

Abstract
CD8 T cells used in adoptive immunotherapy may be manipulated to optimize their effector functions, tissue-migratory properties and long-term replicative potential. We reported that antigen-stimulated CD8 T cells transduced to express an active form of the transcription factor signal transducer and activator of transcription 5 (STAT5CA) maintained these properties upon adoptive transfer. We now report on the requirements of STAT5CA-expressing CD8 T cells for cell survival and proliferation in vivo. We show that STAT5CA expression allows for greater expansion of T cells in vivo, while preserving dependency on T-cell-receptor-mediated tonic stimulation for their in vivo maintenance and return to a quiescent stage. STAT5CA expression promotes the formation of a large pool of effector memory T cells that respond upon re-exposure to antigen and present an increased sensitivity to γc receptor cytokine engagement for STAT5 phosphorylation. In addition, STAT5CA expression prolongs the survival of what would otherwise be short-lived terminally differentiated KLRG1-positive effector cells with up-regulated expression of the senescence-associated p16INK4A transcripts. However, development of a KLRG1-positive CD8 T cell population was independent of either p16INK4A or p19ARF expression (as shown using T cells from CDKN2A−/− mice) but was associated with expression of transcripts encoding p15INK4B, another protein involved in senescence induction. We conclude that T-cell-receptor- and cytokine-dependent regulation of effector T cell homeostasis, as well as mechanisms leading to senescent features of a population of CD8 T cells are maintained in STAT5CA-expressing CD8 T cells, even for cells that are genetically deficient in expression of the tumour suppressors p16INK4A and p19ARF.
Keywords: effector CD8 T cell, gene regulation, immunotherapy, senescence, transcription factor
Introduction
Mechanisms driving CD8 T cell differentiation into effector and memory cells in response to infection have been the subject of intensive studies in the last decade. In particular, while both T-cell receptor (TCR) engagement and cytokine signalling have been shown to regulate the differentiation of naive CD8 T cells into effector T (Teff) cells, chronic antigen stimulation1 or increased inflammatory signals2 promote their terminal differentiation. For instance, although the in vitro expansion phase of antigen-specific T cells allows for the accumulation of large numbers of cells, it appears to irreversibly induce terminally differentiated Teff cells that promptly enter into senescence.1 Similarly, in conditions of chronic inflammation or infection, persistent immune activation accelerates the replicative senescence of T lymphocytes.3 Indeed, a feature common to many cell lineages is that functional differentiation occurs at the expense of their proliferative capacity.4 This knowledge can now be used to manipulate CD8 T cells to increase their potential clinical utility in adoptive transfer therapies.
Loss of CD8 Teff-cell replicative potential has been correlated with up-regulation of killer-cell lectin like receptor G1 (KLRG1),2,5,6 an immunoreceptor tyrosine-based inhibition motif-bearing receptor.7 Additionally, the KLRG1hi CD8 Teff cells showed increased p16INK4A transcripts5 encoded by the CDKN2A locus and controlling cell cycle progression and senescence.8 In contrast, replication competent CD8 T cells with a KLRG1lo phenotype produced efficient recall responses.2,5 It is not clear, however, whether sustained expression of surface KLRG1hi is merely a marker for a population of terminally differentiated effector cells as suggested by the absence of phenotype observed for KLRG1-deficient mice9 or whether the engagement of the molecules may induce negative signalling as suggested for human T cells10 and in certain circumstances for mouse T cells.11
At the molecular level, both the T-Bet transcription factor and γc cytokine signalling appeared to tightly regulate the functional programme of CD8 Teff cells and their proliferative capacities.12,13 Additionally, in different models of acute infection, interleukin-2 (IL-2) via CD25-dependent signalling has been shown to control the sustained differentiation of effector CD8 T cells14 or the development of functional CD8 memory T cells.15 We have reported that expression of an active signal transducer and activator of transcription 5 (STAT5CA) in CD8 T cells mimicked the effect of IL-2 for the sustained expression of effector molecules in vitro16 and in vivo.17 Furthermore, upon adoptive transfer STAT5CA-expressing Teff cells (Teff-STAT5CA) efficiently induced regression of an autochthonous mouse melanoma17 that recapitulates aspects of human melanoma disease.18,19 Indeed, as compared with tumour-specific unmanipulated CD8 Teff cells, Teff-STAT5CA showed an increased capacity to infiltrate the tumour and to maintain a high level of granzyme B expression20 together with augmented production of interferon-γ (IFN-γ) in situ.21 As maintenance of a replicative potential is a pre-requisite for T cells to be efficient in adoptive cell therapy,1 we further characterized the effect of active STAT5 expression in CD8 Teff cells in terms of in vivo cell survival and control of proliferation. We next evaluated how genetic deletion of the CDKN2A locus, thought to control senescence induction, affected the properties of the STAT5CA-expressing Teff cells. Our data showed that STAT5CA-expressing CDKN2A-deficient Teff cells still reach a state of terminal differentiation. These results shed new light on the mechanisms of acquisition of T cell senescent features independent of CDKN2A-encoded cell cycle regulatory proteins p16INK4A and p19ARF.
