

Abstract
The 2′-trifluoromethylthio (2′-SCF3) modification endows ribonucleic acids with exceptional properties and has attracted considerable interest as a reporter group for NMR spectroscopic applications. However, only modified pyrimidine nucleosides have been generated so far. Here, the syntheses of 2′-SCF3 adenosine and guanosine phosphoramidites of which the latter was obtained in highly efficient manner by an unconventional Boc-protecting group strategy, are reported. RNA solid-phase synthesis provided site-specifically 2′-SCF3-modified oligoribonucleotides that were investigated intensively. Their excellent behavior in 19F NMR spectroscopic probing of RNA ligand binding was exemplified for a noncovalent small molecule–RNA interaction. Moreover, comparably to the 2′-SCF3 pyrimidine nucleosides, the purine counterparts were also found to cause a significant thermodynamic destabilization when located in double helical regions. This property was considered beneficial for siRNA design under the aspect to minimize off-target effects and their performance in silencing of the BASP1 gene was demonstrated.
Keywords: fluorine, NMR spectroscopy, oligonucleotides, phosphoramidites, solid-phase synthesis
Introduction
Chemical modification can significantly enrich the structural and functional repertoire of ribonucleic acids and equip them with new fascinating properties.[1–9] Recently, we have reported the original synthesis of 2′-SCF3-modified RNA.[10,11] This modification has considerable potential for broad NMR spectroscopic applications in the nucleic acids field, particularly for probing of RNA–ligand interactions and for monitoring structural rearrangements, both at the secondary and tertiary structure level.[12–21] The main reason for this alluring prospect originates from the very high sensitivity compared to the commonly used single-fluorine labeling patterns that involve mostly 5-fluoro or 2′-fluoro uridines.[13] Surprisingly, we also found that the 2′-SCF3 modification has a significant effect on base-pairing strength when positioned in double helical regions, rendering 2′-SCF3 nucleosides to one of the most destabilizing 2′-modifications known to date.[11,22] Thereby, the extent of thermodynamic destabilization is comparable to that of nucleic acids containing acyclic (“unlocked”) nucleosides.[23] We have previously investigated this effect which is probably due to the strong preference for C2′-endo conformation of the 2′-SCF3 ribose moiety.[11] Nevertheless, all our knowledge stems from 2′-SCF3 uridine and/or 2′-SCF3 cytidine containing RNA exclusively.[10,11] In view of the broad spectrum of applications in chemical biology, we synthesized the novel 2′-SCF3 adenosine and 2′-SCF3 guanosine phoshoramidites, incorporated them into oligoribonucleotides, studied their physicochemical properties and demonstrated their principal potential for siRNA technologies; all of these features are reported in this article.
Results and Discussion
Synthesis of 2′-SCF3 adenosine phosphoramidite (A9)
Our synthetic route began with the simultaneous protection of the 5′ and 3′-hydroxyl groups of commercially available 9-(β-d-arabinofuranosyl)adenine using 1,3-dichloro-1,1,3,3-tetraisopropyl disiloxane (TIPDSCl2) to furnish the nucleoside intermediate A1 (Scheme 1),[24] followed by protection of the exocyclic adenine 6-amino group using N,N-dibutylformamide dimethyl acetal to yield derivative A2. After triflation of the arabinose 2′-OH, compound A3 was treated with potassium thioacetate and 18-crown-6-ether, producing derivative A4. Selective cleavage of the acetyl group under basic conditions gave the precursor thiol A5 in 75 % yield. Only minor amounts (<10 %) of disulfide bridged dimer was observed as a byproduct. The key step, regioselective trifluoromethylation of the thiol group, was achieved in excellent yields using 3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole (Togni’s reagent).[25] Deprotection of the TIPDS moiety of A6 proceeded in a straightforward manner using tetrabutylammonium fluoride (TBAF) and acetic acid. Finally, compound A7 was transformed into the dimethoxytritylated derivative A8, and conversion into the corresponding phosphoramidite A9 was achieved in good yields by treatment with 2-cyanoethyl N,N-diisopropylchlorophosphoramidite. Starting with arabinoadenosine, our route provides A9 in 26 % overall yield in nine steps with seven chromatographic purifications; in total, 0.5 g of A9 was obtained in the course of this study.
Scheme 2.
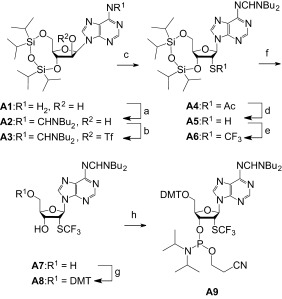
Synthesis of 2′-SCF3 adenosine phosphoramidite A9. Reaction conditions: a) 3.0 equiv N,N-di-n-butylformamide dimethyl acetal, in DMF, RT, 26 h, 96 %; b) 1.5 equiv F3CSO2Cl, 3.0 equiv DMAP, in CH2Cl2, 0 °C, 20 min; c) 1.5 equiv CH3COS−K+, 1.5 equiv 18-crown-6, in toluene, 17 h, 45 °C, 93 % (over two steps); d) 0.1 M NaOH, in EtOH/pyridine/H2O (20:20:1), 0 °C, 10 min, 75 %; e) 1.2 equiv 3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole, in CH2Cl2, −78 °C to RT, 16 h, 85 %; f) 1 M TBAF, 0.5 M CH3COOH, in THF, RT, 1.0 h, 72 %; g) 1.1 equiv DMT-Cl, 0.1 equiv DMAP, in pyridine, RT, 14 h, 87 %; h) 1.5 equiv 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, 10 equiv CH3CH2N(CH3)2, CH2Cl2, RT, 3 h, 73 %.
Synthesis of 2′-SCF3 guanosine phosphoramidite (G7)
The synthesis of 2′-modified guanosine derivatives usually requires protection of the guanine lactam moiety against electrophilic reagents which is often accomplished by the O6-(4-nitrophenyl)ethyl group that is introduced under Mitsunobu conditions.[26,27] Although continuously optimized over the years in our laboratory, we experienced unsatisfying yields for this transformation. Additionally, extensive purification protocols were required rendering such a pathway not very attractive. In the present case, we therefore commenced with the O6-tert-butyl, N2(bis-[tert-butyloxycarbonyl]) (O6-tBu, N2-Boc2) protected 9-(β-d-arabinofuranosyl)guanine derivative G1 (Scheme 2). This compound is readily available in large quantities from 9-(β-d-arabinofuranosyl)guanine, in analogy to a recent report on the synthesis of guanosine-based amphiphiles.[28–30]
Scheme 2.
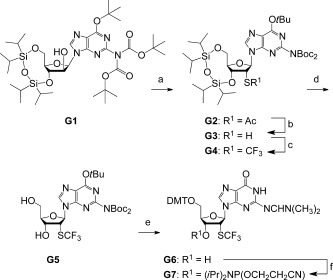
Synthesis of 2′-SCF3 guanosine phosphoramidite G7. Starting compound G1 was obtained according to ref. [27]. Reaction conditions: a) i. 1.5 equiv F3CSO2Cl, 3.0 equiv DMAP, in CH2Cl2, 0 °C, 20 min; ii. 1.5 equiv CH3COS−K+, 1.5 equiv 18-crown-6, 1.5 equiv EtN(iPr)2 in toluene, 16 h, 45 °C, 82 %; b) 1.6 M MeNH2, in EtOH/CH2Cl2 (23:1), 0 °C, 25 min, 85 %; c) 1.2 equiv 3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole, CH2Cl2, −78 °C to RT, 16 h, 73 %; d) 1 M TBAF, in THF, RT, 3.5 h, 81 %; e) i. CF3COOH/CH2Cl2 (1:7), RT, 2.5 h; ii. 3.0 equiv (H3CO)2CHN(CH3)2, in CH3OH, reflux, 6 h; iii. 1.1 equiv DMT-Cl, 0.1 equiv DMAP, in pyridine, RT, 18 h, 48 %; f) 1.5 equiv 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, 10 equiv CH3CH2N(CH3)2, CH2Cl2, RT, 3 h, 72 %.
