

Abstract
Host immune responses must be tightly regulated by an intricate balance between positive and negative signals while fighting pathogens; persistent pathogens may usurp these regulatory mechanisms to dampen host immunity to facilitate survival in vivo. Here we report that Tim-3, a negative signalling molecule expressed on monocytes and T cells, is up-regulated on natural killer (NK) cells in individuals chronically infected with hepatitis C virus (HCV). Additionally, the transcription factor T-bet was also found to be up-regulated and associated with Tim-3 expression in NK cells during chronic HCV infection. MicroRNA-155 (miR-155), an miRNA that inhibits signalling proteins involved in immune responses, was down-regulated in NK cells by HCV infection. This Tim-3/T-bet over-expression and miR-155 inhibition were recapitulated in vitro by incubating primary NK cells or NK92 cell line with Huh-7 hepatocytes expressing HCV. Reconstitution of miR-155 in NK cells from HCV-infected patients led to a decrease in T-bet/Tim-3 expression and an increase in interferon-γ production. Blocking Tim-3 signalling also enhanced interferon-γ production in NK cells by improving signal transducer and activator of transcription-5 phosphorylation. These data indicate that HCV-induced, miR-155-regulated Tim-3 expression regulates NK cell function, suggesting a novel mechanism for balancing immune clearance and immune injury during chronic viral infection.
Keywords: hepatitis C virus, interferon-γ, microRNA-155, natural killer cells, signal transducer and activator of transcription-5, T-cell immunoglobulin and mucin domain protein-3
Introduction
Host immune responses must be tightly regulated by an intricate balance between positive and negative signals to ensure their appropriate onset and termination while fighting pathogens and avoiding autoimmunity; persistent pathogens may usurp these regulatory machineries to dampen host immune responses for their survival in vivo. Hepatitis C virus (HCV), a blood-borne viral infection characterized by a high rate of chronic infection, has evolved multiple strategies to evade host immunity, so becoming an excellent model to study the mechanisms of persistent viral infections.1,2 Although the use of direct antiviral agents has resulted in a significant improvement in the outcome of HCV treatment, this therapeutic cocktail is still in development and already facing new issues such as viral mutation, relapse and re-infection following therapy.3,4 Additionally, the lack of a vaccine for this virus is a major hurdle to control this global infection. The failure to successfully manage this chronic viral infection and to develop an effective vaccine stems from our incomplete understanding of the correlates of host immunity to HCV and of the HCV–host interactions that permit viral persistence.
Natrual killer (NK) cells represent the first line of host defence against invading pathogens. Human NK cells comprise CD56bright and CD56dim subsets, which differ in their maturation status, proliferative capacity, cytotoxic activity and cytokine production.5,6 They generally become activated in an early phase of viral infection, and play an essential role in eliminating HCV-infected cells directly by cytolytic killing and indirectly by secreting cytokines.7,8 They also interact with professional dendritic cells and virus-specific T cells in implementing antiviral immunity.9 The antiviral activity of NK cells is regulated through surface NK receptors, including the inhibitory killer immunoglobulin-like receptors, the leucocyte immunoglobulin-like receptors, cytotoxicity receptors NKp30, NKp44, NKp46, and C-type lectin-like receptors CD94/NKG2, which comprise inhibitory (NKG2A) or activating (NKG2C/NKG2D) isoforms.5,6 Not surprisingly, HCV has evolved multiple strategies to counter-regulate the host's NK cellular responses by altering the expression of these inhibitory and stimulatory receptors, contributing to the pathogenesis and progression of liver disease.10–12 Notably, aberrant NK activities may also contribute towards liver injury.9 Therefore, further studies are required to understand how NK cells are fine-tuned during host defence and in liver injury during HCV infection.
Tim-3, a member of T-cell immunoglobulin and mucin domain proteins (Tim), represents a major mechanism to maintain the balance between positive and negative signals to ensure adequate immune protection against pathogens, and yet prevent over-activation of lymphocytes, and hence immune-injury or autoimmunity.13–15 Compelling evidence is emerging for the role of Tim-3 in peripheral immune tolerance, autoimmune response, anti-tumour and antiviral immune evasion, which has raised the possibility that a therapeutic strategy targeting this inhibitory pathway might be of clinical benefit, including for those with HCV infection.13–15 Although Tim-3 has been identified as an inhibitory receptor preferably expressed on exhausted T helper type 1 cells, its role in innate immune modulation remains less understood. We and others have previously demonstrated that Tim-3 is up-regulated on both monocytes and T cells and negatively regulates innate and adaptive immune responses during HCV infection.16–20 Additionally, Tim-3 has been shown to regulate NK functions in healthy subjects and in chronic viral (HBV, HIV) infections.21–24 The role of Tim-3 expression and control of NK cell functions during HCV infection remains largely unknown.
MicroRNAs (miRNAs or miR) are a class of small, non-coding RNAs that can regulate gene expression through translational repression and have been implicated as negative regulators of innate and adaptive immune responses.25–27 miR-155 has been identified as a key modulator of cell functions in both innate and adaptive immunity.28–35 Recent studies have reported that miR-155 is regulated in monocytes, NK cells and hepatocytes and acts as a positive regulator of inflammation in chronic HCV infection.36–38 Transcription of miR-155 is regulated by nuclear factor-κB (NF-κB), and p300 increased NF-κB-dependent miR-155 expression.37 Over-expression of miR-155 can inhibit cell apoptosis and promote cell proliferation, whereas miR-155 inhibition induces G0/G1 arrest.37 The mechanisms for miR-155 regulation of NK cell function during chronic viral infection, however, remain to be determined.
In this study, we examined Tim-3 and miR-155 expressions in NK cells and assessed their effect on NK interferon-γ (IFN-γ) production in individuals with chronic HCV infection. We found that Tim-3 expression was up-regulated, whereas miR-155 was down-regulated, by HCV infection. Reconstitution of miR-155 or blocking Tim-3 signalling enhanced IFN-γ production in NK cells by improving signal transducer and activator of transcription 5 (STAT-5) phosphorylation. These results suggest that HCV-induced, miR-155-regulated Tim-3 expression negatively regulates NK cell function in chronic viral infection.
Materials and methods
Subjects
The study protocol was approved by the institutional review board of East Tennessee State University and James H. Quillen VA Medical Center (ETSU/VA IRB, Johnson City, TN), which have contributed to a database for the storage of blood samples taken from HCV-infected individuals for the purpose of viral immunology studies. As shown in Table1, the study participants comprised three populations: (i) 36 chronically HCV-infected patients, HCV genotype (70% type 1, 30% type 2 or 3) and viral load (ranging from 12 300 to 500 000 IU/ml) were performed by Lexington VA Medical Center (Lexington, KY), and all subjects were virologically and serologically positive for HCV before the antiviral treatment; (ii) eight HCV participants who achieved a sustained virological response (SVR) following antiviral therapy with pegylated interferon plus ribavirin and/or boceprevir; and (iii) 19 healthy subjects (HS; buffy coat derived from Key Biologics LLC, Memphis, TN) who were negative for HBV, HCV and HIV infection. Written informed consent was obtained from all participants. Most of the study subjects were male. The mean ages of the three populations was comparable (P > 0·05).