Material and methods
Mice
Mice heterozygous for the H-2Ld/P1A35-43-specific TCR-transgene (TCRP1A)17 were kept on the Rag-1−/− B10.D2 background. OT-1 mice specific for H-2Kb/ovalbumin (SIINFEKL) were kept on a Rag-2−/− C57BL/6 background. To obtain CDKN2A−/− mice, Ink4a/Arfflox/flox conditional knock-out mice (which have exons 2 and 3 of the CDKN2A gene flanked by loxP sites18) have been crossed with Cre-deleter mice, both on a B10.D2 background. Rag-1−/− B10.D2 and Rag-2−/− C57BL/6 mice were also used. All these mice were bred in the CIML animal facility. CD3ε−/− C57BL/6 and β2-microglobulin−/− Kb−/− Db−/− CD3ε−/− C57BL/6 mice were purchased from TAAM-CNRS UPS44 (Orleans, France).
Animal experiments respected French and European directives.
Cell preparation
CD8 T cells were prepared from lymph nodes or spleens of TCRP1A Rag-1−/− mice according to standard procedures. When prepared from immunocompetent B10.D2 mice, CD8 T cells were enriched using a Mouse CD8-negative selection kit (Dynal, Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
CD8 T cell activation and retroviral infections
TCRP1A and OT1 CD8 T cells were activated for 72 hr with 10−7 m of P1A35-43 (LPYLGWLVF) and OVA (SIINFEKL) peptides, respectively. Polyclonal CD8 T cells were activated with coated anti-CD3 monoclonal antibody (145·2C.11, 3 μg/ml) and soluble anti-CD28 monoclonal antibody (37·51, 1 μg/ml). Twenty hours (for TCR transgenic T cells) or 40 hr (for polyclonal T cells) after initial stimulation, CD8 T cells were retrovirally transduced as previously described.17,20 Those Teff cell populations were either analysed directly or adoptively transferred to Rag−/− congenic mice.
Cell cycle analysis
Mice were injected intraperitoneally with 1·5 mg bromodeoxyuridine (BrdU; Sigma-Aldrich, St Louis, MO) 16 hr before being killed, and their drinking water was supplemented with BrdU (0·8 mg/ml) diluted in 2% glucose. BrdU/7-aminoactinomycin D staining, performed according to the manufacturer's instructions (BrdU labelling Flow kit; BD Biosciences, San Jose, CA), permits the enumeration of cells that actively synthesize DNA.
Annexin V staining
Allophycocyanin-coupled Annexin V (BD Biosciences) was used according to the manufacturer's protocol.
Flow cytometry
Teff-STAT5CA or Teff cells adoptively transferred in congenic Rag-deficient mice were recovered from recipients' pooled lymph nodes and spleens. Antibodies were from BD Biosciences. Cells (106) were analysed on an LSR2 cytometer (BD Biosciences). Data were analysed using FlowJo (Treestar Inc., Ashland, OR., CA) or Diva (BD Biosciences) software. For intracellular cytokine staining, CD8 T cells were stimulated ex vivo for 4 hr in the presence of monensin (4 μm) and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences). The MitoTracker Green FM probe (50 nm; Molecular Probes, Invitrogen) was used to determine the mitochondrial mass by flow cytometry according to the manufacturer's instructions.
Intracellular phospho-flow stainings
T cells were stimulated for the indicated time with cytokines (50 ng/ml each), fixed with 1·6% paraformaldehyde and permeabilized with methanol. After staining with anti-CD8 and anti-p-Y694-STAT5 monoclonal antibody (BD Biosciences) or anti-total-STAT5a (R&D Systems, Minneapolis, MN), data were collected on an LSR2 561 cytometer (BD Biosciences) and analysed using Cytobank (http://www.cytobank.org). Control fluorescence minus one (FMO) are also acquired for all conditions.
Western blot
After cell lysis in TNE buffer (50 mm Tris–HCl, 1% Nonidet P-40, 20 mm EDTA) supplemented with protease and phosphatase inhibitors, lysates were subjected to immunoblot analysis. Antibodies against pY694-STAT5 (9351) and total STAT5 (9363) were purchased from Cell Signaling (Danvers, MA).
Transcriptome analyses and quantitative RT-PCR
Methods are provided in the Supporting Information.