Therefore, compound G1 was triflated at the arabinose 2′-OH and subsequently treated with potassium thioacetate and 18-crown-6-ether, producing derivative G2. Selective cleavage of the acetyl group under basic conditions gave the precursor thiol G3 in 85 % yield. Then, regioselective trifluoromethylation of the thiol group was achieved using 3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole (Togni’s reagent)[25] and provided derivative G4 in high yields. Deprotection of its TIPDS moiety was straightforward using tetrabutylammonium fluoride (TBAF). Then, a series of transformations starting with tBu and Boc deprotection of nucleoside G5 using trifluoro acetic acid, followed by amidine protection of the exocyclic amino group and dimethoxytritylation of the 5′-OH group, were optimized in a one-pot procedure requiring only a single chromatographic purification of the target compound G6. Conversion into the corresponding phosphoramidite was achieved in good yields by treatment with 2-cyanoethyl N,N-diisopropylchlorophosphoramidite. Our route provides G7 in a 14 % overall yield in six steps with six chromatographic purifications; in total, 0.7 g of G7 was obtained in the course of this study.
We point out that the Boc protection concept proved very convenient and was the key for the high efficiency of the synthesis. Therefore, we currently plan to integrate the Boc approach for the syntheses of other 2′-modified (e.g., 2′-SeCH3 or 2′-N3)[26,27] guanosine building blocks as well.
Synthesis of 2′-SCF3-containing RNA
We used the 2′-O-TOM approach for the solid-phase synthesis of RNA with site-specific 2′-SCF3 adenosine and guanosine modifications.[31,32] Coupling yields of the two novel building blocks were higher than 98 % according to the trityl assay. The oligoribonucleotides were cleaved from the solid support and deprotected using CH3NH2 in ethanol/H2O, followed by treatment with tetrabutylammonium fluoride (TBAF) in tetrahydrofuran (THF). Subsequent size-exclusion chromatography on a Sephadex G25 column removed salts. The RNA sequences were purified by anion-exchange chromatography under strong denaturating conditions (6 M urea, 80 °C). The molecular weights of the purified RNAs were confirmed by liquid chromatography (LC) electrospray ionization (ESI) mass spectrometry (MS). A selection of 2′-SCF3 RNA sequences is listed in the Supporting Information, Table S1. Noteworthy, 2′-SCF3 guanosine (such as the previously investigated 2′-SCF3 uridine and 2′-SCF3 cytidine)[10,11] appeared completely stable under the repetitive oxidative conditions (20 mM aqueous iodine solution) required during RNA solid-phase synthesis for the transformation of PIII into PV, and the subsequent deprotection (Figure 1). Unexpectedly, the 2′-SCF3 adenosine label turned out to be sensitive during the standard deprotection procedure. Only when we added millimolar amounts of threo-1,4-dimercapto-2,3-butandiol (DTT), a high quality of the crude deprotected RNA was achieved as analyzed from the corresponding ion-exchange HPLC traces (Figure 1 B) and mass spectrometric experiments. We hypothesize that possible oxidation products (such as sulfoxides, 2′-SOCF3) of the protected RNA were reduced by this additive, and hence, follow-up side-products that otherwise dominated during RNA deprotection at high pH values (as a result of sulfoxide elimination and successive strand cleavage) could not form any more. This observation is reminiscent of the chemical synthesis of 2′-SeCH3 RNA that we investigated several years ago;[33] for 2′-SeCH3 guanosine-modified RNA, the corresponding oxidation products were analyzed in detail by mass spectrometry, and additionally isolated and quantitatively reduced by DTT.[26] Unfortunately, our attempts to isolate oxidized species of 2′-SCF3 RNA have failed so far.1
Figure 1.
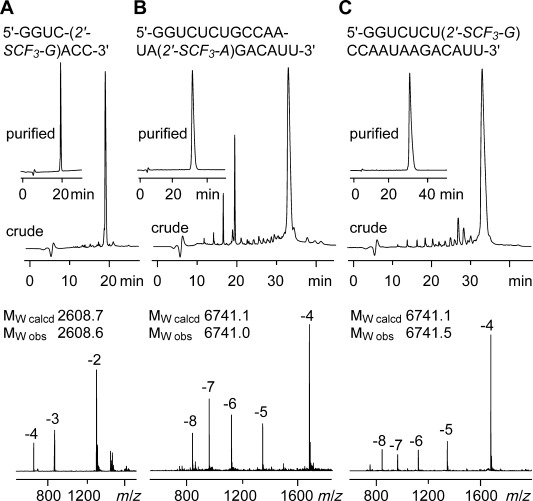
Characterization of 2′-SCF3 modified RNA. Anion-exchange HPLC traces (top) of: A) 8 nt RNA, B) 21 nt RNA, and C) 21 nt RNA, and the corresponding LC-ESI mass spectra (bottom). HPLC conditions: Dionex DNAPac column (4×250 mm), 80 °C, 1 mL min−1, 0–60 % buffer B in 45 min; buffer A: Tris-HCl (25 mM), urea (6 M), pH 8.0; buffer B: Tris-HCl (25 mM), urea (6 M), NaClO4 (0.5 M), pH 8.0. For LC-ESI MS conditions, see the Supporting Information.
Base pairing properties of 2′-SCF3-containing RNA
A single 2′-SCF3 adenosine or 2′-SCF3 guanosine exhibited a pronounced attenuation of RNA duplex stability provided that the modification was located in the Watson–Crick base-pairing region. For instance, UV melting profile analysis of the palindromic RNA 5′-GGUC(2′-SCF3-G)ACC (Figure 2 A) revealed an average decrease of 24 °C in Tm values for RNA concentrations in the micromolar range (ΔG°, −7.8 kcal mol−1; ΔH°, −72.3 kcal mol−1; ΔS°, −216 cal mol−1 K−1), compared to the unmodified counterpart (ΔG°, −15.4 kcal mol−1; ΔH°, −84.8 kcal mol−1; ΔS°, −233 cal mol−1 K−1). As a second example, the hairpin-forming RNA 5′-GAA(2′-SCF3-G)G-GCAA-CCUUCG (Figure 2 B) also showed a decrease (14 °C) of Tm values determined at micromolar RNA concentrations (ΔG°, −5.3 kcal mol−1; ΔH°, −54.7 kcal mol−1; ΔS°, −166 cal mol−1 K−1), compared to the unmodified counterpart (ΔG°, −7.1 kcal mol−1; ΔH°, −52.1 kcal mol−1; ΔS°, −151 cal mol−1 K−1). Likewise, the same hairpin sequence but with 2′-SCF3 adenosine, 5′-GA(2′-SCF3-A)GG-GCAA-CCUUCG (Figure 2 C), was destabilized (ΔG°, −5.5 kcal mol−1; ΔH°, −57.9 kcal mol−1; ΔS°, −176 cal mol−1 K−1).2
Figure 2.