Table 1.
Demographic features of the study subjects
| Group | No. of subjects | Mean age | % of male | % of genotype 1 | HCV RNA (IU/ml) | Medications used for HCV treatment |
|---|---|---|---|---|---|---|
| HCV-infected patients | 36 | 53 (31–69) | 96 | 70 | 23 000–50 000 000 | N/A |
| HCV-treated patients (SVR) | 8 | 53 | 100 | N/A | N/A | PegIFN + RBV ± Boceprevir |
| Healthy subjects | 19 | 47 | 84 | N/A | N/A | N/A |
HCV, hepatitis C virus; N/A, not applicable; PegIFN, pegylated interferon; RBV, ribavirin; SVR, sustained virological response.
Cell isolation and culture
Human peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood of study subjects by Ficoll-density centrifugation with lympho-H (Atlanta Biological, Lawrenceville, GA). Where indicated, CD3− CD56+ NK cells were purified from PBMCs by negative selection according to the manufacturer's instructions (purity > 95%; Miltenyi Biotec Inc., Auburn, CA). The cells were cultured in RPMI-1640 medium containing 200 U/ml IL-2 (eBioscience, San Diego, CA), 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 100 IU/ml penicillin and 2 mm l-glutamine (Thermo Scientific, Logan, UT) at 37° and 5% CO2 atmosphere. The human IL-2-dependent NK cell line NK-92 (ATCC® CRL2407™, Manassas, VA) was maintained in culture Medium minimal essential medium-α (Gibco, Grand Island, NY), supplemented with 12·5% heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA) and 12·5% horse serum, 2 mm l-glutamine and 200 U/ml recombinant human IL-2 (hIL-2_, 0·2 mm inositol (Hoffman-LaRoche, Basel, Switzerland); 0·1 mm 2-mercaptoethanol (Hoffman-LaRoche); 0·02 mm folic acid (Hoffman-LaRoche) per ATCC instructions.
Co-culture of human primary NK cells or NK-92 cells with HCV+ or HCV− Huh-7 hepatocytes
Transfection of Huh-7 hepatocytes (kindly provided by Dr T.J. Liang, Liver Section, NIH/NIDDK) with HCV JFH-1 strain (kindly provided by Dr T. Wakita) was carried out as described previously.16 RNA transfection control as well as non-transfected control was carried out to assess the potential effects of RNA on the co-cultured cells in our preliminary studies. Before the co-culture experiment, HCV+ or HCV− Huh-7 hepatocytes were serum-starved for 18 hr, then activated with recombinant human IFN-γ (rhIFN-γ; (0·1 μg/ml; R&D Systems, Minneapolis, MN) for 48 hr to boost the HCV replication.16 Activated hepatocytes were removed from plates with 0·05% trypsin–EDTA and then plated at 5 × 105 cells/well in a six-well plate. Purified human primary NK cells or NK-92 cells were then incubated with the adherent hepatocytes in RPMI-1640 or minimal essential medium containing 200 U/ml IL-2 (eBioscience) and/or 100 ng/ml IL-18 (MBL Co., Woburn, MA) for an additional 48 hr, and the expression levels of Tim-3, T-bet and IFN-γ were analysed by flow cytometry as described below. MicroRNA from NK cells was extracted 6 hr following co-culture and microRNA155-5p was analysed by real-time PCR as described below.
Flow cytometry
Procedures for detection of cell surface markers and intracellular cytokine staining were performed as described previously.16–19 Briefly, human PBMCs, purified NK cells, or NK-92s (0·2 × 106 per well in a 96-well plate) were stimulated with 10 ng/ml IL-12 (eBioscience) and 100 ng/ml IL-18 (MBL Co.) for 24 hr, followed by 1 μg/ml Brefeldin A (BioLegend, San Diego, CA) 5 hr before harvesting the cells to forbidden cytokine secretion. Cell surface markers were stained with specific conjugated anti-CD3-phycoerythrin, CD56-Peridinin chlorophyll protein 710, Tim-3-allophycocyanin antibodies (eBioscience, F38-2E2). Alexa Fluor 488-conjugated KLRG1 (13F12) was a gift from Dr Hanspeter Pircher. For intracellular staining, the cells were fixed and permeabilized with Inside Stain Kit (Miltenyi Biotec), followed by incubation with conjugated anti-IFN-γ-phycoerythrin (Miltenyi Biotec Inc.), T-bet-Peridinin chlorophyll protein 710. Isotype-matched controls (eBioscience) were used to determine the level of background staining; the fluorescence minus one strategy was used to determine background levels of staining and to adjust multicolour compensation for cell gating. The cells was sorted on an Accuri C6 flow cytometer (BD, Franklin Lakes, NJ) and analysed using flowjo software (7·6·1, Ashland, OR).
Real-time RT-PCR
Total RNA isolated from NK cells with or without IL-2 stimulation was extracted using RNAzol®RT (Molecular Research Center, Inc., Cincinnati, OH). The cDNA was generated according to the manufacturer's recommendations per TaqMan MicroRNA reverse transcription kit using primers specific for miR-155 and U6 as control (TaqMan, Grand Island, NY). Levels of miRNA levels quantified by real-time PCR (4800 PCR machine; Bio-Rad Systems, Hercules, CA) using specific miR-155 and snRU6 primer sets and TaqMan Universal Master Mix (both from Applied Biosystems). Data for miR-155 transcript levels were analysed per comparative CT, normalized to snRU6 levels, and expressed as fold changes using the 2−ΔΔct method.
NK cell transfections
Purified human NK cells from HCV patients or NK-92 cells were transfected with 30 pmol of miR-155 mimic or the negative control (Life Technologies, Grand Island, NY) using the Human NK Nucelofector Kit and Nucleofector I Device (Lonza, Allendale, NJ). Transfection efficiency was approximately 20% for primary NK cells (50% in naive CD4+ T cells and 60% in monocytes) as determined by the transfection of a fluorescently labeled negative miRNA control. Transfection efficiency was also assayed by measuring miR-155 transcript levels following transfection. After transfection, NK cells were cultured in Iscove's modified Eagles's medium medium (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum, 200 U/ml IL-2 for 48 hr, further stimulated with 10 ng/ml IL-12 and 100 ng/ml IL-18 for 24 hr as described above before harvest for Tim-3 and T-bet, IFN-γ measurement.
