

Abstract
Proteinases have been implicated in the mobilization of haematopoietic progenitor cells (HPCs) from the bone marrow (BM). Here, we report the involvement of the plasminogen (Plg) system in the haematopoietic recovery following chemotherapy. By using gene-deficient mice, we found that plasmin and its activators tPA and uPA play a role in the haematopoietic recovery upon delivery of the cytotoxic agent 5-fluoro-uracil (5-FU). The impaired haematopoietic recovery of Plg-deficient (Plg−/−) mice after 5-FU was not rescued by depletion of fibrinogen, indicating that it was not due to defective fibrinolysis. Instead, loss of Plg impaired breakdown of fibronectin, VCAM-1 and laminin-BM matrix proteins involved in adhesion of HPCs to their BM microenvironment and in transendothelial migration of HPCs. These findings provide novel insights in how plasmin regulates haematopoietic recovery upon cytotoxic myeloablation.
Keywords: plasmin, fibrinolysis, extracellular matrix remodelling, bone marrow, progenitors
Introduction
Recent studies reveal that haematopoietic progenitor cells (HPCs) sense signals from the bone marrow (BM) microenvironment [1]. In response to cytotoxic stress, cells in the BM secrete proteinases such as MMP-9 [2, 3]. These enzymes proteolytically inactivate adhesive signals including membrane-bound KitL (mKitL), cKit, SDF-1 and CXCR-4 that anchor HPCs in the BM microenvironment; their inactivation allows HPCs to proliferate and become released in the peripheral blood [2, 3].
Plasminogen (Plg) is activated to plasmin via proteolytic cleavage by two plasminogen activators (PA), i.e. tissue-type PA (tPA) and urokinase-type PA (uPA); the latter binds to membrane-anchored uPAR (uPAR). We recently found that uPAR is a novel adhesive signal for HPCs, and that plasmin inactivates uPAR, thereby enabling mobilization of HPCs from the BM [4]. Besides, plasmin also inactivates another adhesive signal for HPCs, namely mKitL, which could also contribute to the impaired haematopoietic recovery upon loss of Plg [4].
However, we explored in this study whether plasmin might regulate haematopoietic recovery via (yet) additional mechanisms. Indeed, plasmin is capable of cleaving a wide spectrum of substrates, including cell surface receptors, extracellular matrix (ECM) proteins and fibrin (reviewed in [5]). Several of these plasmin targets are present in the BM and regulate haematopoietic recovery. For instance, VCAM-1 and the ECM proteins fibronectin and laminin ensure adhesion of HPCs in the BM microenvironment, whereas their breakdown facilitates HPC migration to the peripheral blood and repopulation of blood cells [2, 6–12]. In addition, fibrinogen supports the proliferation and differentiation of HPCs in vitro[13–15]. In the BM, fibrinogen is localized at the luminal side of sinusoidal vessels, where it might promote maturation of megakaryocytes [16].
It remains unknown whether, in response to cytotoxic stress, plasmin affects haematopoiesis via mechanisms other than cleavage of uPAR and mKitL, for instance via fibrinolysis. This is a relevant question, as health and survival of Plg−/− mice are improved after inter-crossing with mice lacking fibrinogen (Fbg−/−), indicating a critical role of the fibrinolytic system [17]. In addition, based on fibrin(ogen) immunostaining results, a recent paper suggested that fibrin(ogen) might play a role in haematopoi-etic regeneration following chemo-myeloablation, however, without providing functional evidence for a role of fibrinolysis [18]. It also remains outstanding whether plasmin cleaves any of the abovemen-tioned ECM proteins, onto which HPCs adhere and are retained.
Even though administration of exogenous tPA improved haematopoietic recovery [18], it still remains unknown whether the endogenous PA are important in this process and, if so, whether tPA or uPA are relevant. We therefore analysed the role of the PA in the response to cytotoxic stress in further detail. Since initial experiments revealed that loss of tPA or uPA did impair haematopoietic recovery (unlike what has been reported recently [18]), we analysed haematopoietic recovery in mice lacking tPA and/or uPA, and, for reasons of comparison and in order to study the role of fibrinolysis, also in mice lacking Plg, as such and after depletion of fibrinogen.
Experimental methods
Animal studies
Wild-type (WT) mice and mice lacking Plg, uPA, tPA, and both uPA and tPA (all in mixed 75% C57Bl6 25% 129Sv background) were used. For all experiments, age-, gender- and strain-matched mice were used. Mice were maintained in HEPA-filtered IVC units. All experiments were performed according to the guidelines for care and use of laboratory animals approved by the institutional ethical animal care committee. Mice were injected with a bolus i.v. of 5-FU (150 or 200 mg/kg, Fluroblastin®, Pharmacia, Brussels, Belgium). Peripheral blood was repetitively sampled by retro-orbital puncture under light anaesthesia, and full blood counts (EDTA buffered) were determined on a haemocytometer (Cell-Dyn 1300, Abbott, Louvain-la-Neuve, Belgium). Tranexamic acid (Exacyl®, Bournonville, Brussels, Belgium) was administered via osmotic minipumps (1.8 mg/day; Alzet 2001, Charles River, Brussels, Belgium) and via drinking water (20 mg/ml), as described [19]. Ancrod (1 IU; NIBSC, Hertfordshire, UK) was administered via daily i.p. injections, as described [20, 36].