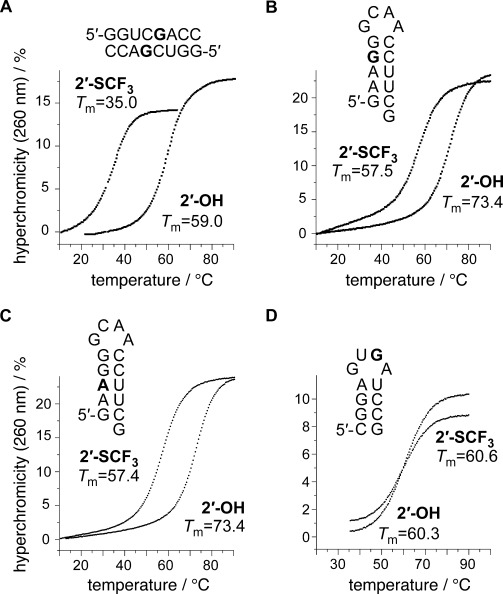
Thermal stabilities of unmodified and 2′-SCF3 modified oligoribonucleotides. UV-melting profiles of A) self-complementary 8 nt RNA, B) 15 nt RNA hairpin, C) 15 nt RNA hairpin, and D) 12 nt RNA hairpin. Conditions: cRNA=8 μM; 10 mM Na2HPO4, 150 mM NaCl, pH 7.0. Nucleotide abbreviations in bold indicate the positions for 2′-SCF3 modification.
The influence of the 2′-SCF3 purine nucleosides on thermodynamic parameters of double helix stability was therefore comparable to the corresponding 2′-SCF3 pyrimidine series investigated previously, for which the destabilization was attributed to the inherent preference for C2′-endo conformation of these nucleosides.[11] To support such a hypothesis also for the purine nucleoside series investigated here, we synthesized the short, single-stranded RNAs, 5′-UU(2′-SCF3-A)GCG, and 5′-UU(2′-SCF3-G)UUU, and determined 3J(H1′-H2′) coupling constants by 2D 1H,1H exclusive correlation spectroscopy (ECOSY) (Figure 3). For 2′-SCF3-adenosine and -guanosine, the values were determined to be 9.7 and 8.0 Hz, respectively, accounting for a population of 96 and 80 % of C2′-endo ribose conformation in single-stranded RNA.3
Figure 3.
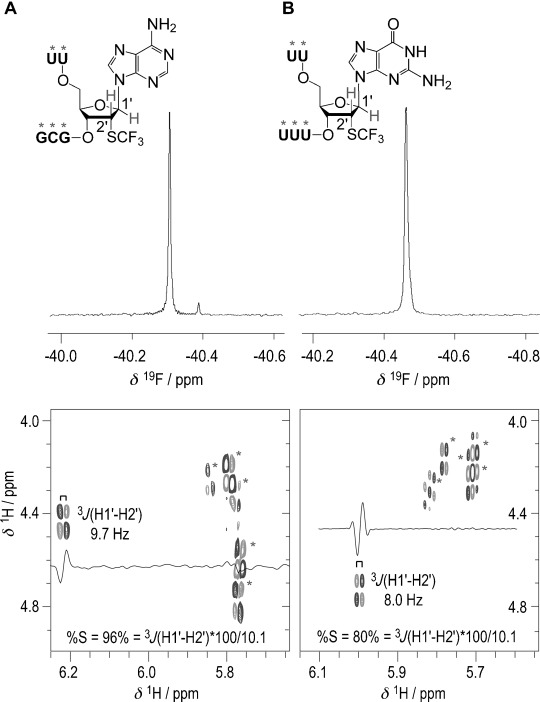
19F and 1H, 1H ECOSY NMR spectra of single-stranded RNAs: A) 5′-UU(2′-SCF3-A)GCG, and B) 5′-UU(2′-SCF3-G)UUU. For the 2′-SCF3 moiety, the 3-bond scalar coupling constants of H1′ and H2′ (3JH1′-H2′) were determined to be 9.7 and 8.0 Hz, respectively. These account for 96 and 80 % C2′-endo (South) populations in C2′/C3′-endo equilibria.[35,36] Note that for pyrimidine nucleosides the C2′-endo (South) population was 100 %.[10,11] Conditions: cRNA=0.3 mM; 25 mM sodium cacodylate, pH 7.0, 298 K.
As a consequence, this observation portends that forcing a 2′-SCF3 nucleoside into a C3′-endo (or C3′-endo-like) ribose pucker, as mandatory for an A-form RNA double helix to avoid steric interference of the 2′-substituent, would introduce an energetic penalty. Importantly, a very recent computational study by Li and Szostak supports this hypothesis.[34] The calculated free energy landscape revealed that the C2′-endo conformation of a single nucleoside within a native A-form RNA duplex is significantly less stable by 6 kcal mol−1 compared to the C3′-endo conformer.[34] This large value accounts for the disruption of the planar base-pair structure (therefore weakening stacking and hydrogen-bonding interactions) if a C2′-endo ribose had to be accommodated into the overall A-form geometry.[34]
We recall that 2′-SCF3 pyrimidine nucleosides exerted only a minor or negligible effect on thermodynamic stability if located in single-stranded regions (such as loops, bulges, or overhangs) next to double helixes.[11] This behavior was also found for the corresponding purine nucleosides investigated here. For instance, the hairpin forming RNA 5′-CGGA-GUGA-UCCG (Tm=60.3 °C; ΔG°, −5.7 kcal mol−1; ΔH°, −54.4 kcal mol−1; ΔS°, −163 cal mol−1 K−1) showed thermodynamic parameters that were comparable to those of the modified counterpart of 5′-CGGA-GU(2′-SCF3-G)A-UCCG (Tm=60.6 °C; ΔG°, −5.7 kcal mol−1; ΔH°, −55.3 kcal mol−1; ΔS°, −166 cal mol−1 K−1; Figure 2 D).
In response to a reviewer’s comment, we point out that the 2′-SCF3 modification is destabilizing also in the context of a DNA double helix. This is not unexpected because the C2′-endo pucker exposes the C2′-substituent in ribose configuration (such as the 2′-SCF3 group used here) to steric hindrance with the phosphate backbone in B-form conformation. In preliminary experiments, we found that the destabilization is of comparable degree for DNA and RNA. For example, the DNA hairpin 5′-CCGGAAGGT-ACGA-ACCTTCCG-3′ melts at a Tm value of 73.8 °C in 10 mM Na2HPO4, 150 mM NaCl, pH 7.0 (ΔG°, −6.8 kcal mol−1; ΔH°, −49.0 kcal mol−1; ΔS°, −142 cal mol−1 K−1) while 5′-CCGGAAGGT-ACGA-ACCUSCF3TCCG-3′ melts 23 degrees lower under the same conditions (Tm=50.7 °C; ΔG°, −3.4 kcal mol−1; ΔH°, −41.9 kcal mol−1; ΔS°, −129.2 cal mol−1 K−1; Supporting Information, Figure S1).
Probing of RNA structures by 19F NMR spectroscopy
To evaluate the applicability, and importantly, the uniformity of the 2′-SCF3 labeling concept not only with respect to pyrimidine but also with respect to purine nucleosides, we demonstrate a single-case study for NMR spectroscopic RNA probing here. Figure 4 A depicts the secondary structure model for the Thermoanaerobacter tengcongensis preQ1 class-I riboswitch.[37–39] This RNA becomes preorganized into a pseudoknot fold when Mg2+ is present at physiological concentrations. The distribution between the stem-loop fold (SL) with an unpaired strand overhang and the more compact RNA pseudoknot (P) was nicely reflected by the two 19F resonances at −40.2 and −40.5 ppm, in 3:7 ratio (Figure 4 B, middle). Ligand addition in fourfold excess resulted in a dominant population of the preQ1-bound RNA complex (C), reflected by a new signal at −40.8 ppm (Figure 4 B, bottom). The simplicity of the population analysis based on 19F spectra becomes obvious from a direct comparison with the corresponding NH imino proton 1H NMR spectra (Figure 4 C). Note that the imino protons exchange with the solvent and therefore result in much weaker intensities for the two dynamic, ligand-unbound RNA conformations (Figure 4 C, top, middle) while signal intensities are higher for the significantly more stable preQ1–aptamer complex (Figure 4 C, bottom), hence impairing an accurate quantification of populations of the coexisting folds.4
Figure 4.