Tim-3 blockade
Purified NK cells from HCV patients and healthy subjects were incubated with 10 μg/ml LEAF™ anti-human Tim-3 antibody and/or anti-human KLRG-1 (3 μg/ml, from Dr Hanspeter Pircher) or control IgG (BioLegend) for 48 hr, followed by stimulation with IL-12 and IL-18 for 24 hr as described above, then subjected to flow cytometric analysis of IFN-γ. For flow cytometric analysis of p-STAT5, NK cells from HCV patients were blocked with LEAF™ anti-human Tim-3 antibody or control IgG (BioLegend) for 54 hr, then stimulated with 100 ng/ml IL-15 (eBioscience) for 18 hr, followed by 1 μg/ml Brefeldin A (BioLegend) 5 hr before harvesting the cells, after which the cells were pulsed with 10 ng/ml IL-15 (eBioscience) for 30 min. The NK cells were fixed and permeabilized with ice-cold 90% methanol, and sequentially incubated with p-STAT5-Peridinin chlorophyll protein 710 (eBioscience) for 1 hr at room temperature. The cells was sorted on an Accuri C6 flow cytometer (BD) and analysed using flowjo software (7.6.1).
Western blot analysis
The Tim-3 or control IgG-treated NK cells from HCV-infected individuals were lysed on ice in RIPA lysis buffer (Boston BioProducts Inc, Ashland, MA) in the presence of protease inhibitors (Thermo Scientific, Rockford, IL). Cell lysates were centrifuged for 10 min at 4°, supernatants were recovered and the protein concentrations were measured by Pierce BCA protein assay kit (Thermo Scientific). Proteins were separated by SDS–PAGE, and transferred to polyvinylidene difluoride membranes. Membranes were blocked with 5% milk, 0·5% Tween-20 in Tris-buffered saline, and incubated with the anti-phosphorylated STAT5 (Cell Signaling Technology, Inc, Danvers, MA). Appropriate horseradish peroxide-conjugated secondary antibody (Cell Signaling) was then used and proteins were detected using an enhanced chemiluminescence assay kit (Amersham, Piscataway, NJ). Membranes were stripped and re-probed with total STAT5 antibody as an internal control (Cell Signaling). Protein bands were captured and quantitatively analysed by a Chemi Doc™ MP Imaging System (Bio-Rad System).
Statistical analysis
Study results are summarized for each group and results are expressed as the mean ± standard deviation (SD). Comparison between two groups was performed by multiple comparisons testing/least significant difference or Tukey's procedure depending on the analysis of variance F test prism software (version 4; GraphPad Software, San Diego, CA) by a non-parametric Mann–Whitney U-test. A pairwise t-test was used to compare the significance of changes in Tim-3 blockade or miR-155 transfection experiments. Correlation between Tim-3 and T-bet expression in NK cells was analysed using a Pearson Correlation program. Values of P < 0·05 were considered significant.
Results
Tim-3 is up-regulated on NK cells, which are significantly reduced in HCV-infected individuals
Compromised NK cell functions have been reported in chronically HCV-infected individuals, whereas normalization of depressed NK activity after antiviral therapy is associated with a low frequency of relapse and improved SVR.10–12 To characterize the effect of HCV infection on NK cells, we first compared the cell frequency of CD3− CD56+ NK cells in the gated lymphocytes in PBMCs from 26 HCV-infected patients, six SVR individuals following antiviral treatment, and six HS. As shown in the representative dot plots and summary data in Fig.(1a,b), CD3− CD56+ NK lymphocyte frequencies in patients with chronic HCV infection were significantly reduced compared with SVR and HS; whereas there was no difference between SVR and HS. These results suggest that HCV infection reduces NK cell numbers, and successful antiviral therapy recovers the HCV-induced NK cell discrepancy.
Figure 1.

Tim-3 is up-regulated on natural killer (NK) cells by hepatitis C virus (HCV) infection. (a) Representative flow cytometric plots for Tim-3 expressions on CD3− CD56bright/dim NK cells from chronically HCV-infected individuals, participants with a sustained virological response (SVR) following antiviral therapy, and healthy subjects (HS). Peripheral blood mononuclear cells (PBMCs) were first gated on lymphocytes, then CD3− CD56+ NK cells, and CD56bright CD56dim subsets. Percentage of cell frequency in the gated area is shown. (b) Summary data showing the percentages of CD3− CD56+ NK cells in the gated lymphocyte populations from three groups of subjects. Each dot represents one individuals and horizontal bar represents mean value. *P < 0·05. (c) Summary data showing the percentages and mean fluorescence intensity (MFI) of Tim-3 expression in total CD3− CD56+ NK cells, CD3− CD56bright and CD3− CD56dim subsets. Each dot represents one individual and horizontal bar represents mean value. *P < 0·05, **P < 0·01. (d) Representative flow cytometric plots, and summary data for Tim-3 and KLRG-1 expressions on CD3− CD56+ NK cells from 18 HCV patients and 6 HS. *P < 0·05.
Human NK cells are divided into two subsets, CD56bright and CD56dim, through their expression levels of CD56. Approximately 90% of peripheral blood NK cells belong to the CD56dim subsets that are mainly cytolytic, and the remaining 10% of NK cells are CD56bright subsets, which primarily produce cytokines.5,6 To determine the effect of Tim-3 on NK cells during HCV infection, we examined Tim-3 expression on gated total NK cells, as well as CD56bright and CD56dim subsets, in PBMCs from 23 chronically HCV-infected patients, eight SVR individuals, and six HS. The representative dot plots and summary data of the percentage as well as the mean fluorescence intensity (MFI) of Tim-3+ cell frequency on total CD56+, CD56bright, CD56dim NK cells are shown in Fig.(1a,c). Of note, Tim-3 expression in chronically HCV-infected individuals was significantly elevated, not only on CD3− CD56+ NK cells, but also on CD3− CD56bright and CD3− CD56dim NK subsets, compared with SVR and HS. Notably, Tim-3 levels on NK cells were decreased in SVR subjects following antiviral therapy, but were not completely restored to the levels seen in HS. This is shown by a residually elevated percentage as well as MFI of Tim-3+ NK cells, albeit the difference of MFI for Tim-3 expression was not statistically significant between SVR and HS. These results indicate that HCV infection up-regulates Tim-3 expression, and antiviral therapy in individuals with SVR reduces Tim-3 expression on NK cells.
We have previously shown that Killer cell lectin-like receptor subfamily G member 1 (KLRG-1), a negative signalling molecule expressed on lymphocytes, is up-regulated on NK cells from HCV-infected patients; however, blocking KLRG-1 signalling only partially restored NK cell functions.39 Here we demonstrated that both KLRG-1 and Tim-3 were up-regulated and could co-express on CD3− CD56+ NK cells from HCV patients versus HS (Fig.1d). Whether these Tim-3 (an exhaustion marker) and KLRG-1 (an aging or senescence marker) double-positive cells present a distinct population of NK cells that may lead to more dysfunction through two separate but cross-talking signalling pathways is under further investigation.
