

Abstract
In humans, the TPα and TPβ isoforms of the thromboxane A2 receptor are transcriptionally regulated by distinct promoters, designated Prm1 and Prm3. Previous investigations identified two upstream repressor regions (URR) 1 and URR2 within Prm1. Herein, it was sought to characterize Prm1, identifying the factor(s) regulating URR1 and URR2 in human erythroleukaemia (HEL) 92.1.7 cells. Genetic reporter assays and 5′ deletions confirmed the presence of URR1 and URR2 but also identified a third repressor, designated RR3, within the proximal ‘core’ promoter. Bioinformatic analysis revealed several GC elements representing putative sites for Egr1/Sp1/Wilms tumour (WT)1 within URR1, URR2 and RR3. While mutation of three GC elements within URR1 and of an adjacent GC element suggested that repressor binding occurs through a cooperative mechanism, repressors binding to the single GC elements within URR2 and RR3 act independently to regulate Prm1. While electrophoretic mobility shift assays and supershift assays demonstrated that each of the GC elements can bind Egr1 and WT1 in vitro, chromatin immunoprecipitations established that WT1 is the factor predominantly bound to each of the repressor regions in vivo. Additionally, ectopic expression of –KTS isoforms of WT1 decreased Prm1-directed gene expression and TPα mRNA expression. Collectively, these data establish WT1 as a critical repressor of Prm1, suppressing TPα expression in the platelet progenitor megakaryoblastic HEL cells.
Keywords: thromboxane receptor, Wilms’ tumour, WT1, gene, transcription
Introduction
The Wilms’ tumour 1 (WT1) gene encodes a zinc finger transcription factor that plays a central role in the development of the genitourinary, haematopoietic and central nervous systems [1, 2]. The discovery that mutation of the WT1 gene is one of the main causes of the paediatric kidney cancer Wilms’ tumour led to its identification as a tumour suppressor protein [3]. More recently, however, the finding that wild-type WT1 is abundantly expressed in a variety of cancers, including those of the breast [4], oesophagus [5] and pancreas [6] has suggested that it may play an oncogenic role in certain instances. Moreover, accumulating evidence implicates WT1 as an important factor associated with both normal and aberrant haematopoiesis (reviewed in [1]).
As well as playing an important role as a transcriptional regulator, the WT1 protein also appears to be involved in post-transcriptional regulation, with specific functions in RNA metabolism and splicing [7, 8], and possibly in translation [9]. Its wide-ranging functions are attributed to the existence of multiple isoforms of the WT1 protein that arise due to alternative splicing or the use of multiple translational start codons within the WT1 gene (reviewed in [10]). The best-characterized isoforms are the +/– exon 5 variants and the +/– KTS isoforms. Alternative splicing at these two sites gives rise to four different protein isoforms, specifically (+/+), (+/–), (–/+) and (–/–) with respect to the presence or absence of exon 5 and KTS sequences, respectively. In general, the –KTS isoforms may repress or activate transcription, while the +KTS isoforms have a reduced ability to bind DNA (reviewed in [1]).
WT1 has four C-terminal Kruppel-like C2H2 fingers that share significant identity with those of early growth response (Egr)1, another zinc finger transcription factor. Moreover, the zinc finger domain of WT1 mediates binding to DNA at the Egr1 consensus sequence 5′GCG(G/T)GGGCG3′, but its affinity for this site is somewhat less than that of Egr1 [11]. Additionally, WT1 has also been reported to bind to another motif, 5′GCGTGGGAGT3′, termed the Wilms' tumour element (WTE) [12]. Depending on the cellular context and/or promoter, WT1 can act as either an activator or a repressor of transcription. For example, overexpression of WT1 in the human erythroleukaemia K562 cell line or in human breast cancer cell lines activated the c-Myc promoter [13], whilst WT1 repressed the same promoter in HeLa cells [14]. WT1 can interact with other DNA-binding transcription factors, including p53 [15], as well as specific co-activators, such as CBP [16], or co-repressors, including BASP1 [17], to regulate transcription. It is thought that it is the nature of binding of these co-factor partners that largely determines whether WT1 functions as a transcriptional activator or repressor.
The prostanoid thromboxane (TX) A2 plays a central role in haemostasis, acting as a potent mediator of platelet aggregation [18], but can induce other diverse cellular responses including constriction of vascular and bronchial smooth muscle cells [18, 19]. Within the kidney, TXA2 induces contraction of glomerular mesangial cells and intrarenal vascular tissue, decreasing glomerular filtration rates [20]. Moreover, in glomerulonephritis, a leading cause of end-stage renal failure, it is the most abundant eicosanoid synthesized in the nephritic glomeruli [21]. Alterations in the levels of TXA2 or the TXA2 receptor (TP) are associated with a variety of vascular disorders [22, 23], inflammatory renal diseases [24] and in renal failure [25].
In humans, TXA2 signals through two isoforms of the TXA2 receptor, termed TP and TP2, that are encoded by a single gene and arise by alternative splicing [26–28]. TPα and TPβ are identical for their N-terminal 328 amino acid residues but differ exclusively in their C-tail domains [27, 28]. TP and TP regulate both common and distinct signalling pathways [26, 29–31] but are subject to entirely different modes of regulation, such as through both agonist-dependent (homologous) desensitization and through intra-molecular cross-talk between other signalling systems [32–36]. Hence, while the functional relevance for the existence of two TP receptors in primates is currently unknown, there is abundant evidence that they have distinct physiologic roles. Moreover, while TP and TP mRNAs are differentially expressed in a range of cell types [37], platelets exclusively express TP [38]. Consistent with this, TP and TP are transcriptionally regulated by distinct promoters, termed Prm1 and Prm3, respectively, within the single TP gene [39, 40]. A recent study aimed at characterizing Prm1 in HEL cells localized the proximal ‘core’ promoter and identified two upstream activator regions (UAR) and two upstream repressor regions (URR) within Prm1 [41]. Therein, it was established that the ‘core’ Prm1 is under the control of the general transcription factors Sp1 and Egr1, as well as the more haematopoietic-specific NF-E2, while GATA-1 and Ets-1 bind and regulate UAR1 (from −7962 to −7717). However, the transacting factors regulating UAR2 (from −7717 to −7504), as well as URR1 (from −8500 to −7962) and URR2 (from −6848 to −6648) remained unidentified.
Hence, the central aim of the current study was to identify the key cis-acting elements and trans-acting factors that mediate repression of Prm1 within the previously identified URR1 and URR2 sequences. Herein, through 5 deletion and genetic reporter assays, a third repressor region, designated RR3, was also unmasked within the proximal ‘core’ Prm1. Amongst the transcription factor binding elements identified in URR1, URR2 and RR3 within Prm1 were multiple GC-rich elements predicted to represent putative WT1/Egr1/Sp1 binding sites. Data herein established that WT1, as opposed to Sp1 or Egr1, is the major transcription factor that binds the three repressor regions under basal conditions in the megakary-oblastic HEL cell line and WT1 binding correlates with transcriptional repression of Prm1 and TPα expression. Considering the reported role of WT1 as a transcriptional repressor of several genes, as well as its role in normal and aberrant haematopoiesis [42, 43] and in renal (dys)function [1, 2], the finding of its regulation of Prm1 and TPα expression may have important clinical implications.
Materials and methods
Materials
pGL3Basic, pRL-Thymidine Kinase (pRL-TK) and Dual Luciferase® Reporter Assay System were obtained from Promega Corporation (Madison, WI, USA). DMRIE-C®, RPMI 1640 culture media and foetal bovine serum (FBS) were from Invitrogen Life Technologies (Carlsbad, CA, USA). Anti-WT1 (sc-192 X), anti-Sp1 (sc-59 X), anti-Egr1 (sc-110 X), normal rabbit IgG (sc-2027) and goat anti-rabbit horseradish peroxidase (sc-2204) were obtained from Santa Cruz Biotechnology. All antibodies used for chromatin immunoprecipitation (ChIP) analysis were ChIP-validated by the supplier (Santa Cruz, CA, USA) and have been widely used in the literature for such analyses.
Construction of luciferase-based genetic reporter plasmids
The plasmid pGL3b:Prm1, containing a Prm1 sequence (2605 bp) from the human TP gene in the pGL3Basic reporter vector, in addition to pGL3b:Prm1B, pGL3b:Prm1BΔGata/Ets, pGL3b:Prm1BΔ, pGL3b:Prm1C, pGL3b:Prm1D and pGL3b:Prm1E have been previously described [41]. Additional 5′ deletion sub-fragments of Prm1 were amplified by the polymerase chain reaction (PCR) using pGL3b:Prm1 as template and subsequently sub-cloned into pGL3Basic. Specifically, for pGL3b:Prm1I, a PCR fragment was generated using the sense primer Kin358 (5′GAGAGGTACCTGAGAGACAGCGGGAGACAGAGAC3′; nucleotides (nu) –6258 to –6235, where the underlined sequence corresponds to a Kpn1 cloning site) and the antisense primer Kin109 (5′AGAGACGCGTCTTCAGAGACCTCATCTGCGGGG3′; complementary to nu –5917 to –5895 of Prm1, where the underlined sequence corresponds to a Mlu1 cloning site). For pGL3b:Prm1J, a fragment was generated using sense primer Kin391 (5′GAGAGGTACCCCTCCATCTGTGTGGGTCCTC 3′; nu –6122 to –6102) and the antisense primer Kin109. The identity and fidelity of all Prm1-derived sub-fragments in the corresponding recombinant pGL3Basic plasmids were verified through DNA sequencing.
Site-directed mutagenesis (SDM)
SDM was carried out using the QuikChange™ method (Stratagene). The identity of the Prm1 elements subjected to SDM and the corresponding plasmids generated, as well as the identity, sequence and corresponding nucleotides of the specific primers used are listed below. In each case, the – designation indicates nucleotides 5′ of the translational ATG start codon (designated +1).
GC at –8345 changed from tgccccCGCCcccac to tgccccTGACcccac using template pGL3b:Prm1 to generate pGL3Basic:Prm1GC*(–8345). Primers used: Kin423 (5′ TGGAAGCTGCCCCTGACCCCACCCAGCTTC 3′) and the complementary oligonucleotide Kin424.
GC at –8281 changed from gcccgGCCCccgccgga to gcccgGTTCccgccgga using template (a) pGL3b:Prm1 to generate pGL3Basic:Prm1GC*(–8281) and (b) pGL3Basic:Prm1GC*(–8345,–7831) to generate pGL3Basic:Prm1GC*(–8345,–8281,–7831). Primers used: Kin478 (5′ CTCCCTGCCCGGTTCCCGCCGGAAACC 3′) and the complementary oligonucleotide Kin479.