Statistical analyses
Analyses were done in Fig. 3 with an unpaired t-test (GraphPad, San Jose, CA) with two-tailed P < 0·05 given as (*); in Figs 6 with a Mann–Whitney test (GraphPad) with two-tailed P < 0·05 given as (*); P < 0·01 given as (**); P < 0·001 given as (***), P < 0·0001 given as (****).
Figure 3.
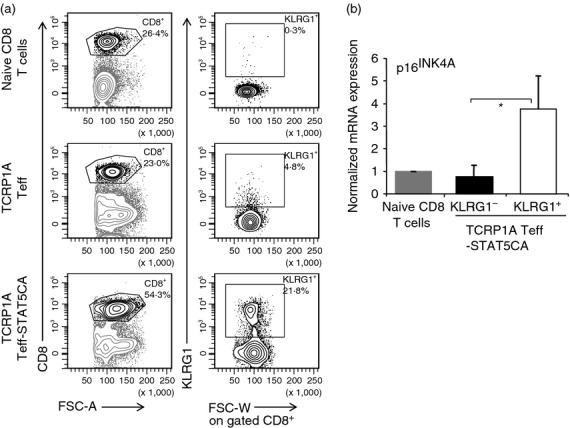
TCRP1A STAT5-expressing effector T cells (Teff-STAT5CA) contain a population of KLRG1-high (KLRG1hi) cells with senescent features. (a, b) TCRP1A Teff cells or Teff-STAT5CA were injected in Rag-1−/− B10.D2 mice and analysed at day 25. Naive CD8 T cells are included as control. (a) The representation of the transferred CD8 T cells in the spleen is shown in dot plots of CD8 expression versus FSC-A (left dot plots). KLRG1 expression versus FSC-W among gated CD8+ T cells is shown (right dot plots) and the % of KLRG1+ cells is reported. (b) TCRP1A Teff-STAT5CA were sorted on the basis of surface expression in KLRG1low and KLRG1hi subsets. Naive TCRP1A T cells were included as control. For all cell types, p16INK4A transcripts were measured by quantitative RT-PCR. Ratios of 2−ΔCt values normalized to that of CD8 naive T cells are shown. The mean ± SD of three independent experiments done in duplicates is reported.
Figure 6.
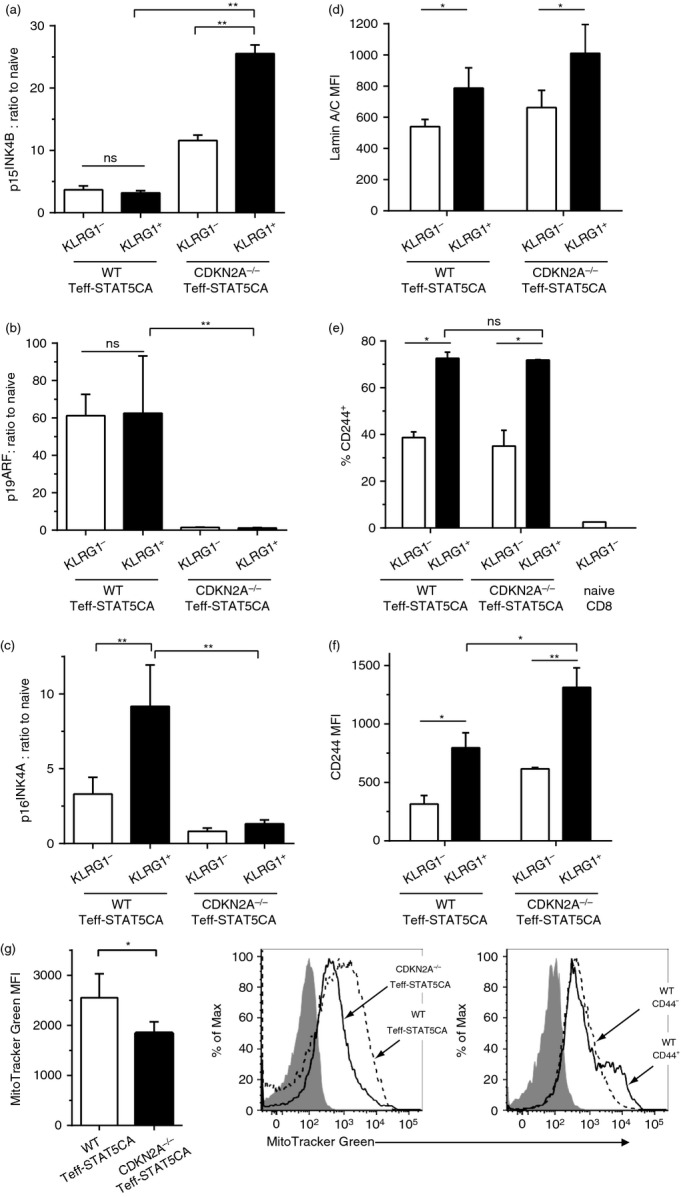
Compensation of CDKN2A deficiency by CDKN2B transcript expression in KLRG1-high (KLRG1hi) STAT5-expressing effector T cells (Teff-STAT5CA) cells that also present altered Lamin A/C and CD244 expression. (a–c) wild-type (WT) or CDKN2A−/− Teff-STAT5CA injected in Rag-1−/− B10.D2 mice were recovered 25 days later from pooled lymph nodes and spleen recipients. (a) CD8 KLRG1lo and KLRG1hi cells were purified and quantitative RT-PCR were performed. Naive CD8 T cells are included as control. Ratios of 2−ΔCt values normalized to that of CD8 naive T cells are shown. The mean ± SD of three independent experiments performed in duplicates is reported. (d–f) Surface (CD8, KLRG1 and CD244) and intracellular (Lamin A/C) stainings were performed on the same cell subpopulation as in (a). For KLRG1hi and KLRG1lo T cell subpopulations, the MFI for Lamin A/C and CD244 stainings are reported as well as the % CD244+ cells (n = 6). (g) Cells stained with the MitoTracker Green were than labelled for CD8 alone or with CD44 (for WT T cells). The MFI for MitoTracker Green are reported for five independent experiments and representative stainings are also shown.