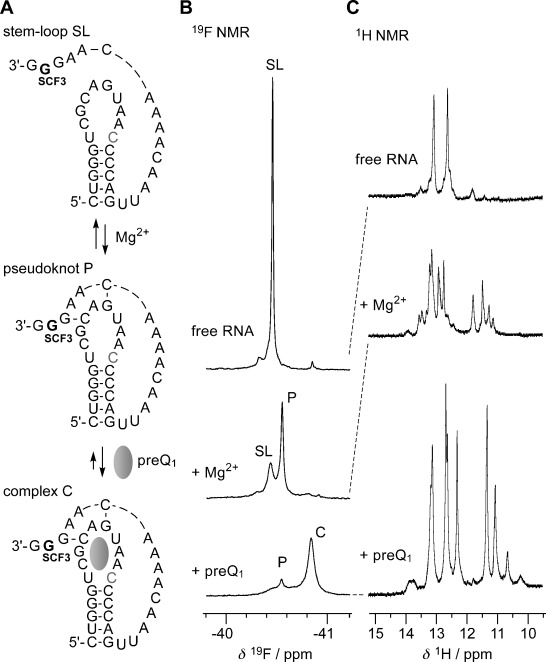
NMR spectroscopic analysis of Mg2+-assisted RNA pseudoknot formation, and subsequent stabilization through binding of a small ligand (Thermoanaerobacter tengcongensis preQ1 class-I riboswitch), using a 2′-SCF3 guanosine label. A) RNA secondary structure model, B) corresponding 19F NMR spectra, and C) imino proton 1H NMR spectra. Conditions: cRNA=0.3 mM, 25 mM sodium cacodylate, pH 7.0, 298 K; additions: cMg2+=2.0 mM; followed by cpreQ1=1.2 mM. The cytosine that forms a Watson–Crick base pair with the preQ1 ligand is highlighted in grey.
As a second example for 19F NMR spectroscopic applications of the novel labels, we analyzed duplex formation by titration and in temperature-dependent manner. The 14 bp RNA contained a single 2′-SCF3 adenosine in the middle region. Its melting temperature was readily obtained. The corresponding set of data is depicted in the Supporting Information, Figure S2.
The NMR analysis presented in Figure 4 together with other examples that we demonstrated previously for the pyrimidine series makes us confident that the 2′-SCF3 label awaits rapid and widespread applications.[10,11] Its performance confirms our original expectations for facile NMR spectroscopic probing of RNA structure rearrangements and RNA–ligand interactions.
We also mention that, to the best of our knowledge, only two other CF3 sensor nucleosides for probing of RNA secondary structures have been reported so far, namely 4′-C-[(4-trifluoromethyl-1H-1,2,3-triazol-1-yl)methyl]thymidine[40] and 5-[4,4,4-trifluoro-3,3-bis(trifluorometh-yl)but-1-ynyl]-2′-deoxyuridine,[41,42] both are sterically more demanding and represent DNA units. Additionally, trifluorothymidine has been analyzed for its NMR spectroscopic properties within DNA.[43]
RNA interference by 2′-SCF3-modified siRNA
As a novel application for the 2′-SCF3 modification, we tested the potential of this modification for gene silencing by small interfering RNA (siRNA). Nucleosides with destabilizing effects on Watson–Crick base pairing are of specific interest for the development of oligonucleotide therapeutics.[2,4,23] Most prominent is the highly flexible unlocked nucleic acid (UNA; or “seconucleoside”) modification.[23] UNA, missing the covalent C2′=C3′ bond of a ribose sugar, is not conformationally restrained, and can be used to influence oligonucleotide flexibility. UNA inserts reduce duplex Tm values by 5 to 10 °C per insert,[23] they facilitate antisense strand selection as the RISC guide, and UNA modifications to the seed region of a siRNA guide strand can significantly reduce off target effects.[44]
The comparable extent of destabilization of UNA and 2′-SCF3 modifications prompted us to explore a potential role of the latter in siRNA approaches. For reasons of comparability, we employed the same model system used previously to knock down the brain acid soluble protein 1 (BASP1) encoding gene by transient siRNA nucleofection in the chicken DF-1 cell line.[45,46] Expression of the BASP1 gene is specifically suppressed by Myc, an evolutionary conserved oncoprotein;[47] conversely, the BASP1 protein is an efficient inhibitor of Myc-induced cell transformation.[46]
We synthesized five siRNA duplexes for the BASP1 target gene with the sequence organization depicted in Figure 5 A (see also the Supporting Information, Table S2), two of them with a single 2′-SCF3 adenosine (A15) or guanosine (G8) in the sense strand, two of them with a single 2′-SCF3 guanosine modification in the antisense strand, very close to (G10) or within (G2) the seed region, and another one with two 2′-SCF3 modifications (G2 and A13) in the antisense strand. We determined the thermodynamic parameters for two of the five modified siRNAs (A15 s and G2 as; seed region) by UV melting profile analysis and—as expected—found significant destabilization compared to the native siRNA (Supporting Information, Figure S3).
Figure 5.
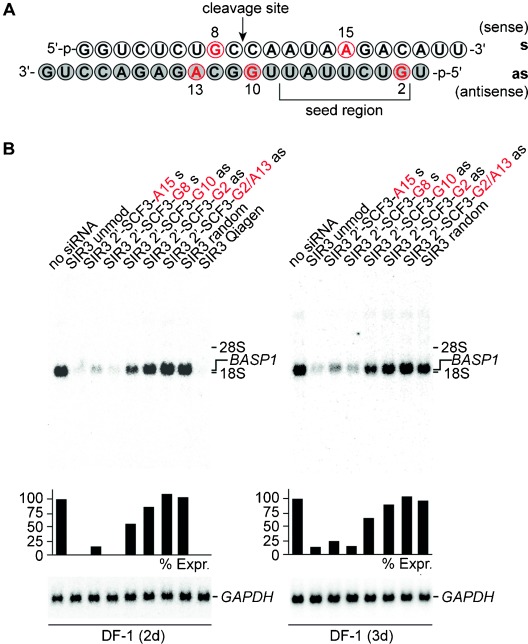
Gene silencing by 2′-SCF3-modified siRNAs. A) Sequence of the brain acid soluble protein 1 gene (BASP1)[46] targeting siRNA duplex used in this study; nucleosides in red indicate positions for 2′-SCF3 modification tested. B) Biological activities of 2′-SCF3-adenosine or 2′-SCF3-guanosine modified siRNAs directed against BASP1 mRNA. Chicken DF-1 cells grown on 60 mm dishes were transiently nucleofected with 0.24 nmol (∼3.0 μg) aliquots of unmodified (SIR) or modified siRNAs (SIR 2′-SCF3) on sense (s) or antisense (as) strands. An equal aliquot of siRNA with a shuffled (random) nucleotide sequence was used as a control. Total RNAs were isolated 2 days (left panel) or 3 days (right panel) after siRNA delivery, and 5 μg aliquots were analyzed by Northern hybridization using a DNA probe specific for the chicken BASP1 gene, and subsequently a probe specific for the housekeeping quail GAPDH gene.[46] The levels [%] of BASP1 expression (Expr.) were determined using a phosphorimager and are depicted as bars relative to mock transfections (no SIR, 100 %). The electrophoretic positions of rRNAs are indicated in the margin. SIR random: 5′-UCUGGGUCUAAGCCAAACAUT/5′-UGUUUGGCUUAGACCCAGAUdG.