GC at –8146 changed from cGGGGGGTgggGGGCGGGGGGCgggccaa to cGGGGGTCgtgGGGTGGATGGCgggccaa using template (a) pGL3b:Prm1 to generate pGL3Basic:Prm1GC*(–8146) and (b) pGL3Basic: Prm1GC*(–8345,–8281,–7831) to generate pGL3Basic: Prm1GC*(–8345,–8281,–8146,–7831). Primers used: Kin510 (5′ GTGCTGGCGGGGGTCGTGGGGCGGGGGGCG 3′) and the complementary oligonucleotide Kin511, as well as Kin512 (5′ GGGGTCGTGGGGTGGATGGCGGGCCAAGAC 3′) and the complementary Kin513.
GC at –7831 changed from tcactGCCCcctcatct to tcactGTCCtctcatct using template (a) pGL3b:Prm1 to generate pGL3Basic:Prm1GC*(–7831), (b) pGL3b:Prm1B to generate pGL3Basic:Prm1BGC*(–7831) and (c) pGL3Basic:Prm1GC*(–8345) to generate pGL3Basic:Prm1GC*(–8345,–7831). Primers used: Kin361 (5′ TCCGTCTCTCACTGTCCTCTCATCTGGAGCCC 3′) and the complementary oligonucleotide Kin362.
GC at –6717 changed from tctgtcctCCCAcccca to tctgtcctATCAcccca using template pGL3Basic:Prm1D to generate pGL3Basic: Prm1DGC*(–6717). Primers used: Kin502 (5′ CATCCCTCTGTCCTATCACCCCACCCCTGG 3′) and the complementary oligonucleotide Kin503.
GC at –6206 changed from cagcggccCCCAcccgt to cagcggccTACAcccgt using template (a) pGL3Basic:Prm1I to generate pGL3Basic:Prm1IGC*(–6206) and (b) pGL3Basic:Prm1DGC*(–6717) to generate pGL3Basic:Prm1DGC*(–6717,–6206). Primers used: Kin506 (5′ GCTGCCAGCGGCCTACACCCGTCCCAGC 3′) and the complementary oligonucleotide Kin507.
Cell culture
HEL 92.1.7 cells were obtained from the American Type Culture Collection and were grown at 37°C in a humid environment with 5% CO2. HEL cells were cultured in RPMI 1640, 10% FBS.
Assay of luciferase activity
HEL cells were co-transfected with the various pGL3Basic-recombinant plasmids, encoding firefly luciferase, along with pRL-TK, encoding renilla luciferase, using DMRIE-C® transfection reagent and assayed for firefly and renilla luciferase 48 hrs later using the Dual-Luciferase Reporter Assay System™, as previously described [44]. Relative firefly to renilla luciferase activities (arbitrary units) were calculated as a ratio and were expressed in relative luciferase units (RLU).
To investigate the effect of overexpression of exon 5 (±) or KTS (±) isoforms of WT1 on Prm1-directed gene expression, HEL cells were co-transfected with either pGL3b:Prm1 or pGL3b:Prm1D (1.5 μg) plus 200 ng of pRL-TK along with either pcDNA3:WT1 (+/+), pcDNA3:WT1 (+/–), pcDNA3:WT1 (–/+), pcDNA3:WT1 (–/–) (0.5 μg), or as a control, pcDNA3 (0.5 μg). Cells were harvested 48 hrs after transfection and assayed for luciferase activity, as above, or subjected to Western blot or RT-PCR analysis.
The plasmids pcDNA3:WT1 (+/+), pcDNA3:WT1 (+/–), pcDNA3:WT1 (–/+) and pcDNA3:WT1 (–/–) were generously donated by Dr. Charles T. Roberts Jr., Oregon National Primate Research Center (OR, USA) and have been previously described [45].
Western blot analysis
The expression of WT1, Sp1 and Egr1 proteins in HEL cells was confirmed by western blot analysis. Briefly, whole cell protein was resolved by SDS-PAGE (10% acrylamide gels) and transferred to polyvinylidene difluoride membrane according to standard methodology. Membranes were screened using anti-WT1, anti-Sp1 or anti-Egr-1 sera in 5% non-fat dried milk in 1× TBS (0.01 M Tris/HCl, 0.1 M NaCl) for 2 hrs at room temperature. Thereafter, membranes were washed and screened using goat anti-rabbit horseradish peroxidase, followed by chemiluminescence detection as previously described [36].
RT-PCR
Total RNA was isolated from HEL 92.1.7 cells (5 × 106 approximately) using TRIzol reagent (Invitrogen Life Technologies). RT-PCR was carried out with DNase 1-treated total RNA using oligonucleotide primers to specifically amplify TPα and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA sequences, as previously described [37]. The following primers were used:
Kin16: TPα forward 5′ GAGATGATGGCTCAGCTCCT 3′,
DT75: TPα reverse 5′ CCAGCCCCTGAATCCTCA 3′,
Kin291: GAPDH forward 5′ CCACAGTCCATGCCATCAC 3′ and
DT92: GAPDH reverse 5′ CATGTGGGCCATGAGGTC 3′.
Electrophoretic mobility shift and supershift assays
Nuclear extract was prepared from HEL cells as previously described [44]. Oligonucleotides corresponding to the sense (5′ end-labelled with biotin) and antisense strands of each probe (90 μm) were annealed by heating at 95°C for 2 min. followed by slow cooling to room temperature. Electrophoretic mobility shift assays (EMSAs) were performed as previously described [41]. The identities and sequences of the forward biotin-labelled oligonucleotide probes are listed below. Sequences of the corresponding non-labelled complementary oligonucleotides are omitted.
GC−8345,–8281 probe (Kin733; 5′[Btn] GAAGCTGCCCCCGCCCCCACCCAGCTTCCTGACTTTGGCTGTGTCCAGAGCTAAGAATAGACGCTCCCTGCCCGGCCCCCGCCGGAAACCG 3′, nu –8350 to –8260 of Prm1),
GC−8146 probe (Kin737; 5′[Btn] GTGCTGGCGGGGGGTGGGGGGCGGGGGGCGGGCCAAGACCGG3′, nu –8153 to –8118 of Prm1),
GC−7831 probe (Kin739; 5′[Btn] TCCTCCGTCTCTCACTGCCCCCTCATCTGGAGCCCCAG 3′, nu –7842 to –7805 of Prm1),
GC−6717 probe (Kin762; 5′[Btn] CACCCCCCATCCCTCTGTCCTCCCACCCCACCCCTGGAAG 3′, nu –6730 to –6691 of Prm1) and
GC−6206 probe (Kin764; 5′[Btn] GCCGCGGGCTGCCAGCGGCCCCCACCCGTCCCAGCTCGGC3′, nu –6218 to –6178 of Prm1).
The sequences of the competitor/non-competitor oligonucleotides used were as follows:
Prm1−8345 competitor (Kin458; 5′ CTGGAAGCTGCCCCCGCCCCCACCCAG 3′, nu –8453 to –8327 of Prm1),
Prm1−8281 competitor (Kin742; 5′ CTCCCTGCCCGGCCCCCGCCGGAAACCGC 3′, nu –8287 to –8259 of Prm1),
Prm1−8146 competitor (Kin745; 5′ GTGCTGGCGGGGGGTGGGGGGCGGGGGGCGGGCCAAGACCGG 3′, nu –8153 to –8112 of Prm1),
Prm1−7831 competitor (Kin798; 5′ TCCTCCGTCTCTCACTGCCCCCTCATCTGGAGCCCCAG 3′, nu –7842 to –7805 of Prm1),
Prm1−6717 competitor (Kin779; 5′ CACCCCCCATCCCTCTGTCCTCCCACCCCACCCCTGGAAG 3′, nu –6730 to –6691 of Prm1),
Prm1−6206 competitor (Kin780; 5′ GCCGCGGGCTGCCAGCGGCCCCCACCCGTCCCAGCTCGGC 3′, nu –6218 to –6179 of Prm1),
WTE consensus (Kin748; 5′ CGAGTGCGTGGGAGTAGAATT 3′),
Sp1 consensus (Kin651; 5′ ATTCGATCGGGGCGGGGCGAGC 3′),
Egr1 consensus (Kin746; 5′ GGATCCAGCGGGGGCGAGCGGGGGCGA 3′) and
non-specific (Kin484; 5′ GGGCCGAGGACAGGTGAAGTGGGGACAG 3′).
For supershift assays, nuclear extract (2.5 μg total protein) was pre-incubated with 4 μg of anti-Sp1, anti-Egr1, anti-WT-1 or anti-cJun sera for 2 hrs at room temperature. Thereafter, the nuclear extract–antibody mixtures were incubated for 20 min. at room temperature with the appropriate biotin-labelled double-stranded probe, as previously described [41].
Chromatin immunoprecipitation assays
ChIP assays were performed using HEL 92.1.7 cells, as previously described [41]. Chromatin samples were immunoprecipitated with 10 μg of anti-WT1, anti-Sp1, anti-Egr1, normal rabbit IgG control antibody or a ‘no antibody’ control. The identities of the primers used for the ChIP PCR reactions, as well as their sequences and corresponding nucleotides within Prm1 are listed below.
Kin462 (5′ CGAGACCCTGCAGGCAGACTGGAG 3′; –8460 to –8437),
Kin463 (5′ GAGATGGGGAAACTGAGGCACAAAG 3′; –8030 to –8006),
Kin468 (5′ GCCTTGCAGAGATGTGGTGAGGC 3′; –7978 to –7973),
Kin467 (5′ GAGGTGAGCTAGGAAGACATCTTG 3′; –7630 to –7607),
Kin233 (5′ GAGAGGTACCGCTCCAAAGCCACCTCCG 3′; –6848 to –6831),
Kin144 (5′ AGAGACGCGTCGCTTCCTCGGGAGCCTCA 3′; –6455 to –6437),
Kin456 (5′ CTTCCCCAGAAGGCTGTAGGGTGTC 3′; –6368 to –6344),
Kin109 (5′ AGAGACGCGTCTTCAGAGACCTCATCTGCGGGG 3′; –5917 to –5895),
Kin364 (5′ TTGGGTCCAGAAGGTCGAGGC 3′; –1081 to –1061) and
Kin365 (5′ GCGAACCAGGGCGAGGC 3′; –711 to –695).
Statistical analysis
Statistical analyses of differences were analysed using the two-tailed Student’s unpaired t-test. All values are expressed as mean ± standard error of the mean (S.E.M.). P-values <0.05 were considered to indicate statistically significant differences and *, **, *** and **** indicate P < 0.05, P < 0.01, P < 0.001 and P < 0.0001, respectively.
Results
Identification of three distinct repressor regions within Prm1 of the human TP Gene
The α and β isoforms of TP are under the transcriptional regulation of Prm1 and Prm3, respectively, within the human TP gene [39]. Prm1 is defined as nucleotides –8500 to –5895 located 5′ of the translation initiation codon within the human TP gene [39]. In a recent study aimed at characterizing Prm1 within the megakaryoblastic HEL 92.1.7 cell line, two URR were identified (Fig. 1A and [41]). However, the factors regulating URR1 (–8500 to –7962) and URR2 (–6848 to –6648) remained to be identified (Fig. 1A and [41]). The aim of the current study was to further characterize Prm1 and to identify the cis-acting elements and trans-acting factors mediating its repression through the aforementioned URR1 and URR2 in HEL cells.
