

Abstract
MSP58, a 58-kD nuclear microspherule protein, is an evolutionarily conserved nuclear protein implicated in the regulation of gene transcription as well as in malignant transformation. An analysis of mRNA expression by real-time PCR revealed that MSP58 was significantly up-regulated in 29% of high-grade glioblastoma tissues as well as in four glioblastoma cell lines. In the present study, we further evaluated the biological functions of MSP58 in U251 glioma cell proliferation, migration, invasion and tumour growth in vivo by specific MSP58 knockdown using short hairpin RNA (shRNA). We found that MSP58 depletion inhibited glioma cell growth, primarily by inducing cell cycle arrest rather than apoptosis. MSP58 depletion also decreased the invasive capability of glioma cells and anchorage-independent colony formation in soft agar. Moreover, suppression of MSP58 expression significantly impaired the growth of glioma xenografts in nude mice. Finally, a cell cycle-associated gene array revealed potential molecular mechanisms contributing to cell cycle arrest in MSP58-depleted glioma cells. In summary, our data highlight the importance of MSP58 in glioma progression and provided a biological basis for MSP58 as a novel candidate target for treatment of glioma.
Keywords: MSP58, RNAi, proliferation, migration, invasion, glioma
Introduction
Gliomas are the most common malignant intracranial tumours arising from the brain or spinal cord tissue in adults. Approximately 3∼4 new glioma cases per 100,000 individuals are diagnosed each year in the United States. Among these, nearly half are high-grade gliomas; half of patients diagnosed with high-grade glioma die within the first year [1]. This type of tumour is characterized by progressive overgrowth of glial tissue, diffuse and relentless invasion that, even with treatment, has a poor prognosis due to recurrence. These features impair the effectiveness of surgical therapy [2]. Current chemo/radiotherapy conditions act sub-lethally but cannot effectively suppress the proliferation of glioma cells. Thus, a deeper understanding of the molecular mechanisms involved in the high rate of proliferation and significant invasion of glioma tumours will allow the development of an adjuvant therapy to improve the current therapy.
The 58-kD microspherule protein MSP58, also known as microspherule protein 1 (MCRS1), was identified initially as interacting partner of nucleolar protein p120, Mi-2β, the transcription factors Daxx and STRA13, and the RNA-binding protein FMR [3–7]. These interactions led to the characterization of MSP58 as a participant in transcriptional regulation. Moreover, TOJ3, an avian homologue of MSP58, was a target of the transcription factor v-Jun. Ectopic expression of TOJ3 in avian fibroblasts led to anchorage-independent growth, strongly suggesting that TOJ3 plays an important role in Jun-induced cell transformation and tumourigenesis [8]. In another study, an isoform of MSP58, p78, was found to bind to centrosomal proteins, such as Nde1 and Su48, and have an essential role in centrosome dynamics. Thus, MSP58 could be a component of the centrosomal protein complex [9]. We have shown previously that MSP58 interacts with N-myc downstream regulated gene 2 (Ndrg2), a protein intimately involved in regulation of proliferation and differentiation, and that the interaction between these two proteins is important in controlling the cell cycle of Hela cell [10]. In addition, ectopic expression of Ndrg2 inhibits glioma cell proliferation [11]. We have also found that MSP58 is expressed in both normal brain tissue and tumour tissue from glioma patients, but its expression is significantly up-regulated in high-grade glioblastoma tissues as well as in the four glioblastoma cell lines we examined. Based upon all these studies, we hypothesized that reducing MSP58 expression may inhibit the malignant behaviour of glioma cells.
RNA interference (RNAi), a novel mechanism of post-transcriptional gene silencing, has been found in many eukaryotes. Recently, small interfering RNA (siRNA) has been shown to attenuate the expression of specific proteins both in vitro and in vivo by sequence-specific, double-stranded RNA (dsRNA) molecules [12, 13]. Chemically synthesized small RNAs targeting the mRNA of interest could produce RNAi, but the effects were transient. The development of siRNA vectors engineered to express short hairpin RNA (shRNA) in mammalian cells has led to extensive gene-specific silencing [14–16]. In the present study, a plasmid expressing an shRNA directed against human MSP58 was generated to block the expression of MSP58 in the human glioma cell line U251. The siRNA-induced suppression of MSP58 significantly inhibited glioma tumour proliferation, invasion as well as migration. Furthermore, we used a cDNA microarray to detect potential signal transduction pathways in which MSP58 could be involved. Our results demonstrated that MSP58 participated in multiple malignant cellular processes of human glioma cells, and suggested a potential use for highly specific RNAi-based gene-silencing therapeutics in gliomas.
Materials and methods
Tissue samples
Intracranial tissue samples were obtained from 29 patients who underwent surgery at the Institute of Neurosurgery of Xi Jing Hospital, the Fourth Military Medical University, Xi'an, China. According to the revised World Health Organization criteria for the central nervous system, 5 patients were classified as Grade II (low-grade astrocytoma), 17 as Grade III (anaplastic astrocytoma) and 4 as Grade IV (glioblastoma), respectively. Three samples from human normal brain tissues were used as control. All of the tumour tissues were obtained at primary resection, and none of the patients had been subjected to chemotherapy or radiation therapy before resection. The samples were snap frozen in liquid nitrogen and stored at −80°C until analysis. The diagnosis of glioma was confirmed histologically in all cases. For the experimental use of the surgical specimens, informed consent was obtained from the patients according to the hospital ethical guidelines.
Quantitative real-time RT-PCR
Total RNA was isolated from human brain and brain tumour tissues or cultured glioma cells using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Total RNA (2 μg) was reverse transcribed with reverse transcriptase (Fermentas Vilnius, Lithuania). The first strand cDNA was used as the template for real-time quantitative PCR analysis. β-actin cDNA was used as an internal control to normalize variances. The real-time quantitative PCR primers were designed using the Primer Express software (Applied Biosystems, Foster City, CA, USA). The sequences and PCR products length were reported in Table 1. The PCR reaction consisted of 12.5 μl of SYBR Green PCR Master Mix, 300 nM of the forward and reverse primers and 1.5 μg template cDNA in a total volume of 25 μl. The thermal cycling conditions were as following: 95°C for 5 min., followed by 45 cycles of 95°C for 30 sec., 60°C for 30 sec. The 2-ΔΔCt equation was applied to calculate the relative expression of the genes in tumour samples versus the median of normal brain tissues, where ΔCt = Ct gene – Ct β-actin, and ΔΔCt =ΔCt tumour − mean ΔCt normal brain tissues, To ensure specificity of the PCR product amplification, the melting curves for standard and sample products were analysed.
Table 1.
Primers for real-time PCR
| Gene name | Oligonucleotide sequence | Product size |
|---|---|---|
| MSP58 | F: AGGCTATTGCAGCCATCCAGAG | 118 bp |
| R: CTGTGCAGCAGGTCCTGGAA | ||
| ATM | F: GGTATGCTATGAGGCTCCTGTTCTG | 192 bp |
| R: GTCGCCAAGGCTGGAATACAA | ||
| p53 | F: ACTAAGCGAGCACTGCCCAAC | 130 bp |
| R: CCTCATTCAGCTCTCGGAACATC | ||
| ATR | F: TCGCCAGTGTATGCTACCAAAGTC | 166 bp |
| R: AGTGGAACGGCAGTAAGCTGATCTA | ||
| Cyclin H | F: GGAGCGATGTCATTCTGCTGAG | 129 bp |
| R: ACCAGGTCGTCATCAGTCCATTC | ||
| Ki67 | F: GAATGAATGCAGAAATCAGCGGTA | 80 bp |
| R: GATCATGGATGACGCTGTGAGAA | ||
| Cyclin G1 | F: CGGCAATTGAAGCATAGCTACTACA | 197 bp |
| R: TGAGACCATCATGCTTATCTCGTG | ||
| E2F2 | F: GTGTGGCTTAGCGCATGTGAA | 129 bp |
| R: GCCACCATGGTCACTGAGGA | ||
| Cyclin G2 | F: AGCTTGCAACTGCCGACTCA | 192 bp |
| R: TCGGCTAGGCATTTAGAAACCAAC | ||
| BRCA1 | F: GCAGTTCTCAAATGTTGGAGTGGA | 89 bp |
| R: CCATGCCCAGGTTTCAAGTTTC | ||
| ANAPC2 | F: GCAACGTGGAGCTGCTGAAG | 188 bp |
| R: CGGCCAGAACTCACTGGACA | ||
| ANAPC4 | F: AGTGCTATTCCCACCCGTACCA | 177 bp |
| R: CTCATCGAGCTCCCATTCATCA | ||
| ANAPC5 | F: AGGATTGCCCAGGAGTCCAAC | 114 bp |
| R: CTTCACAGAATGCTCCAGCAGAAC | ||
| β-actin | F: ATCATGTTTGAGACCTTCAACA | 318 bp |
| R: CATCTCTTGCTCGAAGTCCA |
Cell culture
The human malignant glioma cell line U251, which expresses high levels of MSP58 protein, was obtained from the Institute of Neurosurgery of Xijing Hospital, the Fourth Military Medical University, Xi’an, China. The cells were maintained as adherent monolayer cultures in Dulbecco’s modified Eagle’s medium (DMEM, Gibco/BRL, Grand Island, USA) and supplemented with 10% fetal bovine serum (FBS; Sigma, St Louis, MO, USA).