The modified siRNAs caused complete gene silencing as observed for the non-modified reference duplex only when the modification resided in the sense strand (Figure 5 B). Instead, siRNA activity was impaired for the 2′-SCF3–G10 antisense-modified siRNA. Not unexpectedly, the two siRNAs carrying the 2′-SCF3 modification in the seed region at the critical position 2 of the antisense strand[23] were not active. These results indicate that the 2′-SCF3 modification is a promising tool for the alternative design of siRNAs with reduced off-target effects and warrants comprehensive studies in the future along these lines.[44]
Conclusion
The ribose 2′-trifluoromethylthio group makes ribonucleic acids an attractive reporter for spectroscopic investigations of RNA structure, structural dynamics, folding, and ligand interactions. So far, only RNA with 2′-SCF3 modified pyrimidine nucleosides has been accessible by chemical synthesis. The syntheses of the novel 2′-SCF3 purine nucleoside phosphoramidites and the corresponding RNAs has been demonstrated in this work and significantly expands the scope of applications for this modification. Their excellent behavior in 19F NMR probing of structure preformation and ligand binding was exemplified for the preQ1 class-I riboswitch and for melting of an RNA duplex. Moreover, all 2′-SCF3-modified nucleosides cause thermodynamic destabilization when they reside in double helices. Since this property is reminiscent of “unlocked nucleic acid” (UNA) which is widely used for siRNA technologies to minimize off-target effects,[23,44] we have highlighted the principal potential of 2′-SCF3 RNAs for siRNA design as a promising novel application of this modification.
Experimental Section
O6-tert-Butyl-N,N-bis(tert-butyloxycarbonyl)-3′,5′-O-(1,1,3,3-tetraisopropylsiloxane-1,3-diyl)-2′-acetylthio-2′-deoxyguanosine (G2)
Nucleoside G1 (1.46 g, 1.86 mmol) was dissolved in dichloromethane (25 mL) and DMAP (696 mg, 5.70 mmol) and trifluoromethanesulfonyl chloride (298 μL, 2.80 mmol) was added at 0 °C. After 40 min, the reaction mixture was treated with aqueous NaHCO3 solution (5 %), the organic layer was separated, dried over Na2SO4 and evaporated to yield the 2′-triflated derivative of G1 as a dark-yellow foam under high vacuum. The residue was dissolved in DMF (25 mL) and treated with potassium thioacetate (319 mg, 2.79 mmol), overnight, at ambient temperature. The solvent was distilled under reduced pressure and the crude product was purified by column chromatography on SiO2 (0–4 % CH3OH in dichloromethane v/v) to yield G2 as a brown foam (1.28 g, 1.52 mmol, 82 % over two steps). TLC (3 % CH3OH in CH2Cl2) Rf=0.72. 1H NMR (300 MHz, CDCl3): δ=1.06 (m, 28 H, 2×((CH3)2CH)2Si), 1.39 (s, 18 H, C(2)-N(Boc)2), 1.71 (s, 9 H, C(6)-O-C(CH3)3), 2.27 (s, 3 H, C(2′)-SOAc), 4.04 (m, 3 H, H1-C(5′), H2-C(5′), H-C(4′)), 4.60 (triplettoid, 1 H, H-C(2′)), 4.84 (dd, J=7.31, J=4.73 Hz, 1 H, H-C(3′)), 6.11 (d, J=6.09 Hz, 1 H, H-C(1′)), 8.11 ppm (s, 1 H, H-C(8)); 13C NMR (300 MHz, CDCl3): δ=12.74–13.47 (((CH3)2CH)2Si), 17.00–17.59 (((CH3)2CH)2Si), 27.95 (N((CO)OC(CH3)3)2), 28.43 (O-C(CH3)3), 30.43 (SCOCH3), 50.98 (C(2′), 63.06 (C(5′)), 71.86 (C(3′)), 85.56 (C(4′)), 87.41 (C(1′)), 140.64 ppm (C(8)); ESI-MS (m/z): [M+H]+ calcd for C38H65N5O10SSi2 839.41, found 840.18.
O6-tert-Butyl-N,N-bis(tert-butyloxycarbonyl)-3′,5′-O-(1,1,3,3-tetraisopropylsiloxane-1,3-diyl)-2′-sulfanyl-2′-deoxyguanosine (G3)
Compound G2 (5.04 g, 6.0 mmol) was dissolved in absolute ethanol (70 mL) and CH2Cl2 (3 mL) and was stirred for one hour at 0 °C and then treated with methyl amine (24 mL, 7 M in EtOH). After 25 min the solvents were evaporated under reduced pressure and the crude product was purified by column chromatography on SiO2 (0–2 % CH3OH in dichloromethane v/v) to yield G3 as a red foam (4.05 g, 5.08 mmol, 85 %). TLC (3 % CH3OH in CH2Cl2) Rf=0.79. 1H NMR (300 MHz, [D6]DMSO): δ=1.04 (m, 28 H, 2× (((CH3)2CH)2Si), 1.40 (s, 18 H, C(2)-N(Boc)2), 1.65 (s, 9 H, C(6)-O-C(CH3)3), 2.73 (d, J=8.13 Hz, 3 H, C(2′)-SH), 3.93–4.05 (m, 3 H, H1-C(5′), H2-C(5′), H-C(4′)), 4.35 (dd, 1 H, H-C(2′)), 4.62 (m, 1 H, H-C(3′)), 5.95 (d, J=7.20 Hz, 1 H, H-C(1′)), 8.52 ppm (s, 1 H, H-C(8)); 13C NMR (75 MHz, [D6]DMSO): δ=12.60–13.49 (((CH3)2CH)2Si), 17.33–17.93 (((CH3)2CH)2Si), 27.93 (N((CO)OC(CH3)3)2), 28.57 (O-C(CH3)3), 44.75 (C(2)), 63.62 (C(5′)), 72.84 (C(3′)), 85.67 (C(4′)), 89.96 (C(1′)), 142.99 ppm (C(8)); ESI-MS: (m/z) [M+H]+ calcd for C36H64N5O9SSi2 798.40, found 798.10.