Figure 1.
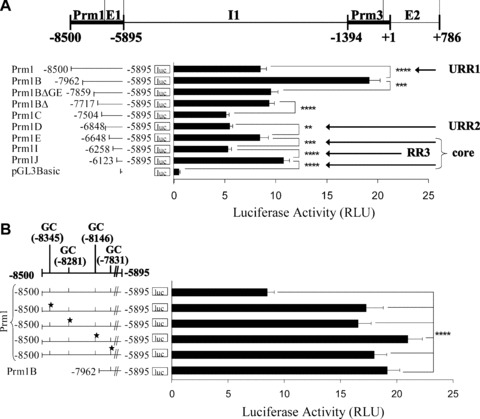
Effect of 5′ deletions on Prm1-directed gene expression and identification of GC elements within the –8500 to –7962 region of Prm1. (A): Schematic of the human TP gene spanning nucleotides –8500 to +786 encoding Prm1 (–8500 to –5895), Prm3, exon (E)1, intron (I)1 and E2, where nucleotide +1 represents the translational start site (ATG). pGL3Basic plasmids (2 μg) encoding Prm1 (–8500 to –5895) or its 5′ deletion fragments Prm1B (–7962), Prm1BΔGata/Ets (–7859), Prm1BΔ (–7717), Prm1C (–7504), Prm1D (–6848), Prm1E (–6648), Prm1I (–6258), Prm1J (–6123) or, as a control, pGL3Basic were co-transfected with pRL-TK into HEL 92.1.7 cells. Mean firefly relative to renilla luciferase activity was expressed in arbitrary relative luciferase units (RLU ± S.E.M.; n= 6). (B), (C) and (D): GC elements containing putative overlapping WT1/Egr1/Sp1 binding sites within Prm1, where the 5′ nucleotide is indicated and the star symbol signifies mutated elements. pGL3Basic plasmids (2 μg) encoding: (B) Prm1, Prm1GC*(–8345), Prm1GC*(–8281), Prm1GC*(–8146), Prm1GC*(–7831) and Prm1B; (C) Prm1, Prm1GC*(–8345), Prm1GC*(–8345,–7831), Prm1GC*(–8345,–8281,–7831) and Prm1GC*(–8345,–8281,–8146,–7831); or (D) Prm1B, Prm1BGC*(–7831) and Prm1BΔ were co-transfected with pRL-TK into HEL cells. Luciferase activity was expressed as mean firefly relative to renilla luciferase activity (RLU ± S.E.M.; n= 8).
Initially, and consistent with previous findings [41], genetic reporter assays and progressive 5′ deletion of nucleotides from –8500 to –6648 to yield the core promoter (Prm1E; Fig. 1A) confirmed the presence of two upstream regions of repression, namely URR1 and URR2. Specifically, deletion of nucleotides from Prm1 (–8500) to generate Prm1B (–7962) yielded a 2.3-fold increase in luciferase expression (P < 0.0001), while 5′ deletion of Prm1D (–6848) to generate Prm1E (–6648) resulted in a 1.5-fold increase in luciferase activity (P= 0.0032). Further 5′ deletion of Prm1E (–6648) to generate Prm1I (–6258) resulted in a 1.6-fold decrease in luciferase expression (P= 0.0002), thereby uncovering an activator region within the ‘core’ Prm1. Consistent with this observation, two functional overlapping Sp1/Egr1 elements have been previously identified within this region, specifically at –6294 and –6278, that mediate activation of Prm1 [41]. However, herein, further 5′ deletion of Prm1I (–6258) to generate Prm1J (–6123) yielded a 2-fold increase in luciferase activity (P < 0.0001) to reveal a third, previously unidentified, repressor region (–6258 to –6123) also located within the ‘core’ Prm1. The luciferase expression directed by Prm1J was substantially higher than that of the empty pGL3Basic vector (Fig. 1A). Consistent with this observation, two functional overlapping Sp1/Egr1 elements within Prm1J, specifically at –6022 and –6007, in addition to an NF-E2 element at –6080 were previously identified, and these elements mediate activation of Prm1 within this core proximal region [41]. Hence, collectively, through these and previous studies, three distinct regions of repression have been identified within Prm1; namely, the two previously identified URR1 (–8500 to –7962) and URR2 (–6848 to –6648) and an additional repressor region, designated RR3, located between –6258 and –6123 within the proximal core promoter.
Identification of multiple GC-enriched elements in the –8500 to –7962 repressor region of Prm1
Bioinformatic analysis [46] to identify transcription factor elements within URR1 revealed three putative GC elements representing putative overlapping WT1/Egr1/Sp1 sites at –8345, –8281 and –8146, where the 5′ nucleotide of each element is indicated (Table 1 and Fig. 1B). SDM of each of these GC elements led to substantial increases in luciferase activity directed by Prm1 (2.0-fold, 1.9-fold and 2.5-fold, respectively; P < 0.0001 in each case; Fig. 1B). A fourth GC element was identified somewhat adjacent to URR1, specifically at –7831 within Prm1B. Mutation of this element also substantially increased luciferase activity directed by Prm1 (2.1-fold; P < 0.0001). These data suggest that the GC elements at –8345, –8281, –8146 and –7831 mediate repression of Prm1.
Table 1.
Consensus sequences for Egr1, WTE and Sp1, as well as sequences of GC elements within Prm1
| Element* | Sequence** |
|---|---|
| Egr1 consensus [12] | 5′ GcGGGGGCG 3′ |
| WTE [12] | 5′ gtgcGTGGGaGtagaat 3′ |
| Sp1 consensus [66] | 5′ gGGGCGGGgc 3′ |
| Prm1−8345 (–) | 5′ ctggGTGGGGGCGGGgGcagctt 3′ |
| Prm1−8281 (–) | 5′ tccgGcGGGGGCCGGgcag 3′ |
| Prm1−8146 (+) | 5′ ggcGGGGGGTGGGGGGCGGGGGGCGGGccaa 3′ |
| Prm1−7831 (–) | 5′ agatGaGGGGGCAgtga 3′ |
| Prm1−6717 (–) | 5′ ccagGGGTGGGGTGGGaGgacaga 3′ |
| Prm1−6206 (–) | 5′ acggGTGGGgGccgctg 3′ |
The + and – designation indicates that elements are found on the sense or antisense strands of Prm1, respectively.
Base pairs underlined denote the core sequences of the elements, while base pairs in capital letters are in positions that exhibit a high conservation profile [46].
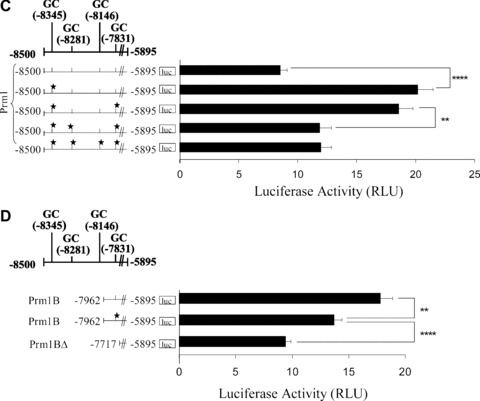
To investigate the combined contribution of GC elements in directing Prm1 activity, the effect of collectively mutating the sites within Prm1 (–8500) was examined (Fig. 1C). Disruption of the GC−7831 element within Prm1GC*(–8345) to generate Prm1GC*(–8345,–7831) did not significantly affect luciferase expression directed by Prm1GC*(–8345) (P= 0.3781). However, disruption of the GC−8281 element in Prm1GC*(–8345,–7831), generating Prm1GC*(–8345,–8281,–7831), yielded a 1.6-fold decrease in luciferase expression compared to that of Prm1GC*(–8345,–7831) (P= 0.0043). Luciferase expression of Prm1GC*(–8345,–8281,–8146,–7831) was not significantly different than that of Prm1GC*(–8345,–8281,–7831) (P= 0.7499). Therefore, generation of Prm1GC*(–8345,–8281,–8146,–7831) from Prm1GC*(–8345) led to an overall 1.7-fold decrease in luciferase expression (P= 0.0024), suggesting that repressor factors rely on a cooperative mechanism of binding to multiple neighbouring GC elements. It is likely that disruption of cooperative binding upon SDM shifts the overall affinity of intact GC elements for activator, rather than for repressor factors. Consistent with this suggestion, disruption of GC−7831 in the Prm1B (–7962) sub-fragment, which does not contain any of the other three GC elements, actually decreased the luciferase activity directed by Prm1B (1.4-fold; P= 0.0004; Fig. 1D). This effect is in contrast to the substantial increase (2.1-fold; P < 0.0001; Fig. 1B) in luciferase expression that occurred upon disruption of the same GC element within Prm1 where the other three GC elements at –8345, –8281 and –8146 were intact (compare Fig. 1B and D).
The expression of WT1, Sp1 and Egr1 in the HEL 92.1.7 cell line was confirmed by immunoblot analysis (Fig. 2). Consistent with previous studies in K562 cells [47], a doublet of WT1 protein at 52/54 kD was detected herein in HEL cells (Fig. 2A). Also consistent with studies in K562 cells [48], an immunoreactive Egr1 band of approximately 82 kD was detected in HEL cells (Fig. 2B), while abundant expression of the ubiquitous Sp1 protein was also confirmed (Fig. 2C). Thereafter, EMSAs were carried out to investigate the presence and identity of nuclear factors capable of binding to the overlapping WT1/Egr1/Sp1 elements at –8345, –8281, –8146 and –7831 in vitro (Fig. 3). It has been well documented that both Egr1 and WT1 proteins can bind to the Egr1 consensus sequence [11], while it has also been suggested that the WTE may act as a specific binding element for WT1 [12]. Moreover, overlapping sites for Sp1 and Egr1/WT1 are frequently found in promoter sequences due to the similarity in their consensus elements [49].
Figure 2.

Expression of WT1, Sp1 and Egr1 proteins in HEL 92.1.7 cells. Immunoblot analysis of WT1 (A), Egr1 (B) and Sp1 (C) expression in HEL cells (60 μg whole cell protein per lane). The positions of the molecular size markers (kD) are indicated to the left, while the sizes of WT1, Sp1 and Egr1 are indicated to the right of the panels, respectively.
Figure 3.
