

Abstract
Transmigration of neutrophils through the microvascular endothelium is a cardinal event of acute inflammation. It has been suggested that gap junctions made of connexin43 (Cx43) may serve as a conducting pathway to spread inflammatory signals within the lung capillary network. To determine whether Cx43 contributes to neutrophil transmigration in vivo, the number of transmigrated neutrophils was monitored in lungs of Cx43 mouse models subjected to inflammation by intratracheal instillations of Pseudomonas aeruginosa lipopolysaccharide (LPS). Cx43 was detected in inflamed lungs independently of neutrophil recruitment, whereas Cx43 up-regulation was not detected in mice genetically protected from inflammation. Mice heterozygous for the Cx43 gene (gja1) showed a 56% (P < 0.01) reduction in airway neutrophil count. In contrast, increased (P < 0.05) neutrophil recruitment in response to LPS was observed in a mouse model expressing a mutant Cx43 with enhanced channel conductivity. In vitro adhesion assays showed that reduced conductivity of Cx43 channels with 43Gap26, a Cx43 blocking peptide, decreased adhesion of neutrophils to endothelial cells. Finally, we found that instillation of 43Gap26 in inflamed lungs reduced neutrophil transmigration by 65% (P < 0.05). These results indicate that inflammatory mediators up-regulate alveolar Cx43 to promote neutrophil recruitment to the airspace. Cx43 may therefore represent a pharmacological target in lung diseases characterized by excessive neutrophil recruitment to the airways.
Keywords: connexin, blocking peptide, lung inflammation, neutrophil recruitment, mouse models
Introduction
Acute lung injury (ALI) is attributable to a severe inflammation that is most commonly caused by sepsis, infection, trauma or acid aspiration. Despite optimal management, ALI remains a clinical syndrome related to a high mortality in both children and adults [1, 2]. Hallmarks of ALI are massive invasion of leukocytes and protein-rich oedema in the interstitial and intra-alveolar spaces leading to impaired gas exchange and increased pulmonary resistance [1]. ALI develops rapidly across the lung vascular surface involving an entire lung or even both lungs [1], but the mechanism underlying the spatial expansion of inflammation is unexplained.
The endothelium plays a pivotal role in the orchestration of lung inflammation and recruitment of leukocytes into the airway lumen. Under normal and pathological circumstances, circulating leukocytes migrate from vessels into tissues by a multistep process involving the sequential activation of adhesion proteins and their ligands on both leukocytes and endothelial cells [3]. The pulmonary circulation is unique in comparison to the systemic circulation in that there are large numbers of marginating leukocytes and migration predominantly occurs from the alveolar capillary network [4]. The role of intercellular communication in the lung capillary bed has recently received increasing attention. Thus, Ca2+-dependent endothelial exocytosis of P-selectin initiates inflammation by establishing leukocyte rolling on the vascular surface [5]. Ca2+-signalling between endothelial cells may contribute to the spatial expansion of the pro-inflammatory response. Applying real-time digital imaging techniques and photo-excited Ca2+ uncaging, Parthasarathi and collaborators observed the propagation of Ca2+ waves between alveolar endothelial cells [6]. This propagation was abolished in mice with endothelial-specific deletion of the gap junction gja1 gene (coding for connexin43 [Cx43]) as compared to wild-type animals [6]. In addition, in vitro studies on primary cultures of porcine microvascular endothelial cells and rat endothelial cell lines involved Cx43 in maintenance of the endothelial barrier function [7]. It was thus proposed that gap junctions may serve as conduits for the spread of pro-inflammatory signals in the lung capillary bed [6]. Whether Cx43 modulates neutrophil transmigration to the airspace during lung inflammation, however, has not been investigated.
To specifically address this question, we evoked lung inflammation by intratracheal (IT) instillation of Pseudomonas aerugi-nosa lipopolysaccharide (LPS) in control mice and in mice where the expression level and/or functional status of Cx43 were altered. We report findings demonstrating the pivotal role of Cx43 in mediating the recruitment of neutrophils. In addition, we show that targeting Cx43 with a blocking peptide reduced neutrophil adhesion to the endothelium in vitro and decreased the number of neutrophils that transmigrated to the alveolar space by 65%, indicating that Cx43 may represent a valuable target to hamper lung inflammation.
Methods
Mice
Mice heterozygous (Cx43+/− mice) for the Cx43 gene (gja1) were previously described [8]. Both wild-type mice and Cx43+/− mice (B6;129S-Gja1m1Kdr/J) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Mice in which the C-terminal region of Cx43 was truncated at K258 (Cx43K258/− mice) were crossed as previously reported [9]. Of note, mice homozygous for the null or truncated allele of Cx43 are lethal [8, 9]. Transgenic mice expressing a human soluble tumour necrosis factor receptor (TNFR)1-IgG3 fusion protein (sTNFR1) and TNF-α knockout mice were also used [10, 11]. All mice were backcrossed >6 times onto a C57BL/6 genetic background. Lung inflammation in these mouse models was performed in series of experiments using littermates of similar age and weight. Cx43+/+ littermates (control mice) responded to IT instillation of LPS similarly than C57BL/6 wild-type mice. Wild-type mice were used in some experiments where indicated in the text. Mice were kept under standard housing conditions with a fixed light/dark cycle.