Results
CD8 Teff cells expressing active STAT5 maintain controlled proliferation upon in vivo transfer
CD8 T cells from TCRP1A transgenic mice22 express a TCR specific for a tumour-associated antigen encoded by cancer-germline gene P1A presented in the context of H-2Ld. Retroviral transduction (see Material and methods) was used to express STAT5CA in antigen-activated TCRP1A cells together with green fluorescent protein (GFP) as a marker (referred to as T eff-STAT5CA), whereas control cells received GFP alone (Teff-GFP). Upon adoptive transfer of 1·5 × 105 sorted GFP+ TCRP1A Teff-GFP or Teff-STAT5CA in congenic Rag-1−/− mice, Teff-STAT5CA accumulated to a greater extent in recipient mice than either TCRP1A Teff-GFP (Fig.1a) or untransduced TCRP1A Teff cells17 (see also Fig.1d). An analysis of cell cycle status of adoptively transferred TCRP1A Teff and Teff-STAT5CA cells showed that 70% of TCRP1A Teff-STAT5CA were in S phase by day 3 after adoptive transfer (Fig.1b) and that they were back in G0/G1 phase by day 14 post-transfer. In comparison, only 20% of untransduced TCRP1A Teff cells were in S phase by day 3 post-transfer.
Figure 1.

TCRP1A STAT5-expressing effector T cells (Teff-STAT5CA) proliferate and have a survival advantage over mock- or un-transduced Teff cells but revert to a quiescent state during long-term maintenance. (a) TCRP1A Teff GFP+ and Teff-STAT5CA GFP+ cells were sorted at day 3 in vitro and 1·5 × 105 cells were injected in congeneic Rag−/− mice. At day 17, the absolute number of CD8+ GFP+ cells in recipient spleen is reported with n = 6 (Teff GFP+) and n = 8 (Teff-STAT5CA GFP+) in two independent experiments. (b) Cell-cycle analyses. Rag-1−/− B10.D2 mice received 2 × 106 or 106 TCRP1A Teff-STAT5CA (for day 3 and day 14 analysis, respectively) or 4 × 106 non-transduced TCRP1A Teff cells. Mice were injected with bromodeoxyuridine (BrdU) 16 hr before analysis. BrdU versus 7-aminoactinomycin D (7-AAD) dot plots (see Materials and methods) are shown for lymph nodes. (c) At 72 hr after their in vitro culture or day 4 after their adoptive transfer into Rag-1−/− B10.D2 mice, TCRP1A Teff-STAT5CA or TCRP1A Teff-GFP were analysed for binding to annexin V. (d) Competitive reconstitution of congeneic hosts demonstrates a higher proliferation/survival of TCRP1A Teff-STAT5CA compared with untransduced Teff cells. Analyses at days 6 and 14 of peripheral leucocytes of Rag-1−/− B10.D2 mice that received 5 × 104 TCRP1A Teff-STAT5CA sorted on GFP+ together with increasing numbers (from 5 × 104 to 5 × 106) of untransduced TCRP1A Teff cells. Percent CD8+ GFP− (untransduced) and CD8+ GFP+ (STAT5CA) Teff cells are reported. (b–d) Stainings are representative of two independent experiments with three or four mice per condition in each. (e) H-2Kb−/− H-2Db−/− β2 m−/− CD3ε−/− B6 or CD3ε−/− B6 mice were injected with 6 × 105 OT-1 Teff-STAT5CA and expansion of CD8 T cells was followed among peripheral leucocytes. Staining representative of at least six independent mice is shown.
