA duplex was diluted in 250 μl of Opti-MEM I Reduced Serum Medium and mixed with the pre-diluted Lipofectamine 2000. The mixture was incubated at RT for 20 min. 50 nmol/l of siRNA was added into each well and incubated at 37°C.
Western blot
Cells were washed twice with phosphate buffered saline and harvested with cold RIPA lysis buffer containing protease inhibitors (PMSF 1 mmol/l, and leupeptin 0.1 g/l). Cell lysates were collected from culture plates using a rubber policeman, and protein collected by centrifugation. Protein concentrations were determined by BCA protein assay (Pierce Biotechnology, Rockford, IL, USA). Forty micrograms of total protein were boiled in 2× loading buffer (0.1 M Tris-Cl, pH 6.8, 4% SDS, 0.2% bromophenyl blue, 20% glycerol) for 10 min., then loaded into Tris-HCl-Polyacrylamide gels and transferred electrophoretically to Immobilon-P membrane (Millipore Corporation, Billerica, MA, USA). Membranes were incubated with primary antibodies and appropriate HRP-secondary antibodies. Membranes were additionally probed with an antibody against β-actin (Santa Cruz Biotechnology) to normalize loading of protein among samples. The secondary antibodies were detected by chemiluminescent agents (Pierce Biotechnology).
Quantitative real-time PCR
Growing MCF-7 cells were harvested after treatment with 100 μmol/l of quercetin for 12 hrs in triplicate, and total RNA was isolated with TRIZOL reagent (Invitrogen). cDNA was generated from 1 μg total RNA using AMV reverse-transcription kit according to the manufacturer’s instructions (Promega Biotechnology, Madison, WI, USA). GRP78 and HSP70 were amplified by real-time PCR using the SybrGreen PCR amplification mix (total volume 25 μl) and 320 nmol/l of primers. β-actin was also amplified as a reference gene. The primer sequences for human GRP78 are as follows: 5′-CTGGGTACATTTGATCTGACT-3′ (forward) and 5′-GCATCTTGGTGGCTTTCCAGC-3′ (reverse). The primers for human HSP70 are as follows: 5′-CTCCAGCATCCGACAAGAAGC-3′ (forward) and 5′-ACGGTGTTGTGGGGGTTCA-3′ (reverse). Relative quantification with the comparative threshold cycle (Ct) was done using the ΔΔCt method. The amount of GRP78 or HSP70 gene normalized to the endogenous reference gene (β-actin) is given by 2−ΔCt, where ΔCt is Ct (GRP78 or HSP70) –Ct (β-actin).
Flow cytometry
Apoptotic cells were determined by the propidium iodide staining and flow cytometry. Briefly, replicate cultures of 1 × 106 cells were plated in cell culture wells. The cells were transfected with siRNA and/or treated with quercetin. Forty-eight hours after the transfection, cells were harvested, washed with PBS and fixed in 70% ethanol for 30 min. at 4°C. The fixed cells were treated with 50 μg/ml RNase A (Sigma Chemical Company, St Louis, MO, USA), stained with 50 μg/ml propidium iodide for 20 min. at 4°C in the dark before flow cytometric analyses. The propidium iodide fluorescence of individual nuclei was measured in the red fluorescence using a flow cytometer (Beckman Coulter Elite, Miami Lakes, FL, USA). Quantification of apoptotic cells was carried out by measurement of sub-G1 DNA content.
Hoechst 33342 staining
Replicate cultures of 1 × 106 cells per well were plated in 24-well plate. The cells were transfected with siRNA and/or treated with quercetin. After a change of fresh medium 24 hrs later, the cells were incubated with 5 μl of Hoechst 33342 solution per well at 37°C for 10 min., followed by observation under a fluorescence microscope. Strong fluorescence can be observed in the nuclei of apoptotic cells, while weak fluorescence was observed in non-apoptotic cells. Quantification of apoptotic cells was performed by taking the images in random fields and counting at least 200 cells in four random fields in each well.
WST1 assay
MCF-7 or T47D cells were plated in 96-well plates at 5000 or 6000 cells per well, respectively. The next day, cells were treated with or without 10 μmol/l EGCG, 50 μmol/l quercetin or both with five to six replicates. After 24 hrs, the cytotoxity was assessed by incubating cells with WST1 reagent (Roche, Indianapolis, IN, USA) for 2 hrs and measuring the absorbance at 450 nm, and at 630 nm as reference, with a microplate reader (Bio-Rad, Hercules, CA, USA).
Colony formation assay
The cells were seeded in six-well plates at 5000 cells per well. The next day, the medium was changed to include quercetin or EGCG. Six or 24 hrs later, the cells were grown in the absence of drug and allowed to form colonies for 14 days. The colonies were stained with 1% methylene blue and then counted. The experiments were done in triplicates and each experiment was reproduced.