Lung inflammation
Mice were anaesthetized with intraperitoneal injection of a ketamine/xylazine solution (8 μl per g). Lung inflammation was induced by a first IT instillation (IT1) of 0.1 μg Pseudomonas aeruginosa LPS (Sigma Co., Saint Louis, MO, USA). Additional (IT2 and IT3) instillations were sometimes performed 120 hrs after each previous IT treatment. Low concentration of LPS was chosen to prevent massive and saturating recruitment of neutrophils to the airspace. Mice were killed before (0 hr) and 6, 24, 48 and 120 hrs after each instillation and subjected to bronchoalveolar lavages (BALs). The cell suspension was then used for neutrophil identification and count. After collecting BALs, lungs were frozen in liquid nitrogen and kept at –80°C for later use. To confirm neutrophil infiltration in Cx43 mouse models subjected to LPS-induced lung inflammation, the activity of myeloperoxydease (MPO), an enzyme specific for neutrophils, was measured in lungs and BALs. As previously described [14], the method was based on the oxydation of tetramethylbenzidine by H2O2 in the presence of MPO. MPO activity was measured after a 5-min. reaction and expressed as U/g of total lung protein.
For some experiments, lungs from wild-type mice were instilled with 25 μg of the mimetic Cx43 peptide 43Gap26 (VCYDKSFPISHVR) or with a control peptide with a non-relevant amino acid sequence (VCYDHYFPISHIR). In a first protocol, 43Gap26 was administrated 12 hrs after IT LPS instillation and the number of neutrophils present in BALs was scored 12 hrs later. In a second protocol, 43Gap26 was administrated at peak of lung inflammation, 24 hrs after LPS treatment, and the number of neutrophils was collected from BALs 24 hrs later. Peptides were either purchased from Alpha Diagnostic (San Antonio, TX, USA) or synthesized at the core facility of the Medical Faculty, Geneva, Switzerland. For neutrophil depletion experiments, mice were subcutaneously injected with 150 μg of an anti-GR1 antibody (AbD Serotec, Düsseldorf, Germany) 24 hrs before LPS instillation. All experiments were carried out according to the Swiss law for protection of animals and with prior approval by local government authorities.
Mouse cytokine antibody array
BALs from Cx43+/+ and Cx43+/− mice were centrifuged and supernatants were applied to a mouse cytokine antibody array (62 cytokines; RayBiotech Inc., Norcross, GA, USA). The array membranes were processed according to the manufacturer’s instructions. Briefly, membranes were exposed to individual or pooled supernatants at room temperature for 2 hrs. Finally, the results with immunoreactivity were assessed and quantified by using a GeneTool Imaging System (Syngene Inc., San Diego, CA, USA) and graphed.
Haematoxylin and eosin staining of lung sections
Lungs were rapidly removed, fixed in 4% formol and embedded in paraffin. They were 5 mm sectioned, haematoxylin and eosin stained and examined by light microscopy.
Western blots
Western blotting of proteins extracted from lungs was performed as previously described [12]. The antibody against Cx43 (dil 1:500) was generated using a 23 amino acid sequence within the cytoplasmic C-term domain of mouse Cx43 (B12-A, Alpha Diagnostic). For this connexin [14], up to three isoforms of 41, 43 and 46 kD (corresponding to the non-phosphorylated, NP or P0, and phosphorylated forms, P1 and P2), are detected. Membranes were stained with Ponceau red then stripped and incubated with β-actin to attest for equal protein loading. Neutrophils of human and mouse origin were also subjected to Western blots for Cx43 detection. Human neutrophils were isolated from buffy coat of healthy volunteers and mouse neutrophils from blood of wild-type mice by density-gradient ultracentrifugation in Ficoll-Plaque, according to standard procedures. The polyclonal antibody (B12-A, Alpha Diagnostic) was used for mouse neutrophils while a monoclonal (dil 1:250) antibody (Transduction Laboratories, Lexington, KY, USA) was used for human neutrophils.
Immunofluorescence
Mouse lungs were instilled and embedded with Tissue-Tek O.C.T. (Hatfield, PA, USA), then snap-frozen in liquid nitrogen. Immunofluorescence of Cx43 was performed on methanol fixed sections, as previously described [12]. Negative controls include staining with second antibody alone or pre-absorption of the first antibody with its corresponding immunogenic peptide (B12-P, Alpha Diagnostic). Sections were treated with DAPI and counter-stained with Evans Blue. Images were taken with LSM 510 META confocal microscope (Zeiss, Thüringen, Germany) and processed using Adobe Photoshop (version 9.0, Adobe Systems Incorporated, San Jose, CA, USA).
Dye coupling and adhesion assays
Dye coupling was performed as previously studied [12] using Lucifer Yellow or neurobiotin as gap junction-permeant tracers on mouse endothelial bEnd.3, type I-alveolar epithelial murine pneumocyte (MLE12) and N2A cell lines stably expressing or not a truncated Cx43 at amino acid 257 [19]. Prior to dye coupling, bEnd.3 and MLE12 cells were treated twice, every 24 hrs, with 0.25 μg/μl of 43Gap26 or control peptide. These same experimental conditions were used for neutrophil adhesion assays onto bEnd.3 and MLE12 cell monolayers. For some experiments, neutrophils were treated with 43Gap26 during their adhesion on untreated cell monolayer. Adhesion was allowed for 3 hrs then cells were rinsed in DPBS and fixed with 4% paraformaldehyde. The number of adherent neutrophils/field was counted with an inverted TMD300 microscope (Nikon AG, Kusnacht, Switzerland) equipped for fluorescence detection using a 20× objective.
Statistics
All values were expressed as mean ± S.E.M., and compared by ANOVA, unless otherwise notified in the text. P < 0.05 was considered significant.
Results
Cx43 contributes to the lung inflammatory response evoked by LPS instillation
The level of Cx43 expression in lungs from Cx43+/+ and Cx43+/− mice was first evaluated by Western blot (Fig. 1A). Using an antibody against its C-terminus, Cx43 exhibited on a 10% SDS-PAGE gel several bands, which is typical for this protein [13]. As described [14], the 43- and 46-kD isoforms, were the most prominently expressed forms in mouse lungs. The specificity of the Cx43 antibody was verified on lung extracts from Cx43K258stop/− mice. As expected, the antibody no longer recognized Cx43 isoforms in the absence of its immunogenic C-terminus motif (Fig. 1A). Western blots on proteins from lungs of Cx43+/− mice showed 61% reduction in Cx43 expression as compared to Cx43+/+ animals (Fig. 1B).