U251 cells were grown at 37°C in a humidified incubator containing 5% carbon dioxide, fed every 2–3 days with complete medium and subcultured when confluence was reached.
Transfection of synthetic siRNA
According to what was described previously [9], target siRNA sequences for MSP58 were synthesis as follows: antisense: GAAGUUCGAUGAUGAGCUG; sense: CAGCUCAUCAUCGAACUUC. The synthetic siRNA duplex oligonucleotides were obtained from Shanghai GenePharma Co., Ltd, Shanghai, China. U251 cells (5 × 104) were seeded into six-well plates. After 24 hrs, cells in each well were transfected with 100 nM siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Construction of shRNA expressing plasmid and stable gene transfection
A pSilencer3.1-H1neo plasmid (Ambion, Inc, Austin, TX, USA) was used to construct the shRNA-expressing vector. The following sequence was used for MSP58: 5′-GATCCGCAGCTCATCATCGAACTTCTTCAAGAGAGAAGTTCGATGATGAGCTGTTTTTTGGAAA-3′ as describe previously [9]. A non-specific siRNA, 5′-GATCCGACTTCATAAGGCGCATGCACTTCAAGAGAGTGCATGCGCCTTATGAAGTCTTTTTTGTCGACA-3′ (ShanghaiGenePharma Co., Ltd, Shanghai, P.R.China.), was used as a negative control. The two resulting plasmids were designated as pSilencer3.1-MSP58 and pSilencer3.1-NC, respectively. U251 cells were transfected with pSilencer3.1-MSP58 and pSilencer3.1-NC using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, and the parental cells were used as controls. Stable cell lines were selected with G418 (800 μg/ml; Sigma) and individual clones were isolated and maintained in medium containing G418 (400 μg/ml). The stable clones were identified as U251-S, U251-S1, U251-S2 (transfected with pSilencer3.1-MSP58), U251-NC (transfected with pSilencer3.1-NC), and U251-H1 neo (transfected with empty vector pSilencer3.1-H1 neo). U251-Smix was the pooled clone generated by pooling U251-S, U251-S1 and U251-S2 together.
Semi-quantitative reverse transcriptase polymerase chain reaction (RT-PCR)
The stable transfected clones and cells transfected with siRNA were harvested for RT-PCR analyses. According to the manufacturer’s instructions, total RNA was extracted with TRIZOL reagent (Invitrogen) and cDNAs were generated from 2 μg of total RNA using reverse transcriptase (Fermentas Vilnius, Lithuania). The primers for human MSP58 were: 5′-ACGCCCTGCTCTACGAT-3′ and 5′-TCATGCCTGTGATGCTGTC-3′; and the predicted PCR product was 483 bp. The primers for GAPDH, used as an internal control, were: 5′-AGGTCCACCACTGACACGTT-3′ and 5′-GCCTCAAGATCATCAGCAAT-3′; and the predicted product was 310 bp. PCR experiments were performed with a final volume of 25 μl, which contained Taq polymerase (Fermentas Vilnius, Lithuania), 1 μl template and 0.75 μl of each primer. The cycling program was performed as follows: 94°C for 5 min. as hot start, 94°C for 30 sec., 55°C for 40 sec., 72°C for 1 min. for 35 cycles and 72°C for 7 min. The PCR product was analysed by electrophoresis in a 1% TAE-agarose gel with ethidium bromide (0.5 mg/ml TE buffer) and photographed using Kodak gel documentation system. Densitometric analysis was performed using Scion Image software (Scion Corporation, Frederick, MD, USA). The inhibitory rates of MSP58 mRNA expression were calculated as follows: [1-(U251-S MSP58 density/U251-S GAPDH density)/(U251 MSP58 density/U251 GAPDH density)]× 100%.
Western blot analysis
The U251 cells were collected at the indicated time points, and washed twice with cold phosphate-buffered saline (PBS) (Gibco/BRL, Grand Island, USA) before being lysed in lysis buffer [50 mM Tris-HCl, 150 mM NaCl, 50 mM EDTA, 1% NonIdet P-40, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), and 2 μg/ml pepstatin A]. The protein samples (20 μg/lane) were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to nitrocellulose membranes (Sigma-Aldrich Co.). After blocking overnight at 4°C in a buffer containing PBS, 0.1% Tween 20 and 5% low fat milk powder, the resulting blots were incubated with a rabbit anti-human MSP58 serum (1:600) as described previously [17], and then with peroxidase-conjugated secondary antibodies (1:4000 dilution; Santa Cruz Biotechnology Inc. Santa Cruz, CA, USA) and an enhanced chemiluminescence’s (ECL) detection solution was applied (Pierce, Rockford, IL, USA). Finally, the blots were stripped as described previously [18]. The membranes were reprobed with a β-actin rabbit polyclonal antibody (1:1000; Boster, Wu Han, China) to normalize for loading and allow comparisons of target protein expression among all groups of cells. Densitometric analysis was performed using Scion Image software and the inhibitory rates of MSP58 protein expression were calculated as described previously for RT-PCR.
Measurement of cell growth by methyl thiazolyl tetrazolium assay (MTT)
U251 cells, both parental and transfected lines, were seeded at a density of 1 × 104 cells/well in 96-well plates containing 0.2 ml DMEM (with 10% FBS) and cultured for 7 days. During this period, the cells were given fresh complete medium every 2–3 days. Six wells from each group were selected randomly for the MTT (Sigma-Aldrich Co.) assay (50 μg for each well) every day. After 4 hrs of incubation, the reaction was stopped by adding 150 μl of dimethyl sulfoxide (DMSO; Sigma-Aldrich Co.) to each well and incubating for 10 min. The percentage of viable cells was determined by measuring the absorbance at 490 nm on a multiscanner reader (TECAN-spectra mini Grodig, Austria). Cell growth curves were drawn by using average absorbance at 490 nm from three independent experiments. Percentage of inhibition was calculated using the formula: % inhibition = 1 − (OD test/OD control) × 100.
Annexin V/propidium iodide (PI) staining assay
Apoptosis was assessed by measuring membrane redistribution of phosphatidylserine with fluorescent Annexin V. Cells were collected and washed twice with PBS, then resuspended in 500 μl of staining solution containing fluorescein isothiocyanate (FITC)-conjugated annexin V antibody (5 μl) (BD PharMingen, San Diego, CA, USA) and propidium iodide (PI, 5 μl of 250 μg/ml stock solution; Sigma-Aldrich Co.). After incubation for 15 min. at room temperature in dark, cells were immediately analysed on a flow cytometer. Data acquisition analysis was performed in a Becton Dickinson (San Jose, CA, USA) FACS Calibur flow cytometer using CellQuest software. Apoptotic cells were double stained with annexin V and PI. The percentage of cells undergoing apoptosis was determined by three independent experiments.
Flow cytometric analysis of the cell cycle
The cells were seeded in 25 ml flasks and incubated until they were 80–85% confluent in DMEM containing 10% FBS. Then the cells were harvested, washed twice with ice-cold PBS, fixed with 70% ethanol overnight at 4°C, washed and resuspended in 100 μl of PBS containing a final concentration of 50 μg/ml RNase A for 30 min. at room temperature. Finally, the cells were stained with 20 μg/ml PI in a final volume of 300 μl for 20 min. DNA content and cell cycle were analysed by flow cytometry (FACSCalibur, Becton Dickinson), using the software MODFIT and CELLQUEST. All of the samples were assayed in triplicate. U87 cells, which were treated with 5 μg/ml Cisplatin (Sigma) for 48 hrs, were used as positive control for apoptosis as described previously [19].