Statistical analysis
One-way ANOVA with least significant difference post hoc test was used to test for the differences in the means of apoptosis rate or colony survival rate. The quantitative real-time PCR data were analysed with REST-XL © version-2, a calculating software for the relative expression in real-time PCR using pair-wise fixed reallocation randomization test [32]. All statistical tests were two-sided, and difference to be considered to be statistically significant when P < 0.05.
Results
Quercetin and HSP70 knockdown induce the UPR
HSP70 is a molecular chaperone for cytosolic protein refolding, renaturation and trafficking. The bioflavonoid quercetin can transcriptionaly repress HSP70 expression. To investigate whether HSP70 down-regulation would trigger the UPR, the effect of quercetin on the unfolded protein responsive protein GRP78 was examined. MCF-7 cells were treated with different doses of quercetin for 24 hrs, and then subjected to Western blot analysis of HSP70 and GRP78. Whereas HSP70 expression was inhibited by quercetin, treatment of MCF-7 cells with quercetin stimulated GRP78 expression in a dose-dependent manner. Similar effects were observed in T47D, a breast cancer cell line, and MDA-MB-435, a cell line that has been used as a representative of breast cancer cells for decades but is shown to be from a melanoma (Fig. 1A). In addition, T47D cells were treated with 100 nM quercetin for 4, 8, 12 and 24 hrs, then subjected to Western blot analysis of HSP70 and GRP78. GRP78 expression was up-regulated after T47D cells were treated with quercetin for 12 hrs, while HSP70 down-regulation was detectable 8 hrs after quercetin treatment (Fig. 1B). Moreover, we used quantitative real-time PCR to determine whether quercetin increased GRP78 expression and down-regulated HSP70 expression at transcription levels. As shown in Fig. 1C, quercetin caused an increase in GRP78 transcription and a decrease in HSP70 transcription.
Figure 1.
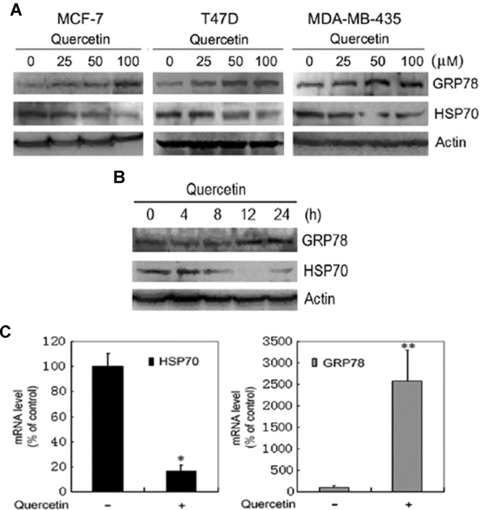
Induction of GRP78 expression and suppression of HSP70 expression by bioflavonoid quercetin. (A) Western blot analysis of GRP78 and HSP70 expression in response to quercetin treatment. MCF-7, T47D and MDA-MB-435 cells were treated with indicated doses of quercetin for 24 hrs. Cell lysates were subjected to SDS-PAGE, and then blotted with anti-GRP78 and anti-HSP70 antibodies. (B) T47D cells were treated with 100 μM quercetin for 4, 8, 12 and 24 hrs. Cell lysates were subjected to SDS-PAGE, and then blotted with anti-GRP78 and anti-HSP70 antibodies. (C) Quantitative real-time PCR analysis of HSP70 and GRP78 expression in response to quercetin treatment of T47D cells. Columns, mean of replicates; bars, SE. *, P= 0.013. **, P= 0.001.
Next, we investigated the effects of quercetin on other unfolded protein responsive proteins including CHOP, spliced XBP-1 and phosphorylated eIF2α. Treatment of T47D cells with quercetin for 6 hrs resulted in XBP-1 splicing in a dose-dependent manner (Fig. 2A). In addition, quercetin induced CHOP/GADD153 expression (Fig. 2B). During the UPR, a transient translation arrest is induced upon phosphorylation of the eukaryotic initiating factor 2α (eIF2α) by PERK. The phosphorylation of eIF2α was observed after treatment of breast cancer cells with quercetin for 1 hr (Fig. 2C). Thus, quercetin is identified as a novel inducer of the UPR.
Figure 2.