The resistance to apoptosis of TCRP1A Teff-STAT5CA was addressed next. After 3 days of in vitro culture, no annexin V binding was detected on any TCRP1A Teff cells (Fig.1c) whether or not they expressed STAT5CA. This result suggested that the T cell subsets did not differ by their sensitivity to antigen-induced cell death in vitro. However, 4 days after in vivo transfer, a marked difference in cell survival was observed. While only 26% of GFP+ TCRP1A Teff-STAT5CA were positive for annexin V, 51·4% of the Teff-GFP population expressed the apoptosis marker. Therefore, experiments conducted on Teff cells at late stages after transfer (refs 17,20 and this study) examine cells that survived this activation-induced cell death. Providing exogenous IL-2 during the in vitro culture did not protect TCRP1A Teff cells from in vivo apoptosis (not shown).
Altogether, both increased initial proliferation and resistance to antigen-induced cell death might contribute to the higher capacity of Teff-STAT5CA compared with Teff cells for host colonization, an observation that was further confirmed by the results of the competitive reconstitution experiments shown in Fig.1d. Differences in apoptosis sensitivity between IL-2-treated and STAT5CA-expressing cells suggest the need for sustained STAT5 activation to mediate cell survival.
We next evaluated the extent to which STAT5CA-expressing T cells remained under the control of TCR signalling.23 Transfer of OT1 Teff-STAT5CA into MHC class I deficient C57BL/6 mice showed that the T cells failed to expand/survive in the absence of self MHC class I expression by the host (Fig.1e). As natural killer(NK) cells from the host MHC class I deficient mice are hyporesponsive,24 this effect cannot be attributed to NK-mediated rejection of the T cells. Rather, this result clearly demonstrated a dependency on ‘tonic’ stimulation23 of the Teff-STAT5CA TCR by MHC class I molecules for their in vivo survival.
Increased sensitivity to IL-2 and IL-15 for STAT5-phosphorylation in long-term in vivo transferred CD8 Teff-STAT5CA that exhibit effector memory characteristics
To evaluate whether expression of STAT5CA provided sustained intrinsic STAT5-phosphorylation or increased sensitivity to signalling by means of γc cytokine receptors, we analysed p-Y694-STAT5 expression in Teff cells and Teff-STAT5CA at different stages in vitro and in vivo.
When stimulated in vitro with antigenic peptide P1A35-43, TCRP1A CD8 T cells produced very small amounts of IL-2 [detected only when autocrine consumption was blocked by anti-CD25 (IL2Rα) monoclonal antibody (Fig.2a)]. Accordingly poor STAT5-Y694 residue phosphorylation (referred as p-Y694) was observed in these conditions. Addition of exogenous IL-2 led to increased p-Y694-STAT5 (Fig.2b,d). We previously showed that expression of STAT5CA could partially mimic the effect of IL-2 on antigen-activated naive CD8 T cells.16 The STAT5CA mutant (initially described as STAT5CA1*625) bears an S710F substitution in the transactivation domain, reducing the sensitivity of STAT5a to phosphatases. Due to an additional substitution (H299R), the mutated protein was shown to be dependent on wild-type STAT5 protein for DNA binding.26 A very low amount of p-Y694-STAT5 was detected in TCRP1A Teff-GFP compared with TCRP1A Teff-STAT5CA, which expressed higher levels of both p-Y694-STAT5 and total STAT5 (Fig.2c,d). Upon in vivo transfer of TCRP1A Teff-STAT5CA into congeneic Rag−/− recipients, the levels of expression of both p-(Y694)-STAT5 and total STAT5 were found to decrease when analysed at day 14 compared with day 5 after injection (Fig.2e).
Figure 2.