Figure 1.

Cx43 expression in Cx43+/+ and Cx43+/− lungs. (A) Cx43 expression was detected by Western blot in Cx43+/+ and Cx43+/− lung homogenates. Using an antibody against the C-terminus of Cx43, three isoforms of 46, 43 and 41 kD were detected; Cx43+/− lungs express 61% less Cx43. The specificity of the Cx43 antibody was verified on lung extracts from Cx43K258stop/− mice. As expected, the antibody no longer recognized the three forms of Cx43 in the absence of its immunogenic C-terminus motif. The 46, 43 and 41 kD forms of Cx43 are indicated at the right of the gel. (B) The Cx43/β-actin signals were quantified from five lungs per condition. (C) Cx43 was detected by Western blot using a polyclonal and a monoclonal antibody in control cells (Cont) from mouse and human origins, respectively. However, no Cx43 expression was seen in murine and human neutrophils. Note that blots were combined from separate gels. **Indicates P < 0.01. D) Cell-to-cell diffusion of neurobiotin was demonstrated in Cx43K258stop-expressing cells but not in the parental N2A cells. Phase-contrast images of the corresponding fields are also shown. Scale bar represents 40 μm.
We next evoked lung inflammation in Cx43+/+ and Cx43+/− mice by IT instillation of low concentration of Pseudomonas aeruginosa LPS. LPS was instilled once (IT1) to induce a first inflammatory response. Figure 2A shows the median number of neutrophils detected in BALs at various time-points after IT instillation of LPS. In the absence of LPS, neutrophils were not observed in BALs. In contrast, the number of neutrophils recruited to the airways increased with time after LPS treatment to peak at 24 hrs. Inflammation resolved within 120 hrs. Although an inflammatory response was evoked in both Cx43+/+ and Cx43+/− mice 24 hrs after LPS instillation, we found that the number of neutrophils recruited to the airway at this time course was markedly lower in Cx43+/− mice (Fig. 2A). After resolution of the inflammatory response (120 hrs after LPS treatment), LPS was administrated a second and a third time to different groups of mice. Figure 2B shows the quantitative analysis for mean neutrophil counts 24 hrs after each IT administration of LPS. Although there was some variability in the average neutrophil number recruited in Cx43+/+ lungs for the three LPS treatments, the amount of neutrophils collected in BALs was systematically lower in Cx43+/− mice. These results are indicative for a correlation between Cx43 expression and the extent of neutrophil recruitment in lungs. On average, neutrophil recruitment was reduced by about 56% in Cx43+/− mice. As shown in Table 1, no change in the number of lymphocytes and macrophages was detected in either Cx43+/+ or Cx43+/− mice 24 hrs after each LPS administration. Morphological changes of lung injury were also evaluated by light microscopy on haematoxylin and eosin stained sections of lung tissues. Cx43+/+ lungs subjected to LPS were characterized by a marked thickening of the alveolar membrane and presence of granulocytes during inflammation. These parameters were less prominent in Cx43+/− mice (Fig. 3A). We have also used the MPO assay to monitor neutrophil infiltration and accumulation in the airspace of LPS instilled mice. As shown in Fig. 3B, MPO activity was reduced (P < 0.05) in BALs of Cx43+/− mice whereas no apparent changes were detected in whole lungs. Finally, the presence of up to 62 pro- and anti-inflammatory cytokines was compared in BALs of Cx43+/+ and Cx43+/− mice 24 hrs after LPS treatment by using an antibody array. We observed decreased levels of several cytokines in BALs, including interleukin (IL)-12p40/p70, IL-6, KC, MIP-2, TIMP1, L-selectin and V-CAM1, from Cx43+/− mice (Fig. 4). These results indicate that decreased expression of Cx43 in Cx43+/− mice limits the transmigration of neutrophils to the airways.
Figure 2.

Lung inflammation is reduced in Cx43+/− mice 24 hrs after IT LPS instillation. (A) Median number of recruited neutrophils (PMN) to the bronchoalveolar space at various time-points after IT LPS instillation in Cx43+/+ and Cx43+/− mice. This number is lower in Cx43+/− mice as compared to Cx43+/+ mice. (B) Average number of neutrophils collected in BALs 24 hrs after IT instillations of LPS. The recruitment of neutrophils was reduced by 64, 49 and 54% in Cx43+/− mice after the first, the second and the third LPS instillation, respectively. Values were calculated from 7–10 mice per condition. *Indicates P < 0.05.
Table 1.
The number of lymphocytes and macrophages (expressed as mean ± S.E.M.) before (0 hr) and 24 hrs after each IT (IT1 +24 hrs, IT2 +24 hrs and IT3 +24 hrs) LPS instillation are given. The number of mice used per condition is given in parenthesis. IT +24-hr values correspond to the mean number of recruited leukocytes for all IT LPS instillations. Small variability in the number of lymphocytes and macrophages were sometimes detected, probably due to the low number of cells found in BALs as compared to neutrophils.