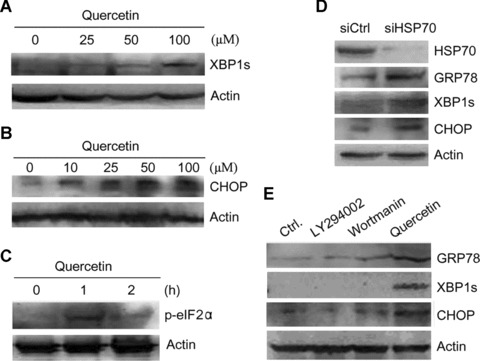
Induction of the unfolded protein response by bioflavonoid quercetin and HSP70 knockdown in T47D cells. (A) Western blot analysis of the expression of spliced XBP-1 (XBP-1s) in response to quercetin treatment. Cells were treated with indicated doses of quercetin for 6 hrs. Cell lysates were subjected to SDS-PAGE, and then blotted with anti-XBP-1 antibody. (B) Western blot analysis of CHOP expression in response to quercetin treatment. Cells were treated with indicated doses of quercetin for 24 hrs. Cell lysates were subjected to SDS-PAGE, and then blotted with anti-CHOP antibody. (C) Western blot analysis of eIF2α phosphorylation in response to quercetin treatment. Cells were treated with quercetin for 1 hr and 2 hrs. Cell lysates were subjected to SDS-PAGE, and then blotted with anti-phosphorylated eIF2α antibody. (D) Cells were transfected with control siRNA (siCtrl) or the siRNA targeting Hsp70 (siHsp70). Cell lysates were subjected to SDS-PAGE, and then blotted with anti-GRP78, anti-CHOP, anti-XBP-1 and anti-HSP70 antibodies. (E) Cells were treated with or without 10 μM LY294002, 200 nM wortmanin and 100 μM quercetin for 24 hrs. Cell lysates were subjected to SDS-PAGE, and then blotted with anti-GRP78, anti-CHOP and anti-XBP-1 antibodies.
Since HSP70 is not the only target of quercetin, we then confirmed that HSP70 down-regulation led to the UPR by suppressing HSP70 expression with siRNA against HSP70. The suppression of HSP70 with siRNA resulted in a substantial decrease in HSP70 levels and significant increase in GRP78 and CHOP levels. In addition, HSP70 knockdown induced XBP-1 splicing (Fig. 2D). Thus, HSP70 down-regulation triggers the UPR. In addition to suppressing HSP70 expression, quercetin can inhibit phosphoinositide 3 kinase (PI3K) signalling. To investigate whether inhibition of PI3K is involved in the induction of UPR by quercetin, we treated T47D cells with quercetin or two PI3K inhibitors, LY294002 and wortmanin, and detected the expression of representative UPR markers, including GRP78, CHOP and spliced XBP-1. Treatment of cells with LY294002 and wortmanin did not stimulate GRP78 and CHOP expression and XBP-1 splicing (Fig. 2E), indicating that quercetin does not induce the UPR by suppressing PI3K/Akt.
HSP70 down-regulation by siRNA or quercetin induces JNK and caspase activation
To investigate whether HSP70 down-regulation by siRNA or quercetin activates pro-apoptotic components of the UPR, the effects of HSP70 down-regulation on the activation of JNK, caspase-4, caspase-7 and caspase-3 were examined. The suppression of HSP70 expression with the use of either siRNA or quercetin resulted in an increase in JNK phosphorylation. In addition, we observed caspase-7, caspase-3, caspase-4 and PARP cleavage in quercetin-treated T47D cells (Fig. 3A). Similar effects were observed in siHsp70-transfected cells (Fig. 3B).
Figure 3.
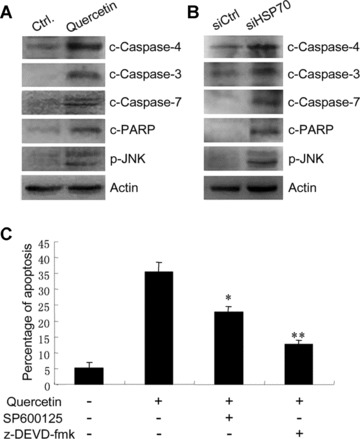
Effects of quercetin and HSP70 knockdown on pro-apoptotic components of the UPR. (A) T47D cells were treated with or without 100 μM quercetin for 24 hrs. Cell lysates were subjected to SDS-PAGE, and then blotted with anti-cleaved caspase-3, -4, -7 and PARP antibodies, and anti-phospho-JNK antibody. (B) T47D cells were transfected with control siRNA (siCtrl) or the siRNA targeting Hsp70 (siHsp70). Cell lysates were subjected to SDS-PAGE, and then blotted with anti-cleaved caspase-3, -4, -7 antibodies and anti-phospho-JNK antibody. (C) T47D cells were treated with or without 100 μM quercetin, 10 μM SP600125 and 50 μM z-DEVD-fmk for 24 hrs. Cells were harvested, fixed with ethanol, stained with propidium iodide and subjected to flow cytometry analysis. Columns, mean of three individual experiments; bars, SE.
Caspase-3 and caspase-7 are two key executor caspases that mediate ER stress-induced apoptosis. To determine whether the activation of JNK and caspases contribute to quercetin-induced apoptosis, the effect of JNK inhibitor, caspase-3 and caspase-7 inhibitor on quercetin-induced apoptosis was examined by flow cytometry. Treatment of T47D cells with JNK inhibitor SP600125 partially blocked quercetin-induced apoptosis. More importantly, treatment of the cells with caspase-3 and caspase-7 inhibitor z-DEVD-fmk inhibited quercetin-induced apoptosis to more dramatic extent (Fig. 3C). These results imply that the activation of pro-apoptotic arms of the UPR may contribute to quercetin-induced apoptosis.