Increased Tim-3 expression is associated with T-bet up-regulation and IFN-γ inhibition in NK cells during HCV infection
T-bet has been shown as transcription factor for Tim-3 in T helper type 1 cells and to control key checkpoints of NK cell maturation for immune responses.40,41 To determine the role of T-bet in Tim-3 regulation, we examined the expression of T-bet, along with Tim-3, in NK cells from HCV-infected patients and HS. As the representative dot plots, summary data of MFI, and correlation analysis shown in Fig.2(a), T-bet was up-regulated during chronic HCV infection and closely associated with the Tim-3 expression level.
Figure 2.

Increased Tim-3 expression is associated with T-bet up-regulation and interferon-γ (IFN-γ) inhibition in natural killer (NK) cells during hepatitis C virus (HCV) infection. (a) Representative flow cytometric dot plots, summary data, and correlation analysis of T-bet and Tim-3 expressions in CD3− CD56+ NK cells from HCV-infected individuals and healthy subjects (HS). Each symbol represents one individual and horizontal bar represents mean value. *P < 0·05, **P < 0·01. (b) Up-regulation of Tim-3 expression on NK cells incubated with HCV-transfected (HCV+) or untransfected (HCV−) Huh-7 hepatocytes. Purified human CD16+ CD56+ NK cells (purity >95%) were co-cultured with HCV+ Huh-7 (red line) and HCV− Huh-7 cells (green line) for 48 hr, followed by flow cytometry analysis (histogram) of Tim-3 expression on NK cells. Summary data from six independent experiments are shown. *P < 0·05. (c) Tim-3 expression on NK92 cells co-cultured with HCV+ Huh-7 and HCV− Huh-7 cells. Medians, 25th and 75th centiles as boxes and 10th and 90th centiles as whiskers are shown for the Tim-3 expression summarized from three independent experiments. *P < 0·05. (d) IFN-γ expression in NK92 cells co-cultured with HCV+ Huh-7 and HCV− Huh-7 cells. Summary data from three independent experiments are shown. *P < 0·05.
The increased Tim-3 expression might be a result, rather than a cause, of NK cell dysregulation during HCV infection. Additionally, the driving force for Tim-3 up-regulation in HCV infection remains to be determined. To further elucidate the role of HCV in the regulation of Tim-3 expression, we employed a newly established cell culture system by transfecting Huh-7 hepatocytes with the HCV-JFH-1 strain in vitro to mimic the in vivo setting of early HCV infection. The expression of HCV in this cell co-culture system has been described previously.16 To this end, purified CD56+ or CD16+ CD56+ NK cells from HS were co-cultured with HCV-transfected or untransfected Huh-7 hepatocytes for 48 hr, followed by flow cytometric analysis for Tim-3 expression. In line with the data observed in natural HCV infection, HCV-expressing Huh-7 cells significantly enhanced Tim-3 expression in co-cultured CD56+ (data not shown) or CD16+ CD56+ NK cells (Fig.2b). The results are consistent with reports by us and other investigators using this short-term co-culture system to study the effects of HCV on human primary cells; these studies demonstrated that monocytes, NK cells and T cells co-cultured with HCV+ Huh7 cells express higher levels of negative signalling molecules, such as programmed death-1 and Tim-3, but lower levels of type I cytokines, such as IL-12, IL-2 and IFN-γ, compared with those co-cultured with HCV− Huh7.16,17,39,42–44
In addition to primary NK cells, we also examined the expression of Tim-3 and T-bet in NK92 cells co-cultured with HCV+/− hepatocytes. As shown in Fig.2(c), probably because of the difference between proliferating cell line and primary NK cells, T-bet was highly expressed in the IL-2/IL-18-stimulated NK92 cells and so no difference was identified between HCV-positive and HCV-negative cultures. However, Tim-3 was found to be up-regulated, whereas IFN-γ was inhibited, significantly, in NK92 cells co-cultured with Huh-7 cells expressing HCV (Fig.2d), suggesting that HCV may induce Tim-3 expression to inhibit NK cell function upon activation.
miR-155 is down-regulated in NK cells from chronically HCV-infected individuals
miR-155 has been shown to be regulated in monocytes, NK cells, and hepatocytes and functions as a positive regulator of inflammation in chronic HCV infection.36–38 To further determine the effect of miR-155 in NK cells during HCV infection, we measured miR-155 levels in CD3− CD56+ NK cells isolated from PBMCs of 19 chronically HCV-infected individuals and eight HS, using real-time RT-PCR. As shown in Fig.3(a; left panel), miR-155 expression in NK cells from HCV-infected patients was down-regulated, almost 10-fold, when compared with that in NK cells from HS. As NF-κB-dependent miR-155 expression has been shown to be a feedback mechanism preventing cell over-activation,28 we also examined miR-155 expression in NK cells stimulated with rhIL-2. As shown in Fig.3(a; right panel), miR-155 levels were fivefold down-regulated in HCV patients versus HS following IL-2 stimulation.
Figure 3.
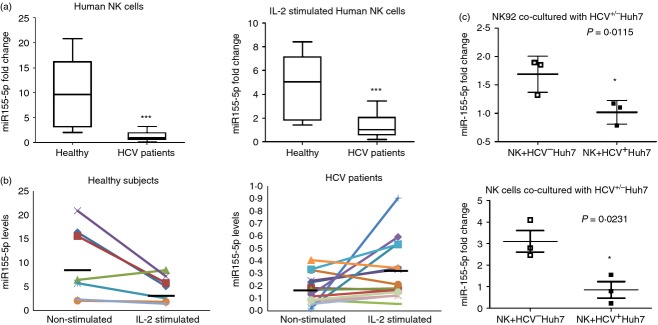
MicroRNA-155 (miR-155) is down-regulated in natural killer (NK) cells from individuals chronically infected with hepatitis C virus (HCV). (a) CD3− CD56+ NK cells were purified from peripheral blood mononuclear cells (PBMCs) isolated from 19 HCV-infected individuals and eight healthy subjects using magnetic beads. MicroRNAs (miRs) were isolated from NK cells, with or without interleukin-2 (IL-2) stimulation, using the mirVanaTM miR isolation kit. The miR-155 levels were measured by RT-PCR using specific Taqman assays, normalized to snRU6 internal control, and quantified with the 2−ΔΔct relative quantification method. Medians, 25th and 75th centiles as boxes and 10th and 90th centiles as whiskers are shown for the miR-155 fold changes ***P < 0.001. (b) Relative miR-55 expression level, normalized by U6 (2−ΔΔCt), in NK cells from healthy subjects (HS) and HCV subjects before and after IL-2 stimulation are shown. The horizontal bars represent median values. (c) NK92 cells and primary NK cells were co-cultured with HCV+ Huh-7 and HCV− Huh-7 cells. The miR-155 levels were quantified with the 2−ΔΔct. Summary data from three independent experiments are shown. *P < 0·05.