Monolayer wound-healing assay
Wound-healing assay was performed as previously described with minor modification [20, 21]. U251, U251-H1 neo, U251-NC, U251-S and U251-Smix cells were grown in 60-mm cell culture plates containing DMEM with 10% FBS. After 90% confluence was reached, the medium was replaced with FBS-free media for 24 hrs. A sterile 200-μl pipette tip was used to create a wound in the monolayer by scraping. The cells were washed with PBS and grown in FBS-free media for 24 hrs. The wounds were observed under a phase contrast microscope (model BX2, Olympus, Tokyo, Japan). The images were analysed by digitally drawing lines and averaging the position of the migrating cells at the wound edges using Scion Image software. The distance of cell migration was calculated by subtracting the distance between the lesions edges at 24 hrs from the distance measured at 0 hr. The width of the scratch was measured at 0 and 24 hrs post-treatment. The migration distance in the wound was calculated according to the following formula: cell-free area at 0 hr – cell-free area at 24 hrs. Experiments were repeated thrice in duplicate with comparable results.
Transwell invasion assay
The invasiveness of cells was measured by using the Matrigel invasion assay as described previously [22]. Briefly, Transwell insert chambers (Becton Dickinson) with 8 μm pore filters were coated with a final concentration of 1 mg/ml of Matrigel (Becton Dickinson, Bedford, MA, USA). Cells at a density of 1 × 104 cells/ml were seeded in the upper chambers with 200 μl serum-free DMEM and the lower wells were filled with 500 μl DMEM with 10% FBS as an inducer of cell migration. Cells were allowed to migrate for 24 hrs. Cells on the filter were fixed by replacing the culture medium in the bottom and top of the chamber with 4% formaldehyde. Cells that remained on the upper surface of the filter were removed using a cotton swab. After fixing for 15 min. at room temperature, the chambers were rinsed in PBS and stained with haematoxylin and eosin (H&E) for 5 min. The cells that migrated onto the lower surface of the filter were examined by microscope after mounting on a slide. A total of six random high-power microscopic fields (HPF) (100× magnification) per filter were photographed and the numbers of cells were counted.
Plate colony formation assay
For colony formation assays, 1 × 103 cells were seeded into 60 mm dishes with 5 ml of DMEM supplemented with 10% FBS and 400 μg/ml G418. After 14 days, the resulting colonies were rinsed with PBS, fixed with methanol at −20°C for 5 min., and stained with Giemsa (Sigma-Aldrich Co.) for 20 min. Only the colonies clearly visible (diameter >50 μm) were counted.
Soft-Agar Assay
Cells (1 × 104) were added to 3 ml of DMEM (supplemented with 10% FBS) with 0.3% agar and layered onto 6 ml of 0.5% agar beds in 60-mm dishes. Cells were cultured for 2 weeks, after which colonies were photographed. Colonies larger than 50 μm in diameter were counted as positive for growth. Assays were conducted in duplicate in three independent experiments.
In vivo experiment
A total of 32 female BALB/c-nu/nu mice weighing 15–18 g and 5 weeks of age were purchased from the Shanghai SLAC Laboratory Animal Company, Ltd. The nude mice were maintained in pathogen-free conditions at 26°C, at 70% relative humidity and under a 12-hr light/dark cycle. All animal experiments complied with the international guidelines for the care and treatment of laboratory animals. The mice were divided randomly into four groups. U251, U251-H1 neo, U251-NC and U251-S cells were harvested and counted. The cells (1 × 107) were suspended in 0.2 ml of normal saline, and then inoculated subcutaneously into the right flank of nude mice, which led to palpable nodules on day 4. The tumour growth was measured with callipers every 4 days from day 4 to 24. As reported previously [23, 24], tumour volumes in mice were measured with a slide calliper and recorded using the formula: volume = axb2/2, where a stands for the larger, whereas b stands for the smaller of the two dimensions.
Immunohistochemistry
Tumour tissues from nude mice were collected on day 24, excised and fixed with 10% formalin, embedded in paraffin. For immunohistochemistry, 5-μm thick tissue sections were cut, dewaxed in xylene and rehydrated. To perform Ki67 staining, the slides were incubated with 1% bovine serum albumin in PBS at room temperature for 1 hr for blocking, then stained with primary antibodies against Ki-67 (Neomarkers, Fremont, CA, USA) (1:100 dilution) at temperature for 1 hr, subsequently washed three times with PBS to remove excess primary antibody and then incubated with anti-mouse HRP conjugate IgG (1: 500 dilution) for 1 hr at room temperature. Finally, the slides were washed three times, incubated with DAB peroxidase substrate (Sigma) and covered with glass cover slips. The staining results were observed in a brightfield microscope.
Terminal deoxynucleotidyltransferase-mediated UTP end labelling (TUNEL) assay
The slides were prepared as described above in part of ‘immunohistochemistry’. TUNEL assay was conducted by using a TUNEL detection kit according to the manufacturer’s instruction (Roche Applied Science, Penzberg, Germany). Briefly, sections were incubated with 20 μg/ml proteinase K for 15 min. at room temperature and then washed with PBS. Endogenous peroxidase was blocked by 3% H2O2 in methanol for 30 min. at RT. Sections were incubated with 50 μl of TUNEL reaction mixture for 60 min. at 37°C in a humidified atmosphere in the dark. 50 μl converted-POD was added, and sections were incubated at 37°C for 30 min. Colour was developed after the addition of DAB (Sigma). A negative control, in which terminal transferase was not added, was included, in addition to a positive control consisting of cells treated with 10 μg/μl DNase I.
cDNA array analysis
Total RNA was extracted from U251-NC and the U251-S transfectants using TRIZOL reagent (Invitrogen), and two micrograms of total RNA were reverse-transcribed to generate cDNA probes. cDNA microarray analysis was performed to analyse the cell cycle-specific signalling pathway by GEArray according to a manufacturer’s instructions, and mRNA levels were quantitated by GEArrayAnalyzer software (SuperArray Inc., Bethesda, MD, USA). The relative mRNA expression of cell cycle-associated genes was normalized to the signals derived from four different housekeeping genes on the same membrane and expressed in arbitrary units, calculated with the following formula: (cell cycle signal − background signal)/(GAPDH signal − background signal). For the cDNA array expression analysis, a 2-fold change in gene expression between control and treated cultures was considered significant.
Statistical analysis
All experiments were performed in triplicate, and standard deviations were calculated. All statistical analysis was performed using SPSS (version 10.0; SPSS, Chicago, IL, USA). Comparisons among all groups were performed with a one-way ANOVA test. P value of less than 0.05 was considered statistically significant.
Results
MSP58 mRNA overexpression in glioma tissue samples and glioma cell lines
To evaluate the expression levels of MSP58 in low-grade glioma (grade II, astrocytoma: n= 5), other high-grade glioma tissue samples (grade III, anaplastic astrocytoma: n= 17; grade IV, glioblastoma: n= 4) and normal adult brain tissues (n= 3), real-time quantitative PCR was performed. The values in each sample were standardized for sample to sample variations using β-actin as normalization. Glial neoplasms demonstrated increased expression levels of MSP58 mRNA relative to the average level of MSP58 examined in pooled mRNA from three normal brain tissue samples (Fig. 1A). Separate real-time PCR for MSP58 in these three separate specimens concurred within 5% of each other. Among these 26 glioma tissue samples, the five low-grade astrocytomas displayed only slight MSP58 mRNA induction compared with normal brain, whereas analysis of 21 high-grade glioma samples showed an average 2.78-fold overexpression of MSP58 mRNA compared with normal brain. The expression of MSP58 mRNA was observed with 2- to 18.5-fold increase in 29% of the high-grade glioma samples (Grade III and IV). Thus, it appears that MSP58 mRNA is overexpressed in many advanced glial neoplasms, which was also confirmed in several high-grade glioma-derived cell lines, such as U87, U251, BT325 and SHG44 (Fig. 1B). MSP58 mRNA was 3.5-fold overexpressed in U251 cells and 2- to 3.2-fold overexpressed in other three glioma cell lines.
Figure 1.