Abrogation of GRP78 induction sensitizes breast cancer cells to quercetin
Since GRP78 represents a pro-survival arm of the UPR, the induction of GRP78 by quercetin may contribute to increased resistance to quercetin-induced tumour cells death. To test whether breast cancer cells depend on GRP78 for protection against quercetin-mediated cell death, breast cancer cells were transfected with siRNA against GRP78 (siGRP78) or the negative control siRNA (siCtrl), followed by treatment with or without quercetin. Hoechst 33342 staining was done to evaluate the drug sensitivity of the siGRP78-transfected cells. The results demonstrated that abrogation of GRP78 induction caused a significant increase in quercetin-induced apoptosis in both T47D and MCF-7 cell lines (Fig. 4A and B). An in parallel Western blot analysis of protein lysates from untreated cells transfected with siCtrl and siGRP78 confirmed that GRP78 expression was significantly inhibited by siGRP78 (Fig. 4B). Moreover, colony survival assays were done to evaluate the quercetin sensitivity in siRNA-transfected MCF-7 cells. The results showed that knockdown of GRP78 caused a statistically significant decrease in colony survival in quercetin-treated cells, while it did not cause significant decrease in colony survival in DMSO-treated cells (Fig. 4C). These data indicate that suppression of GRP78 expression may lead to increased sensitivity of breast cancer cells to quercetin.
Figure 4.
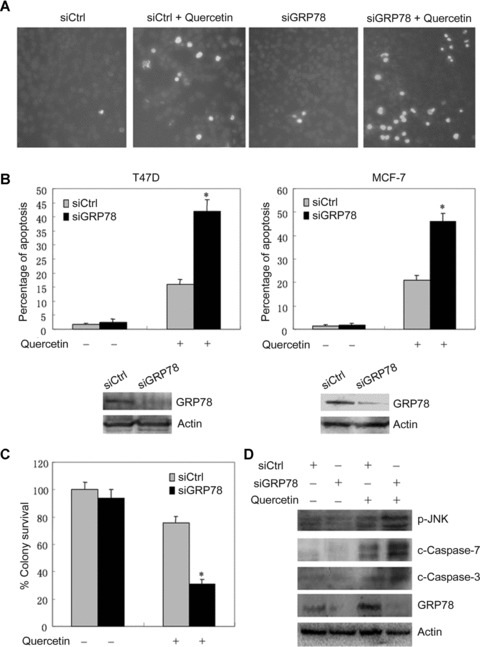
Effects of GRP78 knockdown on quercetin-induced cell death and the activation of caspase-3, -7 and JNK. (A) T47D cells were transfected with siCtrl or siGRP78. Twenty-four hours later, the cells were treated with or without 50 μM quercetin for another 24 hrs and stained with Hoechst 33342. The apoptotic cells with strong fluorescence were observed under fluorescent microscopy. (B) The apoptotic cells in four randomized fields were counted. The percentage of apoptotic T47D cells was plotted. In addition, the effects of GRP78 knockdown on quercetin-treated MCF-7 cells death were shown. The columns represent the mean of triplicate wells, and the bars represent the SE. *, P < 0.001. In parallel, untreated T47D and MCF-7 cells transfected with siGRP78 and siCtrl were harvested and the protein lysates were analysed by Western blot with antibodies against GRP78 and actin. A representative of three experiments is shown. (C) 5000 MCF-7 cells per well were seeded in six-well plates. Twenty-four hours later, the cells were transfected with siCtrl or siGRP78, followed by treatment with 30 μM quercetin for 6 hrs. The cells were allowed to form colonies for 14 days. The columns represent the mean of triplicate tests, and the bars represent the SE. *, P < 0.001. (D) T47D cells were transfected with siCtrl or siGRP78. Twenty-four hours later, the cells were treated with or without 50 μM quercetin for another 24 hrs. Cells were harvested and the protein lysates were subjected to SDS-PAGE, and then blotted with anti-cleaved caspase-3, -7, anti-phospho-JNK and anti-GRP78 antibodies.
In addition, we examined the effect of GRP78 knockdown on quercetin-induced activation of caspase-3, caspase-7 and JNK. Whereas GRP78 knockdown did not induce the activation of caspase-3, caspase-7 and JNK in non-treated cells, it caused a significant increase in quercetin-induced activation of caspase-3, caspase-7 and JNK (Fig. 4D). These data imply that suppression of the pro-survival molecule GRP78 may lead to an increase in the activation of pro-apoptosis molecules by quercetin.