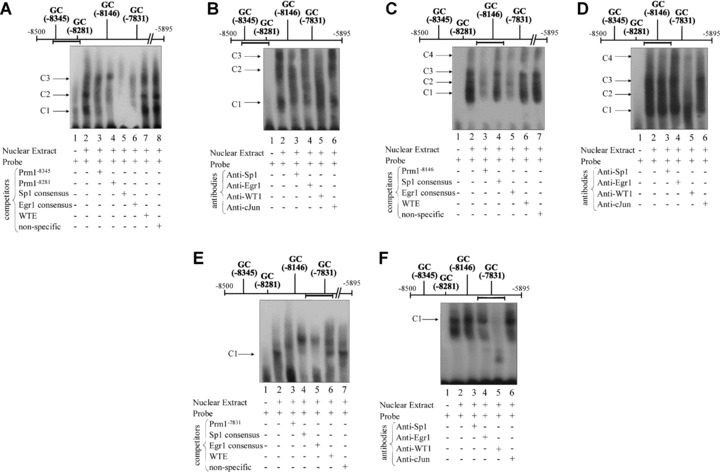
Nuclear factor binding to 5′ GC elements within Prm1 in vitro. EMSAs (A, C, E) and supershift assays (B, D, F) using nuclear extract from HEL cells and biotin-labelled double-stranded probes encoding GC−8345, GC−8281 (A and B), GC−8146 (C and D) and GC−7831 (E and F). In each case, the horizontal bar indicates the relative position of the probe within Prm1. (A, C and E): Nuclear extract was pre-incubated with the vehicle (−) or with excess non-labelled competitor oligonucleotides (+) prior to addition of the relevant probe. (B, D and F): Nuclear extract was pre-incubated with anti-Sp1, anti-Egr1, anti-WT1 or, as a negative control, anti-cJun prior to addition of the relevant probe. Images are representative of three independent experiments.
Incubation of biotin-labelled oligonucleotide probes encoding GC−8345 and GC−8281 (Fig. 3A and B), GC−8146 (Fig. 3C and D) and GC−7831 (Fig. 3E and F) with nuclear extract prepared from HEL cells resulted in the appearance of a number of protein-DNA complexes. Specifically, incubation of the probe encoding both GC−8345 and GC−8281 elements with nuclear extract generated three main complexes, C1-C3 (Fig. 3A). C1 and C2 were partially competed by non-labelled competitors containing either the specific GC−8345, GC−8281, consensus Sp1 or Egr1 sequences (Fig. 3A, lanes 3–6, respectively). C3 was efficiently competed by consensus Sp1 or Egr1 sequences but not by GC−8345 or GC−8281 sequences. The WTE sequence or a non-specific competitor based on a random sequence within the TP gene failed to compete with C1, C2 or C3 complexes (Fig. 3A, lanes 7 and 8, respectively). Therefore, it seems that complexes C1 and C2 consist of Sp1, Egr1 and/or WT1 proteins bound to the GC−8345 and GC−8281 elements in vitro, whereas C3 may consist of Sp1, Egr1 and/or WT1 bound to an unidentified element within the probe (Fig. 3A). Thereafter, in order to investigate the specific nature of the factors that bind to GC−8345 or GC−8281 in vitro, supershift assays were carried out. While no supershifted complex was observed upon addition of an anti-WT1 antibody, both C2 and C3 were substantially reduced following its addition. The reduction in C2 and C3 despite the absence of a supershifted complex is thought to be due to aggregation of specific antibody-protein complexes and failure of these large complexes to enter the gel. Moreover, addition of anti-Sp1 or anti-Egr1 antibodies led to reductions in all three complexes but without the appearance of supershifted complexes per se. Addition of an anti-cJun antibody, which acted as a negative control, did not have any substantial effects on binding patterns to the probe. Therefore, these data indicate a specific role for WT1, Sp1 and Egr1 binding to GC−8345 and/or GC−8281 in vitro (Fig. 3B).
Incubation of the GC−8146 probe with nuclear extract generated four main complexes, C1-C4 (Fig. 3C). All four complexes were efficiently competed by non-labelled competitors containing the GC−8146 or consensus Egr1 sequences, while none of the four complexes were competed by the consensus Sp1, WTE or non-specific oligonucleotide sequences. Despite the fact that none of the four complexes were competed by the WTE sequence, addition of an anti-WT1 antibody substantially reduced C2, C3 and C4, indicating a role for WT1 binding to the probe. Moreover, addition of an anti-Egr1 antibody appeared to increase formation of higher complexes, suggesting that Egr1 may also possibly play a role in binding to GC−8146 in vitro. However, addition of an anti-Sp1 antibody or an anti-cJun antibody, which acted as a negative control, did not have any substantial effects on binding patterns to the probe (Fig. 3D). Thus, it is indicated that WT1, and possibly Egr1, bind to the GC−8146 element in vitro.
Incubation of the GC−7831 probe with nuclear extract generated a single complex C1 and a diffuse faster migrating complex (Fig. 3E) that were competed by a non-labelled competitor containing the GC−7831 element, or by consensus Sp1 or Egr1 sequences. C1 was competed to a much lesser extent by the WTE sequence, but was not competed by the non-specific oligonucleotide. Therefore, C1 is likely to consist of Sp1, Egr1 and/or WT1 proteins complexed to the GC−7831 element. It was notable, however, that a second slower-migrating complex appeared where the main complex C1 was competed by non-labelled competitors. Thus, it is indicated that an increase in binding of unidentified protein(s) to an element within the probe may occur when the protein(s) involved in the formation of C1 are unavailable for binding to the probe. Thereafter, pre-incubation of nuclear extract with an anti-WT1 antibody prevented the generation of C1, suggesting a role for WT1 binding to GC−7831 despite the fact that no supershift complex was observed. Pre-incubation of nuclear extract with an anti-Egr1 antibody reduced C1 to a lesser extent, while addition of an anti-Sp1 antibody or an anti-cJun antibody, which acted as a negative control, did not have any substantial effect on formation of C1 (Fig. 3F). Overall, these EMSA data (Fig. 3) indicate that GC−8345, GC−8281, GC−8146 and GC−7831 elements have a sequence capacity to bind Egr1 and WT1 isoform(s), while GC−8345 and GC−8281 also have a capacity to bind Sp1.
Thereafter, to investigate whether endogenous Sp1, Egr1 and/or WT1 can actually directly bind to chromatin encoding Prm1 in vivo, ChIP assays were carried out on chromatin extracted from HEL cells (Fig. 4). PCR analysis using primers to amplify the 5′ Prm1 repressor region, specifically from –8460 to –8006 and containing GC elements at –8345, –8281 and –8146, resulted in amplification of DNA recovered from both the input chromatin and from an anti-WT1 immunoprecipitate, but not from anti-Sp1, anti-Egr1 or control IgG precipitates (Fig. 4A). Furthermore, PCR analysis using primers specific to the adjacent Prm1 region, specifically from –7978 to –7607 and containing the GC element at –7831, also resulted in amplification of DNA recovered from the input chromatin, anti-WT1 and, to a much lesser extent, from anti-Egr1 immunoprecipitates. However, these primers failed to amplify DNA from anti-Sp1 or control IgG precipitates (Fig. 4B). These data provide evidence that WT1, but not Sp1, occupies element(s) within the Prm1 region between –8460 and –7607 of chromatin in HEL cells in vivo, while Egr1 occupies elements within this region to a much lesser extent than WT1. Conversely, as controls, primers specific to the –1081 to –695 region of Prm3 of the TP gene resulted in generation of an amplicon from the input chromatin, but not from Sp1, Egr1 or control IgG precipitates (Fig. 4C).
Figure 4.
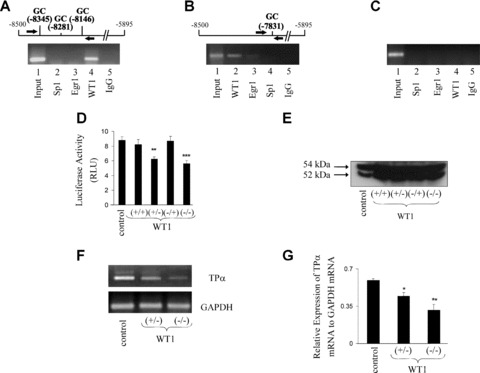
Chromatin immunoprecipitation (ChIP) analysis of WT1, Sp1 and/or Egr1 binding to the 5′ region of Prm1 and effect of overexpression of WT1 on Prm1-directed luciferase expression. (A)– (C): ChIP analysis of WT1, Sp1 and/or Egr1 binding to Prm1 in HEL 92.1.7 cells. Schematic of Prm1 and primers (arrows) used in the PCR to amplify the –8460 to –8006 (A) or the –7978 to –7607 (B) regions of Prm1 using either input chromatin or chromatin extracted from anti-WT1, anti-Egr1, anti-Sp1 or, as a control, normal rabbit IgG immunoprecipitates. Primers to detect a region of Prm3 (–1081 to –695; C) from input chromatin, anti-Sp1, anti-Egr1, anti-WT1 or normal rabbit IgG precipitates were used as a negative control. Images are representative of three independent experiments. (D) and (E): HEL cells were transiently co-transfected with either 0.5 μg of pcDNA3 (control) or 0.5 μg of recombinant pcDNA3 encoding (+/+), (+/–), (–/+) or (–/–) isoforms of WT1, along with pGL3b:Prm1 (1.5 μg) plus pRL-TK (200 ng). Cells were either assayed 48 hrs after transfection for mean luciferase activity (RLU ± S.E.M.; n= 9) or subjected to Western blot analysis (40 μg whole cell protein per lane). The size of WT1 isoforms are indicated to the left of the panel. (F) and (G): RT-PCR analysis using primers to amplify TPα and GAPDH mRNA sequences from total RNA isolated from HEL cells following transfection with 0.5 μg of pcDNA3 (control) or 0.5 μg of recombinant pcDNA3 encoding either the (+/–) or (–/–) isoform of WT1. Densitometric analysis (G) was carried out to assess the relative expression of TPα mRNA to GAPDH mRNA in HEL cells transfected with pcDNA3 (control) or recombinant pcDNA3 encoding either the (+/–) or (–/–) isoform of WT1.
Hence, to expand these studies, the effect of ectopic expression of WT1 on Prm1-directed reporter gene expression and TPα mRNA was investigated (Fig. 4D–G). The four main isoforms of WT1, specifically (+/+), (+/–), (–/+) and (–/–) with respect to the presence or absence of exon 5 and KTS sequences, respectively, were over-expressed in HEL cells (Fig. 4E) and the effects on Prm1-directed luciferase activity were investigated. While immunoblot analysis confirmed overexpression of WT1 (Fig. 4E), the (+/–) and (–/–) isoforms led to 1.4-fold (P= 0.0013) and 1.7-fold (P= 0.0007) reductions in Prm1-directed luciferase expression, respectively, while neither the (+/+) nor (–/+) isoforms had any significant effect (P= 0.4817 and P= 0.8881; Fig. 4D). Consistent with this, RT-PCR confirmed that ectopic expression of the transcriptionally active (+/–) and (–/–) isoforms both reduced TPα mRNA expression with no substantial changes in GAPDH expression (Fig. 4F). Moreover, densitometric analysis revealed that ectopic expression of (+/–) and (–/–) isoforms led to significant decreases in TPα mRNA expression relative to GAPDH mRNA expression, compared to HEL cells transfected with a control plasmid (P= 0.0167, P= 0.0080; Fig. 4G). Taken together, these data indicate that –KTS isoforms of WT1 mediate repression of Prm1 and TPα expression and considering the data from mutational, EMSA and ChIP analyses, it appears that WT1 exerts this repression by binding to GC elements at –8345, –8281, –8146 and –7831.