To further illustrate the effect of IL-2 stimulation on miR-155 expression in NK cells, we plotted the trend of miR-155 expression in NK cells with and without IL-2 stimulation in HCV patients and HS, respectively. Interestingly, although the miR-155 expression in NK cells from HS decreased upon IL-2 stimulation, which is consistent with the notion of miR-155 being a brake for cell activation,16 its expression was increased in NK cells from chronically HCV-infected individuals following IL-2 stimulation (Fig.3b). Again, to determine the role of HCV in regulation of miR-155, we measured its levels in purified primary NK cells or NK92 cell line incubated with Huh-7 hepatocytes with or without HCV infection. As shown in Fig.3(c), miR-155 levels declined in both NK cells co-cultured with HCV+ Huh-7 cells compared with those in HCV− cultures. These findings suggest that miR-155 is down-regulated in NK cells from patients with HCV infection and that their response to IL-2 stimulation is different to that observed in NK cells from HS.
Reconstitution of miR-155 in NK cells enhances IFN-γ production by inhibiting Tim-3/T-bet expression
To better understand the role of miR-155 in NK cell function, we reconstituted miR-155 by transfecting NK cells from HCV-infected patients with miR-155 mimic or negative control, and then measured intracellular IFN-γ expression by flow cytometric analysis. We first examined the miR transfection efficiency by the methods to be used (Lonza Human NK Nucelofector Kit and Nucleofector I Device), and found efficiency of approximately 20% for primary NK cells, as determined by the transfection of a fluorescently labelled negative miRNA control. Transfection efficiency was also assayed by measuring miR-155 transcript levels using real-time PCR and a nearly fivefold increase of miR-155 levels was found over negative control in NK cells transfected by miR-155-5p (Fig.4a). Reconstitution of miR-155 in NK cells from HCV patients resulted in an increase of IFN-γ levels compared with cells transfected with negative control (Fig.4b). Additionally, reconstitution of miR-155 in NK92 cells significantly reduced Tim-3 and T-bet expression, and enhanced IFN-γ expression (Fig.4c). These results suggest that miR-155 positively regulates IFN-γ expression by NK cells, probably via the Tim-3 pathway.
Figure 4.

Reconstitution of microRNA-155 (miR-155) in natural killer (NK) cells enhances interferon-γ (IFN-γ) production by inhibiting Tim-3/T-bet. (a) Purified CD3− CD56+ NK cells from hepatitis C virus (HCV) infected subjects were transfected by miR-155 mimic and negative control with a Lonza Human NK Nucelofector Kit and Nucleofector I Device, followed by real-time RT-PCR assay for miR-155 levels to determine the transfection efficiency. (b) Purified CD3− CD56+ NK cells from HCV subjects were transfected by miR-155 mimic and negative control and incubated in the presence of interleukin-2 (IL-2) for 48 hr, followed by flow cytometric analysis for intracellular IFN-γ production. Representative dot plots and summary data from three independent experiments are shown. (c) NK92 cells were transfected by miR-155 mimic and negative control and incubated with HCV+ Huh-7 cells in the presence of IL-2 for 48 hr, followed by flow cytometric analysis for Tim-3, T-bet, and IFN-γ expression. Representative dot plots and summary data from three independent experiments are shown. *P < 0·05.
Tim-3 blockade reinvigorates NK cells by correcting the impaired STAT-5 phosphorylation in HCV infection
Given the fact that high levels of Tim-3 expression are associated with an impaired IFN-γ production by NK cells in the setting of chronic HCV infection, we next sought to determine whether blockade of Tim-3 signalling may improve IFN-γ production by NK cells from HCV-infected patients. To this end, purified CD3− CD56+ NK cells from chronically HCV-infected patients and HS were incubated with anti-Tim-3 or control IgG antibodies for 48 hr, followed by rhIL-2/IL18 stimulation for an additional 24 hr, and flow cytometric analysis of IFN-γ production. As the representative dot plots show in Fig.5(a), blockade of Tim-3 signalling in NK cells from chronically HCV-infected individuals increased IFN-γ expression by CD3− CD56+ NK cells from HCV-infected patients, but not HS, when compared with the cells treated with isotype IgG control, indicating that blockade of Tim-3 reverses the functional defects associated with over-expression of this receptor in the setting of chronic viral infection. The question remains as to whether this is due to removal of a negative signal generated by Tim-3/ligand interactions or a positive signal generated by antibody interaction with this receptor. As Tim-3 and KLRG-1 were co-expressed on NK cells (Fig.1d), we also tried to simultaneously block both Tim-3 and KLRG-1 signalling with their specific antibodies; unfortunately, no further improvement of IFN-γ production by NK cells was observed in dual blockade compared with single blockade alone (data not shown). Nevertheless, these findings support the notion that Tim-3 negatively controls IFN-γ production by NK cells during HCV infection.
Figure 5.

Tim-3 blockade improves natural killer (NK) cell functions via enhancing signal transducer and activator of transcription 5 (STAT-5) phosphorylation. (a) CD3− CD56+ NK cells were purified from hepatitis C virus (HCV) -infected patients and healthy subjects, incubated with anti-Tim-3 or control IgG antibodies for 48 hr, followed by stimulation with interleukin-12 (IL-12) and IL-18 for 24 hr, then subjected to flow cytometric analysis of intracellular IFN-γ expression level. Representative dot plots (left) and the summary (right) percentage of IFN-γ production from four healthy subjects and five HCV patients are shown. **P < 0·01. (b) Purified NK cells from five HCV-infected individuals were treated with anti-Tim-3 or control IgG antibodies for 54 hr, then stimulated with IL-15 for 18 hr and pulsed with IL-15 for 30 min. Representative dot plots measuring p-STAT5 production are shown on the left, and the summary average percentage of p-STAT5 production are shown on the right. **P < 0·01. (c) Western blot analysis for phosphorylation of STAT-5 protein. Total STAT-5 was used as protein loading control. Densitometry data from three independent experiments using samples from nine HCV-infected subjects are shown. **P < 0·01.
Recent studies have shown that STAT-5 phosphorylation plays a pivotal role in cytokine responses and normal NK functions.45,46 To determine the underlying mechanism for correction of NK cell function by blocking Tim-3 signalling, we further investigated the phosphorylation of STAT-5 in NK cell activation with cytokine stimulation following Tim-3 blockade by flow cytometry Western blot. As shown in Fig.(5b,c), blockade of Tim-3 signalling in purified CD3− CD56+ NK cells from chronically HCV-infected individuals significantly enhanced STAT-5 phosphorylation when compared with cells treated with control IgG. These results are reproducible in independent experiments using purified NK cells from multiple HCV-infected individuals (n = 5 and n = 6, respectively), indicating an inhibitory role for Tim-3 in the regulation of human NK cell functions via the Jak/STAT pathway.