Synergistic promotion of breast cancer cells death by EGCG and quercetin
The above-mentioned results imply that down-regulation of GRP78 by compounds directed against GRP78 expression or activity could lead to increased sensitivity of breast cancer cells to quercetin. Previous studies have identified the major component of green tea, EGCG, as a natural compound that directly interacts with the ATP-binding domain of GRP78, blocks its interaction with procaspase-7 and suppresses the protective function of GRP78 [29]. To determine whether the combination of EGCG and quercetin, two natural compounds, can synergistically promote breast cancer cells apoptosis, the effect of EGCG, quercetin alone and combination on apoptosis was examined by Hoechst 33342 staining in MCF-7 and T47D cells. Whereas EGCG alone did not induce cell death, the combination of EGCG and quercetin resulted in significantly more cell death than quercetin alone in both cell lines (Fig. 5). Similar results were obtained when we used WST1 assays to measure the cytotoxity induced by treatment with EGCG, quercetin or both (Fig. 6).
Figure 5.
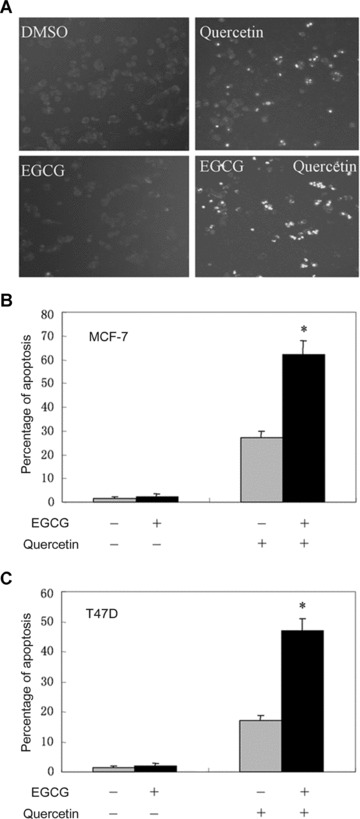
Treatment with EGCG sensitizes breast cancer cells to quercetin. (A) MCF-7 cells were treated with or without 10 μM EGCG and 50 μM quercetin for 24 hrs and stained with Hoechst 33342. The apoptotic cells with strong fluorescence were observed under fluorescent microscopy. (B) The apoptotic cells in four randomized fields were counted. The percentage of apoptotic MCF-7 cells was plotted. The columns represent the mean of triplicate wells, and the bars represent the SE. (C) T47D cells were treated with or without 10 μM EGCG and 50 μM quercetin for 24 hrs and stained with Hoechst 33342. The percentage of apoptotic cells was plotted. Columns, mean of triplicate wells. Bars, SE. *, P < 0.001. A representative of three experiments is shown.
Figure 6.
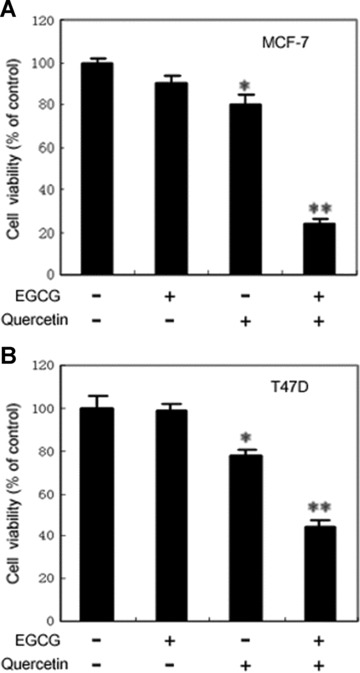
Cytotoxity assay of breast cancer cells treated with EGCG, quercetin, or both. (A) MCF-7 cells were plated in 96-well plates at 5000 cells per well. The next day, the cells were treated with 10 μM EGCG, 50 μM quercetin or both. Control cells were treated with the vehicle, DMSO. Twenty-four hours later, the cytotoxity was assessed by incubating cells with WST1 reagent for 2 hrs and measuring the absorbance at 450 nm, and at 630 nm as reference, with a microplate reader. These experiments were carried out with five to six replicates. Columns, mean expressed as the percentage of absorbance relative to values from vehicle-treated controls. Bars, SE. Co-treatment with EGCG and quercetin resulted in a drastic reduction in the number of viable cells, compared with treatment with EGCG or quercetin alone for which minimal effect was observed. *, P < 0.05; **, P < 0.001, compared with control. (B) T47D cells were plated in 96-well plates at 6000 cells per well. The next day, the cells were treated with 10 μM EGCG, 50 μM quercetin or both. Control cells were treated with the vehicle, DMSO. After 24 hrs, the cytotoxity was assessed by WST1 assays. Columns, mean expressed as the percentage of absorbance relative to values from vehicle-treated controls. Bars, SE. *, P < 0.05; **, P < 0.001, compared with control.