| 0 hr | IT1 +24 hrs | IT2 +24 hrs | IT3 +24 hrs | IT +24 hrs | P** | |
|---|---|---|---|---|---|---|
| Lymphocytes | ||||||
| Cx43+/+ | 49’991 ± 32’452 (7) | 30’135 ± 8’230 (8) | 44’507 ± 11’481 (9) | 119’494 ± 36’583 (7) | 61’588 ± 13’693 (24) | N.S. |
| Cx43+/− | 13’740 ± 6’055 (7) | 33’173 ± 13’546 (8) | 77’634 ± 17’108 (10) | 60’308 ± 5’583 (8) | 58’622 ± 8’520 (26) | <0.05 |
| P* | N.S. | N.S. | N.S. | N.S. | N.S. | |
| Macrophages | ||||||
| Cx43+/+ | 127’041 ± 26’094 (7) | 95’033 ± 16’956 (8) | 129’507 ± 18’242 (9) | 116’023 ± 7’375 (7) | 114’083 ± 9’268 (24) | N.S. |
| Cx43+/− | 158’400 ± 82’227 (7) | 125’100 ± 25’149 (8) | 170’022 ± 28’969 (10) | 238’133 ± 27’529 (8) | 177’157 ± 17’773 (26) | N.S. |
| P* | N.S. | N.S. | N.S. | <0.01 | N.S. | |
Indicates P between Cx43+/+ and Cx43+/− mice.
Indicates P between LPS-treated and control conditions. N.S.: not significant.
Figure 3.
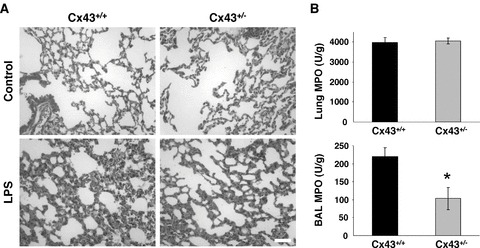
Morphology and neutrophil infiltration in Cx43+/− mice 24 hrs after LPS instillation. (A) haematoxylin and eosin stained lung sections from Cx43+/+ and Cx43+/− mice. Before the induction of inflammation (control), mice showed identical morphologies in terms of their alveolar membranes (upper panels). However, 24 hrs after LPS instillation, a thickening of the alveolar membrane and presence of granulocytes in Cx43+/− mice were less prominent than in Cx43+/+ mice. (B) Activity of MPO, an enzyme specific for neutrophils, was determined in whole lung extract (top panel) and in BALs (bottom panel) of Cx43+/+ and Cx43+/− mice. *Indicates P < 0.05.
Figure 4.

Antibody array showing the relative expression of cytokines in BALs from Cx43+/+ and Cx43+/− mice 24 hrs after LPS instillation. Several cytokines were decreased in Cx43+/− mice. The figure is representative of experiments confirmed on pooled materials from three Cx43+/+ and three Cx43+/− mice.
Inflammatory mediators induce Cx43 expression in alveolar septa
The effect of LPS instillation on total Cx43 expression in lungs of Cx43+/+ and Cx43+/− mice was evaluated by Western blot. However, we did not detect significant changes in the Cx43/β-actin ratio after LPS treatment (0.69 ± 0.03 and 0.28 ± 0.07 24 hrs after LPS instillation as compared to the control values 0.79 ± 0.04 and 0.29 ± 0.07 for Cx43+/+ and Cx43+/−, respectively; n= 3), a finding that was not unexpected regarding that Cx43 is also markedly expressed in smooth muscles. The detection by Western blot of small changes in the structural cells of inflamed lungs might therefore be impaired by the massive expression of Cx43 in smooth muscle cells. To specifically localize Cx43 in the lungs, we therefore performed immunofluorescence experiments. Interestingly, we detected weak or no staining for Cx43 in alveolar septa of lung cryostat sections from untreated Cx43+/+ mice (Fig. 5A). This was, however, in sharp contrast with the immunodetection of Cx43 in alveolar septa from mice subjected to LPS. Indeed, the typical punctuate fluorescent staining of Cx43 was abundantly evident in alveolar septa after IT instillation of LPS for 24 hrs (Fig. 5B). The characteristic histology of the lungs and the close apposition of epithelial and endothelial cell membranes in the alveolar septa did not allow for the identification of Cx43-expressing cell types at the resolution provided in cryostat sectioning, also not when endothelial or epithelial cell markers were used. Typical Cx43 immunostaining was observed within the lung cryosection on smooth muscle cells surrounding bronchi (Fig. 5D and E). The specificity of the Cx43 staining was confirmed by pre-absorption of the Cx43 antibody with its corresponding immunogenic peptide; under these conditions, no staining was detected in both alveolar septa and smooth muscles of inflamed lungs (Fig. 5C and F). Altogether, these results indicate that Cx43 is detected in alveolar septa during the inflammatory response evoked by LPS.
Figure 5.

Cx43 localization in alveolar septa during inflammation. Images of alveoli (A, B, C) and bronchi (D, E, F) from mice subjected (B, E, C, F) or not (A, D) to a 24 hrs IT LPS instillation are shown. An abundant staining for Cx43 in alveolar septa was observed after LPS instillation (B, arrows) as compared to untreated animals (A). The specificity of the staining was evaluated by pre-absorption of the antibody with its corresponding immunogenic peptide; under these conditions, Cx43 was no longer observed in alveolar septa and in smooth muscle cells of inflamed airways (C, F). The images are representative from six untreated and eight LPS-treated mice. The Cx43 staining is in green. Sections were counter-stained with Evans blue, in red. Scale bar represents 15 μm.