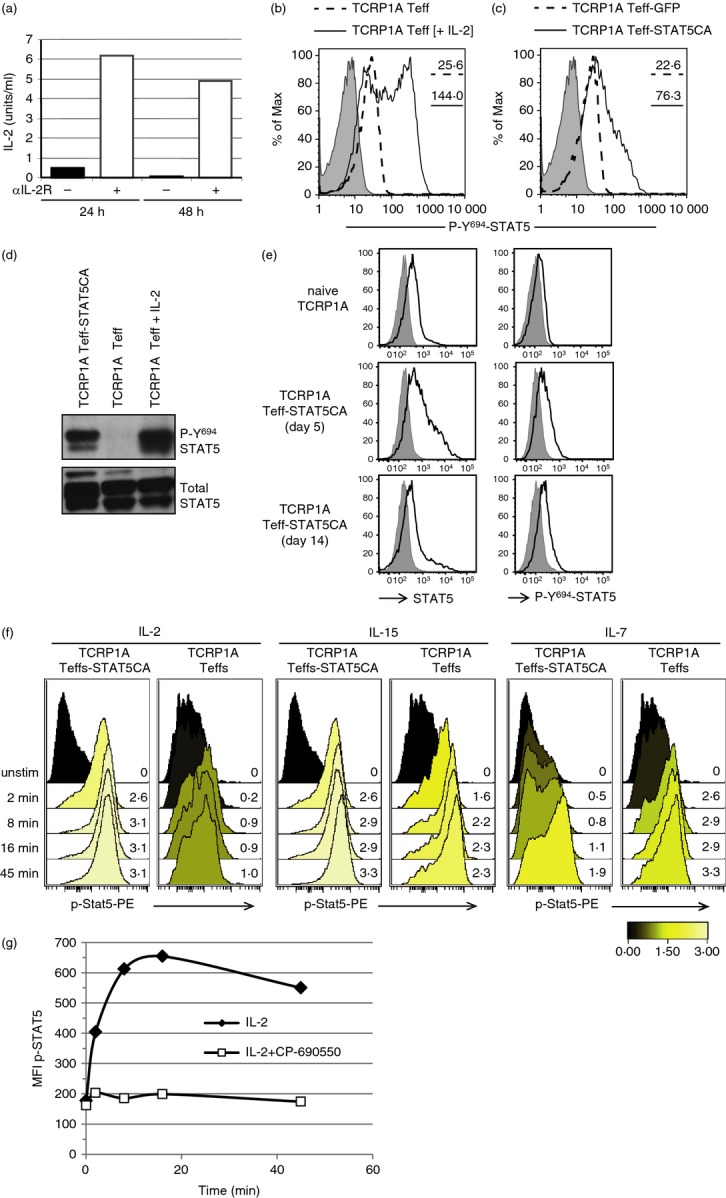
Response to γc cytokines for STAT5 phosphorylation in long-term in vivo transferred CD8 STAT5-expressing effector T cells (Teff-STAT5CA). (a–e) TCRP1A CD8 Teff cells produce limited amounts of interleukin-2 (IL-2) inducing limited p-Y694-STAT5. (a) TCRP1A CD8 T cells were cultured with peptide P1A35-43 in the presence or absence of anti-CD25 blocking antibody (PC61). IL-2 contained in culture supernatant was measured at 24 and 48 hr. (b, d) TCRP1A CD8 T cells were cultured during 72 hr with peptide P1A35-43 in the presence or absence of exogenous IL-2 (10 U/ml). (b) p-Y694-STAT5 was analysed by flow cytometry [dotted line, no added IL-2; full line, exogenous IL-2; grey, fluorescence minus one (FMO control)] and by Western blot (d), which also revealed total STAT5. (c) TCRP1A CD8 T cells were stimulated by peptide P1A and transduced to express STAT5CA (Teff-STAT5CA) or GFP in control (Teff-GFP). (c) p-Y694-STAT5 was analysed by flow cytometry (dotted line: Teff-GFP; full line: Teff-STAT5CA; grey: FMO control). (a–c) Results are representative of three independent experiments done in triplicates. TCRP1A Teff-STAT5CA have also been analysed by Western blot (d) one representative result is shown out of three. (e) Analyses by flow cytometry of total STAT5 and p-Y694-STAT5 on splenic B10.D2 CD8 TC (top); TCRP1A Teff-STAT5CA recovered from the spleen of Rag-1−/− B10.D2 mice adoptively transferred 5 days (middle) or 14 days (lower) earlier. Analysis on the two latter subsets was performed on gated CD8+ GFP+ Teff cells. FMO control stainings are shown in grey. Results are representative of two independent experiments with four mice per condition. (f, g) Analyses of cytokine responsiveness of TCRP1A Teff-STAT5CA. (f) TCRP1A Teff-STAT5CA or Teff cells were transferred into Rag-1−/− B10.D2 mice. At day 45, T cells recovered from pooled lymph nodes and spleen were briefly stimulated with cytokines and stained for CD8 and p-Y694-STAT5, the latter being shown in histograms. The response is reported with a colour code as an ArcSinh ratio (activated/unstimulated) of median fluorescence intensities. Staining representative of three different experiments are shown. (g) Same as (f); T cells were stimulated with IL-2 in the presence or absence of the JAK3 inhibitor, CP-690550 (33 nm) and stained for CD8 and p-Y694-STAT5. The raw median of fluorescence intensity is reported. Note that incubation with CP-690550 did not change the level of p-Y694-STAT5 in unstimulated (0 min) TCRP1A Teff-STAT5CA. One representative result is shown out of two.