FACS analysis
Murine BMCs were filtered through a 40 or 70 μm nylon mesh (Falcon, VWR, Haasrode, Belgium). To quantify Lin−Sca1+ BMCs, we combined magnetic bead depletion (EasySep, Stem Cell Technologies, Grenoble, France) with flow cytometry. Staining was performed with PE- or FITC-labelled anti-mouse antibodies against the following antigens: Sca-1, BrdU (all from BD Biosciences, Erembodegem, Belgium), and cKit (eBioscience, Halle-Zoersel, Belgium). Control stainings included appropriate isotype control antibodies. Non-specific binding was prevented by addition of mouse serum (DakoCytomation, Heverlee, Belgium), as alternative for Fc-receptor block. Apoptosis was analysed using TUNEL staining (fluorescein in situ cell death detection kit, Roche, Brussels, Belgium), as described previously [21]. For cell cycle analysis, BM cells were stained with primary antibodies, fixed in 70% ethanol and treated with propidium iodide (PI/RNAse, BD Biosciences), as previously described [2]. To determine HSC proliferation, mice were injected i.p. with BrdU (1 mg) every 8 hrs and the number of BrdU+ Lin−Sca1+ BMCs were quantified by flow cytometry, as described previously [22].
Immunohistochemistry
Mice were killed by cervical dislocation, the femurs were removed, fixed in 2% paraformaldehyde in phosphate buffer solution (PBS) for 24 hrs, and decalcified in 0.5 M EDTA solution for 8 days. After dehydration and paraffin embedding, 10 μm longitudinal sections were prepared on Superfrost Plus slides. Immunohistochemistry was performed using antibodies against tPA, uPA (both from Santa Cruz, Boechout, Belgium), laminin and fibronectin (both from Sigma, Bornem, Belgium), and fibrin (ogen) (Nordic, Tilburg, Netherlands). Specificity for PA staining was performed with the use of deficient mice. Corresponding secondary antibodies labelled with HRP or biotin (for signal amplification via TSA (Perkin Elmer, Zaventem, Belgium) or via Vectastain ABC kit (Vector Laboratories, Brussels, Belgium) were used. For light microscopy, sections were developed with 3,3’-diaminobenzidine (DAB, Sigma) as a chromogen substrate and counterstained with Harris Haematoxilin. For fluorescence imaging, sections were counterstained with DAPI and mounted with Vectashield (Vector Laboratories). Analysis was performed on a Zeiss Axioplan2 connected to a 3CCD video camera (DXC-93OP, Sony), and KS300 software (Zeiss, Zaventem, Belgium). Laminin or fibronectin immunopositivity was quantified by integration of the stained area and expressed as the ratio of the immunopositive area over the BM matrix and near the endosteum, respectively.
ELISA, reagents, protease activity and Western blotting
BM extracellular fluid (BM plasma) from mice was obtained as described previously [23]. Briefly, femurs were flushed with PBS; after centrifugation, the supernatant was collected and frozen for analysis. Total protein amounts on BM plasma samples were determined with the BCA protein analysis kit (Perbio, Erembodegem, Belgium), to control for loading. Murine sVCAM-1 protein levels were quantified in BM plasma samples using a commercially available ELISA (R&D Systems, Abingdon, UK), which uses antibodies specifically raised against sVCAM-1 according to manufacturer. Western blotting was performed on reduced BM plasma samples, which were neutralized with a cocktail of protease inhibitors (Complete Inhibitor + EDTA; Roche). ECM components were detected with rabbit antibodies against fibronectin and laminin (both from Sigma), appropriate secondary HRP-labelled antibodies (DakoCytomation) and the ECL detection system (Amersham Biosciences, Diegem, Belgium). Notably, we used an affinity purified rabbit anti-laminin polyclonal antibody from Sigma, which specifically recognizes (vascular) basement membrane laminins including the beta1 and gamma1 chain, and its plasmin cleavage products ([24, 25] and instructions of the manufacturer). We used an affinity purified rabbit anti-fibronectin polyclonal antibody from Sigma, which specifically recognizes fibronectin and its plasmin cleavage products ([25] and instructions of the manufacturer).
In vitro cleavage experiments
For studying in vitro cleavage of membrane-bound VCAM-1, we used the mouse BM cell line OP9, which expressed VCAM-1 as determined on flow cytometry (not shown). Cells were seeded in 48-well plate (2.5 × 105 cells per well), allowed to adhere, and starved overnight in serum-free medium. Thereafter, cells were stimulated with human active plasmin (1 nM), diluted in serum-free medium, and incubated at 37°C for 5 hrs. This protocol yielded reproducible plasmin activity (assayed by S-2403) without affecting cellular viability (not shown). Levels of VCAM-1 in cell lysates and conditioned medium were quantified using commercially available ELISAs (R&D Sytems).
Statistics
We used SPSS v. 11.0 for statistical calculations. Unless stated otherwise, data (mean ± S.E.M.) were statistically analysed by an unpaired Student’s t-test. To determine the genotypic differences in white blood cell (WBC) counts after 5-FU, an ANOVA for repeated measurements was used, complemented with Dunnett’s multiple comparison t-test (correction for multiple testing using a single control group) to identify statistically significant genotypic differences at each individual time point. Cox regression was used to analyse the genotypic differences in survival after 5-FU. P < 0.05 was considered statistically significant.