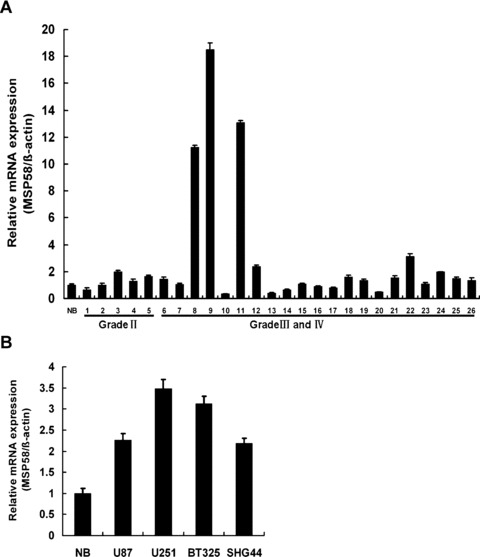
mRNA expression level of MSP58 in human glioma tissues and glioma cell lines. (A) Quantitation of MSP58 mRNA expression in various glial tissues using real-time PCR. Brain tumors were grouped according to the grade of malignancy: grade II, low grade astrocytoma (n = 5); grade III, anaplastic astrocytoma (n = 17); grade IV, glioblastoma (n = 4). RNA derived from three human normal brain tissues (NB, average MSP58 expression in three normal brain samples) was used as a control (n = 3). Total RNA was extracted from various glial tissuses and analyzed by quantitative PCR for MSP58 mRNA. Results were normalized relative to the amount of β-actin mRNA. MSP58 expression of various glial tissues was further normalized to that of normal brain samples. (B) Quantitation of MSP58 mRNA expression in different glioma cell lines. Results were normalized relative to the amount of β-actin mRNA. Samples were run in triplicate. Values were mean ± SD of three independent experiments.
Stable knowdown of MSP58 using shRNA in U251 cells
To study the role of MSP58 in malignant process of glioma, MSP58 shRNA-stable clones were generated. Briefly, the DNA oligonucleotides specific for MSP58 and negative control were generated and ligated into the pSilencer3.1-H1 neo plasmid, referred to hereafter as pSilencer3.1-MSP58 and pSilencer3.1-NC, respectively. U251 cells were transfected with these constructs and individual clones were selected by G418. Knockdown of MSP58 expression was detected by Western blot. Three pSilencer3.1-MSP58–transfected clones (U251-S, U251-S1 and U251-S2) showed a significant decrease in MSP58 expression when compared with the pSilencer3.1-NC-transfected clones (U251-NC) (Fig. 2A). Similar result was observed in another pSilencer3.1-MSP58–transfected clone U251-Smix, which was the pooled clone of clone U251-S, U251-S1 and U251-S2 (Fig. 2B). Among three individual clones, U251-S was used for the further investigation. To eliminate the possibility that results could be due to a clonal artifact, the pooled clones, U251-Smix was also used in the subsequent experiment. The inhibitory rate of MSP58 expression in U251-S cells was confirmed by RT-PCR and Western blot, whereas there was no obvious difference among U251-NC, U251-H1 neo (U251 cells transfected with the empty vector) and parental U251 cells (P > 0.05) (Fig. 2C and E). These results demonstrate that pSilencer3.1-MSP58 successfully reduced MSP58 expression in human glioma U251 cells.
Figure 2.
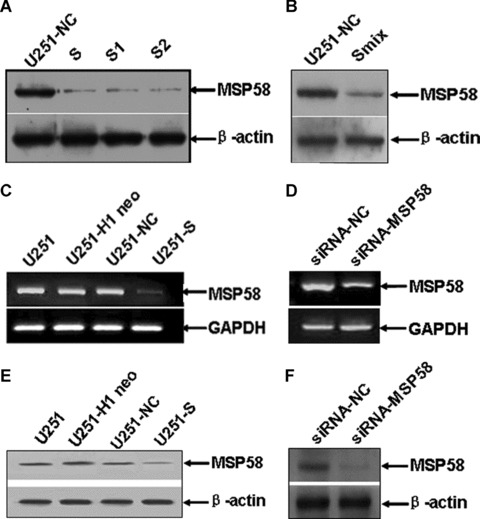
RNA interference reduced expression of MSP58 in U251 cells. (A) Down-regulation of MSP58 protein expression in clone U251-S1, S2 and S compared with control-transfected clone. (B) Down-regulation of MSP58 protein expression in clone U251-Smix. Clone U251-S1, S2 and S were pooled together to produce a mixture clone U251-Smix. (C) Down-regulation of MSP58 mRNA expression in clone U251-S was confirmed by semi-quantitative RT-PCR (C) and Western blot (E). (D) MSP58 was knocked down by transient transfection with the synthetic MSP58-targeting siRNA oligonucleotides. The expression of MSP58 in siRNA-treated U251 cells was confirmed by semi-quantitative RT-PCR (D) and Western blot (F). GAPDH was also amplified as an internal control for the RT-PCR, and β-actin levels were examined to show that similar amounts of cell lysate used in the Western Blot. U251: the parental cells; U251-H1 neo: U251 cells transfected with empty vector pSilencer3.1-H1 neo; U251-NC: U251 cells transfected with vector pSilencer3.1-NC; U251-S, S1, S2 and Smix: different U251 clones transfected with pSilencer3.1-MSP58. siRNA-NC: the transient transfectants with a negative siRNA duplex; siRNA-MSP58: the transient transfectants with a MSP58 specific siRNA duplex.
Down-regulation of MSP58 inhibited growth of U251 cells in vitro
To investigate the possible role of MSP58 in U251 cell growth, we first analysed cellular growth of U251-S cells and U251-Smix clones. The cell growth curve showed that the growth of U251-S cells and U251-Smix were inhibited notably in a time-dependent manner (Fig. 3A). The inhibitory rate was calculated as described in ‘Materials and methods’, which were 58.2% (±2.2%, P < 0.05) for U251-S cells and 48.2% (±1.8%, P < 0.05) for U251-Smix at day 7, respectively. However, there was no significant difference in cell growth among U251-NC, U251-H1 neo, and the parental U251 cells (P < 0.05; Fig. 3A). To address whether the decreased cell number was due to apoptosis induced by MSP58-targeting shRNA, we compared apoptosis in U251-S and U251-Smix cells to apoptosis occurring in U251 control cells. There is no difference in the apoptosis rate in U251-S and U251-Smix when compared with control cells (Fig. 3C). Here a well-characterized model for drug-induced apoptosis was used as a positive control. As shown in Fig. 3C, Cisplatin (5 μg/ml, 48 hrs) induced apoptosis on 24.5% of U87 cells [19]. To explore the potential contribution of MSP58 shRNA to cell cycle progression, flow cytometry was used to evaluate the cell cycle distribution. The results showed that U251-S cells accumulated in the G0/G1 phase (72.6 ± 1.9%; P < 0.05), but the cell numbers in the G2/M phase were reduced sharply (5.3 ± 2.6%; P < 0.05). The similar cell distribution of cell cycle was observed in U251-Smix cells with 69.1 ± 4.1% cells accumulated in G0/G1 phase, whereas 4.6 ± 2.7% cells distributed in G2/M phase. There was no obvious difference in cell cycle distribution among U251-NC, U251-H1 neo, and parental U251 cells (P < 0.05; Fig. 3B).
Figure 3.
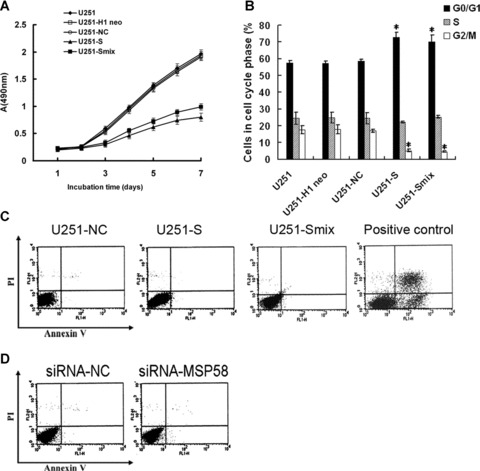
RNAi-mediated downregulation of MSP58 inhibited growth of U251 cells in vitro. (A) Cell growth curve. Parental U251 cells, vector control U251 cells (U251-H1 neo and U251-NC), and MSP58 shRNA-stable cells (clone U251-S and U251-Smix) were seeded into 96-well tissue culture plates. Cell viability was measured using the MTT assay. Cell growth curves were determined by reading the absorbance at 490 nm on a microreader. (B) Cell cycle distribution. Parental U251 cells, vector control U251 cells (U251-H1 neo and U251-NC), and MSP58 shRNA-stable cells (clone U251-S and U251-Smix) were stained with propidium iodide. The DNA content of the cells was analyzed by FACS. (C) Cell apoptosis in shRNA stably transfected U251 cells. Vector control U251 cells (U251-NC) and MSP58 shRNA-stable clones (U251-S, U251-Smix) were stained by AnnexinV/propidium iodide and analyzed by FACS to detect apoptosis. U87 cells treated by cisplatin (5 μg/ml, 48h) were used as positive control. (D) Cell apoptosis in siRNA-treated U251 cells. U251 cells were transfected transiently with non-silencing control siRNA or siRNA targeting MSP58. 48 hours later, apoptosis was analyzed by FACS. Values are mean ± SD of three independent experiments. *P < 0.05 versus U251, U251-H1 neo, and U251-NC control cells.