In addition, we used colony formation assay to test the benefit of concomitant administration of EGCG and quercetin in inhibiting breast cancer cells survival in a relatively longer term. After transfection with siRNA, the cells were treated with or without 10 or 30 μmol/l quercetin. After a change of fresh medium 24 hrs later, the cells were allowed to form colonies for 14 days in the absence of drug. EGCG at the tested concentration did not affect the colony survival of MCF-7 cells by itself. However, the combination of EGCG and quercetin resulted in significantly less colonies than quercetin alone (Fig. 7). Whereas quercetin at 10 μmol/l concentration did not cause cell death by itself, the combination of 10 μmol/l quercetin and 10 μmol/l EGCG led to significant less colonies. The combination of 10 μmol/l EGCG and 30 μmol/l quercetin achieved much stronger inhibition in colony survival. No colonies survived after MCF-7 cells were treated with 20 μmol/l EGCG and 30 μmol/l quercetin simultaneously (data not shown). These results indicate that combination of drugs capable of GRP78 suppression could sensitize breast cancer cells to quercetin.
Figure 7.
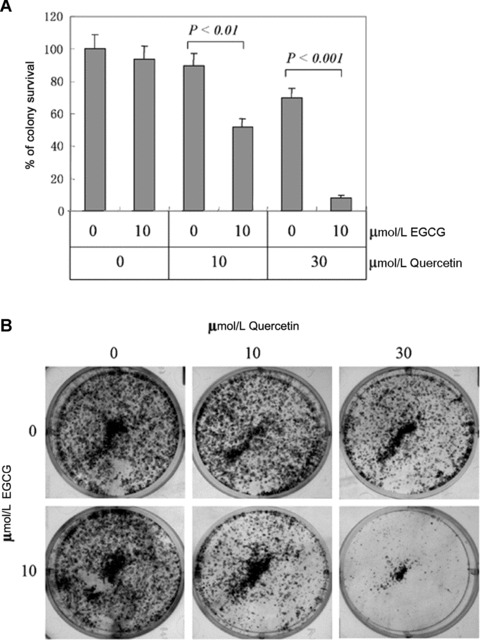
Effects of EGCG and quercetin on colony survival. (A) 5000 MCF-7 cells per well were seeded in six-well plates. Twenty-four hours later, the cells were treated with quercetin in the absence or presence of indicated concentration of EGCG for 24 hrs. The cells were allowed to form colonies for 14 days. The percentage of colony survival was plotted against the quercetin and EGCG dosage. Columns, mean value of triplicate tests; bars, SE. (B) Pictures of the colonies from all treatment groups. A representative of two independent experiments is shown.
Discussion
HSP70 belongs to a family of proteins that play diverse roles in many different cellular processes, including protein folding, trafficking and the assembly/disassembly of multi-proteins complexes [4]. Previous studies have established a role for HSP70 in potentiating signal transduction and in protecting cells from stresses-induced death [15, 33]. HSP70 up-regulation is observed in a variety of aggressive tumours. The connection between HSP70 and the oncogenic potential of tumour cells suggests that HSP70 may be a potential molecular target for cancer therapy or chemoprevention [25]. The bioflavonoid quercetin could suppress HSP70 expression. Quercetin displays anti-tumour activities in several types of cancer [28, 34], but the exact mechanisms underlying these activities remains incompletely understood. This study has shown that HSP70 down-regulation with both quercetin and siRNA resulted in the activation of UPR, with both the pro-survival arm and the pro-apoptotic arm being involved.
The UPR is generally triggered by disruption in ER homeostasis. ER resides in the secretory pathway and is responsible for transferring secreted and transmembrane proteins to the golgi or other cellular compartments. Also, a primary function of the ER is to assist newly synthesized proteins refold into native conformation. To achieve correct folding and maturation, secreted proteins must translocate into the ER to undergo several post-translational modifications, including glycosylation and disulfide binding [35]. The quality of proteins in the ER is tightly controlled by resident ER chaperone and folding enzymes [36, 37]. Proteins that do not mature properly are retrotranslocated to the cytosol for degradation by the 26S proteasome [38]. HSP70 and HSP90 are cytosolic chaperones that associate with denatured or partially unfolded proteins to prevent them from further denaturation. In this study, we have shown that depletion of HSP70 in breast cancer cells induces the UPR. These results are consistent with the recent observations that inhibition of HSP90 leads to the UPR in multiple myeloma [39]. Thus, both HSP70 and HSP90 are essential for ER homeostasis.
Previous studies have demonstrated that depletion of HSP70 could activate a tumour-specific death program. Many and often controversial mechanisms underlying the antiapoptotic effect of HSP70 were proposed. It has been reported that the recruitment of procaspase-9 to the Apaf-1 apoptosome can be inhibited by HSP70 [40]. However, another report indicated that HSP70 may inhibit apoptosis upstream of mitochondria and not through interaction with Apaf-1 [41]. In addition, it has been shown that HSP70 may function upstream of the caspase cascade by inhibiting the release of cytochrome c from the mitochondria [42]. Therefore, it seems that the mechanisms underlying the antiapoptotic effect of HSP70 are cell-type specific or context-dependent. In this study, we have shown that HSP70 down-regulation resulted in the activation of several key pro-apoptotic molecules in the UPR pathways, suggesting that HSP70 may be required to maintain ER homeostasis in cancer cells. However, HSP70 down-regulation also results in the up-regulation of molecular chaperone GRP78, which represents a pro-survival arm of the UPR.