We next evaluated whether in spite of low intrinsic STAT5-phosphorylation, long-term in vivo transferred TCRP1A Teff-STAT5CA presented increased responsiveness to γc-cytokines. Given the differential expression of γc-cytokine receptors on STAT5CA-expressing CD8 Teff cells that exhibited a CD122hi, CD25med, CD127lo phenotype compared with untransduced CD8 Teff cells with CD122hi, CD25−, CD127hi expression,17 we measured the basal and induced p-Y694-STAT5 levels in response to IL-2, IL-7 and IL-15 (Fig.2f). Although weak and intermediate levels of phosphorylation were reached in control Teff-GFP stimulated by IL-2 or IL-15, respectively, both cytokines induced high levels of p-Y694-STAT5 in Teff-STAT5CA. Of note, this latter induction of p-Y694-STAT5 was completely abolished in the presence of JAK3 inhibitor CP-690550 (Fig.2g), indicating its dependency on cytokine receptor engagement. In agreement with their low CD127 expression17 (see also Fig. 4d in the following section), Teff-STAT5CA retained moderate STAT5 activation in response to IL-7 (Fig.2f) compared with the higher response of CD127hi Teff-GFP.
Figure 4.

Increased accumulation of KLRG1-high (KLRG1hi) STAT5-expressing effector T cells (Teff-STAT5CA) in the absence of CDK2NA expression. Wild-type (WT) or CDK2NA−/− CD8 Teff-STAT5CA were injected in Rag-1−/− B10.D2 mice. Thirty days later, CD8 T cells recovered from pooled lymph nodes and spleens were analysed. (a–c) The representation of the transferred CD8 T cells in the spleen is shown in dot plots of CD8 expression versus SSC-A (left dot plots). KLRG1 expression versus FSC-A among gated CD8+ T cells is shown (right dot plots) and the % of KLRG1+ cells is reported. (b) % and absolute numbers of KLRG1+ cells are reported. (c) the MFI for KLRG1 staining is reported. (d, e) Cells were stained for CD8 and activation markers: KLRG1 versus CD127 (left) and CD25 (right) plots are shown. Naive CD8 T cells are included as control. Percent positive cells are reported. Data are representative of four independent experiments. The MFI for CD25 staining on KLRG1+ cells is reported in (e).
The increased sensitivity of Teff-STAT5CA to IL-15 may contribute to their long-term survival in vivo as suggested by the role of IL-15 for the maintenance of memory CD8 T cells.27 Indeed, transfer of Teff-STAT5CA from OT-1 TCR transgenic mice in IL-15-deficient, compared with IL-15-proficient Rag−/− C57BL/6 mice led to decreased expansion/survival of the transferred T cells (results not shown).
The fact that Teff-STAT5CA returned to a quiescent state 14 days after adoptive transfer and were dependent on both TCR ‘tickling’ and IL-15 for their long-term maintenance (previous section) suggested that those cells behaved as memory CD8 T cells. We therefore analysed their secondary responses both in in vitro and in vivo restimulation assays. TCRP1A Teff-STAT5CA transferred in Rag-1−/− B10.D2 mice were recovered from the recipients' spleens 40 days later and stimulated ex vivo. Recovered CD8 T cells efficiently produced IFN-γ in response to P1A+ tumour cells, but failed to do so in presence of P1A-negative cells (see Supplementary material, Fig. S1a, upper part). The P1A− and P1A+ tumour lines express similar levels of MHC I molecules (not shown) and had similar stimulatory capacity when loaded with exogenously added P1A peptide (see Supplementary material, Fig. S1a, lower part). Interestingly, although TCRP1A Teff-STAT5CA behave quite similarly to TCRP1A Teff cells or control memory CD44+ T cells from immunized mice in a 4 hr assay, they showed a more efficient IFN-γ production in a shorter (1 hr) test (see Supplementary material, Fig. S1b). To analyse their in vivo secondary response, TCRP1A Teff-STAT5CA were purified from spleens of adoptively transferred Rag-1−/− B10.D2 recipients and re-injected in Rag-1−/− B10.D2 mice inoculated with P1A+ or P1A− tumour cells 1 week earlier. Efficient expansion of the transferred Teff-STAT5CA was observed in the peripheral blood of recipients bearing P1A+ tumours (P511 mastocytoma or T429 melanoma) (see Supplementary material, Fig. S1c), but not in P1A-negative tumour-bearing hosts. All together, these experiments showed that adoptively transferred Teff-STA5CA maintained long-term inducible and specific effector memory functions.