Identification of GC elements in the –6848 to –6648 and –6258 to –6123 repressor regions of Prm1
Amongst the transcription factor binding elements identified within URR2 located between –6848 and –6648 (Fig. 1A), was a putative GC element at –6717 predicted to represent a putative overlapping site for WT1/Egr1/Sp1. Additionally, bioinformatic analysis of the ‘core’ repressor region, referred to as RR3 and located between –6258 and –6123 of Prm1, also revealed a GC element, specifically at –6206 (Table 1). Hence, SDM was used to disrupt the putative GC−6717 and GC−6206 elements within either Prm1D (–6848) or Prm1I (–6258; Fig. 5). Mutation of the GC−6717 element within Prm1D resulted in a 4.8-fold increase in luciferase expression compared to that of the wild-type Prm1D (P < 0.0001). Mutation of the GC−6206 element within Prm1I led to a 1.3-fold increase in luciferase expression (P= 0.0083; Fig. 5A). Mutation of the same GC−6206 element within the Prm1D sub-fragment led to a 3-fold increase in luciferase activity compared to that of the wild-type Prm1D (P < 0.0001; Fig. 5B). To investigate the combined contribution of GC−6717 and GC−6206 elements in directing Prm1 activity, the effect of collectively mutating these elements within Prm1D was examined (Fig. 5B). The luciferase activity directed by Prm1DGC*(–6717,–6206), in which both GC elements at –6717 and –6206 were mutated, was significantly higher than that of either Prm1DGC*(–6717), in which the –6717 element alone was mutated, or Prm1DGC*(–6206), in which the –6206 element alone was mutated (P < 0.0001 in each case). Hence, collectively, these data indicate that GC elements at –6717 and –6206 bind factors that act independently to mediate repression of Prm1.
Figure 5.
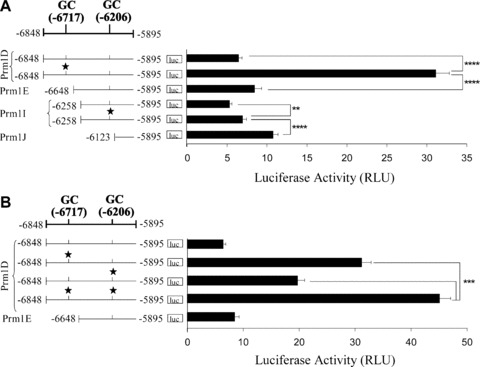
Identification of GC elements within Prm1 regions from –6848 to –6648 and –6258 to –6123. (A) and (B): Schematic of the Prm1 region from –6848 to –5895 and the relative positions of GC elements containing putative overlapping WT1/Egr1/Sp1 binding sites, where the 5′ nucleotide is indicated and the star symbol signifies mutated elements. Recombinant pGL3Basic encoding: (A) Prm1D (–6848), Prm1DGC*(–6717), Prm1E (–6648), Prm1I (–6258), Prm1IGC*(–6206) or Prm1J (–6123); or (B) Prm1D, Prm1DGC*(–6717), Prm1DGC*(–6206), Prm1DGC*(–6717,–6206) or Prm1E were co-transfected with pRL-TK into HEL 92.1.7 cells. Luciferase activity was expressed as mean firefly relative to renilla luciferase activity (RLU ± S.E.M.; n= 9).
Thereafter, EMSAs were employed to confirm the presence of nuclear factors capable of binding to the GC−6717 element in vitro. Incubation of a GC−6717 probe with nuclear extract prepared from HEL cells generated a single DNA – protein complex, C1 (Fig. 6A). C1 was efficiently competed by a non-labelled competitor containing the GC−6717 sequence, and by consensus Sp1 or Egr1 sequences, but was not competed by WTE or non-specific sequences. Despite the fact that C1 was not competed by the WTE sequence, pre-incubation of nuclear extract with anti-WT1 appeared to lead to the formation of a weak supershift complex, as well as preventing formation of C1. This suggested a role for WT1 binding to the GC−6717 probe. Moreover, addition of an anti-Egr1 antibody reduced C1 to a lesser extent, indicating that Egr1 can also bind to the GC−6717 element in vitro. However, addition of an anti-Sp1 antibody or an anti-cJun antibody did not have any substantial effects on binding to the probe (Fig. 6B). Thus, it is suggested that C1 consists of complexes of Egr1 and WT1 proteins bound to the GC−6717 probe. To investigate whether Sp1, Egr1 and/or WT1 can bind to chromatin encoding –6848 to –6648 region of Prm1 in vivo, ChIP assays were carried out using chromatin extracted from HEL cells (Fig. 6C). PCR analysis using primers specific to this region of Prm1 and containing the GC element at –6717 generated amplicons from input chromatin, anti-WT1 and, to a much lesser extent, from anti-Egr1 immunoprecipitates, but not from anti-Sp1 or the control IgG precipitates. These data provide evidence that WT1, and to a lesser extent, Egr1 occupy element(s) within the –6848 to –6648 region of Prm1 in vivo.
Figure 6.
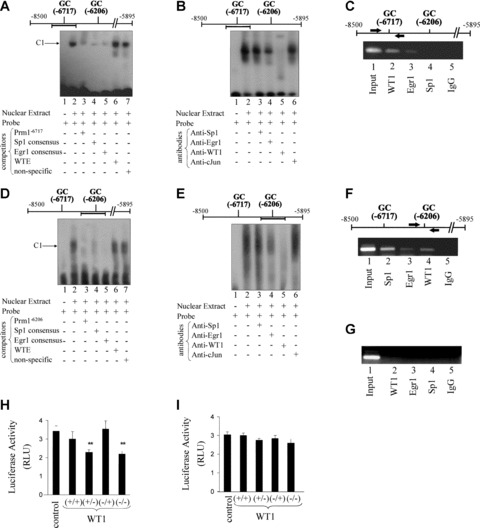
Nuclear factor binding to GC−6717 and GC−6206 elements within Prm1 in vitro and in vivo and effect of overexpression of WT1 on Prm1D and Prm1I-directed luciferase expression. (A, B, D, E): EMSAs (A and D) and supershift assays (B and E) using nuclear extract from HEL cells and a biotinylated double-stranded probe encoding (A and B) the Prm1 GC−6717 element and (D and E) the Prm1 GC−6206 element, where the location of the specific probe within Prm1 is indicated by the horizontal bar. (A) and (D): Nuclear extract was pre-incubated with vehicle (–) or excess non-labelled competitor oligonucleotides (+) before addition of the specific probe. One complex, C1, was observed in each case. (B) and (E): Nuclear extract was pre-incubated with anti-Sp1, anti-Egr1, anti-WT1 or, as a negative control, anti-cJun prior to addition of the relevant probe. The images are representative of three independent experiments. (C, F and G): chromatin immunoprecipitation (ChIP) analysis and schematic of Prm1 and primers (arrows) used in the PCR to amplify the –6848 to –6437 (C) and the –6368 to –5895 (F) regions of Prm1 from input chromatin or from chromatin extracted from anti-WT1, anti-Egr1, anti-Sp1 or, as a control, normal rabbit IgG immunoprecipitates, as indicated. Primers to detect a region of Prm3 (–1081 to –695; G) from input chromatin, anti-WT1, anti-Egr1, anti-Sp1 or normal rabbit IgG precipitates were used as a negative control. The images are representative of three independent experiments. (H and I): HEL cells were transiently co-transfected with either 0.5 μg of pcDNA3 (control) or 0.5 μg of recombinant pcDNA3 encoding (+/+), (+/–), (–/+) or (–/–) isoforms of WT1, along with pRL-TK (200 ng) and 1.5 μg of either pGL3b:Prm1D (H) or pGL3b:Prm1I (I). Cells were assayed for 48 hrs after transfection for mean luciferase activity (RLU ± S.E.M.; n= 9). The asterisks (*) indicate that overexpression of WT1 significantly reduced Prm1D-directed luciferase expression in HEL cells, where ** indicates P < 0.01.
EMSAs were also employed to investigate the presence of nuclear factors capable of binding to the GC−6206 element in vitro. Incubation of a GC−6206 probe with nuclear extract generated a single diffuse complex C1 that was efficiently competed by a non-labelled competitor containing the GC−6206 sequence, or by consensus Sp1 or Egr1 sequences, but not by WTE or non-specific sequences (Fig. 6D). Thereafter, pre-incubation of nuclear extract with an anti-WT1 antibody prevented formation of C1, indicating that WT1 binds to the GC−6206 element. Moreover, addition of an anti-Egr1 antibody also reduced C1, indicating that it may also bind to the probe. However, addition of an anti-Sp1 antibody or an anti-cJun antibody did not have any substantial effects on binding to the probe (Fig. 6E). Thus, C1 is likely to consist of Egr1 and WT1 proteins bound to the GC−6206 probe. To investigate whether Sp1, Egr1 and/or WT1 can bind to the proximal Prm1 (from –6320 to –5895) in vivo, ChIP assays were carried out (Fig. 6F). PCR generated amplicons consisting of Prm1 sequences between –6368 and –5895 from input chromatin, anti-WT1, anti-Sp1 and anti-Egr1 immunoprecipitates but not from the control IgG precipitate. Consistent with the latter findings, it has previously been established that both Sp1 and Egr1 bind to this region (–6368 to –5895) of Prm1 in vivo, where binding occurs at overlapping Sp1/Egr1 elements at –6294, –6278, –6022 and –6007 within the proximal Prm1 [41]. Herein, evidence is also presented that WT1 binds to the proximal Prm1 in vivo, and that binding of WT1 is likely to occur at the GC element at –6206, while Egr1 may also bind to this element. Conversely, as controls, primers specific to the –1081 to –695 region of Prm3 of the TP gene resulted in generation of an amplicon from the input chromatin, but not from Sp1, Egr1 or control IgG precipitates (Fig. 6G).
Thereafter, the effects of ectopic expression of WT1 on luciferase activity directed by Prm1D (–6848) and Prm1I (–6258) were investigated. The (+/+), (+/–), (–/+) and (–/–) isoforms of WT1 were over-expressed in HEL cells. The (+/–) and (–/–) isoforms reduced Prm1D-directed luciferase activity by 1.5-fold (P= 0.0074) and 1.6-fold (P= 0.0082), respectively. However, neither the exon 5(+)/KTS(+) nor exon 5(–)/KTS(+) isoforms had a significant effect on luciferase activity directed by Prm1D (P= 0.4525 and P= 0.8236, respectively; Fig. 6H). None of the four isoforms of WT1 had a significant effect on Prm1I-directed luciferase expression (P= 0.8833, P= 0.1722, P= 0.4767 and P= 0.1437; Fig. 6I). Collectively, data generated from mutational analysis, EMSAs, ChIP analysis and overexpression studies indicate that –KTS isoforms of WT1 bind to elements within URR2 (from –6848 to –6648) and RR3 (from –6258 to –6123) and act independently to repress Prm1 activity.