Because the cells we used were stable transfectants, we wished to confirm that this effect was due to depletion of MSP58 but not random gene insertion or long-term changes induced by altered MSP58 gene expression. Therefore, we synthesized siRNA oligonucleotides against MSP58 (siRNA-MSP58) and negative control siRNA (siRNA-NC), which have been published previously [9]. These siRNAs were transiently transfected into the glioma cell line U251. To evaluate the knockdown effects of siRNA on expression of MSP58, mRNA and protein extracted from U251 cells transiently transfected with either siRNA-NC or siRNA-MSP58 were analysed by RT-PCR and Western blot. As shown in Fig. 2D and F, siRNA-MSP58 successfully knocked down MSP58 expression, whereas siRNA-NC did not. Next, we found that, consistent with the stable transfectants, transient knockdown of MSP58 did not induce apoptosis in U251 cells (Fig. 3D). We therefore concluded that MSP58 might participate in the regulation of cell cycle and the inhibited proliferation but not apoptosis caused the reduced cell number in U251 cells with MSP58 expression reduced by RNAi.
Loss of MSP58 expression abrogated anchorage-dependent and independent colony formation in U251 cells
To determine the effects of MSP58 suppression on tumourigenic processes in U251 cells, we examined the anchorage-dependent plate colony formation of U251-S and U251-Smix cells, as well as the control U251 cells. Compared with control cells, 1.6- and 1.64-fold fewer clones were observed in U251-S and U251-Smix cells, respectively (Fig. 4A–D). We then evaluated the effect of MSP58 suppression on anchorage-independent colony formation in soft agar as an additional assessment of tumourigenicity in vitro. MSP58 down-regulation significantly impaired anchorage-independent colony growth, with both colony number and size much reduced. In contrast, no loss of colony-forming ability was observed in either U251-NC or U251-H1 neo cells compared with parental U251cells (Fig. 4E and F). Collectively, our results indicated that inhibition of MSP58 expression impaired the colony-formation ability of U251 cells in vitro.
Figure 4.
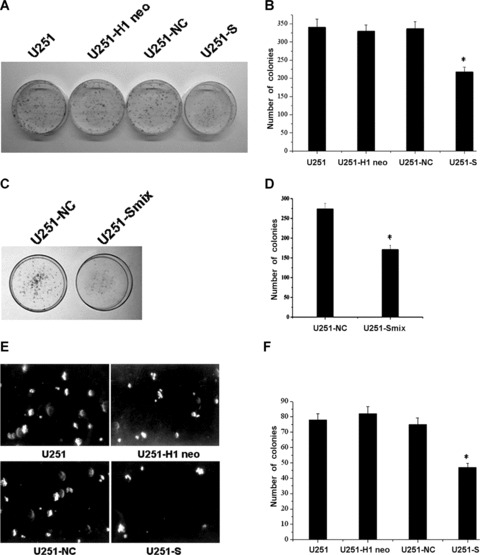
MSP58 depletion suppressed both anchorage-dependent and independent colony formation. (A, B) Equal numbers of U251-S cells, Parental U251 cells and vector control U251 cells (U251-H1 neo and U251-NC) were seeded onto 60mm dish. After 14 days, the cells were fixed and stained with Giemsa (A). The average number of colonies formed in three independent experiments was quantitated (B). (C, D) Plate colony formation ability of clone U251-Smix was detected (C) and the average number of colonies formed in three independent experiment were calculated (D). (E, F) Equal numbers of U251-S cells, parental U251 cells and vector control U251 cells (U251-H1 neo and U251-NC) were plated in 0.3% soft agar and cultured for 14 days. Colony formation was photographed under microscope (E) and scored (F). Clone numbers were mean ± SD of three independent experiments. *P < 0.05 versus U251, U251-H1 neo and U251-NC control cells.
Effects of MSP58 suppression on migration and invasion of U251 cells
Wound healing involves a number of processes, including migration and the establishment of cell polarity. We performed a wound-healing assay to study whether siRNA-mediated inhibition of MSP58 could influence migration of U251 cells. As shown in Fig. 5A and C, the migration of U251-S and U251-Smix cells declined notably. In U251, U251-H1 neo, U251-NC, U251-S and U251-Smix cells, the migration distances were 611 ± 21 μm, 584 ± 18 μm, and 619 ± 22 μm, 326 ± 15 μm and 345 ± 20 μm, respectively. There were no obvious differences in migration among parental U251, U251-H1 neo and U251-NC cells (Fig. 5B and D).
Figure 5.
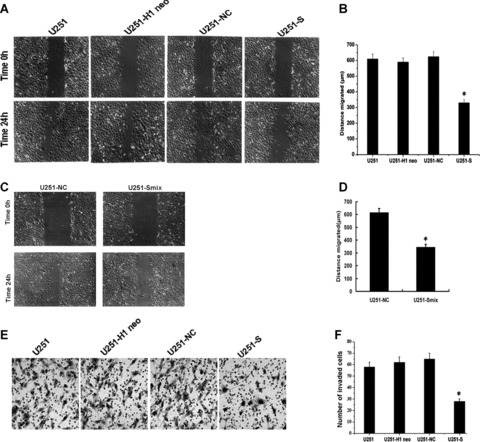
MSP58 suppression modulated migration and invasion in U251 cells. (A) Effect of MSP58 depletion on cell migration. Monolayer of U251, U251-H1 neo, U251-NC and U251-S cells were mechanically wounded with a pipette tip. Repair of the lesion by cell migration was photographed 24 h later. (B) Quantification of cell migration. The distance of cell migration was calculated as described in “Materials and Methods”. Data were obtained from three independent experiments and presented as mean ± SD. (C) Representative photographs of wound-healing assay at time 0 and 24 hours in U251-NC and U251-Smix cells. (D) Quantification of U251-Smix cell migration compared to U251-NC. (E) MSP58 shRNA diminished cell invasion of U251 cells. Each of the indicated U251 cell types was assayed for cell invasion using transwell tissue culture dishes (8 μm pore size). (F) The average cell counts of invading cells from 6 high power fields (HPFs). Values were the mean ± SD obtained from three independent experiments. *P < 0.05 versus U251, U251-H1 neo, and U251-NC control cells.
Since tumour cell invasion is an important feature of glioma cells, we next examined whether stable shRNA expression decreases cell invasion. The Matrigel invasion assay revealed that MSP58 depletion significantly suppressed the invasiveness of U251 cells (Fig. 5E). The average count of cells that crossed a matrigel-coated membrane in six high power fields was 58.0 ± 4.1 for the parental U251 group, 62.0 ± 4.8 for the U251-H1 neo group, 65.0 ± 5.1 for the U251-NC group and 28.0 ± 2.3 for the U251-S group (Fig. 5F). The number of the invaded cells declined notably in U251-S cells (P < 0.01) compared with the control groups, among which no obvious difference in invasion was observed (P > 0.05). These data demonstrate that MSP58 inhibition suppresses cell migration and invasion.
Stable knockdown of MSP58 inhibited tumour growth in vivo
To test the effect of decreased MSP58 levels in glioma cells in vivo, we extended our study to evaluate the effect of MSP58 RNAi on tumour growth. Parental U251, U251-H1 neo, U251-NC and U251-S cells were injected into nude mice (Fig. 6A). Figure 6B showed the time course of the growth of tumours initiated by different U251 transfectants. By day 24, mice injected with U251-S cells showed a statistically significant decrease in average tumour size compared with all other groups (Fig. 6C). The tumour of each mouse was removed on day 24 and weighed. MSP58 RNAi significantly decreased the solid tumour mass compared with the control groups (Fig. 6D). These data demonstrate that inhibition of MSP58 expression suppresses tumour formation in vivo.