The detection of Cx43 may result directly from LPS-induced signalling via its cell surface receptors or indirectly via mediators of the inflammatory cascade initiated by LPS. Unfortunately, the use of toll-like receptor-deficient mice will not allow discriminating between the two possibilities because of toll-like receptor-independent effects of LPS on initiating the inflammatory cascade [15]. To address this question, we performed experiments in mice protected from inflammation either by overexpression of the sTNFR1 mice, neutralizing both TNF-α and lymphotoxin-α, or by knock-out of the TNF-α gene (TNF-α−/− mice) while toll-like receptor expression is unaffected. In these mice, the number of neutrophils collected in BALs 24 hrs after LPS instillation was reduced by 80% as compared to controls (225’310 ± 100’092 cells, n= 4). In contrast to Cx43+/+ mice, sTNFR1 and TNF-α−/− mice showed discrete or no staining of Cx43 in alveolar septa 24 hrs after IT instillation of LPS (Fig. 6A, B and C). These results suggest that Cx43 in alveolar septa is regulated by mediator(s) of the inflammatory cascade initiated by TNF-α in mice subjected to IT instillation with LPS. This idea was further substantiated in experiments whereby LPS was instilled in Cx43+/+ mice that were depleted of neutrophils by subcutaneous injection of an antibody against GR1. Under these conditions, the number of neutrophils collected in BALs of wild-type mice 24 hrs after LPS treatment was strongly diminished (72’399 ± 38’164 neutrophils, n= 8). Interestingly, the antibody treatment did not prevent the detection of Cx43 in alveolar septa (Fig. 6D), indicating that Cx43 signals in LPS-treated lungs do not originate from neutrophils. Furthermore, staining with markers of macrophages (CD68) or myofibroblasts (smooth muscle actin) showed no correlation between these cell types and the Cx43 staining (data not shown). These results therefore indicate that Cx43 is altered in structural cells such as pneumocytes and/or endothelial cells during inflammation.
Figure 6.

Induction of Cx43 expression in alveoli is mediated by pro-inflammatory mediators. Cx43 is immunolocalized in lung alveolar septa of Cx43+/+ mice instilled with LPS for 24 hrs, whether or not neutrophils were depleted by subcutaneous injection of anti-GR1 antibody (A, D, arrows). In contrast, transgenic mice overexpressing the soluble form of the TNF-α receptor 1 (B) or knock-out mice for the TNF-α gene (C), showed discrete or no Cx43 staining in alveoli. The images were representative of 4 mice invalidated for TNF-α and 3 mice treated with anti-GR1. The Cx43 staining is in green. Nuclei were stained with DAPI (blue); sections were counter-stained with Evans blue (red). Scale bar represents 15 μm.
Cx43-mediated cell-to-cell communication regulates neutrophil adhesion in vitro
The amount of Cx43 expressed in alveolar septa controls the recruitment of neutrophils to the airways as concluded from the experiments on Cx43+/− mice. Both alveolar epithelial and endothelial cells may contribute to LPS-induced lung inflammation. To further investigate the mechanisms that link Cx43 to neutrophil recruitment, and hence to inflammation, we used MLE12 and endothelial (bEnd.3) cell lines treated or not with 43Gap26, a specific Cx43 blocking peptide, or a control peptide. Importantly, bEnd.3 cells express all three endothelial connexins observed in situ (Cx43, Cx40 and Cx37) [16] and MLE12 cells only express Cx43. The 43Gap26 peptide is analogue to part of the amino acid sequence of the first extracellular loop of Cx43 and is known to inhibit Cx43-mediated cell-to-cell communication by preventing the docking of hemichannels [6, 17]. As expected, 43Gap26 efficiently decreased the spread of Lucifer Yellow in MLE12 and bEnd.3 cells (Fig. 7A and C). In the presence of 43Gap26, the number of Lucifer Yellow labelled cells was reduced (P < 0.01) to 1.6 ± 0.3 (n= 8) in bEnd.3 cells and to 1.25 ± 0.25 (n= 8) in MLE12, cells as compared to values of 4 ± 0.6 (n= 7) and 8.9 ± 0.7 (n= 8) in bEnd.3 and MLE12 cells treated with control peptide, respectively. We next determined whether reduced Cx43-mediated cell-to-cell communication by 43Gap26 affected the adhesion of activated neutrophils to a monolayer of either MLE12 or bEnd.3 cells. We observed that adhesion of neutrophils was significantly decreased in both cell types treated with 43Gap26 as compared to those exposed to control peptide. This decreased adhesion represented 14% (P < 0.05) on MLE12 and 40% (P < 0.01) on bEnd.3 cells (Fig. 7B and D). In contrast, incubation of neutrophils with 43Gap26 during the adhesion period to prevent the formation of putative gap junctions between the leukocytes and untreated endothelial cells did not change the number of adherent cells (3.9 ± 0.3 cells/field as compared to 4.1 ± 0.3 cells/field in the absence of 43Gap26, n= 4). Of note, no Cx43 was detected by Western blots in neutrophils whatever their human or mouse origin (Fig. 1C). These results indicate that a change in Cx43-mediated intercellular communication in epithelial and endothelial cell lines modulates neutrophil adhesion in vitro and thus could represent a mechanism to regulate their recruitment to the airways in vivo.
Figure 7.

Treatment of murine cell lines with 43Gap26 blocking peptide reduced neutrophil adhesion. Lucifer Yellow injections on pneumocyte (MLE12) and endothelial (bEnd.3) cell monolayers are shown in (A) and (C), respectively. Images are representative from three individual experiments. Both cells lines showed a decreased coupling when treated with 43Gap26 as compared to the control peptide condition. Neutrophil adhesion on MLE12 and bEnd.3 cell monolayers is presented in (B) and (D), respectively. Data are shown as percent of the number of neutrophils (PMN) that adhered under control conditions. Neutrophils adhered 14% (P < 0.05) and 40% (P < 0.01) less on MLE12 and bEnd.3, respectively, as compared to conditions with control peptide. Data are representative of four to six experiments. *Indicates P < 0.05; ** indicates P < 0.01.