Based on the above-mentioned findings, abrogation of GRP78 induction may be a strategy to sensitize cancer cells to bioflavonoid quercetin. Indeed, this study clearly demonstrates that down-regulation of GRP78 by siRNA synergistically promotes quercetin-induced breast cancer cells death. Although down-regulation of HSP70 leads to up-regulation of GRP78, down-regulation of GRP78 does not result in up-regulation of HSP70 (data not shown). Further, we demonstrate that the abrogation of GRP78 induction results in increased activation of caspase-3, caspase-7 and JNK by quercetin. Hence when the pro-survival GRP78 is compromised, the balance between pro-survival arms and pro-apoptosis branches will shift in favour of cell death. Thus, the synergistic promotion of cancer cells death can be achieved by combining quercetin with GRP78 knockdown. Although depletion of GRP78 alone could not induce cancer cells apoptosis, it may be an important adjunct in chemotherapy or biotherapy. Thus, the development of small molecule inhibitors that specifically suppress GRP78 induction or its activity is justified. Recently, EGCG, a major component of green tea, has been identified as a natural GRP78 inhibitor [29]. Treatment with EGCG sensitizes malignant glioma cells to temozolomide [29]. In this study, we showed that treatment with EGCG had similar effects as GRP78 knockdown. Breast cancer cells treated with the combination of EGCG and quercetin exhibited much less survival than cells treated with quercetin alone. Thus, in our study, we present an approach for increasing breast cancer cell sensitivity to quercetin.
Quercetin can exert antagonistic effects on oestrogen receptor function and thus act as a pure anti-oestrogen [43]. GRP78, on the other hand, can protect breast cancer cells against oestrogen starvation-induced apoptosis by inhibiting endoplasmic reticulum BIK [44]. In view of these findings, combination of quercetin with GRP78 inhibitor such as EGCG might become an important adjunct in hormonal treatment of breast cancer. In addition, the combined use of quercetin and EGCG may increase the effectiveness of chemoprevention. It is well recognized that resistance to apoptosis is a major cause of poor responsiveness to cancer therapy. An exciting outcome from our study is the observation that sub-optimal doses of both quercetin and EGCG can synergize to induce cell death and increase the effectiveness of treatment. Moreover, co-administration with low doses may be advantageous since quercetin has poor solubility and high concentration found to be active in vitro may often not be achieved in vivo[45]. Altogether, our studies indicate that combinational administration of compounds that inhibit GRP78 and HSP70 may present a novel approach to the prevention and treatment of cancer.
Acknowledgments
This work was supported in part by grant 30672360 from the National Natural Science Foundation of China and China Medical Board.
References
- 1.Sherman M, Multhoff G. Heat shock proteins in cancer. Ann N Y Acad Sci. 2007;1113:192–201. doi: 10.1196/annals.1391.030. [DOI] [PubMed] [Google Scholar]
- 2.Morimoto RI, Milarski KL. Expression and function of vertebrate Hsp70 genes. In: Morimoto RI, Tissieres A, Georgopoulos C, editors. Stress proteins in biology and medicine. New York: Cold Spring Harbor Laboratory; 1990. pp. 323–59. [Google Scholar]
- 3.Young RA, Elliot TJ. Stress proteins, infection, and immune surveillance. Cell. 1989;59:5–8. doi: 10.1016/0092-8674(89)90861-1. [DOI] [PubMed] [Google Scholar]
- 4.Ang D, Liberek K, Skowyra D, et al. Biological role and regulation of the universally conserved heat shock proteins. J Biol Chem. 1991;266:24233–6. [PubMed] [Google Scholar]
- 5.Gething M-J, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 6.Rothman JE. Polypeptide chain binding proteins: catalysts of protein folding and related processes in cells. Cell. 1989;59:591–601. doi: 10.1016/0092-8674(89)90005-6. [DOI] [PubMed] [Google Scholar]
- 7.Jakob U, Gaestel M, Engel K, et al. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268:1517–20. [PubMed] [Google Scholar]
- 8.Tavaria M, Gabriele T, Kola I, et al. A hitchhiker’s guide to the human Hsp70 family. Cell Stress Chaperones. 1996;1:23–8. doi: 10.1379/1466-1268(1996)001<0023:ahsgtt>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen B, Piel WH, Gui L, et al. The Hsp90 family of genes in the human genome: insights into their divergence and evolution. Genomics. 2005;86:627–37. doi: 10.1016/j.ygeno.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 10.Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther. 2004;3:1021–30. [PubMed] [Google Scholar]
- 11.Ralhan R, Kaur J. Differential expression of Mr 70, 000 heat shock protein in normal, premalignant, and malignant human uterine cervix. Clin Cancer Res. 1995;1:1217–22. [PubMed] [Google Scholar]
- 12.Santarosa M, Favaro D, Quaia M, et al. Expression of heat shock protein 72 in renal cell carcinoma: possible role and prognostic implications in cancer patients. Euro J Cancer. 1997;33:873–7. doi: 10.1016/s0959-8049(97)00002-6. [DOI] [PubMed] [Google Scholar]