Discussion
In humans, TXA2 signals through two isoforms of its cognate GPCR, termed TPα and TPβ[26–28]. Imbalances in the levels of TXA2 and TP have been implicated in a number of vascular and pulmonary disorders [22, 23], as well as in inflammatory renal diseases and in renal failure [24, 25]. Since distinct promoters control transcription of TPα and TPβ[39, 40], identification of the factors regulating Prm1 and Prm3 may lead to a greater understanding of the relative extent to which TPα and TPβ contribute to such vascular and renal pathologies. Previous investigations established that AP1 and Oct-2 are the key factors regulating basal expression of Prm3 in HEL cells [44]. Moreover, specific peroxisome proliferator-activated receptor (PPAR)γ ligands [50, 51] suppressed Prm3 but had no effect on Prm1-directed luciferase expression. Recently, a study aimed at characterizing Prm1 identified the transcription factors Sp1, Egr1 and NF-E2 as having a central role in regulation of the Prm1 ‘core’ promoter under basal conditions in HEL cells. That study also identified two UAR (UAR1 and UAR2) and two URR (URR1 and URR2) within Prm1 [41]. Characterization of UAR1, from –7962 to –7717, revealed central roles for GATA-1 and Ets-1 in the upstream activation of Prm1 in HEL cells. However, the trans-acting factors regulating UAR2 (from –7717 to –7504) as well as URR1 (from –8500 to –7962) and URR2 (from –6848 to –6648) remained to be identified. In the current study, genetic reporter and 5′ deletion analyses revealed a third, previously unidentified repressor region, herein designated RR3 (from –6258 to –6123), within the core Prm1. Hence, the central aim of the current study was to identify the key factors that mediate repression of Prm1 within HEL cells, focusing on the identification of those elements and factors regulating URR1, URR2 and RR3 sequences.
Amongst the transcription factor elements identified by bioinformatic analysis of URR1 [46] were multiple GC-rich elements containing putative WT1 binding sites. WT1 mediates repression of several gene promoters, including the Egr1 promoter [15, 52], IGFI receptor [53], IGFII [54] and PDGF-A [55], as well as mediating auto-repression of its own gene [56]. Additionally, WT1 is thought to be an important factor in the regulation of haematopoiesis. Although it is highly expressed in a subset of CD34+ progenitors, it is down-regulated early in the course of differentiation of these cells [43]. Additionally, WT1 mRNA is down-regulated during induction of erythroid and megakaryocytic differentiation of the K562 cell line [57]. Recently, –KTS isoforms of WT1 have been confirmed to act as transcriptional regulators during haematopoiesis, where they activate transcription of the erythropoietin receptor [58]. Moreover, increased expression of WT1 has been reported to occur in acute human leukaemias [59].
Considering the function of WT1 as a transcriptional repressor in many cases, as well as its role in haematopoietic differentiation, it was sought to determine whether WT1 can act as a repressor of Prm1 in HEL cells. Mutation of GC-rich elements containing putative overlapping WT1/Egr1/Sp1 binding sites, specifically at –8345, –8281, –8146 and –7831, alleviated repression of Prm1. Despite the indication that these GC elements mediate repression of Prm1, collective mutation of the sites resulted in de-activation of the promoter. As outlined in the model presented in Fig. 7, these mutational analyses suggest that repressor factor(s) normally bind to neighbouring GC elements at –8345, –8281, –8146 and –7831 in a cooperative manner (Fig. 7B) and it is suggested that mutation of any of these GC elements by SDM disrupts cooperative binding, thereby alleviating repression of Prm1. In the absence of repressor binding to the remaining intact sites, it is proposed that these elements may now have an increased affinity for factors, such as Egr1 or Sp1, that mediate activation, as opposed to WT1-mediated repression of Prm1 (Fig. 7C). Therefore, it is suggested that disruption of remaining elements results in de-activation of the promoter (Fig. 7D), leading to the overall decrease in luciferase expression upon generation of Prm1GC*(–8345,–8281, –8146,–7831) from Prm1GC*(–8345) (Fig. 1C). Evidence for this proposed model of cooperative binding comes from further studies whereby disruption of GC−7831 in the Prm1B (–7962) sub-fragment, which does not contain any of the other four GC elements, actually decreased the luciferase activity directed by Prm1B (Fig. 1D). This effect is in contrast to the substantial increase in luciferase expression that occurred upon disruption of the same GC element within Prm1, where the other three GC elements at –8345, –8281 and –8146 were intact (Fig. 1B). The contrasting outcomes of disrupting the same element in two distinct Prm1 fragments with different 5′ sequences highlights the influence of cooperation among specific factors on binding to local promoter elements within Prm1.
Figure 7.
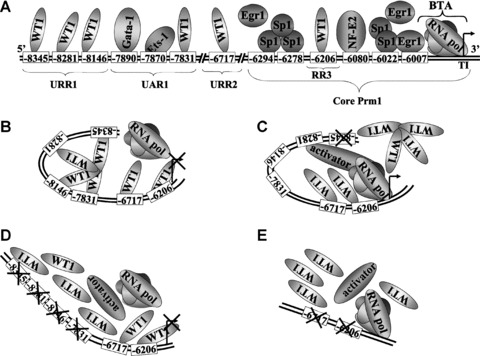
Proposed model for WT1-mediated repression of Prm1 in HEL 92.1.7 cells. (A): Schematic representation of the relative positions of functional binding elements within Prm1 (not drawn to scale), as well as binding of the basal transcription apparatus (BTA) to the transcription initiation site. Overlapping Sp1/Egr1 elements at –6294, –6278, –6022 and –6007, as well as an NF-E2 element at –6080, located within the ‘core’ proximal promoter, direct efficient basal activity of Prm1 in megakaryoblastic HEL cells. Additionally, GATA-1 and Ets-1 bind elements at –7890 and –7870, respectively, within UAR1 to increase Prm1 activity in HEL cells [41]. The data herein indicate that WT1 binds to GC elements within URR1, specifically at –8345, –8281 and –8146, as well as elements at –7831 within UAR1, –6717 within URR2 and –6206 within RR3, to repress Prm1 activity. (B, C, D and E): Proposed model for WT1- mediated repression of Prm1 in HEL cells. It is suggested that WT1 overcomes competition from other factors, such as Egr1 and Sp1, by binding cooperatively to neighbouring GC elements at –8345, –8281, –8146 and –7831 and independently to GC elements at –6717 and –6206 to mediate repression of Prm1-directed transcription by the basal transcription apparatus in HEL cells (B). Mutation of any of the upstream GC elements at –8345, –8281, –8146 and –7831 by site-directed mutagenesis interferes with cooperation among WT1 proteins binding to these elements, thereby inhibiting WT1 binding and alleviating repression of Prm1. In the absence of repressor binding to the remaining intact sites, these elements may now have a higher affinity for activating factors (C). Disruption of remaining upstream GC elements blocks the binding of activators and results in de-activation of the promoter (D). Furthermore, mutation of GC elements at –6717 and –6206 in Prm1D (–6848) alleviates repression of Prm1 (E).
EMSAs using the Egr1 consensus sequence as a non-labelled competitor suggested that each of the four aforementioned GC elements has a sequence capacity to bind Egr1 and/or WT1 (Table 2), since both Egr1 and WT1 proteins have been widely reported to bind to the consensus Egr1 sequence [11]. However, none of the complexes that bind to the four GC elements at –8345, –8281, –8146 and –7831 were competed by the WTE sequence, an element reported to be selectively bound by WT1 [12]. Despite this finding, supershift assays strongly indicated that WT1 binds to GC−8345, GC−8281, GC−8146 and GC−7831, as well as indicating a role for Egr1 binding to these elements in vitro (Table 2). Collectively, these data suggested that the latter four GC elements may have a binding affinity for WT1 isoforms that bind to Egr1 consensus elements but not to the specific WTE sequence. With respect to the latter WTE, while it has been reported to act as an effective competitor for WT1 binding within certain promoters, in the case of the aforementioned GC elements within URR1, or indeed within URR2 and RR3, it is evident that it does not act as an effective competitor. The reason for this apparent discrepancy is unclear but may, for example, reflect differences in WT1 isoform binding specificity and/or other cell-type specific effects. Not surprisingly, EMSAs and supershift assays indicated that GC−8345 and GC−8281 also have a sequence capacity to bind Sp1 (Table 2), since overlapping sites for Sp1 and Egr1/WT1 are frequently found in promoter sequences due to the similarity in their consensus elements [49]. Interestingly, it has previously been reported that Sp1 binding to the GC element at –8345 mediates increased Prm1 activity in response to phorbol 12-myristate 13-acetate in K562 cells [60]. Therefore, it is suggested that this element may play a diverse role in Prm1 regulation. Herein, ChIP analysis indicated that endogenous WT1, and to a lesser extent Egr1, but not Sp1, are bound in vivo to the Prm1 region (from –8460 to –7607) of chromatin extracted from HEL cells. Moreover, ectopic overexpression of –KTS isoforms of WT1 led to modest, but significant, decreases in Prm1-directed luciferase expression and in TPα mRNA expression. It is likely that only modest reductions in Prm1-directed luciferase expression were seen due to the already abundant endogenous expression of WT1 in HEL cells, and it is likely that overexpression of –KTS isoforms may have led to greater reductions in Prm1-directed gene expression if the total amount of transfected DNA herein was not limited by the luciferase-based reporter assay itself. Moreover, since the transcriptional effects of WT1 may be dependent on synergistic activity of more than one isoform of the protein, data from overexpression studies may not reflect the true extent of repression of Prm1 activity by WT1 [61]. Collectively, these data indicate that WT1 is the repressor factor that binds to the GC elements at –8345, –8281, –8146 and –7831. It is proposed that WT1 overcomes competition from other factors, such as Egr1 and/or Sp1, by a cooperative method of binding that relies on multiple neighbouring GC elements within Prm1 to exert its repression (Fig. 7 and Table 2). Similar models have been reported for the IGFII [54] and PDGF-A [55] gene promoters, where maximal repression by WT1 was dependent upon multiple WT1 binding sites. Other transcription factors, including the glucocorticoid receptor [62] and RUNX1 [63], have also been reported to employ a similar model for regulation of promoters that contain multiple binding elements for the specific transcription factor. It is proposed that formation of homodimers or multimers of these proteins bring together distant chromatin sites by looping to facilitate changes in transcriptional activity.
Table 2.
Factors that may compete for binding to specific GC elements within Prm1
| Element | Factors that bind to GC elements within Prm1* |
|---|---|
| GC−8345 | Sp1, Egr1 and WT1 |
| GC−8281 | Sp1, Egr1 and WT1 |
| GC−8146 | Egr1 and WT1 |
| GC−7831 | Egr1 and WT1 |
| GC−6717 | Egr1 and WT1 |
| GC−6206 | Egr1 and WT1 |
Determined from EMSA, antibody supershift assays and chromatin immunoprecipitations.