Results
Expression of PA in the BM
Proteinases have been implicated in the haematopoietic recovery of 5-fluorouracil (5-FU)-induced myeloablation [2]. To evaluate a possible role of the Plg system, we determined the expression pattern and levels of its components in the BM of WT mice. Integrin expression in stem cells from bone marrow and adipose tissue during chondrogenic differentiation Initial immuno-histochemical experiments revealed that uPA was expressed in stromal and haematopoietic cells in steady-state conditions and, even stronger so after 200 mg/kg 5-FU (Fig. 1A). Expression of tPA was largely, though not exclusively, confined to endothelial sinusoids in steady-state conditions (Fig. 1A). After 200 mg/kg 5-FU, a weaker tPA signal was detected in these cells, presumably because endothelial cells were destroyed by 5-FU (not shown) [26]; interestingly, however, tPA staining was stronger in cells lining the endosteum (putatively identified as osteoblasts) after 5-FU (Fig. 1A). We next analysed, by casein zymography, whether activity levels of tPA and uPA in the BM plasma of WT mice changed during haematopoietic recovery in response to 5-FU. As a result of the BM damage, tPA and uPA levels were reduced at 7 days after 200 mg/kg 5-FU (Fig. 1B). Indeed, compared with day 0, both tPA and uPA activity were reduced at 7 days after 5-FU (in artificial units: tPA: 90 ± 10 at 0 days versus 30 ± 20 at 7 days; uPA: 300 ± 10 at 0 days versus 160 ± 60 at 7 days; n= 4: P≤ 0.05). However, during the recovery period from 10–14 days onwards, tPA and, especially, uPA levels were elevated above steady-state (Fig. 1B).
Figure 1.
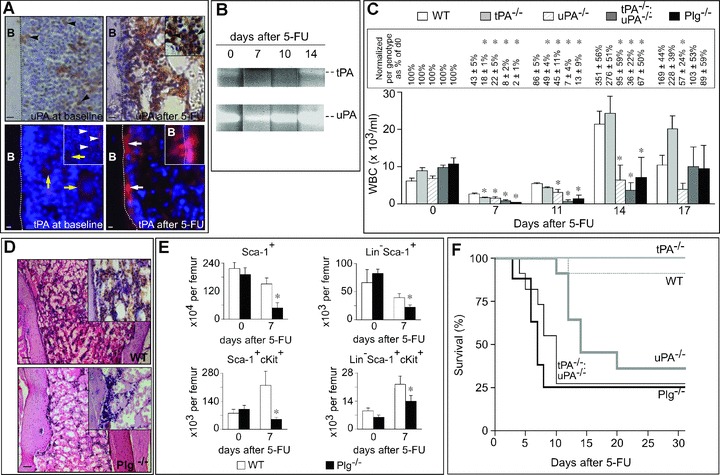
Impaired haematopoietic recovery after 5-FU in Plg−/− mice. (A) Immunostaining for uPA and tPA on longitudinal sections through the femur of WT mice. uPA immunostaining (brown) in steady-state conditions (upper left) and after 5-FU (upper right) revealed that uPA was expressed by stromal and haematopoietic cells (black arrowheads, and inset). Note the enhanced expression of uPA in BM stroma after 5-FU. Immunostaining for tPA (red) in steady-state conditions (lower left) revealed expression of tPA predominantly in sinusoids (white arrowheads; inset) and in megakaryocytes (yellow arrows). In contrast, 5-FU (lower right) increased tPA expression in cells lining the endosteum (white arrows); high magnification imaging showed that cells lining the endosteum (DAPI nuclear counterstaining; blue) indeed expressed tPA (inset). The dotted line demarcates the border between the cortical bone (left) and BM (right). Magnification bars: 50 μm. (B) Casein zymography, revealing an initial reduction (7 days) and subsequent increase (14 days) in tPA and uPA activity (evidenced by the lysis and clearing of the gel) in BM plasma of WT mice after 5-FU. Each BM plasma sample loaded on the gel contains an equal amount of protein. Representative example of 4 experiments. (C) White blood cell (WBC) counts in WT, uPA−/−, tPA−/−, tPA−/−uPA−/− and Plg−/− mice at the indicated days after 5-FU (n= 8–12). P < 0.05 for all genotypes versus WT (anova). *: P < 0.05 versus WT per time point (Dunnett). The values (%) above the bars in the text box indicate the percentage reduction as compared to the respective steady-state value for each genotype. (D) Representative pictures from BM sections (H&E) of WT (upper) and Plg−/− (lower) mice at day 7 after 5-FU (200 mg/kg i.v.), revealing impaired repopulation of the bone marrow in Plg−/− mice. Note the hypocellular BM in Plg−/− mice. Magnification bars: 100 μm. (E) The number of HPCs in the BM of WT and Plg−/− mice in steady-state conditions and at 7 days after 5-FU (200 mg/kg i.v.). Upper: Sca-1+ and Lin−Sca-1+ HPCs. Lower: Sca-1+cKit+, Lin−Sca-1+cKit+ HPCs. *: P < 0.05 versus WT (n= 3–8). (F) Survival of WT, uPA−/−, tPA−/−, tPA−/−uPA−/− and Plg−/− mice after 200 mg/kg 5-FU (n= 8–12). P < 0.05 for Plg−/−, tPA−/−uPA−/− and uPA−/− versus WT (Cox regression).