Alteration of Cx43-mediated cell-to-cell communication modulates neutrophil recruitment in vivo
The C-terminus of Cx43 acts as a ball-and-chain to close the channel pore [18]. Truncation of the C-terminus domain is known to slow transitions between open and closed states, thus increasing channel conductivity [19]. The truncated Cx43 channel showed permeability to gap junction-permeant tracer such as neurobiotin when expressed in communication-deficient N2A cells. The incidence of coupling was 53% (n= 17 injections) for Cx43K258stop-expressing cells as compared to 0% (n= 7) for parental N2A cells (Fig. 1D). To evaluate whether enhanced Cx43-mediated cell-to-cell communication modulates the extent of the inflammatory response in vivo, mice heterozygous for a Cx43 lacking its cytoplasmic C-terminus (Cx43K258stop/− mice) were subjected to IT instillation of LPS. Cx43K258stop/− mice responded 3.9 folds higher (P < 0.01) to LPS instillation in comparison to Cx43+/− mice, pointing to the importance of Cx43 gating in the mechanism regulating neutrophil recruitment (Fig. 8A). They also showed increased (P < 0.05) neutrophil recruitment as compared to their Cx43K258stop/+ littermates (Fig. 8A). Finally, repetitive IT instillation of LPS could not be performed on Cx43K258stop/− mice because of high mortality; mortality rate was 28.6% in Cx43K258stop/− mice at IT1 +24 hrs as compared to 5% in Cx43+/+ and Cx43+/− mice. Altogether, these results show that expression of a mutant Cx43 channel that cannot close is associated with enhanced recruitment of neutrophils to the airspace and point to Cx43 as a molecule that contributes to the propagation of inflammation within the lung.
Figure 8.

Cx43 conductivity modulates lung inflammation in mice instilled with LPS. (A) The number of neutrophils (PMN) was monitored 24 hrs after IT instillation of LPS in mice expressing a truncated allele of Cx43 (Cx43K258stop/− mice). The response was significantly increased in Cx43K258stop/− mice 24 hrs after LPS treatment (n= 8 mice) as compared to Cx43+/− mice or their Cx43K258stop/+ littermates (n= 8 mice). Pooled values from 24–26 Cx43+/+ and Cx43+/− animals taken from Fig. 2 are shown for comparison. *Indicates P < 0.05; **indicates P < 0.01. (B) Treatment of inflamed lungs with 43Gap26 blocking peptide reduced neutrophil recruitment to the airspace. Wild-type mice that were subjected to LPS for 12 or 24 hrs were then treated with an IT instillation of NaCl, a control peptide, or the 43Gap26 blocking peptide for an additional 12 or 24 hrs, respectively. Although instillation of NaCl or control peptide did not affect the number of neutrophils present in lungs 24 and 48 hrs after LPS treatment, neutrophil count was reduced by 60% (P < 0.01) and 70% (P < 0.05), 24 and 48 hrs after LPS instillation, respectively, in mice that received the 43Gap26 peptide instillation. Bar indicates mean value.
To investigate the therapeutic possibility of anti-Cx43 treatment to modulate neutrophil recruitment in vivo, we performed IT instillation of 43Gap26 during the course and at peak of lung inflammation. In the first protocol, 43Gap26 was administrated 12 hrs after LPS instillation and neutrophils were counted in BALs 12 hrs later. In a second protocol, 43Gap26 was administrated at peak of inflammation, 24 hrs after LPS treatment, and neutrophil count determined 24 hrs later. The effect of 43Gap26 was compared with that of NaCl or control peptide. As shown in Fig. 8B, the number of neutrophils collected in BALs of wild-type mice subjected to NaCl (open squares) 24 and 48 hrs after LPS treatment was unaffected by the IT instillation of the control peptide (closed squares). In contrast, mice challenged with the 43Gap26 peptide (open circles) showed a marked reduction in the number of neutrophils recruited to the airways, representing a decrease of 60% (P < 0.01) at the peak of inflammation. These results were also confirmed by the MPO assay. MPO activity was markedly reduced (P < 0.05) in BALs of mice treated with 43Gap26 (120 ± 30 U/g, n= 5) as compared to wild-type (220 ± 25 U/g, n= 7) and Cx43K258stop/− (290 ± 60 U/g, n= 5) mice. In addition, administration of 43Gap26 at peak of inflammation accelerates by 70% (P < 0.05) the resolution of the inflammatory response (Fig. 8B). These results indicate that targeting Cx43 protects against recruitment of neutrophils in the airspace.
Discussion
The initiation, maintenance and resolution of pulmonary innate responses depend upon intercellular communication via release of cytokines and chemokines, ensuring interaction between different cell types present in the lungs, including leukocytes, alveolar epithelial and endothelial cells. Using several mouse models, we demonstrate here that the gap junction protein Cx43 in alveolar septa plays a central role in regulating neutrophil recruitment to the airways, thereby contributing as a modulator of lung inflammation.
Connexins form gap junction channels with distinct unitary conductance, molecular permeability and gating. The opening and closing of gap junction channels respond to changes in the cytoplasmic ion composition, post-translational modification such as phosphorylation (both referred to as chemical gating), and changes in the difference of membrane potential between cells. The cytoplasmic tail of Cx43 plays a critical role in these regulations, and truncation of the Cx43 C-terminus creates gap junction channels unable to close in response to chemical and voltage gating [18, 19]. Although mouse models knock-out for Cx43 (Cx43−/−) or homozygous for a truncated Cx43 at amino acid 257 (Cx43K258stop/K258stop) die shortly after birth [8, 9], mice harbouring heterozygous expression of wild-type and mutant Cx43 are viable. Thus, C-terminal truncation of Cx43 has recently been shown to change number, size and localization of cardiac gap junction plaques [20]. Interestingly, Cx43+/− and Cx43K258stop/− mice showed opposite responses in terms of LPS-evoked lung inflammation as compared to Cx43+/+ animals. Thus, mice expressing the non-regulatable Cx43 showed enhanced recruitment of neutrophils to the airways and mortality in response to LPS. In contrast, LPS-induced neutrophil recruitment was markedly reduced in Cx43+/− mice, the lungs of which express 61% less Cx43. These in vivo observations indicate that Cx43 contributes to the inflammatory lung phenotype.