Bioinformatic analysis [46] of the two remaining repressor regions within Prm1, namely URR2, located between –6848 and –6648, and RR3, located between –6258 and –6123 within the proximal ‘core’ promoter, also revealed putative GC elements in both cases. Mutational analysis of the GC elements in both regions, specifically at –6717 and –6206, indicated that they both mediate repression of Prm1. EMSAs and supershift assays to analyse WT1/Egr1/Sp1 binding to the –6717 and –6206 elements in vitro revealed that these elements had a sequence capacity to be bound by Egr1 and WT1 (Table 2). Moreover, ChIP analysis revealed WT1 as the predominant protein bound to the –6848 to –6648 region in vivo, as well as indicating that Egr1 can bind to this region to a much lesser extent. ChIP analysis also revealed a role for WT1 binding to the ‘core’ Prm1. We have previously reported binding of both Sp1 and Egr1 to this ‘core’ region in vivo, specifically at overlapping Sp1/Egr1 elements at –6294, –6278, –6022 and –6007 [41]. Data herein suggest that WT1 also binds to the proximal Prm1, and it is likely that binding of WT1 occurs at the GC−6206 element in RR3. Moreover, overexpression of the –KTS isoforms, specifically (+exon 5/–KTS) and (–exon 5/–KTS), repressed luciferase activity directed by Prm1D (–6848). In contrast to the cooperative and co-dependent manner in which WT1 binds to GC−8345, GC−8281, GC−8146 and GC−7831, it seems that WT1 binds to the –6717 and –6206 elements independently to mediate repression of Prm1 (Fig. 7).
In the current study, it was sought to identify the key cis-acting elements and trans-acting factors of the three distinct repressor regions, URR1 (from –8500 to –7962), URR2 (from –6848 to –6648) and RR3 (from –6258 to –6123). Herein, it is reported that the repression exerted within each of the three regions in the megakaryoblastic HEL 92.1.7 cell line is largely attributable to the zinc finger transcription factor WT1. Considering the importance of TXA2 and TP within the kidney (reviewed in [20]), together with this novel role for WT1 as a repressor of Prm1, it is possible that WT1 may play a role in regulation of Prm1 and TPα expression in the renal system. Although further studies are required to investigate this hypothesis, it is suggested that abnormal Prm1 activity and TPα expression due to aberrant transcriptional regulation by WT1 may contribute to pathologies of several diseases, including Wilms’ tumour, inflammatory renal diseases and renal failure. Moreover, WT1 triggers lineage-specific differentiation of human primary haematopoietic progenitor cells [64] and WT1 mRNA is down-regulated during induction of erythroid and megakaryocytic differentiation of the K562 cell line [57]. Therefore, down-regulation of WT1 may act to increase Prm1 activity, thereby increasing TPα expression, during megakaryocytic differentiation of HEL cells. Interestingly, WT1 is a downstream target of nitric oxide signalling and WT1 represses matrix metalloproteinase-9 through a nitric oxide sensitive pathway [65]. More specifically, it is thought that nitric oxide promotes translocation of WT1 from the nucleus to the cytoplasm, thereby interfering with its transcriptional activity [65]. Notably, it has been previously established that TPα also undergoes nitric oxide mediated desensitization involving direct nitric oxide/cGMP-dependent protein kinase G phosphorylation at Ser331 within its unique C-tail domain [35]. Collectively, those latter studies and findings herein suggest that nitric oxide may regulate TPα expression and signalling by a complex mechanism involving its regulation of Prm1-directed TPα transcription by WT1 and its regulation of TPα-mediated intracellular signalling by nitric oxide/cGMP-dependent protein kinase G. NF-E2, GATA-1 and Ets-1 were previously identified as key regulators of Prm1 during megakaryocytic differentiation [41]. Collectively, the data from this and previous studies suggest that combinatorial gene regulation by WT1, GATA-1, Ets-1 and NF-E2 may be critical for regulation of TPα expression during different stages of megakaryocytic differentiation.
Acknowledgments
This work was supported by The Wellcome Trust and The Health Research Board (Ireland).
References
- 1.Ariyaratana S, Loeb DM. The role of the Wilms tumour gene (WT1) in normal and malignant haematopoiesis. Expert Rev Mol Med. 2007;9:1–17. doi: 10.1017/S1462399407000336. [DOI] [PubMed] [Google Scholar]
- 2.Roberts SG. Transcriptional regulation by WT1 in development. Curr Opin Genet Dev. 2005;15:542–7. doi: 10.1016/j.gde.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Haber DA, Buckler AJ, Glaser T, et al. An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms’ tumor. Cell. 1990;61:1257–69. doi: 10.1016/0092-8674(90)90690-g. [DOI] [PubMed] [Google Scholar]
- 4.Silberstein GB, Van Horn K, Strickland P, et al. Altered expression of the WT1 Wilms’ tumor suppressor gene in human breast cancer. Proc Natl Acad Sci USA. 1997;94:8132–7. doi: 10.1073/pnas.94.15.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oji Y, Yano M, Nakano Y, et al. Overexpression of the Wilms’ tumor gene WT1 in esophageal cancer. Anticancer Res. 2004;24:3103–8. [PubMed] [Google Scholar]
- 6.Oji Y, Nakamori S, Fujikawa M, et al. Overexpression of the Wilms’ tumor gene WT1 in pancreatic ductal adenocarcinoma. Cancer Sci. 2004;95:583–7. doi: 10.1111/j.1349-7006.2004.tb02490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies RC, Calvio C, Bratt E, et al. WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev. 1998;12:3217–25. doi: 10.1101/gad.12.20.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladomery M, Sommerville J, Woolner S, et al. Expression in Xenopus oocytes shows that WT1 binds transcripts in vivo, with a central role for zinc finger one. J Cell Sci. 2003;116:1539–49. doi: 10.1242/jcs.00324. [DOI] [PubMed] [Google Scholar]
- 9.Bor YC, Swartz J, Morrison A, et al. The Wilms’ tumor 1 (WT1) gene (+KTS isoform) functions with a CTE to enhance translation from an unspliced RNA with a retained intron. Genes Dev. 2006;20:1597–608. doi: 10.1101/gad.1402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hohenstein P, Hastie ND. The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet. 2006:R196–201. doi: 10.1093/hmg/ddl196. 15 Spec No 2: [DOI] [PubMed] [Google Scholar]
- 11.Rauscher FJ, 3rd, Morris JF, Tournay OE, et al. Binding of the Wilms’ tumor locus zinc finger protein to the EGR-1 consensus sequence. Science. 1990;250:1259–62. doi: 10.1126/science.2244209. [DOI] [PubMed] [Google Scholar]
- 12.Nakagama H, Heinrich G, Pelletier J, et al. Sequence and structural requirements for high-affinity DNA binding by the WT1 gene product. Mol Cell Biol. 1995;15:1489–98. doi: 10.1128/mcb.15.3.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han Y, San-Marina S, Liu J, et al. Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene. 2004;23:6933–41. doi: 10.1038/sj.onc.1207609. [DOI] [PubMed] [Google Scholar]
- 14.Hewitt SM, Hamada S, McDonnell TJ, et al. Regulation of the proto-oncogenes bcl-2 and c-myc by the Wilms’ tumor suppressor gene WT1. Cancer Res. 1995;55:5386–9. [PubMed] [Google Scholar]
- 15.Maheswaran S, Park S, Bernard A, et al. Physical and functional interaction between WT1 and p53 proteins. Proc Natl Acad Sci USA. 1993;90:5100–4. doi: 10.1073/pnas.90.11.5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang W, Lee SB, Palmer R, et al. A functional interaction with CBP contributes to transcriptional activation by the Wilms tumor suppressor WT1. J Biol Chem. 2001;276:16810–6. doi: 10.1074/jbc.M009687200. [DOI] [PubMed] [Google Scholar]
- 17.Carpenter B, Hill KJ, Charalambous M, et al. BASP1 is a transcriptional cosuppressor for the Wilms’ tumor suppressor protein WT1. Mol Cell Biol. 2004;24:537–49. doi: 10.1128/MCB.24.2.537-549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 19.Dorn GW, 2nd, Becker MW, Davis MG. Dissociation of the contractile and hypertrophic effects of vasoconstrictor prostanoids in vascular smooth muscle. J Biol Chem. 1992;267:24897–905. [PubMed] [Google Scholar]
- 20.Nasrallah R, Clark J, Hebert RL. Prostaglandins in the kidney: developments since Y2K. Clin Sci. 2007;113:297–311. doi: 10.1042/CS20070089. [DOI] [PubMed] [Google Scholar]
- 21.Lianos EA, Bresnahan BA. Effect of thromboxane A2 inhibition and antagonism on prostaglandin and leukotriene synthesis in glomerular immune injury. J Lab Clin Med. 1999;134:478–82. doi: 10.1016/s0022-2143(99)90169-5. [DOI] [PubMed] [Google Scholar]
- 22.Fitzgerald DJ, Roy L, Catella F, et al. Platelet activation in unstable coronary disease. N Engl J Med. 1986;315:983–9. doi: 10.1056/NEJM198610163151602. [DOI] [PubMed] [Google Scholar]
- 23.Hirsh PD, Hillis LD, Campbell WB, et al. Release of prostaglandins and thromboxane into the coronary circulation in patients with ischemic heart disease. N Engl J Med. 1981;304:685–91. doi: 10.1056/NEJM198103193041201. [DOI] [PubMed] [Google Scholar]
- 24.Coffman TM, Spurney RF, Mannon RB, et al. Thromboxane A2 modulates the fibrinolytic system in glomerular mesangial cells. Am J Physiol. 1998;275:F262–9. doi: 10.1152/ajprenal.1998.275.2.F262. [DOI] [PubMed] [Google Scholar]
- 25.Lariviere R, Moreau C, Rodrigue ME, et al. Thromboxane blockade reduces blood pressure and progression of renal failure independent of endothelin-1 in uremic rats. Prostaglandins Leukot Essent Fatty Acids. 2004;71:103–9. doi: 10.1016/j.plefa.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 26.Hirata M, Hayashi Y, Ushikubi F, et al. Cloning and expression of cDNA for a human thromboxane A2 receptor. Nature. 1991;349:617–20. doi: 10.1038/349617a0. [DOI] [PubMed] [Google Scholar]