Loss of Plg or its activators impairs haematopoietic recovery
We first characterized the haematopoietic response to 5-FU in WT mice and in mice lacking plasminogen (Plg−/−), which are unable to generate plasmin [27]. To avoid any confounding interpretation due to the progressive development of morbidity in Plg−/− mice beyond 16 weeks of age, we consistently used 9- to 12-week-old healthy Plg−/− mice without overt signs of morbidity. No major haematopoietic defects were found in these mice in steady-state conditions (not shown). Administration of 200 mg/kg 5-FU i.v. to WT mice resulted in an initial reduction in WBC counts on day 7, followed by a rebound leukocytosis by day 14, which normalized at day 17 (Fig. 1C). By contrast, 5-FU induced a much more profound leukopenia in Plg−/− mice on day 7 and 11, without rebound leukocytosis at all on day 14 (Fig. 1C), indicating that plasmin participates in myeloid recovery after 5-FU. Similar data were obtained when WT mice were treated with the plasmin inhibitor tranexamic acid (Fig. S1). Microscopic analysis revealed a more severe BM aplasia in Plg−/− than WT mice, 7 days after 5-FU (Fig. 1D). Also, FACS analysis for Sca-1+, Sca-1+cKit+, Lin−Sca-1+ or Lin−Sca1+cKit+ cells revealed that the number of HPCs was significantly reduced in the BM of Plg−/− mice after 5-FU (Fig. 1E). This deficit was, however, not caused by genotypic differences of HPC counts in the BM in steady-state conditions (Fig. 1E). Additional experiments revealed that the reduced HPC counts in the BM of Plg−/− mice after 5-FU was at least in part explained by impaired HPC proliferation (Fig. S2). Also, up to 75% of Plg−/− mice died already from day 4 after 5-FU onwards, whereas only 10% of WT mice succumbed, and only at later times (Fig. 1F). As found previously with this mouse model [2], the WBC counts normalized in the few surviving Plg−/− mice (and tPA−/−:uPA−/− mice; see below) at 17 days after 5-FU (Fig. 1C). Thus, plasmin proteolysis plays a role in the haematopoietic recovery in response to 5-FU.
We also analysed the response to 5-FU in mice lacking uPA (uPA−/−), tPA (tPA−/−), or both activators (tPA−/−uPA−/−). No major haematopoietic defects were found in these mice in steady-state conditions (not shown). Compared with WT mice, WBC counts in tPA−/−uPA−/− mice were significantly lower at day 7 and 11 after administration of 200 mg/kg 5-FU, and failed to become compensatorily up-regulated by day 14 (Fig. 1C). WBC counts were also lower in uPA−/− and tPA−/− mice at 7 days after 5-FU (Fig. 1C) and WBC recovery remained impaired in uPA−/− mice (Fig. 1C). In contrast to WT mice, up to 75% of tPA−/−:uPA−/− mice died already from day 4 after 5-FU onwards, whereas a large fraction (60%) of uPA−/− mice died at 10 days after 5-FU (Fig. 1F). tPA−/− mice survived (Fig. 1F), presumably because loss of tPA promotes megakaryopoiesis after 5-FU (Note S1). Thus, uPA and tPA play a role in haematopoietic recovery after cytotoxic therapy.
Plasmin stimulates haematopoietic recovery, independently of fibrinolysis
To investigate whether plasmin stimulates haematopoietic recovery by promoting fibrinolysis (which is defective in Plg−/− mice [5, 17, 27]), we performed the following experiments. We first analysed by immunostaining the expression pattern of fibrin(ogen) in the BM in steady-state conditions and after 5-FU. When using an antibody that recognizes both fibrinogen and fibrin, a fibrin(ogen) immunoreactive signal was detected mainly at the luminal side of sinusoidal BM vessels in steady-state conditions (Fig. 2A), consistent with previous reports [16]. After 5-FU, fibrin(ogen) immunoreactivity was also detectable in the BM matrix (Fig. 2A), likely caused by extravasation of plasma proteins through damaged vessels following cytotoxin exposure [26]. A similar pattern of fibrin(ogen) staining was observed in Plg−/− mice (Fig. 2A). Consistent with previous findings [27], we did not detect intravascular fibrin clots nor excessive deposition of fibrin(ogen) in the BM of Plg−/− mice (Fig. 2A, Fig. S3).
Figure 2.
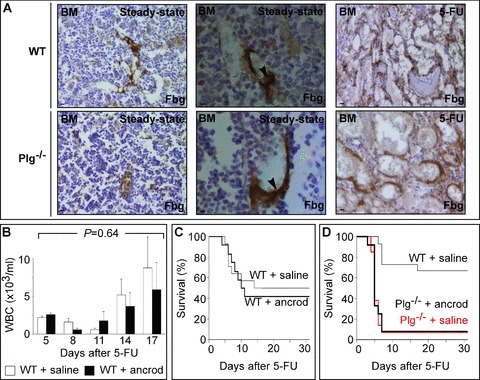
Role of fibrin(ogen) and fibrinolysis in haematopoietic recovery after 5-FU. (A) Immunostaining for fibrin(ogen) (brown) on longitudinal sections through the femur of WT (upper) and Plg−/− (lower) mice. Note that no genotypic differences were found. Left column: in steady-state conditions, fibrin(ogen) was predominantly expressed at the luminal side of sinusoidal BM vessels. Middle column: in steady-state conditions, we did not observe intravascular thrombi. Right column: after 5-FU, expression of fibrin(ogen) increased in the BM matrix, likely because of extravasation of plasma fibrinogen through the dilated leaky sinusoids. Magnification bars: 50 μm in outer panels and 10 μm in middle panels. (B) WBC counts in WT mice, treated with daily i.p. injections of vehicle or ancrod, at the indicated days after 5-FU (200 mg/kg i.v.). P= NS versus vehicle (ANOVA); P= NS versus vehicle per time point (n= 10–13). (C) Compared with vehicle, survival of ancrod-treated WT mice after 5-FU (200 mg/kg i.v.) was similar. Please note that, compared to Fig. 1F, the survival of control mice was reduced, likely because of the daily i.p. injections. P= NS versus vehicle (Cox regression; n= 10–13). (D) Compared with WT mice, survival of vehicle- or ancrod-treated Plg−/− mice after 5-FU (150 mg/kg i.v.) was reduced. Please note the similar survival curves for vehicle- and ancrod-treated Plg−/− mice. P < 0.05 versus WT (Cox regression; n= 10–12).