O6-tert-Butyl-N,N-bis(tert-butyloxycarbonyl)-3′,5′-O-(1,1,3,3-tetraisopropylsiloxane-1,3-diyl)-2′-trifluoromethylthio-2′-deoxyguanosine (G4)
Nucleoside G3 (562 mg, 0.704 mmol) was dissolved in dichloromethane (12 mL) and cooled to −78 °C. To this solution, 3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole (Togni reagent, 279 mg, 8.45 mmol) was added as solid and the mixture was stirred, overnight. Within this time, the reaction mixture was slowly allowed to warm to room temperature. The solvent was removed and the crude product was purified by column chromatography on SiO2 (0–1.5 % CH3OH in dichloromethane) yielding G4 as a yellow foam (444 mg, 0.513 mmol, 73 %). TLC (6 % CH3OH in CH2Cl2 v/v) Rf=0.89. 1H NMR (300 MHz, CDCl3): δ=1.08 (m, 28 H, 2×((CH3)2CH)2Si), 1.36 (s, 18 H, C(2)-N(Boc)2), 1.72 (s, 9 H, C(6)-OC(CH3)3), 4.03 (m, 3 H, H1-C(5′), H2-C(5′), H-C(4′)), 4.89 (m, 2 H, H-C(3′), H-C(2′)), 6.22 (d, J=6.39 Hz, 1 H, H-C(1′)), 7.98 ppm (s, 1 H, H-C(8)); 13C NMR (75 MHz, CDCl3): δ=13.47–13.54 (((CH3)2CH)2Si), 16.95–17.61 (((CH3)2CH)2Si), 27.94 (N((CO)OC(CH3)3)2), 28.38 (O-C(CH3)3), 49.76 (C(2′), 63.42 (C(5′)), 73.13 (C(3′)), 85.81 (C(4′)), 90.37 (C(1′)), 141.66 ppm (C(8)); 19F NMR (565 MHz, CDCl3): δ=−39.35 ppm; ESI-MS (m/z): [M+NEt3+H]+ calcd for C43H78F3N6O9SSi2 967.50, found 967.30.
O6-tert-Butyl-N,N-bis(tert-butyloxycarbonyl)-2′-trifluoromethylthio-2′-deoxyguanosine (G5)
Compound G4 (908 mg, 1.05 mmol) was added to a solution of TBAF in THF (1 M; 5 mL) and was stirred at room temperature for 3 h. After that time, the solvent was removed and the crude product was purified by column chromatography on SiO2 (0–1.5 % CH3OH in dichloromethane) yielding G5 as a yellow foam (528 mg, 0.847 mmol, 81 %). TLC (6 % CH3OH in CH2Cl2 v/v) Rf=0.49. 1H NMR (300 MHz, [D6]DMSO): δ=1.34 (s, 18 H, C(2)-N(Boc)2), 1.65 (s, 9 H, C(6)-OC(CH3)3), 3.57, 3.69 (m, 2 H, H1-C(5′), H2-C(5′)), 4.09 (triplettoid, 1 H, H-C(4′)), 5.05 (triplettoid, 1 H, H-C(3′)), 4.73, 4.76 (dd, J=5.04, 4.91 Hz, 1 H, H-C(2′)), 5.19 (t, J=5.55 Hz, 1 H, HO-C(5′)), 6.32 (d, J=8.79 Hz, 1 H, H-C(1′)), 6.55 (d, J=5.37 Hz, 1 H, HO-C(3′)), 8.67 ppm (s, 1 H, H-C(8)); 13C NMR (75 MHz, [D6]DMSO): δ=27.92 (N((CO)OC(CH3)3)2), 28.37 (O-C(CH3)3), 50.57, 74.14 (C(2′), C(3′)), 63.20 (C(5′)), 88.91 (C(4′)), 91.75 (C(1′)), 130.22 (q, J=307.00 Hz, CF3), 143.03 ppm (C(8)); 19F NMR (565 MHz, CDCl3) δ=−40.00 ppm; ESI-MS (m/z): [M+H]+ calcd for C25H37F3N5O8S 624.23, found 624.29.
N2-(N,N-Dimethylformimidamide)-2′-trifluoromethylthio-5′-O-(4,4′-dimethoxytriphenylmethyl)-2′-deoxyguanosine (G6)
Nucleoside G5 (262 mg, 0.420 mmol) was dissolved in dry dichloromethane (6.7 mL), treated with trifluoroacetic acid (670 μL), and stirred at room temperature. Within few minutes, the yellow solution turned red. After 2.5 h, a second portion of trifluoroacetic acid (300 μL) was added and stirring of the solution was continued for two more hours. The reaction was stopped by addition of isopropanol (4 mL). The reaction mixture was evaporated, co-evaporated three times with isopropanol and subsequently exposed to high vacuum for 30 min. To the solid residue, methanol (10 mL) and N,N-dimethylformamide dimethyl acetal (1.5 mL, 11.3 mmol) was added. The reaction mixture was refluxed for 6 h, then evaporated to dryness, and extensively dried under high vacuum. Then, 4,4′-dimethoxytrityl chloride (156 mg, 0.460 mmol), and 4-(dimethylamino)-pyridine (approximately 5 mg) were dissolved in pyridine (2 mL) and added to the solid from above, yielding a suspension that was stirred for 18 h. Methanol (1 mL) was added, the solvents evaporated and the residue co-evaporated with methanol. The crude product was purified by column chromatography on SiO2 (0.5–3 % CH3OH in dichloromethane containing 0.5 % triethylamine) yielding G6 as yellow foam (186 mg, 0.202 mmol, 48 %). TLC (6 % CH3OH in CH2Cl2 v/v) Rf=0.16. 1H NMR (300 MHz, [D6]DMSO): δ=2.99 (s, 6 H, N(CH3)2), 3.42 (s, 2 H, H1-C(5′), H2-C(5′)), 3.74 (s, 6 H, 2×ar-O-CH3), 4.34 (triplettoid, 1 H, H-C(4′)), 4.47, 4.50 (dd, J=5.13 Hz, J=4.19 Hz, 1 H, H-C(2′)), 4.78 (triplettoid, 1 H, H-C(3′)), 6.78 (d, J=8.37 Hz, 1 H, H-C(1′)), 6.76–7.40 (m, 13 H, H-C(ar)), 7.71 (s br, 1 H, H-N(1)), 8.13 (s, 1 H, H-C(8)), 8.51 ppm (s, 1 H, H-C=N-C(6)); 13C NMR (75 MHz, [D6]DMSO): δ=35.23, 41.27 (N(CH3)2), 52.35 (C(2′), 63.93 (C(5′)), 72.92 (C(3′)), 85.84 (C(4′)), 89.95 (C(1′)), 113.42, 127.15–130.22(C(ar)), 149.13 ppm (C(8)); 19F NMR (565 MHz, [D6]DMSO): δ=−73.51 ppm; ESI-MS (m/z): [M+NEt3+H]+ calcd for C41H50F3N7O6S 825.35, found 826.13.