Discussion
Chronic HCV infection is a world-wide infectious disease, leading to chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. Though the pathogenesis of HCV infection remains only partly elucidated, it has become evident that, in addition to viral mutational escape, the dysregulation of innate to adaptive immune responses plays a major role in viral persistence and disease progression.1,2 The mechanisms involved in the HCV chronicity include dysfunction of monocyte and dendritic cells, impairment or depletion of NK cells, suppression of CD4+ and CD8+ effector T cells, and accumulation of Th17 cells and Foxp3+ regulatory T cells.1,2 As NK cells comprise the first line of host defence against invading pathogens, reduction of NK cell frequency and cytokine secretion may contribute significantly to the impaired cellular immune response and virus persistence in HCV infection. Despite intensive research on the regulation of NK cell frequency, phenotype and function during HCV infection,10–12 the exact mechanisms for contraction of NK cells with impaired cell function in the setting of HCV infection are not completely understood.
Natrual killer cells are evolved to recognize pathogens via cellular receptors expressed in immune cells and induce a wide spectrum of cytokine gene expressions, which in turn initiate and shape the inflammatory and adaptive immune responses in eliminating the invading pathogens. Meanwhile, pathogen-mediated host immune responses are double-edged swords, as aberrant activation of their signalling can be harmful, causing the pathological manifestations of inflammatory or autoimmune disorders. Hence, host immune responses must be tightly regulated by elaborate mechanisms to control their onset and termination. On the other hand, pathogens prone to persistent or latent infection, such as HCV and HIV, have developed multi-layer strategies to evade or subvert the host immune responses for their survival in vivo. The mechanisms for this immune evasion remain to be fully understood.
Tim-3 is a type 1 membrane protein with a structurally conserved immunoglobulin variable (IgV) domain and mucin stalk that connects to an intracellular tail.13–15 Whereas Tim-3 has been identified as an inhibitory receptor preferably expressed on exhausted T helper type 1 cells, its role in innate immune modulation remains poorly understood. In this study, our results indicate that Tim-3 plays a pivotal role in the control of NK cell function during HCV infection. HCV appears to induce Tim-3 expression on NK cells to disrupt NK IFN-γ production, as blocking Tim-3 signalling reverses HCV-induced NK cell dysfunction. It is not clear, however, how the anti-Tim-3 monoclonal antibody treatment improves NK cell function. We suspect that either Tim-3 ligands are also expressed on NK cells and anti-Tim-3 monoclonal antibody blocks the binding sites; or antibody ligation of Tim-3 itself delivers positive signalling and reverses the unresponsiveness of NK cells from virally infected individuals. As Tim-3 ligands are up-regulated on HCV-infected hepatocytes and antigen-presenting cells,44,47 we believe that interference of the interaction of NK Tim-3 with its ligands on accessory cells, rather than a direct activation of NK cells, is likely to be the mechanism for NK functional recovery by anti-Tim-3 treatment. Indeed, our preliminary data revealed that NK cells treated with anti-Tim-3 alone without rhIL-2 stimulations failed to secrete IFN-γ, suggesting that the blocking antibody inhibits negative signalling by Tim-3 rather than directly activating NK cells; however, removing a brake without a driving force, i.e. IL-2 stimulation, yields little and its effect requires additional positive signals generated by cytokine stimulation to drive cell proliferation. Although the reduced NK numbers in the peripheral blood could represent sequestration of NK in the liver or elsewhere, not necessarily reduced numbers overall, given the unique functions of NK cells as the initial defence against intruding pathogens, identifying factors such as Tim-3 that control NK IFN-γ production is critical to understanding innate immune regulation and to improving immunotherapy of chronic viral infection.
MicroRNAs are an important class of small (18–25 nucleotides), non-coding RNAs that can regulate gene expression through translational repression or target mRNA degradation.25–27 More than 700 miRNAs have been identified in mammals; some of them are widely expressed whereas others exhibit only limited developmental stage-, tissue- or cell-type-specific expression patterns, and many of these miRNAs are involved in diverse biological processes, such as cytokine expression and cell differentiation.25–27 As an ever-evolving strategy, viruses may be able not only to modulate cellular miRNA levels but also to interfere with the overall miRNA biogenesis. In particular, miR-155 has been shown to be up-regulated in monocytes and hepatocytes and acts as a positive regulator of inflammation in chronic HCV infection.36–38 Additionally, miR-155 expression has been shown to regulate the expansion and functional activation of NK cells in both mouse and humans.48–50 Despite these observations, little is known regarding the mechanisms for miR-155 regulation of NK cell function during chronic HCV infection.
Our study demonstrates that HCV-induced, miR-155-modulated Tim-3 up-regulation plays a key role in regulating NK cell functions via a STAT-5 signalling pathway during chronic infection. By means of computational miRNA target prediction algorithms, a wide spectrum of potential targets of miR-155 has been identified. Hence, miR-155, like many other miRNAs, may target genes that could be involved in regulation of multiple independent cell signalling processes, such as the STAT family. We have previously shown that HCV inhibits immune responses by regulating STAT-1, STAT-3 and STAT-5 expressions.16–19 Here, we further demonstrated that HCV regulates the STAT5/T-bet/Tim-3 pathway via an miR-155-modulated signalling mechanism. Whereas our study does not identify the direct target of miR-155, the present data suggest a role for miR155 in regulating inflammatory cytokine production in NK cells by regulating the Tim-3 pathway, potentially balancing immune clearance and immune injury during chronic viral infection.
Notably, miR-155 expression in NK cells from HCV-infected and uninfected individuals responds differently to IL-2 stimulation, in that miR-155 levels were decreased in HS but increased in HCV-infected individuals following IL-2 stimulation. This may have resulted from a paradoxical regulatory effect of HCV and inflammatory cytokines on miR-155 expression in NK cells. For healthy NK cells, baseline high levels of miR-155 may function as a gate to maintain homeostasis. This is consistent with the notion of miR-155 being a brake for cell activation.16 Following IL-2 stimulation, miR-155 trends down, and hence the brake is released to allow for cell activation, proliferation and IFN-γ production. For NK cells in the setting of chronic HCV infection, in contrast, the baseline level of miR-155 is already down-regulated by inflammatory cytokines in vivo, and so Tim-3 expression is up-regulated and IFN-γ is inhibited. Following ex vivo IL-2 stimulation and NF-κB activation, the expression of miR-155 is feedback-induced to prevent cell over-activation, perhaps explaining the differential expression of miR-155 in NK cells we observed in HCV-infected versus uninfected individuals in response to IL-2 stimulation.
It may be that HCV-mediated decline of miR-155 fine-tunes the NF-κB and cytokine signalling pathways rather than totally blocking these signals. Nevertheless, the counteracting effects of miR-155 on NF-κB and cytokine signalling may balance immune signalling, fine-tuning immune-mediated viral clearance and host injury. We suggest that the activation of STAT-5 is kept in check by Tim-3, which is double-checked by miR-155 to ensure appropriate immune responses in vivo (Fig.6). We conclude that miR-155 may regulate Tim-3/T-bet/STAT-5 signalling and cytokine expression in NK cells, potentially balancing immune clearance and immune injury during chronic viral infection.