We then analysed whether depletion of fibrinogen would rescue the haematopoietic recovery defect in Plg−/− mice (as one would expect in case impaired fibrinolysis contributes to the impaired haematopoietic recovery in Plg−/− mice), but first assessed whether depletion of fibrinogen itself would alter haematopoietic recovery in response to 5-FU. We therefore treated WT mice with ancrod – a snake venom that depletes fibrinogen in blood and tissues by more than 90%[20, 36]. Initial studies revealed that, consistent with previous studies [20, 36], daily intraperitoneal injection of 1 IU ancrod effectively reduced the levels of fibrinogen in blood and BM plasma below the detection limit, but did not affect peripheral blood parameters including WBC and platelet counts (not shown). Overall, depletion of fibrinogen did not alter the haematopoietic recovery of WT mice in response to 5-FU (200 mg/kg) (n= 10; P= 0.64 by anova; Fig. 2B). Ancrod did also not affect the survival of WT mice after 5-FU (Fig. 2C). We then treated Plg−/− mice with ancrod and analysed their response to 5-FU. Ancrod did not alter the impaired haematopoietic recovery and survival after 5-FU (150 mg/kg) in Plg−/− mice (Fig. 2D and not shown). Hence, the haematopoietic recovery defect in Plg−/− mice was likely not attributable to excessive fibrin depositions, and fibrinolysis appears not to be required for haematopoietic recovery.
Plasmin remodels the BM microenvironment after 5-FU
Since plasmin did not regulate haematopoietic recovery through fibrinolysis, we explored alternative mechanisms. Haematopoietic recovery after 5-FU requires breakdown of ECM proteins such as fibronectin and VCAM-1, onto which HPCs adhere [7], or laminin around BM sinusoidal vessels [28, 29], which impedes transendothelial migration and mobilization of HPCs [8–10]. Degradation of these ECM proteins facilitates the release and migration of HPCs, required for repopulating the BM and peripheral blood [2, 6–12]. Plasmin-mediated matrix breakdown facilitates cell migration in diverse conditions [30, 31], whereas plasmin is capable of cleaving fibronectin, laminin and VCAM-1 in vitro ([32]; Note S2). Since reduced degradation of these ECM proteins in Plg−/− mice might therefore contribute to their impaired haematopoietic recovery after 5-FU, we analysed whether plasmin was involved in the degradation of fibronectin, laminin and VCAM-1 in the BM after 5-FU in vivo.
Since it is unknown whether 5-FU alters the amount, deposition and localization of fibronectin, laminin and VCAM-1 in the BM microenvironment, we first performed immunostaining for fibronectin and laminin on BM sections, and measured by ELISA sVCAM-1 levels in BM plasma of WT mice after 200 mg/kg 5-FU. In steady-state conditions, fibronectin was detected near the endosteum, whereas laminin was predominantly found around sinusoidal vessels, as reported previously [28] (Fig. 3A). At day 7 after 5-FU, the fibronectin-immunoreactive signal near the endosteum and the laminin-positive signal around the sinusoidal vessels were much weaker (Fig. 3A), but became nearly normal again by 14 days after 5-FU (Fig. 3A). sVCAM-1 levels in the BM plasma became transiently increased by ∼1.6-fold on day 7 after 5-FU (% of baseline levels: 157 ± 18% at 7 days and 85 ± 14% at 10 days after 5-FU; n= 4; P < 0.05 on day 7). Hence, administration of 5-FU appears to result in the degradation of fibronectin, laminin and VCAM-1 in the BM microenvironment.
Figure 3.
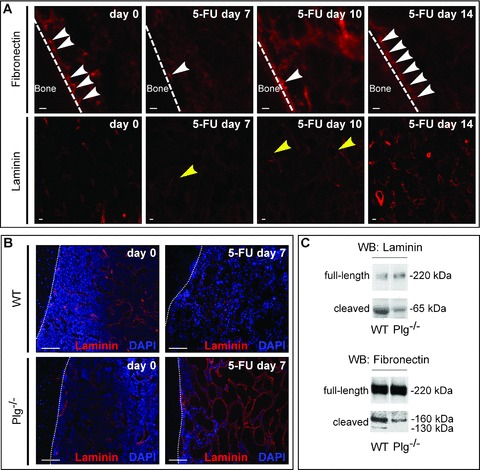
Impaired ECM remodelling after 5-FU in Plg−/− mice. (A) Immunostaining (red) of fibronectin (upper) and laminin (lower) on longitudinal sections through the femur of WT mice at different time points after 5-FU (200 mg/kg i.v.). Upper: Note the loss of fibronectin expression near the endosteum (white arrow heads) at 7 and 10 days after 5-FU. The dotted line demarcates the border between the cortical bone (left) and BM (right). Lower: Note the loss of laminin expression around the sinusoids (yellow arrow heads) at 7 and 10 days after 5-FU. Magnification bars: 10 μm. (B) Laminin immunostaining (red) and nuclear DAPI staining (blue) of longitudinal sections through the femur, revealing a comparable pattern of laminin immunoreactivity around sinusoids in WT (upper left) and Plg−/− (lower left) mice in steady-state conditions. At 7 days after 5-FU, laminin is more abundant around and in the interstitial space between the vascular sinusoids in Plg−/− (lower right) than WT (upper right) mice. The dotted line demarcates the border between the cortical bone (left) and BM (right). Magnification bars: 100 μm. (C) Immunoblots, showing less degradation of fibronectin and laminin in BM plasma samples of Plg−/− than WT mice after 5-FU. The apparent molecular weights of the degradation products of fibronectin and laminin, cleaved by plasmin, are consistent with previous reports [25, 32]. Laminin: intact beta/gamma form (220 kD), plasmin cleavage product (65 kD); fibronectin: intact monomer (220 kD); plasmin cleavage product (160 and 130 kD).