N2-[(Dimethylamino)methylene]-2′-trifluoromethylthio-5′-O-(4,4′-dimethoxytriphenylmethyl)-2′-deoxyguanosine 3′-O-((2-cyanoethyl) N,N-diisopropylphosphoramidite) (G7)
Nucleoside G6 (167 mg, 0.230 mmol) was dissolved in absolute dichloromethane (2.0 mL) and ethyldimethyl amine (175 μL, 1.612 mmol) and stirred for 15 min before 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (111 mg, 0.469 mmol) was added dropwise. The reaction was monitored by thin layer chromatography which showed almost complete reaction after 1.5 h, more 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (40 mg, 0.169 mmol) was added slowly. After a total reaction time of 2.5 h, the reaction was quenched by the addition of CH3OH (50 μL) and stirring was continued for seven more minutes before the solvents were evaporated and dried under high vacuum to give a slightly yellow foam. The crude product was subjected to column chromatographic purification on SiO2 (ethylacetate/hexane 4:6 to 100 % ethylacetate, then 1 % CH3OH in ethylacetate; all eluents contained 0.5 % triethylamine) yielding G7 as white foam (154 mg, 0.166 mmol, 72 %). TLC (6 % CH3OH in CH2Cl2+1 % triethylamine v/v) Rf=0. 35. 1H NMR (300 MHz, CDCl3): δ=1.08–1.26 (m, 12 H, PN(CH(CH3)2)2), 2.35, 2.63 (2×m, 4 H, 2×OCH2CH2CN), 3.05 (m, 12 H, 2×N(CH3)2), 3.62 (m, 2 H, OCH2CH2CN), 3.92 (m, 2 H, OCH2CH2CN), 3.78 (s, 6 H, 2×ar-O-CH3), 4.32, 4.41 (2×s, 2 H, 2×H-C(4′)), 4.51–4.78 (m 4 H, 2×H-C(3′), 2×H-C(2′)), 6.17, 6.23 (2×d, J=9.15, 8.88 Hz); 6.80–7–39 (m, 13 H, H-C(ar)), 7.73, 7.76 (2×s, 2 H, 2×H-C(8)), 8.46, 8.51 (2×s, 2 H, 2×H-C=N-C(6)), 9.35 ppm (s br, H-N(1)); 13C NMR (75 MHz, CDCl3): δ=20.24–20.63 (CH2CN), 24.72 (N(CH3)2, PN(CH(CH3)2)2), 43.46–43.67 (O-CH2CH2-CN), 55.40 (O-CH3), 57.98–63.58 (C(5′), O-CH2CH2-CN), 75.10; 74.87 (C(3′)), 75.91, 76.11 (C(2′)), 84.71, 84.98 (2× C(4′)), 87.07, 87.21 (2×C(1′)); 113.50, 127.30, 128.22, 130.18 ppm (C(ar)), 158.24 H-C=N); 19F NMR (565 MHz, CDCl3): δ=−39.64, −39.82 ppm; 31P NMR (121 MHz, CDCl3): δ=151.27, 152.47 ppm; ESI-MS (m/z): [M+H]+ calcd for C44H53F3N8O7PS 925.34, found 925.25.
Solid-phase RNA synthesis
All oligonucleotides were synthesized on a 1.0 μmol scale using an Applied Biosystems ABI392, following standard synthesis protocols. Detritylation (2 min): 4 % (v/v) dichloroacetic acid in 1,2-dichloroethane. Coupling (3 min): 120 μL phosphoramidite (0.1 M) in acetonitrile plus 360 μL 5-(benzylthio)-1H-tetrazole in acetonitrile (0.30 M) as activator. Capping (2×0.5 min): 1:1 (v/v) Cap A/Cap B, Cap A: 0.2 M phenoxyacetic anhydride in dry THF, Cap B: 1-methyl imidazole (0.2 M) and 2,4,6-trimethylpyridine (0.2 M) in dry THF. Oxidation (1 min): 20 mM iodine in 7:2:1 (v/v/v) THF/pyridine/water. Phosphoramidite and activator solutions were dried over activated molecular sieves (3 Å), overnight. All sequences were synthesized trityl-off.
Deprotection of 2′-SCF3-containing RNA
The solid support was transferred into a screw-capped Eppendorf tube and 1.5 mL of a 1:1 mixture of solution of methylamine in ethanol (33 %) ammonia and aqueous methylamine (40 %) was added and the reaction proceeded at room temperature for 4 to 6 h with occasional shaking. IMPORTANT: For deprotection of 2′-SCF3 adenosine containing RNA, the deprotection solution additionally contained 150 mM of threo-1,4-dimercapto-2,3-butandiol (DTT).[26,33] The suspension was filtered and all volatiles evaporated. The residue was dissolved in 1.0 mL of 1 M tetrabutylammonium fluoride trihydrate in THF and kept at 37 °C for 12 h. The reaction was quenched by addition of 1.0 mL of triethylammonium bicarbonate buffer (1 M, pH 7.4) and the organic solvent was evaporated. The solution was subjected to size-exclusion chromatography on an Amersham HiPrep 26/10 desalting column (2.6×10 cm; Sephadex G25). The crude RNA was eluted with H2O, dried, and redissolved in 1 mL H2O.
Analysis and purification of 2′-SCF3-containing RNA
Analysis of crude products was performed by anion-exchange HPLC on a Dionex DNAPac-100 column (4×250 mm) at 80 °C. Flow rate: 1 mL min−1; eluant A: 25 mM Tris-HCl (pH 8.0), 6 M urea; eluant B: 25 mM Tris-HCl (pH 8.0), 6 M urea, 500 mM NaClO4; gradient: 0–40 % B in A within 25 min; UV detection at 260 nm. Crude products were purified on a semipreparative Dionex DNAPac-100 column (9×250 mm) at 80 °C. Flow rate: 2 mL min−1; gradient: 3–17 % B in A within 15 min for oligonucleotides <10 nt; 24–35 % B in A within 18 min for oligonucleotides >10 nt; UV detection at 260 nm. Fractions containing the oligonucleotide were diluted with an equal volume of triethylammonium bicarbonate buffer (100 mM, pH 7.4) and loaded on an equilibrated C18 SepPak Plus cartridge (Waters), washed with water and eluted with water/acetonitrile (1:1, v/v). Purified fractions were evaporated and redissolved in 1.0 mL water. The RNA yield was determined as units of optical density at 260 nm by UV spectroscopy (Implen NanoPhotometer) at room temperature. The product quality and purity was verified by anion-exchange chromatography on an analytical column (vide supra).
RNA interference and Northern analysis
Lyophilized synthetic siRNA duplexes were dissolved, annealed, and delivered into chicken DF-1 cells by electroporation as described.[45] Total RNA isolation, and analysis of gene silencing by Northern hybridization using specific 32P-radiolabelled DNA probes for detection of BASP1 and GAPDH mRNAs were done as described previously.[45,46]
Acknowledgments
We thank Katja Fauster for providing the 2′-SCF3 DNA data set. M.K. is an ESR fellow of the EU FP7 Marie Curie ITN RNPnet program (289007). Funding by the Austrian Science Foundation FWF (I1040 to R.M., I844 and P26550 to C.K., P23652 to K.B.) is acknowledged.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201500415.