- 27.Raychowdhury MK, Yukawa M, Collins LJ, et al. Alternative splicing produces a divergent cytoplasmic tail in the human endothelial thromboxane A2 receptor. J Biol Chem. 1994;269:19256–61. [PubMed] [Google Scholar]
- 28.Raychowdhury MK, Yukawa M, Collins LJ, et al. Alternative splicing produces a divergent cytoplasmic tail in the human endothelial thromboxane A2 receptor. J Biol Chem. 1995;270:7011. doi: 10.1074/jbc.270.12.7011. [DOI] [PubMed] [Google Scholar]
- 29.Hirata T, Ushikubi F, Kakizuka A, et al. Two thromboxane A2 receptor isoforms in human platelets. Opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J Clin Invest. 1996;97:949–56. doi: 10.1172/JCI118518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinsella BT. Thromboxane A2 signalling in humans: a ‘tail’ of two receptors. Biochem Soc Trans. 2001;29:641–54. doi: 10.1042/0300-5127:0290641. [DOI] [PubMed] [Google Scholar]
- 31.Vezza R, Habib A, FitzGerald GA. Differential signaling by the thromboxane receptor isoforms via the novel GTP-binding protein, Gh. J Biol Chem. 1999;274:12774–9. doi: 10.1074/jbc.274.18.12774. [DOI] [PubMed] [Google Scholar]
- 32.Kelley-Hickie LP, Kinsella BT. Homologous desensitization of signalling by the beta (beta) isoform of the human thromboxane A2 receptor. Biochim Biophys Acta. 2006;1761:1114–31. doi: 10.1016/j.bbalip.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 33.Kelley-Hickie LP, O’Keeffe MB, Reid HM, et al. Homologous desensitization of signalling by the alpha (alpha) isoform of the human thromboxane A2 receptor: a specific role for nitric oxide signalling. Biochim Biophys Acta. 2007;1773:970–89. doi: 10.1016/j.bbamcr.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parent JL, Labrecque P, Orsini MJ, et al. Internalization of the TXA2 receptor alpha and beta isoforms. Role of the differentially spliced cooh terminus in agonist-promoted receptor internalization. J Biol Chem. 1999;274:8941–8. doi: 10.1074/jbc.274.13.8941. [DOI] [PubMed] [Google Scholar]
- 35.Reid HM, Kinsella BT. The alpha, but not the beta, isoform of the human thromboxane A2 receptor is a target for nitric oxide-mediated desensitization. Independent modulation of Tp alpha signaling by nitric oxide and prostacyclin. J Biol Chem. 2003;278:51190–202. doi: 10.1074/jbc.M309314200. [DOI] [PubMed] [Google Scholar]
- 36.Walsh MT, Foley JF, Kinsella BT. The alpha, but not the beta, isoform of the human thromboxane A2 receptor is a target for prostacyclin-mediated desensitization. J Biol Chem. 2000;275:20412–23. doi: 10.1074/jbc.M907881199. [DOI] [PubMed] [Google Scholar]
- 37.Miggin SM, Kinsella BT. Expression and tissue distribution of the mRNAs encoding the human thromboxane A2 receptor (TP) alpha and beta isoforms. Biochim Biophys Acta. 1998;1425:543–59. doi: 10.1016/s0304-4165(98)00109-3. [DOI] [PubMed] [Google Scholar]
- 38.Habib A, FitzGerald GA, Maclouf J. Phosphorylation of the thromboxane receptor alpha, the predominant isoform expressed in human platelets. J Biol Chem. 1999;274:2645–51. doi: 10.1074/jbc.274.5.2645. [DOI] [PubMed] [Google Scholar]
- 39.Coyle AT, Miggin SM, Kinsella BT. Characterization of the 5′ untranslated region of alpha and beta isoforms of the human thromboxane A2 receptor (TP). Differential promoter utilization by the TP isoforms. Eur J Biochem. 2002;269:4058–73. doi: 10.1046/j.1432-1033.2002.03098.x. [DOI] [PubMed] [Google Scholar]
- 40.Nusing RM, Hirata M, Kakizuka A, et al. Characterization and chromosomal mapping of the human thromboxane A2 receptor gene. J Biol Chem. 1993;268:25253–9. [PubMed] [Google Scholar]
- 41.Gannon AM, Kinsella BT. Regulation of the Human Thromboxane A2 Receptor Gene by Sp1, Egr1, NF-E2, GATA-1 and Ets-1 in Megakaryocytes. J Lipid Res. 2008;49:2590–604. doi: 10.1194/jlr.M800256-JLR200. [DOI] [PubMed] [Google Scholar]
- 42.Hosen N, Sonoda Y, Oji Y, et al. Very low frequencies of human normal CD34+ haematopoietic progenitor cells express the Wilms’ tumour gene WT1 at levels similar to those in leukaemia cells. Br J Haematol. 2002;116:409–20. doi: 10.1046/j.1365-2141.2002.03261.x. [DOI] [PubMed] [Google Scholar]
- 43.Maurer U, Brieger J, Weidmann E, et al. The Wilms’ tumor gene is expressed in a subset of CD34+ progenitors and downregulated early in the course of differentiation in vitro. Exp Hematol. 1997;25:945–50. [PubMed] [Google Scholar]
- 44.Coyle AT, Kinsella BT. Characterization of promoter 3 of the human thromboxane A receptor gene. A functional AP-1 and octamer motif are required for basal promoter activity. Febs J. 2005;272:1036–53. doi: 10.1111/j.1742-4658.2004.04538.x. [DOI] [PubMed] [Google Scholar]
- 45.Tajinda K, Carroll J, Roberts CT., Jr Regulation of insulin-like growth factor I receptor promoter activity by wild-type and mutant versions of the WT1 tumor suppressor. Endocrinology. 1999;140:4713–24. doi: 10.1210/endo.140.10.7065. [DOI] [PubMed] [Google Scholar]
- 46.Quandt K, Frech K, Karas H, et al. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–84. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobashi Y, Kudoh T, Ishidate T, et al. The Wilms tumor protein is persistently associated with the nuclear matrix throughout the cell cycle. Mol Cell Biochem. 1997;171:121–6. doi: 10.1023/a:1006884527897. [DOI] [PubMed] [Google Scholar]
- 48.Cheng T, Wang Y, Dai W. Transcription factor egr-1 is involved in phorbol 12-myristate 13-acetate-induced megakaryocytic differentiation of K562 cells. J Biol Chem. 1994;269:30848–53. [PubMed] [Google Scholar]
- 49.Huang RP, Fan Y, Ni Z, et al. Reciprocal modulation between Sp1 and Egr-1. J Cell Biochem. 1997;66:489–99. [PubMed] [Google Scholar]
- 50.Coyle AT, O’Keeffe MB, Kinsella BT. 15-deoxy Delta12,14-prostaglandin J2 suppresses transcription by promoter 3 of the human thromboxane A2 receptor gene through peroxisome proliferator-activated receptor gamma in human erythroleukemia cells. FEBS J. 2005;272:4754–73. doi: 10.1111/j.1742-4658.2005.04890.x. [DOI] [PubMed] [Google Scholar]
- 51.Coyle AT, Kinsella BT. Synthetic peroxisome proliferator-activated receptor gamma agonists rosiglitazone and troglitazone suppress transcription by promoter 3 of the human thromboxane A2 receptor gene in human erythroleukemia cells. Biochem Pharmacol. 2006;71:1308–23. doi: 10.1016/j.bcp.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 52.Madden SL, Cook DM, Morris JF, et al. Transcriptional repression mediated by the WT1 Wilms tumor gene product. Science. 1991;253:1550–3. doi: 10.1126/science.1654597. [DOI] [PubMed] [Google Scholar]
- 53.Damon SE, Plymate SR, Carroll JM, et al. Transcriptional regulation of insulin-like growth factor-I receptor gene expression in prostate cancer cells. Endocrinology. 2001;142:21–7. doi: 10.1210/endo.142.1.7890. [DOI] [PubMed] [Google Scholar]
- 54.Drummond IA, Madden SL, Rohwer-Nutter P, et al. Repression of the insulin-like growth factor II gene by the Wilms tumor suppressor WT1. Science. 1992;257:674–8. doi: 10.1126/science.1323141. [DOI] [PubMed] [Google Scholar]
- 55.Gashler AL, Bonthron DT, Madden SL, et al. Human platelet-derived growth factor A chain is transcriptionally repressed by the Wilms tumor suppressor WT1. Proc Natl Acad Sci USA. 1992;89:10984–8. doi: 10.1073/pnas.89.22.10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rupprecht HD, Drummond IA, Madden SL, et al. The Wilms’ tumor suppressor gene WT1 is negatively autoregulated. J Biol Chem. 1994;269:6198–206. [PubMed] [Google Scholar]
- 57.Phelan SA, Lindberg C, Call KM. Wilms’ tumor gene, WT1, mRNA is down-regulated during induction of erythroid and megakaryocytic differentiation of K562 cells. Cell Growth Differ. 1994;5:677–86. [PubMed] [Google Scholar]
- 58.Kirschner KM, Hagen P, Hussels CS, et al. The Wilms’ tumor suppressor Wt1 activates transcription of the erythropoietin receptor in hematopoietic progenitor cells. FASEB J. 2008;22:2690–701. doi: 10.1096/fj.07-097576. [DOI] [PubMed] [Google Scholar]
- 59.Miwa H, Beran M, Saunders GF. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia. 1992;6:405–9. [PubMed] [Google Scholar]
- 60.D’Angelo DD, Oliver BG, Davis MG, et al. Novel role for Sp1 in phorbol ester enhancement of human platelet thromboxane receptor gene expression. J Biol Chem. 1996;271:19696–704. doi: 10.1074/jbc.271.33.19696. [DOI] [PubMed] [Google Scholar]
- 61.Rae FK, Martinez G, Gillinder KR, et al. Anlaysis of complementary expression profiles following WT1 induction versus repression reveals the cholesterol/fatty acid synthetic pathways as a possible major target of WT1. Oncogene. 2004;23:3067–79. doi: 10.1038/sj.onc.1207360. [DOI] [PubMed] [Google Scholar]
- 62.Adams M, Meijer OC, Wang J, et al. Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol Endocrinol. 2003;17:2583–92. doi: 10.1210/me.2002-0305. [DOI] [PubMed] [Google Scholar]
- 63.Li D, Sinha KK, Hay MA, et al. RUNX1-RUNX1 homodimerization modulates RUNX1 activity and function. J Biol Chem. 2007;282:13542–51. doi: 10.1074/jbc.M700074200. [DOI] [PubMed] [Google Scholar]
- 64.Ellisen LW, Carlesso N, Cheng T, et al. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 2001;20:1897–909. doi: 10.1093/emboj/20.8.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marcet-Palacios M, Ulanova M, Duta F, et al. The transcription factor Wilms tumor 1 regulates matrix metalloproteinase-9 through a nitric oxide-mediated pathway. J Immunol. 2007;179:256–65. doi: 10.4049/jimmunol.179.1.256. [DOI] [PubMed] [Google Scholar]
- 66.Rajput B, Shaper NL, Shaper JH. Transcriptional regulation of murine beta1,4-galactosyltransferase in somatic cells. Analysis of a gene that serves both a housekeeping and a mammary gland-specific function. J Biol Chem. 1996;271:5131–42. doi: 10.1074/jbc.271.9.5131. [DOI] [PubMed] [Google Scholar]