We then morphometrically quantified the amount of immunoreactive fibronectin and laminin in the BM of Plg−/− mice at day 7 after 200 mg/kg 5-FU. No genotypic differences were detected in baseline conditions (Fig. 3B; not shown). Substantially larger deposits of fibronectin were detected in Plg−/− mice at day 7 after 5-FU (% fibronectin+ area: 24 ± 7% in Plg−/− mice versus 7 ± 2% in WT mice; n= 4; P < 0.05). Laminin was also more abundant in 5-FU-treated Plg−/− mice (laminin+ area: 11.8 ± 3.8% in Plg−/− mice versus 4.5 ± 1.8% in WT mice; n= 4; P < 0.05; Fig. 3B), suggesting that loss of plasmin impaired degradation of these ECM proteins. Immunoblotting confirmed that fibronectin and laminin in BM plasma samples were degraded less in Plg−/− mice at day 7 after 5-FU (Fig. 3C). ELISA measurements further demonstrated that, in contrast to WT mice, sVCAM-1 levels failed to increase in the BM plasma of Plg−/− mice on day 7 after 5-FU (% of baseline levels: 97 ± 18% in Plg−/− mice versus 157 ± 18% in WT mice; n= 4; P < 0.05). Thus, during 5-FU recovery, plasmin breaks down various ECM proteins, which anchor HPCs in the BM microenvironment.
Discussion
This study highlights a role for the Plg system in the haematopoietic recovery after myeloablation. In response to 5-FU, plasmin and the PA play a role in the restoration of WBC counts. Plasmin regulates haematopoietic recovery after 5-FU via several mechanism, i.e. by stimulating the proliferation of chemoresistant HPCs, and by promoting proteolytic breakdown of adhesive ECM proteins in the BM microenvironment. Notably, however, the haematopoietic effect of plasmin was independent of fibrinolysis.
Administration of 5-FU causes several changes in the BM microenvironment that affect proliferation, migration and mobilization of HPCs, and the repopulation of the BM and blood [2, 11]. In the early phase after 5-FU, HPCs are released from their BM microenvironment by inactivation of adhesive signals, resulting in the proliferation and differentiation of HPCs [2, 11]. Our data indicate that plasmin participates in the proliferation of HPCs in response to 5-FU; we presume that this mitogenic effect is, at least in part, secondary to the fact that plasmin inactivates adhesive signals for HPCs (though we obviously cannot exclude additional mechanisms). Apart from cleaving the membrane-associated form of uPAR (a novel adhesive signal for HPCs [4]), plasmin also regulates the cleavage of the adhesive signal mKitL into sKitL [18] (and unpublished findings), which is known to promote HPC proliferation and migration [2, 11]. The present findings now reveal that plasmin also stimulates the breakdown of VCAM-1 and fibronectin, onto which HPCs adhere in the BM microenvironment [7], and which would facilitate de-adhesion and migration of HPCs [6, 7]. Moreover, plasmin promotes the degradation of laminins, which are present around BM sinusoidal vessels [28, 29]; since these ECM proteins impede transendothelial migration and mobilization of HPCs [8–10], their breakdown by plasmin would also enhance haematopoietic recovery. Hence, plasmin promotes haematopoietic recovery after myeloablation, at least in part, via breakdown of adhesive signals and migration barriers for HPCs.
We also explored whether the role of plasmin in haematopoietic recovery relied on fibrinolysis. Indeed, Plg−/− mice suffer considerable morbidity and mortality, associated with excessive intra- and extravascular deposition of fibrin [5, 17, 27]. Notably, health and survival of Plg−/− mice were substantially improved on a fibrinogen-deficient background, illustrating the importance of the fibrinolytic system in health and disease [17]. A recent paper suggested that plasmin-mediated fibrinolysis might play a role in haematopoietic regeneration following myeloablation, but did not provide any functional evidence for this hypothesis [18]. However, our study indicates that plasmin does not regulate haematopoietic recovery after 5-FU via fibrinolysis. A role of plasmin, independently of fibrinolysis, has been previously documented in other conditions [31, 33].
Our data are consistent with a recent study by Heissig et al., documenting an impaired recovery and survival of Plg−/− mice after 5-FU [18]. Our results differ, however, from their findings in other aspects. For instance, Heissig et al. show that administration of exogenous tPA, via conversion of mKitL into sKitL (in part independently of plasmin generation), stimulates haematopoietic regeneration, but also mention (as data not shown) that endogenous tPA and uPA are redundant [18]. In contrast, our data clearly show that single loss of uPA as well as loss of tPA alone each impairs haematopoietic recovery. We do not have a precise explanation for the differences between both studies, and can only speculate that differences in experimental conditions may contribute. Whatever the explanation, a role for uPA might not be unexpected, given its up-regulated expression in the BM after myeloablation and well-known prime role in tissue remodelling in various pathological conditions [20, 31].
In conclusion, plasmin and both plasminogen activators play a role in haematopoietic recovery following cytotoxic therapy, in part, through remodelling of the BM matrix but independent of fibrinolysis.