Figure 6.
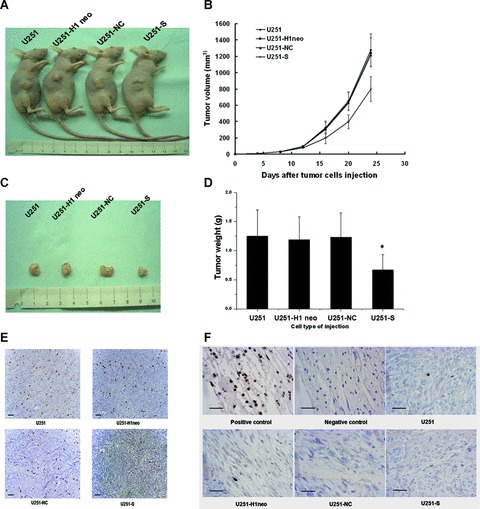
MSP58 shRNA decreased tumorgenicity in nude mice. (A) BALB/c-nu/nu mice were injected subcutaneously with 1 × 107 of the indicated control cells or U251-S cells, respectively. Representative tumor formation was photographed 24 days after injection. (B) Tumor size was determined by measuring the tumor volume every four days from day 4 to 24 days after injection. (C) Tumors were excised and photographed 24 days after injection. (D) Tumor weight of mice 24 days after injection. Values are mean ± SD obtained from three independent experiments. *P < 0.05 versus U251, U251-H1 neo, and U251-NC control cells. (E) Immunohistochemical analysis of Ki67 antigen expression in tumors of nude mice. Number of Ki67 positive cells were evaluated and compared among different groups: U251, U251-H1, U251-NC and U251-S (Bars, 100 μm). (F) TUNEL staining of tumors from nude mice. The excised tumors from different groups (U251, U251-H1, U251-NC and U251-S) were fixed, sectioned and examined for apoptosis by TUNEL staining. The positive control reflected treatment of the section by DNase I prior to staining. The slide without terminal transferase treatment was used as negative control (Bars, 50 μm).
To determine whether tumour growth inhibition is caused by apoptosis, we performed a TUNEL assay to assess the number of apoptotic cells in tumour from nude mice. This assay detects free ends that are generated by apoptotic DNA cleavage and results in brown staining of apoptotic cells. As shown in Fig. 6F, compared with vehicle- treated controls, there was not a significant increase in the number of apoptotic cells. We additionally assessed the expression of Ki67 antigen, a generally used proliferation marker. As shown in Fig. 6E, the Ki67 was down-regulated in tumour of U251-S cells injected nude mice. By contrast, Ki67 positive cells were not significantly changed either in tumour of U251-H1 neo and U251-NC cells injected mice or in tumour of U251 cells treated mice. We conclude that the main mechanism of MSP58 RNAi-induced growth inhibition in vitro and in vivo is due to of cell cycle arrest, rather than apoptosis.
Gene expression analysis of MSP58-suppressed U251 cells
Our results and a previous study support a role for MSP58 in cell cycle regulation. To further explore the molecular mechanisms of growth inhibition caused by MSP58 suppression, we used a focused DNA microarray to assess the mRNA expression levels of cell cycle-related genes. Different gene expression profiles of the U251-S and U251-NC cells were observed (Fig. 7A). MSP58 inhibition induced up-regulation of tumour suppression genes such as BRCA1 and p53, and, conversely, proliferation factor, such as E2F2, were down-regulated as well as the proliferation markers Ki67. Marked changes were also observed in cell cycle-associated genes, including ATM, ATR, Cyclin G1, Cyclin G2, Cullin 4A and Cullin 5, which are involved intimately in the G1-S phase transition. These alterations were consistent with MSP58 shRNA-mediated inhibition of the proliferation of U251 cells. Moreover, we found that inhibition of MSP58 concomitantly up-regulates a set of genes essential for the G2/M phase transition, including anaphase-promoting complex subunit 2 (ANAPC2), ANAPC4 and ANAPC5, which is consistent with decreased cell numbers in the G2/M phase in U251-S cells. These results suggest that the proliferation-inhibition effect of MSP58 shRNA on U251 cells is, at least in part, through the regulation of cell cycle.
Figure 7.
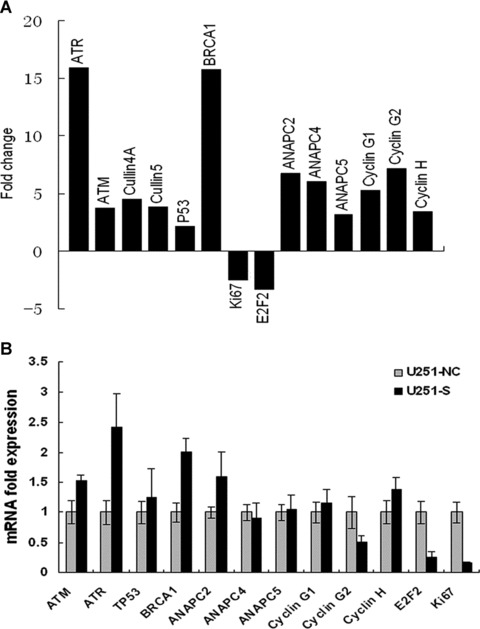
mRNA expression levels of cell cycle-related genes in MSP58 depleted U251 cells. (A) cDNA array analysis demonstrated differentially expressed cell cycle-related genes in MSP58 depleted U251 cells. Total RNA (2 μg) from U251-NC or U251-S cells was reverse transcribed to cDNA in the presence of Biotin-16-UTP. Labeled cDNA was hybridized to cell cycle-specific gene expression array membranes. After normalization of the mRNA levels of a given gene to the average value of an internal control on the same membrane, gene-expression levels in MSP58-depleted cells were calculated in arbitrary units divided by the values from control cells. A two-fold change of gene expression comparing U251-S to U251-NC cells was considered significant. (B) Microarray results were validated by real-time RT-PCR. Total RNA was extracted from U251-NC or U251-S glioma cells, and real-time quantitative RT-PCR was performed to confirm the change of different cell cycle-related gene obtained by cDNA array analysis. mRNA expression of various genes was normalized to β-actin mRNA expression. Results were expressed as mean ± SD from three independent experiments, each performed in duplicate.
To validate expression data obtained in the microarray analysis, the expression changes of some genes listed in Fig. 7A were tested by quantitative real-time PCR. As shown in Fig. 7B, quantitative real-time PCR results partly confirmed the regulation pattern observed on the arrays. Consistent with microarray results, expression of ATM and ATR were 1.5- and 2.4-fold greater in U251-S cells (Fig. 7B) than in control U251-NC cells, respectively. With the inhibition of MSP58 expression, BRCA1 level was 2-fold higher, whereas ANAPC2 was only 1.54-fold higher in U251-S cells in comparison to the U251-NC cells. Although the exact fold induction of these genes varied somewhat between the array and the quantitative real-time PCR, it was in the same tendency. In addition, the down-regulated genes, both Ki67 and E2F2, behaved relatively similar to what was observed in microarray analysis. In particular, Ki67 were down-regulated 5.17-fold in U251-S cells than in control cells. Expressions of other gene, such as Cyclin G1, Cyclin G2, ANAPC4 and ANAPC5, were also evaluated, and no obvious differences were found for expression of these genes between the test and control group regardless of inhibition of MSP58 in U251-S cells. Based on these data, further investigation will be performed to explore the excise mechanism of MSP58 involving in cell cycle regulation.
Discussion
MSP58 was identified as an oncogene due to its involvement in pathway of c-jun induced fibroblast cell transformation, as well as its physical interaction with the PTEN tumour suppressor, where PTEN inhibited the transformation potency of MSP58 [25]. Moreover, we found higher expression of MSP58 in nearly 29% of high-grade glioma samples as well as four glioma cell lines compared with normal brain, which suggests involvement of this gene in glioma tumourgenesis (Fig. 1). In the present study, we examined the role of MSP58 in cellular proliferation inhibition and other cellular malignant progression of glioma cells by depleting of MSP58 with a specific RNAi in U251 cells (Fig. 2). We demonstrated that depletion of MSP58 inhibited the U251 glioma cell proliferation. cDNA array analysis was also performed to try to explain the molecular mechanism under its effect on cell proliferation inhibition.