References
- [1].Phelps K, Morris A, Beal PA. ACS Chem. Biol. 2012;7:100–109. doi: 10.1021/cb200422t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Deleavey GF, Damha MJ. Chem. Biol. 2012;19:937–954. doi: 10.1016/j.chembiol.2012.07.011. [DOI] [PubMed] [Google Scholar]
- [3].Wachowius F, Höbartner C. ChemBioChem. 2010;11:469–480. doi: 10.1002/cbic.200900697. [DOI] [PubMed] [Google Scholar]
- [4].Shukla S, Sumaria CS, Pradeepkumar PI. ChemMedChem. 2010;5:328–349. doi: 10.1002/cmdc.200900444. [DOI] [PubMed] [Google Scholar]
- [5].El-Sagheer AH, Brown T. Proc. Natl. Acad. Sci. USA. 2010;107:15329–15334. doi: 10.1073/pnas.1006447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kiviniemi A, Virta P, Lönnberg H. Bioconjugate Chem. 2010;21:1890–1901. doi: 10.1021/bc100268w. [DOI] [PubMed] [Google Scholar]
- [7].Willibald J, Harder J, Sparrer K, Conzelmann KK, Carell T. J. Am. Chem. Soc. 2012;134:12330–12333. doi: 10.1021/ja303251f. [DOI] [PubMed] [Google Scholar]
- [8].Helm M, Alfonzo JD. Chem. Biol. 2014;21:174–185. doi: 10.1016/j.chembiol.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xu J, Springstubbe D, Rublack N, Appel B, Müller S. Chem. Today. 2012;30:42–44. [Google Scholar]
- [10].Fauster K, Kreutz C, Micura R. Angew. Chem. Int. Ed. 2012;51:13080–13084. doi: 10.1002/anie.201207128. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- [11].Košutić M, Jud L, Da Veiga C, Frener M, Fauster K, Kreutz C, Ennifar E, Micura R. J. Am. Chem. Soc. 2014;136:6656–6663. doi: 10.1021/ja5005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moumné R, Pasco M, Prost E, Lecourt T, Micouin L, Tisné C. J. Am. Chem. Soc. 2010;132:13111–13113. doi: 10.1021/ja1037885. [DOI] [PubMed] [Google Scholar]
- [13].Cobb S, Murphy C. J. Fluorine Chem. 2009;130:132–143. [Google Scholar]
- [14].Hennig M, Scott LG, Sperling E, Bermel W, Williamson JR. J. Am. Chem. Soc. 2007;129:14911–14921. doi: 10.1021/ja073825i. [DOI] [PubMed] [Google Scholar]
- [15].Olejniczak M, Gdaniec Z, Fischer A, Grabarkiewicz T, Bielecki L, Adamiak RW. Nucleic Acids Res. 2002;30:4241–4249. doi: 10.1093/nar/gkf541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hammann C, Norman DG, Lilley DMJ. Proc. Natl. Acad. Sci. USA. 2001;98:5503–5508. doi: 10.1073/pnas.091097498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chu WC, Horowitz J. Nucleic Acids Res. 1989;17:7241–7252. doi: 10.1093/nar/17.18.7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Puffer B, Kreutz C, Rieder U, Ebert MO, Konrat R, Micura R. Nucleic Acids Res. 2009;37:7728–7740. doi: 10.1093/nar/gkp862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luy B, Merino JP. J. Biomol. NMR. 2001;20:39–47. doi: 10.1023/a:1011210307947. [DOI] [PubMed] [Google Scholar]
- [20].Reif B, Wittmann V, Schwalbe H, Griesinger C, Worner K, Jahn-Hoffmann K, Engels J, Bermel W. Helv. Chim. Acta. 1997;80:1952–1971. [Google Scholar]
- [21].Kreutz C, Kählig H, Konrat R, Micura R. Angew. Chem. Int. Ed. 2006;45:3450–3453. doi: 10.1002/anie.200504174. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006;118 [Google Scholar]
- [22].Egli M, Minasov G, Tereshko V, Pallan PS, Teplova M, Inamati GB, Lesnik EA, Owens SR, Ross BS, Prakash TP, Manoharan M. Biochemistry. 2005;44:9045–9057. doi: 10.1021/bi050574m. [DOI] [PubMed] [Google Scholar]
- [23].Campbell MA, Wengel J. Chem. Soc. Rev. 2011;40:5680–5689. doi: 10.1039/c1cs15048k. [DOI] [PubMed] [Google Scholar]
- [24].Gruen M, Becker C, Beste A, Siethoff C, Scheidig AJ, Goody RS. Nucleosides Nucleotides. 1999;18:137–151. [Google Scholar]
- [25].Matoušek V, Pietrasiak E, Schwenk R, Togni A. J. Org. Chem. 2013;78:6763–6768. doi: 10.1021/jo400774u. [DOI] [PubMed] [Google Scholar]
- [26].Moroder H, Kreutz C, Lang K, Serganov A, Micura R. J. Am. Chem. Soc. 2006;128:9909–9918. doi: 10.1021/ja0621400. [DOI] [PubMed] [Google Scholar]
- [27].Fauster K, Hartl M, Santner T, Aigner M, Kreutz C, Bister K, Ennifar E, Micura R. ACS Chem. Biol. 2012;7:581–589. doi: 10.1021/cb200510k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Simeone L, Milano D, De Napoli L, Irace C, Di Pascale A, Boccalon M, Tecilla P, Montesarchio D. Chem. Eur. J. 2011;17:13854–13865. doi: 10.1002/chem.201101827. [DOI] [PubMed] [Google Scholar]
- [29].Wang R-W, Gold B. Org. Lett. 2009;11:2465–2468. doi: 10.1021/ol9007537. [DOI] [PubMed] [Google Scholar]
- [30].Sikchi SA, Hultin PG. J. Org. Chem. 2006;71:5888–5891. doi: 10.1021/jo060430t. [DOI] [PubMed] [Google Scholar]
- [31].Pitsch S, Weiss PA, Jenny J, Stutz A, Wu X. Helv. Chim. Acta. 2001;84:3773–3795. [Google Scholar]
- [32].Micura R. Angew. Chem. Int. Ed. 2002;41:2265–2269. doi: 10.1002/1521-3773(20020703)41:13<2265::AID-ANIE2265>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002;114 [Google Scholar]
- [33].Höbartner C, Rieder R, Kreutz C, Puffer B, Lang K, Polonskaia A, Serganov A, Micura R. J. Am. Chem. Soc. 2005;127:12035–12045. doi: 10.1021/ja051694k. [DOI] [PubMed] [Google Scholar]
- [34].Li L, Szostak JW. J. Am. Chem. Soc. 2014;136:2858–2865. doi: 10.1021/ja412079b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Haziri AI, Leumann CJ. J. Org. Chem. 2012;77:5861–5869. doi: 10.1021/jo300554w. [DOI] [PubMed] [Google Scholar]
- [36].Altona C, Sundaralingam MJ. J. Am. Chem. Soc. 1973;95:2333–2344. doi: 10.1021/ja00788a038. [DOI] [PubMed] [Google Scholar]
- [37].Roth A, Winkler WC, Regulski EE, Lee BWK, Lim J, Jona I, Jona I, Barrick JE, Ritwik A, Kim JN, Welz R, Iwata-Reuyl D, Breaker RR. Nat. Struct. Mol. Biol. 2007;14:308–317. doi: 10.1038/nsmb1224. [DOI] [PubMed] [Google Scholar]
- [38].Jenkins JL, Krucinska J, McCarty RM, Bandarian V, Wedekind JE. J. Biol. Chem. 2011;286:24626–24637. doi: 10.1074/jbc.M111.230375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Santner T, Rieder U, Kreutz C, Micura R. J. Am. Chem. Soc. 2012;134:11928–11931. doi: 10.1021/ja3049964. [DOI] [PubMed] [Google Scholar]
- [40].Granqvist L, Virta P. J. Org. Chem. 2014;79:3529–3536. doi: 10.1021/jo500326j. [DOI] [PubMed] [Google Scholar]
- [41].Kiviniemi A, Virta P. J. Am. Chem. Soc. 2010;132:8560–8562. doi: 10.1021/ja1014629. [DOI] [PubMed] [Google Scholar]
- [42].Barhate NB, Barhate RN, Cekan P, Drobny G, Sigurdsson ST. Org. Lett. 2008;10:2745–2747. doi: 10.1021/ol800872a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gmeiner WH, Pon RT, Lown JW. J. Org. Chem. 1991;56:3602–3608. [Google Scholar]
- [44].Vaish N, Chen F, Seth S, Fosnaugh K, Liu Y, Adami R, Brown T, Chen Y, Harvie P, Johns R. Nucleic Acids Res. 2011;39:1823–1832. doi: 10.1093/nar/gkq961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aigner M, Hartl M, Fauster K, Steger J, Bister K, Micura R. ChemBioChem. 2011;12:47–51. doi: 10.1002/cbic.201000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hartl M, Nist A, Khan MI, Valovka T, Bister K. Proc. Natl. Acad. Sci. USA. 2009;106:5604–5609. doi: 10.1073/pnas.0812101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hartl M, Mitterstiller A-M, Valovka T, Breuker K, Hobmayer B, Bister K. Proc. Natl. Acad. Sci. USA. 2010;107:4051–4056. doi: 10.1073/pnas.0911060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.