Cx43 was markedly detected in the structural cells of the alveolar septa of mice subjected to IT instillation of LPS. LPS-induced modulation of Cx43 expression was regulated by pro-inflammatory mediators, as concluded from our experiments in two mouse models unable to use TNF-α. This conclusion is in agreement with a previous report whereby sepsis was induced by intraperitoneal injection of LPS in rats [21]. Neutrophil depletion experiments and Western blots further showed that the mechanism linking LPS to Cx43 expression in alveolar septa is independent of the recruitment of the leukocytes to the lungs. These results indicate that enhanced Cx43 expression in alveolar septa is part of the inflammatory response, an effect which is likely to be accompanied by important changes in gap junctional intercellular communication (GJIC). However, there is ample in vitro evidence that GJIC is reduced by pro-inflammatory molecules such as LPS, TNF-α or IL-1β in a variety of cell systems, although these observations were obtained using supramaximal concentrations of mediators [22–24]. Possibly, the up-regulation of Cx43 in alveolar septa is unique to the lungs. Another possibility is that GJIC is tightly regulated during the inflammatory response, the increase in Cx43 expression being compensated by a decrease in Cx43 channel conductivity. The latter hypothesis would explain the increased inflammatory response observed in Cx43K258stop/− lungs in response to LPS treatment. Indeed, without its C-terminus, the mutant Cx43 channel cannot be regulated, possibly leading to exaggerated intercellular propagation of inflammatory signals.
Calcium signalling in endothelial and alveolar epithelial cells is involved in numerous functions, including expression of adhesion molecules necessary for the recruitment and transmigration of leukocytes [3, 6]. In keeping with this idea, we have found that decreased Cx43-mediated GJIC with 43Gap26 in mouse pneumocyte and endothelial cell lines was associated with significant and marked reduction of neutrophil adhesion to their surface. The latter effect was more prominent in endothelial cells, an observation in agreement with the report of Parthasarathi and collaborators, showing the Cx43-dependent propagation of Ca2+ waves between microvascular endothelial cells in the intact-blood perfused mouse and rat lungs [6]. Interestingly, IT instillation of the 43Gap26 peptide was also efficient in vivo by reducing neutrophil recruitment to the alveolar space by 65%. Although peptides not modified for protracted action may be short lived, the efficiency of the 43Gap26 peptide in vivo may be explained by the accessibility of the peptide to small gap junctions between endothelial cells or to slow docking of unpaired connexons during inflammation. Altogether, these results are consistent with the idea that Cx43 channels may serve as conduits for the spread of pro-inflammatory signals within the lung alveolar network, leading to enhanced neutrophil adhesion and transmigration across the endothelial-alveolar barrier.
Increasing evidence converges to the concept that GJIC contributes to the severity of the inflammatory response. For example, deletion of Cx32 converted a mild reversible form of acute pancreatitis into a severe disease with increased necrosis, oedema and inflammation of the mouse exocrine pancreas [25]. In another study, forced expression of connexins in astrocytes protected against cell injury induced by a variety of cell stress [26], whereas focal brain ischemia in mice lacking Cx43 was associated with increased apoptosis and inflammation [27]. Moreover, the progression of atherosclerosis as well as restenosis after balloon-induced carotid injury was dramatically reduced in a mouse model heterozygous for a Cx43 null mutation [28, 29]. In contrast, deletion of Cx40 appeared not to contribute to the development of lung inflammation in mice subjected to intranasal instillation of LPS or abdominal infection caused by cecal ligation and perforation [14]. Here we show that Cx43 in structural cells of the lung is implicated in the interaction between neutrophil and the endothelial-alveolar barrier. Inhibition of Cx43 did not only attenuate the development of the inflammatory response, but also promoted its resolution when 43Gap26 was administrated at peak of inflammation. Because of its role on neutrophil adhesion without altering the initial pro-inflammatory signalling headed by TNF-α and leukocyte chemotaxis, Cx43 may represent a relevant pharmacological target to modulate inflammation in acute or chronic lung diseases characterized by excessive neutrophil recruitment to the airway.
Acknowledgments
We thank Isabelle Scerri, Tecla Dudez, Isabelle Roth, Marc Bacchetta and Sophie Crespin for their invaluable help during the preparation of this manuscript. We are indebted to Prof. Klaus Willecke (Institut für Genetik, University of Bonn, Germany) for providing breeding pairs of Cx43K258stop mice. This work was supported by grants from the Swiss National Science Foundation (310000–107846, 3100A0–118196 and PP00A-116897), Vaincre la Mucoviscidose and the Schweizerische Gesellschaft für Cystische Fibrose.