Acknowledgments
The authors thank A. Carton, L. Frederix, B. Hermans, A. Manderveld, K. Maris, S. Meynen, J. Souffreau, S. Terclavers, B. Van Hoef, P. Van Wesemael, B. Vanwetswinkel, J. Verelst for assistance. This work was supported by grants from the Fund for Scientific Research – Flanders (FWO G.0121.02 and G.0209.07), the Belgian Science Policy (project no. IAP-P5/02), and an unrestricted Bristol-Myers-Squibb grant to P.C. M.T. is a research fellow of the Flanders Institute for the Promotion of Innovation by Science and Technology (IWT) and the Fund for Scientific Research – Flanders (FWO).
Disclosures
The authors declare that they have nothing to disclose.
Supporting Information
Note S1: Compared with WT mice, tPA-/- miceexhibited an impaired recovery of WBCs at day 7 but normal survivalafter 200 mg/kg 5-FU. We observed that, at 7 days after 5-FU, theBM of tPA-/- mice contained increased numbers of(activated) megakaryocytes, and that recovery of platelets in theperipheral blood was accelerated (data not shown). Further evidencethat megakaryocytes might modulate the course of haematopoieticrestoration in tPA-/- mice following 5-FU is suggestedby the observation that inhibition of megakaryocyte recovery byanti-CD144 antibodies, as used previously [12], impaired therecovery of WBCs and platelets after 5-FU in of tPA-/- mice, and reduced their survival (data not shown). These data will be presented in detail elsewhere.
Note S2: As VCAM-1 is sensitive to cleavage by serineproteases such as thrombin and neutrophil elastase [34, 35], weinvestigated whether plasmin might cleave membrane-bound VCAM-1in vitro. To this end, we tested the effect of plasmin oncultured OP9 stromal cells, which expressed VCAM-1 as examined byflow cytometry (not shown). Compared with control, plasminincreased the levels of sVCAM-1 in the conditioned medium by∼1.7-fold, as measured by ELISA. Consistent herewith, plasminreduced the levels of VCAM-1 on cell extracts by ∼15% (measured byELISA), indicating that plasmin cleaves VCAM-1 in vitro.
Figure S1: Effect of tranexamic acid in WT mice after5-FU. (A, B) Compared with vehicle, administration oftranexamic acid (TA) via osmotic minipumps and drinkingwater [19] from day 0 onwards impaired the WBC recovery (A) andsurvival (B) of WT mice after 5-FU (250 mg/kg i.v.). Data areexpressed as percentage versus steady-state. These resultsmight be clinically relevant because an anti-fibrinolytic agent issometimes given alongside cytotoxic drugs. P < 0.05versus vehicle (n = 10; anova). *: P< 0.05 versus vehicle-treated WT mice per time point (A).P < 0.05 versus vehicle (Cox regression; B).
Figure S2: Plasmin regulates HPC proliferation after5-FU. To study the role of plasmin on HPC proliferation after 5-FU,WT and Plg-/- mice were challenged with one injection of200 mg/kg 5-FU together with 3 times daily administration of 1mg/kg BrdU i.p. [22], and the number ofBrdU+Lin-Sca-1+ HPCs in the BM wasquantified by flow cytometry 2 days later. Compared with WT mice,the fraction of BrdU+Lin-Sca-1+cells was significantly reduced in Plg-/- mice (Fig.S2A; *: P < 0.05 versus WT; n = 3--4).Consistent herewith, up to ∼85% fewer HPCs were in S-phase of thecell cycle in Plg-/- than WT mice on day 7 after 5-FU(Fig. S2B; *: P < 0.05 versus WT; n =3--4). However, the number of HPCs in S-phase was similar in WT andPlg-/- mice in steady-state conditions (not shown). Asthe fraction of TUNEL-positive Lin-Sca-1+cells on day 2 after 5-FU was similar in WT and Plg-/-mice (Fig. S2C; P = NS versus WT; n = 3--4),the impaired haematopoietic recovery and survival ofPlg-/- mice appears to be caused by decreased proliferation and not increased apoptosis of HPCs.
Figure S3: Positive control for the histological presenceof microthrombi. As positive control, a section of WT mouseinjected with LPS in the footpad to induce inflammatory thrombosisshowed a highly organized intravascular thrombus containingfibrin(ogen) and platelets. Magnification bars: 10 μm.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
References
- 1.Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heissig B, Hattori K, Dias S, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109:625–37. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levesque JP, Hendy J, Takamatsu Y, et al. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111:187–96. doi: 10.1172/JCI15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tjwa M, Sidenius N, Moura R, et al. Membrane-anchored uPAR regulates the quiescence, maintenance, engraftment and mobilization of hematopoietic progenitors. J Clin Invest. 2009;119:1008–18. doi: 10.1172/JCI36010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collen D. Ham-Wasserman lecture: role of the plasminogen system in fibrin-homeostasis and tissue remodeling. Hematology (Am Soc Hematol Educ Program) 2001:1–9. doi: 10.1182/asheducation-2001.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Craddock CF, Nakamoto B, Elices M, et al. The role of CS1 moiety of fibronectin in VLA mediated haemopoietic progenitor trafficking. Br J Haematol. 1997;97:15–21. doi: 10.1046/j.1365-2141.1997.d01-2120.x. [DOI] [PubMed] [Google Scholar]
- 7.Scott LM, Priestley GV, Papayannopoulou T. Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol Cell Biol. 2003;23:9349–60. doi: 10.1128/MCB.23.24.9349-9360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voermans C, Rood PM, Hordijk PL, et al. Adhesion molecules involved in transendothelial migration of human hematopoietic progenitor cells. Stem Cells. 2000;18:435–43. doi: 10.1634/stemcells.18-6-435. [DOI] [PubMed] [Google Scholar]