We found that specifically reducing MSP58 by RNAi negatively affected tumour cell growth in vitro (Fig. 3A). The reduced growth was the result of inhibited proliferation, not apoptosis (Fig. 3C). Because this effect could be due to long-term changes in gene expression, we also examined apoptosis in U251 cells transiently transfected with a synthetic siRNA and got the same result (Fig. 3D). These results are consistent with a previous study that overexpression of p78, an isoform of MSP58, increased cell growth and the rate of proliferation, whereas disruption of p78 function delayed entrance into mitotic prometaphase [9]. As an isoform of MSP58, p78 has considerable homology with MSP58 at the amino acid level, and, in almost all cases, is functionally redundant. An other earlier study reported the interaction between MSP58 and p120, a proliferation-related protein expressed at high levels in most human malignant tumour cells [25], suggesting that MSP58 is involved in cell proliferation. Moreover, MSP58 is localized primarily in the nucleolus and up-regulates ribosomal gene transcription [4]. We proposed a hypothesis that inhibition of MSP58 blocked the synthesis and maturation of rRNA in glioma cells and sequencially induced nucleolar stress which caused cell cycle arrest. In agreement with this idea that ribosomal RNA synthesis and cell cycle arrest are coupled processes, we showed that MSP58 depletion reduced U251 cell proliferation and led to G1 phase cell cycle arrest (Fig. 3B). Similar results have been obtained with other nucleolar proteins [26–28]. Together with our previous study that MSP58 interacts with candidate tumour suppressor Ndrg2 [10], which has a similar function to the PTEN tumour suppressor, reduces the S phase cell accumulation in HeLa cells caused by MSP58 overexpression, all these data confirmed function of MSP58 in cell cycle modulation.
To further define the potential molecular mechanism underlying cell cycle arrest, we analysed genes potentially regulated by MSP58 shRNA using a cell cycle-associated gene array. Among these changed genes, tumour suppression gene, BRCA1, is a known negative regulators of cell growth [29, 30]. In accordance with these observations, we found down-regulation of some proliferation-related genes, such as E2F2 and Ki67, which are used as proliferation markers in a variety of tumour types [31–35]. Some cell cycle-related genes, including ATM, ATR, were also increased by MSP58 shRNA treatment, all of which are known to either arrest the cell cycle or inhibit the growth of different cancer cells [36, 37]. Moreover, increased expression of ANAPC2, which is responsible for the G2/M transition [38], could explain the decrease in cell percent in the G2/M phase in U251-S cells. Although, we favour the idea that depletion of MSP58 caused the cell cycle arrest by inducing nucleolar stress, we cannot role out the possibility that divergent functions might be attributed to the protein. The temporal expression profile showed that MSP58 is an S-phase-specific protein, which increased during S-phase, and decreased when the cells progressed into the G2/M phase, suggesting a potential role for MSP58 in other cellular process, such as DNA replication.
Glioma cell invasion and migration are characteristic features that distinguish them from normal cells. We therefore proposed that, as an oncoprotein, MSP58 might contribute to these tumour-specific behaviours in glioma cells. Interestingly, cell migration and invasion are inhibited in MSP58-depleted glioma cells (Fig. 5). Cell invasion is the result of degradation of extracellular matrix (ECM) proteins by specific proteases. More than one study has reported that the matrix metalloproteinase (MMP) family is activated to promote tumour cell invasion and angiogenesis [39, 40]. Further studies are needed to determine the molecular mechanism where MSP58 shRNA suppresses U251 cell invasiveness.
Our results also showed that suppression of MSP58 affected the transformed morphology of the glioma cells. The inhibition of anchorage-dependent and -independent growth were observed clearly in U251-S cells compared with the control cells, which suggests that depletion of MSP58 could enhance mitogenic or survival signals (Fig. 4). Consistent with these results, a previous study showed that ectopic expression of the TOJ3 gene in quail or chicken embryo fibroblasts induced anchorage-independent growth, providing evidence that the immediate activation of TOJ3 in fibroblasts contributed to cell transformation. These results, together with the high levels of MSP58 expression found in glioma tissue, demonstrate function of MSP58 in cancer development.
Since we proved that MSP58 is important in different malignant processes involved in glioma progression in vitro, we further investigated the ability of MSP58 to affect the growth of glioma tumour xenografts in vivo. MSP58 depletion did impair the growth of glioma tumour xenografts. Given MSP58 inhibition did not induce apoptosis but specifically inhibited proliferation in vitro and in vivo (Figs 3 and 6), the inhibition of glioma growth resulting from MSP58 reduction in vivo is most likely due to decreased cell number as the result of inhibited proliferation rather than apoptosis.
Since MSP58 is up-regulated in some glioma tumours and depletion of MSP58 in U251 cells suppressed multiple tumour-specific behaviours, MSP58 could be an intriguing candidate molecular target for glioma therapy. RNAi was first discovered in the nematode Caenorhabditis elegans [41] and is conserved in a variety of organisms including plants, Drosophila and mammals. Recently, it has been developed as a viable and more effective alternative to antisense- and ribozyme-based techniques for gene silencing, offering great potential in cancer therapeutics [42]. One advantage of RNAi in therapy is that the RNAi, once formed inside a cell, can be stable for up to a few weeks in some cell types [43]. In this study, we specifically knocked down expression of MSP58 by shRNA, and consequently impaired all the malignant processes involved in glioma progression. These results provide the biological basis for inhibition of MSP58 using RNAi as a novel therapeutic approach for glioma.
To conclude, we have shown in this study that targeted MSP58 inhibition by specific RNAi prevents proliferation and suppresses multiple tumourigenic properties of glioma cells both in vitro and in vivo. These results demonstrate great potential for developing highly specific RNAi-mediated down-regulation of MSP58 as a candidate therapeutic target for glioma.
Acknowledgments
The authors thank Prof. E.W. Khandjian (Département de biologie médicale, Faculté de médecine, Université Laval, Québec, Canada) for kindly providing anti MSP58 polyclonal serum. This work was supported by the National High Technology Research and Development Program of China (863 Program) (No. 2006AA02Z194), the Program for Scholars and Innovative Research Team in University (No. PCSIRT0459) and the National Natural Science Foundation of China (No. 30700918, 30670452; Key Program, No. 30830054) and Natural Science Foundation of Shaanxi Province of China (No., 2008K09-09).