Figure 6.
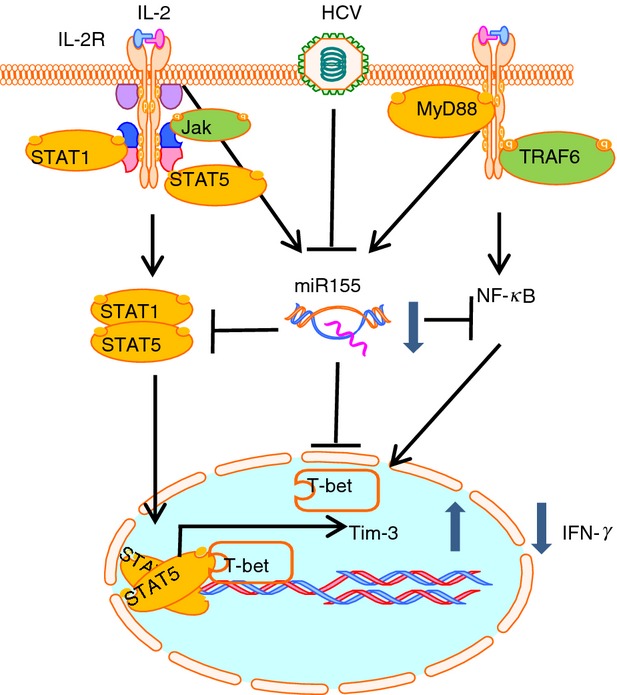
Proposed model for hepatitis C virus (HCV)-induced, microRNA-155 (miR-155) -modulated Tim-3 expression in regulation of interferon-γ (IFN-γ) production by natural killer (NK) cells during chronic viral infection. HCV infection inhibits nuclear factor-κB (NF-κB)-dependent miR-155 expression in NK cells, which in turn up-regulates Tim-3 expression through transcription factor T-bet. Tim-3 feedback suppresses cytokine-stimulated STAT-5 phosphorylation, so inhibiting IFN-γ production.
Acknowledgments
This work was supported by an NIH NIDDK grant to ZQY/JPM (R01DK093526), an NIH NIAID grant to ZQY/JPM (R01AI114748), and an NIAID grant to JPM/ZQY (1R15AI103828-01). Dr YQ Cheng is a visiting scholar, partially supported by Beijing 302 Hospital, Beijing, China. Yun Zhou, a joint PhD student, was supported in part by the China Scholarship Council (CSC 201306590012). We greatly appreciate the support from Dr T. Wakita, Department of Virology II, NIH of Japan, for transferring the HCV JFH-1 strain through an MTA; and Dr T.J. Liang, Liver Section, NIH/NIDDK, for sending Huh-7 cells. We also acknowledge Dr Hanspeter Pircher, Department of Immunology, University of Freiburg, Freiburg, Germany, for generously providing KLRG1 detection and blockade antibodies. This publication is the result of work supported with resources and the use of facilities at the James H. Quillen Veterans Affairs Medical Center. The contents in this publication do not represent the views of the Department of Veterans Affairs or the United States Government.
Disclosures
The authors declare no competing interests of this work.
References
- Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity. 2014;40:13–24. doi: 10.1016/j.immuni.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen HR. Emerging concepts in immunity to hepatitis C virus infection. J Clin Invest. 2013;123:4121–30. doi: 10.1172/JCI67714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au JS, Pockros PJ. Novel therapeutic approaches for hepatitis C. Clin Pharmacol Ther. 2014;95:78–88. doi: 10.1038/clpt.2013.206. [DOI] [PubMed] [Google Scholar]
- Manns MP, von Hahn T. Novel therapies for hepatitis C – one pill fits all? Nat Rev Drug Discov. 2013;12:595–610. doi: 10.1038/nrd4050. [DOI] [PubMed] [Google Scholar]
- Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–40. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197–223. doi: 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Alvarez F. Role of NK and NKT cells in the immunopathogenesis of HCV-induced hepatitis. J Leukoc Biol. 2004;76:743–59. doi: 10.1189/jlb.0304197. [DOI] [PubMed] [Google Scholar]
- Golden-Mason L, Rosen HR. Natural killer cells: primary target for hepatitis C virus immune evasion strategies? Liver Transpl. 2006;12:363–72. doi: 10.1002/lt.20708. [DOI] [PubMed] [Google Scholar]
- Kanto T, Hayashi N. Innate immunity in hepatitis C virus infection: interplay among dendritic cells, natural killer cells and natural killer T cells. Hepatol Res. 2007;37:S319–26. doi: 10.1111/j.1872-034X.2007.00236.x. [DOI] [PubMed] [Google Scholar]
- Nattermann J, Feldmann G, Ahlenstiel G, Langhan B, Spengler U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut. 2006;55:869–77. doi: 10.1136/gut.2005.076463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden-Mason L, Cox AL, Randall JA, Cheng L, Rosen HR. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology. 2010;52:1581–9. doi: 10.1002/hep.23896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmann KA, Bjokstrom NK, Veber H, et al. Interferon-α-induced TRAIL on natural killer cells is associated with control of hepatitis C virus infection. Gastroenterology. 2010;138:1885–97. doi: 10.1053/j.gastro.2010.01.051. [DOI] [PubMed] [Google Scholar]
- Sánchez-Fueyo A, Tian J, Picarella D, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4:1093–101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- Sabatos CA, Chakravarti S, Cha E, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4:1102–10. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- Jin HT, Anderson AC, Tan WG, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. PNAS. 2010;107:14733–8. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ma CJ, Wu XY, Moorman JP, Yao ZQ. A crucial role for Tim-3 in negative regulation of monocyte IL-12 production in chronic hepatitis C infection. PLoS One. 2011;6:e19664. doi: 10.1371/journal.pone.0019664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Shi L, Ma CJ, et al. Differential regulation of IL-12/IL-23 by Tim-3 drives TH17 cell development during HCV infection. J Virol. 2013;87:4372–83. doi: 10.1128/JVI.03376-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CJ, Wang JM, Ying RS. Enhanced virus-specific CD8+ T cell responses by Listeria monocytogenes-infected dendritic cells in the context of Tim-3 blockade. PLoS One. 2014;9:e87821. doi: 10.1371/journal.pone.0087821. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman JP, Wang JM, Zhang Y, et al. Tim-3 controls regulatory and effector T cell balance during HCV infection. J Immunol. 2012;189:755–66. doi: 10.4049/jimmunol.1200162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden-Mason L, Palmer BE, Kassam N, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. 2009;83:9122–30. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason MK, Lenvik TR, McCullar V, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon γ production in response to galectin-9. Blood. 2012;119:3064–72. doi: 10.1182/blood-2011-06-360321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndhlovu LC, Lopez-Verges S, Barbour JD, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119:3734–43. doi: 10.1182/blood-2011-11-392951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y, Hou N, Meng J, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol. 2010;52:322–9. doi: 10.1016/j.jhep.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Finney CA, Ayi K, Wasmuth JD, Sheth PM, Kaul R, Loutfy M. Kain KC. Serghides L. HIV infection deregulates Tim-3 expression on innate cells: combination antiretroviral therapy results in partial restoration. J Acquir Immune Defic Syndr. 2013;63:161–7. doi: 10.1097/QAI.0b013e318285cf13. [DOI] [PubMed] [Google Scholar]