- 13.Jaattela M. Escaping cell death: survival proteins in cancer. Exp Cell Res. 1999;248:30–43. doi: 10.1006/excr.1999.4455. [DOI] [PubMed] [Google Scholar]
- 14.Hannun Y. Apoptosis and the dilemma of cancer chemotherapy. Blood. 1997;89:1845–53. [PubMed] [Google Scholar]
- 15.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocrine Rev. 1997;18:306–60. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 16.Smith DF, Whitesell L, Nair SC, et al. Progesterone receptor structure and function altered by geldanamycin, an hsp90-binding agent. Mol Cell Biol. 1995;15:6804–12. doi: 10.1128/mcb.15.12.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blagosklonny MV. Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002;16:455–62. doi: 10.1038/sj.leu.2402415. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y, Lindquist S. Heat-shock protein Hsp90 governs the activity of pp60 (v-src) kinase. Proc Natl Acad Sci USA. 1993;90:7074–8. doi: 10.1073/pnas.90.15.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munro S, Pelham HRB. An Hsp70-like protein in the ER: identity with the 78 kd glucose regulated protein and immunoglobulin heavy chain binding proteins. Cell. 1986;46:291–300. doi: 10.1016/0092-8674(86)90746-4. [DOI] [PubMed] [Google Scholar]
- 20.Lee AS. Coordinated regulation of a set of genes by glucose and calcium ionophores in mammalian cells. Trends Biochem. 1987;12:20–3. [Google Scholar]
- 21.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–53. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez1 PM, Tabbara SO, Jacobs LK, et al. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat. 2000;59:15–26. doi: 10.1023/a:1006332011207. [DOI] [PubMed] [Google Scholar]
- 23.Pyrko P, Schönthal AH, Hofman FM, et al. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–16. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 24.Dong D, Ni M, Li J, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 25.Murakami A, Ashida H, Terao J. Multitargeted cancer prevention by quercetin. Cancer Lett. 2008;269:315–25. doi: 10.1016/j.canlet.2008.03.046. [DOI] [PubMed] [Google Scholar]
- 26.Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–30. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 27.Hostein I, Robertson D, DiStefano F, et al. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61:4003–9. [PubMed] [Google Scholar]
- 28.Wei YQ, Zhao X, Kariya Y, et al. Induction of apoptosis by quercetin: involvement of heat shock protein. Cancer Res. 1994;54:4952–7. [PubMed] [Google Scholar]
- 29.Ermakova SP, Kang BS, Choi BY, et al. (−)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006;66:9260–9. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
- 30.Reddy RK, Mao C, Baumeister P, et al. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 31.Lee E, Nichols P, Spicer D, et al. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006;66:7849–53. doi: 10.1158/0008-5472.CAN-06-1660. [DOI] [PubMed] [Google Scholar]
- 32.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garrido C, Brunet M, Didelot C, et al. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- 34.Aghdassi A, Phillips P, Dudeja V, et al. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007;67:616–25. doi: 10.1158/0008-5472.CAN-06-1567. [DOI] [PubMed] [Google Scholar]
- 35.Elligaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science. 1999;286:1882–8. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- 36.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–9. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 37.Brodsky JL, Werner ED, Dubas ME, et al. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem. 1999;274:3453–60. doi: 10.1074/jbc.274.6.3453. [DOI] [PubMed] [Google Scholar]
- 38.Jarosch E, Taxis C, Volkwein C, et al. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–9. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- 39.Davenport EL, Moore HE, Dunlop AS, et al. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110:2641–9. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]
- 40.Beere HM, Wolf BB, Cain K, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 41.Steel R, Doherty JP, Buzzard K, et al. Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1. J Biol Chem. 2004;279:51490–9. doi: 10.1074/jbc.M401314200. [DOI] [PubMed] [Google Scholar]
- 42.Klein SD, Bröne B. Heat-shock protein 70 attenuates nitric oxide-induced apoptosis in RAW macrophages by preventing cytochrome c release. Biochem J. 2002;362:635–41. doi: 10.1042/0264-6021:3620635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miodini P, Fioravanti L, Di Fronzo G, et al. The two phyto-oestrogens genistein and quercetin exert different effects on oestrogen receptor function. Br J Cancer. 1999;80:1150–5. doi: 10.1038/sj.bjc.6690479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–40. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 45.Scholz S, Williamson G. Interactions affecting the bioavailability of dietary polyphenols in vivo. Int J Vitam Nutr Res. 2007;77:224–35. doi: 10.1024/0300-9831.77.3.224. [DOI] [PubMed] [Google Scholar]