- 9.Qian H, Tryggvason K, Jacobsen SE, et al. Contribution of alpha6 integrins to hematopoietic stem and progenitor cell homing to bone marrow and collaboration with alpha4 integrins. Blood. 2006;107:3503–10. doi: 10.1182/blood-2005-10-3932. [DOI] [PubMed] [Google Scholar]
- 10.Selleri C, Ragno P, Ricci P, et al. The metastasis-associated 67-kDa laminin receptor is involved in G-CSF-induced hematopoietic stem cell mobilization. Blood. 2006;108:2476–84. doi: 10.1182/blood-2005-11-012625. [DOI] [PubMed] [Google Scholar]
- 11.Hattori K, Heissig B, Wu Y, et al. Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med. 2002;8:841–9. doi: 10.1038/nm740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avecilla ST, Hattori K, Heissig B, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004;10:64–71. doi: 10.1038/nm973. [DOI] [PubMed] [Google Scholar]
- 13.Hatzfeld JA, Hatzfeld A, Maigne J. Fibrinogen and its fragment D stimulate proliferation of human hemopoietic cells in vitro. Proc Natl Acad Sci USA. 1982;79:6280–4. doi: 10.1073/pnas.79.20.6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou YQ, Levesque JP, Hatzfeld A, et al. Fibrinogen potentiates the effect of interleukin-3 on early human hematopoietic progenitors. Blood. 1993;82:800–6. [PubMed] [Google Scholar]
- 15.Weinstein R, Riordan MA, Wenc K, et al. Dual role of fibronectin in hematopoietic differentiation. Blood. 1989;73:111–6. [PubMed] [Google Scholar]
- 16.Larson MK, Watson SP. Regulation of proplatelet formation and platelet release by integrin alpha IIb beta3. Blood. 2006;108:1509–14. doi: 10.1182/blood-2005-11-011957. [DOI] [PubMed] [Google Scholar]
- 17.Bugge TH, Kombrinck KW, Flick MJ, et al. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996;87:709–19. doi: 10.1016/s0092-8674(00)81390-2. [DOI] [PubMed] [Google Scholar]
- 18.Heissig B, Lund L, Akiyama H, et al. The plasminogen fibrinolytic pathway is required for hematopoietic regeneration. Cell Stem Cell. 2007;1:658–70. doi: 10.1016/j.stem.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hattori N, Degen JL, Sisson TH, et al. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J Clin Invest. 2000;106:1341–50. doi: 10.1172/JCI10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suelves M, Vidal B, Serrano AL, et al. uPA deficiency exacerbates muscular dystrophy in MDX mice. J Cell Biol. 2007;178:1039–51. doi: 10.1083/jcb.200705127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inoue A, Seidel MG, Wu W, et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell. 2002;2:279–88. doi: 10.1016/s1535-6108(02)00155-1. [DOI] [PubMed] [Google Scholar]
- 22.Passegue E, Wagers AJ, Giuriato S, et al. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–94. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 24.Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–25. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 25.Ledoux D, Papy-Garcia D, Escartin Q, et al. Human plasmin enzymatic activity is inhibited by chemically modified dextrans. J Biol Chem. 2000;275:29383–90. doi: 10.1074/jbc.M000837200. [DOI] [PubMed] [Google Scholar]
- 26.Kopp HG, Avecilla ST, Hooper AT, et al. Tie2 activation contributes to hemangiogenic regeneration after myelosuppression. Blood. 2005;106:505–13. doi: 10.1182/blood-2004-11-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ploplis VA, Carmeliet P, Vazirzadeh S, et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation. 1995;92:2585–93. doi: 10.1161/01.cir.92.9.2585. [DOI] [PubMed] [Google Scholar]
- 28.Nilsson SK, Debatis ME, Dooner MS, et al. Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J Histochem Cytochem. 1998;46:371–7. doi: 10.1177/002215549804600311. [DOI] [PubMed] [Google Scholar]
- 29.Gu Y, Sorokin L, Durbeej M, et al. Characterization of bone marrow laminins and identification of alpha5-containing laminins as adhesive proteins for multipotent hematopoietic FDCP-Mix cells. Blood. 1999;93:2533–42. [PubMed] [Google Scholar]
- 30.Carmeliet P, Moons L, Lijnen R, et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet. 1997;17:439–44. doi: 10.1038/ng1297-439. [DOI] [PubMed] [Google Scholar]
- 31.Heymans S, Luttun A, Nuyens D, et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–42. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 32.Liotta LA, Goldfarb RH, Brundage R, et al. Effect of plasminogen activator (urokinase), plasmin, and thrombin on glycoprotein and collagenous components of basement membrane. Cancer Res. 1981;41:4629–36. [PubMed] [Google Scholar]
- 33.Carmeliet P, Moons L, Herbert JM, et al. Urokinase but not tissue plasminogen activator mediates arterial neointima formation in mice. Circ Res. 1997;81:829–39. doi: 10.1161/01.res.81.5.829. [DOI] [PubMed] [Google Scholar]
- 34.Levesque JP, Takamatsu Y, Nilsson SK, et al. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood. 2001;98:1289–97. doi: 10.1182/blood.v98.5.1289. [DOI] [PubMed] [Google Scholar]
- 35.Barthel SR, Johansson MW, Annis DS, et al. Cleavage of human 7-domain VCAM-1 (CD106) by thrombin. Thromb Haemost. 2006;95:873–80. [PubMed] [Google Scholar]
- 36.Vidal B, Serrano AL, Tjwa M, et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22:1747–52. doi: 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item