- Beaulieu AM, Bezman NA, Lee JE, Matloubian M, Sun JC, Lanier LL. MicroRNA function in NK-cell biology. Immunol Rev. 2013;253:40–52. doi: 10.1111/imr.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RP, Leong JW, Fehniger TA. MicroRNA regulation of natural killer cells. Front Immunol. 2013;4:1–12. doi: 10.3389/fimmu.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava S, Mukherjee A, Ray RB. Hepatitis C virus infection, microRNA and liver disease progression. World J Hepatol. 2013;5:479–86. doi: 10.4254/wjh.v5.i9.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaravelu R, Russell RS, Tyrrell DL, Pezacki JP. Hepatitis C virus and microRNAs: miRed in a host of possibilities. Curr Opin Virol. 2014;7C:1–10. doi: 10.1016/j.coviro.2014.03.004. [DOI] [PubMed] [Google Scholar]
- O'Connell RM, Taganov KD, Boldin MP, et al. MicroRNA-155 is induced during the macrophage inflammatory response. PNAS. 2006;104:1604–9. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RP, Leong JW, Schneider SE, Keppel CR, Germino E, French AR, Fehniger TA. MicroRNA deficient NK cells exhibit decreased survival but enhanced function. J Immunol. 2012;188:3019–30. doi: 10.4049/jimmunol.1102294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang Y, Sun Q, Yan J, Huang J, Zhu S, Yu J. Identification of microRNA transcriptome involved in human natural killer cell activation. Immunol Lett. 2012;143:208–17. doi: 10.1016/j.imlet.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Lind EF, Elford AR, Ohashi PS. Micro-RNA 155 is required for optimal CD8+ T cell responses to acute viral and intracellular bacterial challenges. J Immunol. 2013;190:1210–6. doi: 10.4049/jimmunol.1202700. [DOI] [PubMed] [Google Scholar]
- Dudda JC, Salaun B, Ji Y, et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity. 2013;38:742–53. doi: 10.1016/j.immuni.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta R, Chen L, Ciariariello D, et al. miR-155 regulates IFN-γ production in natural killer cells. Blood. 2012;119:3478–85. doi: 10.1182/blood-2011-12-398099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Hou J, Lin L, et al. Inducible microRNA-155 feedback promotes type I IFN signaling in antivial innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. 2010;185:6226–33. doi: 10.4049/jimmunol.1000491. [DOI] [PubMed] [Google Scholar]
- Bala S, Tilahun Y, Taha O, Alao H, Kodys K, Catalano D, Szabo G. Increased microRNA-155 expression in the serum and peripheral monocytes in chronic HCV infection. J Transl Med. 2012;10:151. doi: 10.1186/1479-5876-10-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wei W, Cheng N, Wang K, Li B, Jiang X, Sun S. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology. 2012;56:1631–40. doi: 10.1002/hep.25849. [DOI] [PubMed] [Google Scholar]
- El-Ekiaby N, Hamdi N, Negm M, Ahmed R, Zekri AR, Esmat G, Abdelaziz AI. Repressed induction of interferon-related microRNAs miR-146a and miR-155 in peripheral blood mononuclear cells infected with HCV genotype 4. FEBS Open Bio. 2012;2:179–86. doi: 10.1016/j.fob.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Cheng YQ, Shi L, Ying RS, Wu XY, Li GY, Moorman JP, Yao ZQ. KLRG1 negatively regulates natural killer (NK) cell functions through Akt pathway in individuals with chronic hepatitis C. J Virol. 2013;87:11626–36. doi: 10.1128/JVI.01515-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AC, Lord GM, Dardalhon V, Lee DH, Sabatos-Peyton CA, Glimcher LH, Kuchroo VK. T-bet, a Th1 transcription factor regulates the expression of Tim-3. Eur J Immunol. 2010;40:859–66. doi: 10.1002/eji.200939842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Linsten T, Reiner SL. The transcription factor T-bet and Eomes control checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67. doi: 10.1016/j.immuni.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JC, Lim JB, Park JH, Lee JM. Cell-to-cell contact with hepatitis C virus-infected cells reduces functional capacity of natural killer cells. J Virol. 2011;85:12557–69. doi: 10.1128/JVI.00838-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder KA, Stapleton SN, Gallant ME, Russell RS, Grant MD. Hepatitis C virus-infected cells downregulate NKp30 and inhibit ex vivo NK cell functions. J Immunol. 2013;191:3308–18. doi: 10.4049/jimmunol.1300164. [DOI] [PubMed] [Google Scholar]
- Ji XJ, Ma CJ, Wang JM, Wu XY, Niki T, Hirashima M, Moorman JP, Yao ZQ. Hepatitis C virus induces CD4+CD25+Foxp3+ regulatory T cell development through the Tim-3/Gal-9 pathway. Eur J Immunol. 2012;43:458–67. doi: 10.1002/eji.201242768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong JW, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA, Fehniger TA. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transplant. 2014;20:463–73. doi: 10.1016/j.bbmt.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JX, Li P, Liu D, et al. Critical role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity. 2012;36:586–99. doi: 10.1016/j.immuni.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolina JS, Braciale TJ, Hahn YS. Liver-primed CD8+ T cells suppress antiviral adaptive immunity through galectin-9-independent T-cell immunoglobulin and mucin 3 engagement of high-mobility group box 1 in mice. Hepatology. 2014;59:1351–65. doi: 10.1002/hep.26938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta R, Chen L, Costinean S, et al. Overexpression of miR-155 causes expansion, arrest in terminal differentiation and functional activation of mouse natural killer cells. Blood. 2013;121:3126–34. doi: 10.1182/blood-2012-12-467597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawislak CL, Beaulieu AM, Loeb GB, et al. Stage-specific regulation of natural killer cell homeostasis and response against viral infection by microRNA-155. Proc Natl Acad Sci USA. 2013;110:6967–72. doi: 10.1073/pnas.1304410110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RP, Fogel LA, Leong JW, et al. MicroRNA-155 tunes both the threshold and extent of NK cell activation via targeting of multiple signaling pathways. J Immunol. 2013;191:5904–13. doi: 10.4049/jimmunol.1301950. [DOI] [PMC free article] [PubMed] [Google Scholar]