References
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–49. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.MacCallum NS, Evans TW. Epidemiology of acute lung injury. Curr Opin Crit Care. 2005;11:43–9. doi: 10.1097/00075198-200502000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Ley K, Laudanna C, Cybulsky MI, et al. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–89. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 4.Wagner JG, Roth RA. Neutrophil migration mechanisms, with an emphasis on the pulmonary vasculature. Pharmacol Rev. 2000;52:349–74. [PubMed] [Google Scholar]
- 5.Ichimura H, Parthasarathi K, Issekutz AC, et al. Pressure-induced leukocyte margination in lung postcapillary venules. Am J Physiol Lung Cell Mol Physio. 2005;289:407–12. doi: 10.1152/ajplung.00048.2005. [DOI] [PubMed] [Google Scholar]
- 6.Parthasarathi K, Ichimura H, Monma E, et al. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J Clin Invest. 2006;116:2193–200. doi: 10.1172/JCI26605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagasawa K, Chiba H, Fujita H, et al. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J Cell Physiol. 2006;208:123–32. doi: 10.1002/jcp.20647. [DOI] [PubMed] [Google Scholar]
- 8.Reaume AG, De Sousa PA, Kulkarni S, et al. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–4. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 9.Maass K, Ghanem A, Kim JS, et al. Defective epidermal barrier in neonatal mice lacking the C-terminal region of connexin43. Mol Biol Cell. 2004;15:4597–608. doi: 10.1091/mbc.E04-04-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia I, Miyazaki Y, Araki K, et al. Transgenic mice expressing high levels of soluble TNF-R1 fusion protein are protected from lethal septic shock and cerebral malaria, and are highly sensitive to Listeria monocytogenes and Leishmania major infections. Eur. J. Immunol. 1995;25:2401–7. doi: 10.1002/eji.1830250841. [DOI] [PubMed] [Google Scholar]
- 11.Marino MW, Dunn A, Grail D, et al. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci USA. 1997;94:8093–8. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiszniewski L, Sanz J, Scerri I, et al. Functional expression of connexin30 and connexin31 in the polarized human airway epithelium. Differentiation. 2007;75:382–92. doi: 10.1111/j.1432-0436.2007.00157.x. [DOI] [PubMed] [Google Scholar]
- 13.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J. Cell Biol. 1991;115:1357–74. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rignault S, Haefliger J-H, Waeber B, et al. Acute inflammation decreases the expression of connexin40 in mouse lung. Shock. 2007;28:78–85. doi: 10.1097/shk.0b013e3180310bd1. [DOI] [PubMed] [Google Scholar]
- 15.Ramphal R, Balloy V, Huerre M, et al. TLRs 2 and 4 are not involved in hypersusceptibility to acute Pseudomonas aeruginosa lung infections. J Immuno. 2005;175:3927–34. doi: 10.4049/jimmunol.175.6.3927. [DOI] [PubMed] [Google Scholar]
- 16.Kwak BR, Pepper MS, Gros DB, et al. Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol Biol Cell. 2001;12:831–45. doi: 10.1091/mbc.12.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boitano S, Evans WH. Connexin mimetic peptides reversibly inhibit Ca2+ signaling through gap junctions in airway cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:623–30. doi: 10.1152/ajplung.2000.279.4.L623. [DOI] [PubMed] [Google Scholar]
- 18.Thomas MA, Huang S, Cokoja A, et al. Interaction of connexins with protein partners in the control of channel turnover and gating. Biol Cell. 2002;94:445–56. doi: 10.1016/s0248-4900(02)00015-1. [DOI] [PubMed] [Google Scholar]
- 19.Moreno AP, Chanson M, Anumonwo J, et al. Role of the carboxyl terminal of connexin43 in transjunctional fast voltage gating. Circ Res. 2002;90:450–7. doi: 10.1161/hh0402.105667. [DOI] [PubMed] [Google Scholar]
- 20.Maass K, Shibayama J, Chase SE, et al. C-terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circ Res. 2007;101:1283–91. doi: 10.1161/CIRCRESAHA.107.162818. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Cobo M, Gingalewski C, Maio AD. Expression of the connexin43 gene is increased in the kidneys and the lungs of rats injected with bacterial lipopolysaccharide. Shock. 1998;10:97–102. doi: 10.1097/00024382-199808000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Sáez JC, Brañes MC, Corvalán LA, et al. Gap junctions in cells of the immune system: structure, regulation and possible functional roles. Braz J Med Biol Res. 2000;33:447–55. doi: 10.1590/s0100-879x2000000400011. [DOI] [PubMed] [Google Scholar]
- 23.De Maio A, Vega VL, Contreras JE. Gap junctions, homeostasis, and injury. J Cell Physiol. 2002;191:269–82. doi: 10.1002/jcp.10108. [DOI] [PubMed] [Google Scholar]
- 24.Chanson M, Derouette JP, Roth I, et al. Gap junctional communication in tissue inflammation and repair. Biochim Biophys Acta. 2005;1711:197–207. doi: 10.1016/j.bbamem.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Frossard JL, Rubbia-Brandt L, Wallig MA, et al. Severe acute pancreatitis and reduced acinar cell apoptosis in the exocrine pancreas of mice deficient for the Cx32 gene. Gastroenterol. 2003;124:481–93. doi: 10.1053/gast.2003.50052. [DOI] [PubMed] [Google Scholar]
- 26.Lin JHC, Yang J, Liu S, et al. Connexin mediates gap junction-independent resistance to cellular injury. J Neurosci. 2003;23:430–41. doi: 10.1523/JNEUROSCI.23-02-00430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakase T, Sohl G, Theis M, et al. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am J Pathol. 2004;164:2067–75. doi: 10.1016/S0002-9440(10)63765-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwak BR, Veillard N, Pelli G, et al. Reduced connexin43 expression inhibits atherosclerotic lesion formation in low-density lipoprotein receptor–deficient mice. Circulation. 2003;107:1033–9. doi: 10.1161/01.cir.0000051364.70064.d1. [DOI] [PubMed] [Google Scholar]
- 29.Chadjichristos CE, Matter CM, Roth I, et al. Reduced connexin43 expression limits neointima formation after balloon distension injury in hypercholesterolemic mice. Circulation. 2006;113:2835–43. doi: 10.1161/CIRCULATIONAHA.106.627703. [DOI] [PubMed] [Google Scholar]