References
- 1.Hess KR, Broglio KR, Bondy ML. Adult glioma incidence trends in the United States, 1977–2000. Cancer. 2004;101:2293–9. doi: 10.1002/cncr.20621. [DOI] [PubMed] [Google Scholar]
- 2.Kaba SE, Kyritsis AP. Recognition and management of gliomas. Drugs. 1997;53:235–44. doi: 10.2165/00003495-199753020-00004. [DOI] [PubMed] [Google Scholar]
- 3.Davidovic L, Bechara E, Gravel M, et al. The nuclear microspherule protein 58 is a novel RNA-binding protein that interacts with fragile X mental retardation protein in polyribosomal mRNPs from neurons. Hum Mol Genet. 2006;15:1525–38. doi: 10.1093/hmg/ddl074. [DOI] [PubMed] [Google Scholar]
- 4.Shimono K, Shimono Y, Shimokata K, et al. Microspherule protein 1, Mi-2beta, and RET finger protein associate in the nucleolus and up-regulate ribosomal gene transcription. J Biol Chem. 2005;280:39436–47. doi: 10.1074/jbc.M507356200. [DOI] [PubMed] [Google Scholar]
- 5.Ivanova AV, Ivanov SV, Lerman ML. Association, mutual stabilization, and transcriptional activity of the STRA13 and MSP58 proteins. Cell Mol Life Sci. 2005;62:471–84. doi: 10.1007/s00018-004-4423-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin DY, Shih HM. Essential role of the 58-kDa microspherule protein in the modulation of Daxx-dependent transcriptional repression as revealed by nucleolar sequestration. J Biol Chem. 2002;277:25446–56. doi: 10.1074/jbc.M200633200. [DOI] [PubMed] [Google Scholar]
- 7.Ren Y, Busch RK, Perlaky L, et al. The 58-kDa microspherule protein (MSP58), a nucleolar protein, interacts with nucleolar protein p120. Eur J Biochem. 1998;253:734–42. doi: 10.1046/j.1432-1327.1998.2530734.x. [DOI] [PubMed] [Google Scholar]
- 8.Bader AG, Schneider ML, Bister K, et al. TOJ3, a target of the v-Jun transcription factor, encodes a protein with transforming activity related to human microspherule protein 1 (MCRS1) Oncogen. 2001;20:7524–35. doi: 10.1038/sj.onc.1204938. [DOI] [PubMed] [Google Scholar]
- 9.Hirohashi Y, Wang Q, Liu Q, et al. p78/MCRS1 forms a complex with centrosomal protein Nde1 and is essential for cell viability. Oncogene. 2006;25:4937–46. doi: 10.1038/sj.onc.1209500. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Liu J, Li X, et al. The physical and functional interaction of NDRG2 with MSP58 in cells. Biochem Biophys Res Commun. 2007;352:6–11. doi: 10.1016/j.bbrc.2006.10.141. [DOI] [PubMed] [Google Scholar]
- 11.Deng Y, Yao L, Chau L, et al. N-Myc downstream-regulated gene 2 (NDRG2) inhibits glioblastoma cell proliferation. Int J Cancer. 2003;106:342–7. doi: 10.1002/ijc.11228. [DOI] [PubMed] [Google Scholar]
- 12.Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 13.Wianny F, Zernicka-Goetz M. Specific interference with gene function by double-stranded RNA in early mouse development. Nat Cell Biol. 2000;2:70–5. doi: 10.1038/35000016. [DOI] [PubMed] [Google Scholar]
- 14.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 15.Paddison PJ, Caudy AA, Bernstein E, et al. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–58. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kappler M, Bache M, Bartel F, et al. Knockdown of survivin expression by small interfering RNA reduces the clonogenic survival of human sarcoma cell lines independently of p53. Cancer Gene Ther. 2004;11:186–93. doi: 10.1038/sj.cgt.7700677. [DOI] [PubMed] [Google Scholar]
- 17.Davidovic L, Bechara E, Gravel M, et al. The nuclear microspherule protein 58 is a novel RNA-binding protein that interacts with fragile X mental retardation protein in polyribosomal mRNPs from neurons. Hum Mol Genet. 2006;15:1525–38. doi: 10.1093/hmg/ddl074. [DOI] [PubMed] [Google Scholar]
- 18.Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–21. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 19.Kondo S, Barna BP, Morimura T, et al. Interleukin-1 beta-converting enzyme mediates cisplatin-induced apoptosis in malignant glioma cells. Cancer Res. 1995;55:6166–71. [PubMed] [Google Scholar]
- 20.Mitra A, Fillmore RA, Metge BJ, et al. Large isoform of MRJ (DNAJB6) reduces malignant activity of breast cancer. Breast Cancer Res. 2008;10:R22. doi: 10.1186/bcr1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khwaja FW, Duke-Cohan JS, Brat DJ, et al. Attractin is elevated in the cerebrospinal fluid of patients with malignant astrocytoma and mediates glioma cell migration. Clin Cancer Res. 2006;12:6331–6. doi: 10.1158/1078-0432.CCR-06-1296. [DOI] [PubMed] [Google Scholar]
- 22.Valster A, Tran NL, Nakada M, et al. Cell migration and invasion assays. Methods. 2005;37:208–15. doi: 10.1016/j.ymeth.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Jin N, Chen W, Blazar BR, et al. Gene therapy of murine solid tumors with T cells transduced with a retroviral vascular endothelial growth factor–immunotoxin target gene. Hum Gene Ther. 2002;13:497–508. doi: 10.1089/10430340252809793. [DOI] [PubMed] [Google Scholar]
- 24.Koga S, Hirohata S, Kondo Y, et al. A novel telomerase-specific gene therapy: gene transfer of caspase-8 utilizing the human telomerase catalytic subunit gene promoter. Hum Gene Ther. 2000;11:1397–406. doi: 10.1089/10430340050057477. [DOI] [PubMed] [Google Scholar]
- 25.Okumura K, Zhao M, Depinho RA, et al. Cellular transformation by the MSP58 oncogene is inhibited by its physical interaction with the PTEN tumor suppressor. Proc Natl Acad Sci USA. 2005;102:2703–6. doi: 10.1073/pnas.0409370102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frescas D, Guardavaccaro D, Bassermann F, et al. JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature. 2007;450:309–13. doi: 10.1038/nature06255. [DOI] [PubMed] [Google Scholar]
- 27.Pestov DG, Strezoska Z, Lau LF. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: effects of nucleolar protein Bop1 on G(1)/S transition. Mol Cell Biol. 2001;21:4246–55. doi: 10.1128/MCB.21.13.4246-4255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyoshi M, Okajima T, Matsuda T, et al. Bystin in human cancer cells: intracellular localization and function in ribosome biogenesis. Biochem J. 2007;404:373–81. doi: 10.1042/BJ20061597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elmariah SB, Huse J, Mason B, et al. Multicentric glioblastoma multiforme in a patient with BRCA-1 invasive breast cancer. Breast J. 2006;12:470–4. doi: 10.1111/j.1075-122X.2006.00307.x. [DOI] [PubMed] [Google Scholar]
- 30.Yacoub A, Mitchell C, Hong Y, et al. MDA-7 regulates cell growth and radiosensitivity in vitro of primary (non-established) human glioma cells. Cancer Biol Ther. 2004;3:739–51. doi: 10.4161/cbt.3.8.968. [DOI] [PubMed] [Google Scholar]
- 31.Hashiba T, Izumoto S, Kagawa N, et al. Expression of WT1 protein and correlation with cellular proliferation in glial tumors. Neurol Med Chir. 2007;47:165–70. doi: 10.2176/nmc.47.165. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Wang Y, Wang Y, et al. Expression of EphA2 in human astrocytic tumors: correlation with pathologic grade, proliferation and apoptosis. Tumour Biol. 2007;28:165–72. doi: 10.1159/000103010. [DOI] [PubMed] [Google Scholar]
- 33.Nakagawa N, Akai F, Fukawa N, et al. Early effects of boron neutron capture therapy on rat glioma models. Brain Tumor Pathol. 2007;24:7–13. doi: 10.1007/s10014-007-0214-4. [DOI] [PubMed] [Google Scholar]
- 34.Preusser M, Wolfsberger S, Czech T, et al. Survivin expression in intracranial ependymomas and its correlation with tumor cell proliferation and patient outcome. Am J Clin Pathol. 2005;124:543–9. doi: 10.1309/PP2G5GAAFKV82DTG. [DOI] [PubMed] [Google Scholar]
- 35.Gomez-Manzano C, Mitlianga P, Fueyo J, et al. Transfer of E2F-1 to human glioma cells results in transcriptional up-regulation of Bcl-2. Cancer Res. 2001;61:6693–7. [PubMed] [Google Scholar]
- 36.Luciani MG, Campregher C, Gasche C. Aspirin blocks proliferation in colon cells by inducing a G1 arrest and apoptosis through activation of the checkpoint kinase ATM. Carcinogenesis. 2007;28:2207–17. doi: 10.1093/carcin/bgm101. [DOI] [PubMed] [Google Scholar]
- 37.Park I, Avraham HK. Cell cycle-dependent DNA damage signaling induced by ICRF-193 involves ATM, ATR, CHK2, and BRCA1. Exp Cell Res. 2006;312:1996–2008. doi: 10.1016/j.yexcr.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 38.Peters JM. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell. 2002;9:931–43. doi: 10.1016/s1097-2765(02)00540-3. [DOI] [PubMed] [Google Scholar]
- 39.Belien AT, Paganetti PA, Schwab ME. Membrane-type 1 matrix metalloprotease (MT1-MMP) enables invasive migration of glioma cells in central nervous system white matter. J Cell Biol. 1999;144:373–84. doi: 10.1083/jcb.144.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roomi MW, Ivanov V, Kalinovsky T, et al. Inhibition of glioma cell line A-172 MMP activity and cell invasion in vitro by a nutrient mixture. Med Oncol. 2007;24:231–8. doi: 10.1007/BF02698045. [DOI] [PubMed] [Google Scholar]
- 41.Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 42.Zhang YC, Taylor MM, Samson WK, et al. Antisense inhibition: oligonucleotides, ribozymes, and siRNAs. Methods Mol Med. 2005;106:11–34. [PubMed] [Google Scholar]
- 43.Dykxhoorn DM, Lieberman J. Knocking down disease with siRNAs. Cell. 2006;126:231–5. doi: 10.1016/j.cell.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]