A fraction of CD8 Teff-STAT5CA express the inhibitory receptor KLRG1 and elevated levels of p16INK4A transcripts
Replicative senescence has been reported to occur in terminally differentiated CD8 Teff cells that are characterized by the sustained expression of the KLRG1 receptor.28 These cells were further shown to express elevated levels of the p16INK4A (and p19ARF) transcripts5 and a cell autonomous role for p16INK4A in T cell aging was later demonstrated.29
As long-term transferred Teff-STAT5CA maintained an effector phenotype17 and also displayed a more sustained proliferation (Fig.1a), we wondered whether a fraction of those T cells could achieve a state of terminal differentiation with KLRG1 expression. Although a majority of TCRP1A Teff-STAT5CA remained KLRG1lo (Fig.3a), a significant fraction (about 20%) of KLRG1hi cells was detected in STAT5CA-expressing TCRP1A Teff cells. By contrast, unmanipulated TCRP1A Teff cells contained only a small population (4·8%) of weakly KLRG1-positive cells and naive CD8 T cells were all negative for that marker (Fig.3a). Sorted KLRG1lo and KLRG1hi Teff-STAT5CA, expressed low and high levels, respectively, of p16INK4A transcripts (Fig.3b). It is important to stress that KLRG1hi cells were only observed in CD8 T cells expressing STAT5CA and never in control Teff cells. This observation suggests that the sustained (i) proliferation of Teff-STAT5CA and/or (ii) STAT5 activity within these cells was required to generate/maintain such KLRG1hi CD8 T cells.
A fraction of Teff-STAT5CA are KLRG1hi even when genetically deficient for the CDK2NA locus
Having shown that the increased proportion of KLRG1hi cells among CD8 Teff-STAT5CA correlated with their elevated expression of p16INK4A transcripts, we asked whether the corresponding genetic ablation would prevent the generation of the KLRG1hi subpopulation. The CDKN2A/B loci encode two INK4 members (p16INK4A and p15INK4B) as well as an unrelated alternate open reading frame (ARF) protein (p14ARF in humans and p19ARF in mice). All three gene products are tumour suppressor genes and control cellular senescence. Loss of p16INK4A/p14ARF is a common genetic alteration in melanoma-prone families.30 We here use CDKN2A−/− mice as an experimental model aimed at mimicking the extreme case of defects in both p16INK4A and p19ARF expression to test the long-term effect of the combination of STAT5CA expression and CDKN2A deletion in CD8 T cells upon their adoptive transfer in mice. Polyclonal CD8 T cells originating from wild-type (WT) or CDKN2A−/− mice were purified, activated by anti-CD3/CD28 and transduced to express STAT5CA. After 96 hr of culture, the resulting CD8 Teff-STAT5CA were adoptively transferred into congenic Rag-1−/− hosts.
As with transgenic TCRP1A T cells, expression of STAT5CA in polyclonal CD8 T cells led to an accumulation of KLRG1hi cells (Fig.4a,b). Surprisingly, a more pronounced accumulation of KLRG1hi CD8 T cells was detected when STAT5CA was expressed in CDKN2A−/− compared with WT T cells (Fig.4a,b). The level of surface expression of KLRG1 was also higher on the CDKN2A-deficient T cells (Fig.4c). These data demonstrated that genetic ablation of CDKN2A did not prevent, but even enhanced, differentiation and maintenance of KLRG1hi CD8 Teff-STAT5CA.
Survival of KLRG1lo central memory T cells and KLRG1hi terminal Teff cell differentiation, have been shown to be promoted, respectively, by CD127 (IL-7Rα) and CD25 (IL-2Rα) -mediated signals.31,32 We therefore characterized the STAT5CA-expressing KLRG1hi cells for their expression of cytokine receptors. Both WT and CDKN2A−/− Teff-STAT5CA had a CD127low phenotype irrespective of their KLRG1lo or KLRG1hi status (Fig.4d). The expression of CD25 was slightly higher, however, on the KLRG1-positive CDKN2A-deficient T cells (Fig.4e). Whether a direct link exists between the slightly higher CD25 and KLRG1 expression of CDKN2A-deficient T cells is unknown.
We next examined the capacity of CDKN2A proficient or deficient STAT5CA-expressing CD8 T cells to produce IFN-γ in secondary stimulations, depending on KLRG1 expression. Around 50% of WT Teff-STAT5CA produced IFN-γ upon stimulation, irrespective of their KLRG1 status (Fig.5a). In comparison, the CDKN2A−/− Teff-STAT5CA response was more heterogeneous as 30–50% of them secreted IFN-γ, without any significant difference depending on their KLRG1 status. However, the KLRG1hi cells produced lower levels of IFN-γ than their KLRG1lo counterparts (Fig.5b), a characteristic that was more pronounced for the CDKN2A−/− than for the WT cells. The significance of this latter observation remains to be determined. In correlation with the reduced responsiveness of KLRG1hi T cells, we noticed that those cells displayed reduced TCR surface expression (Fig.5c).
Figure 5.
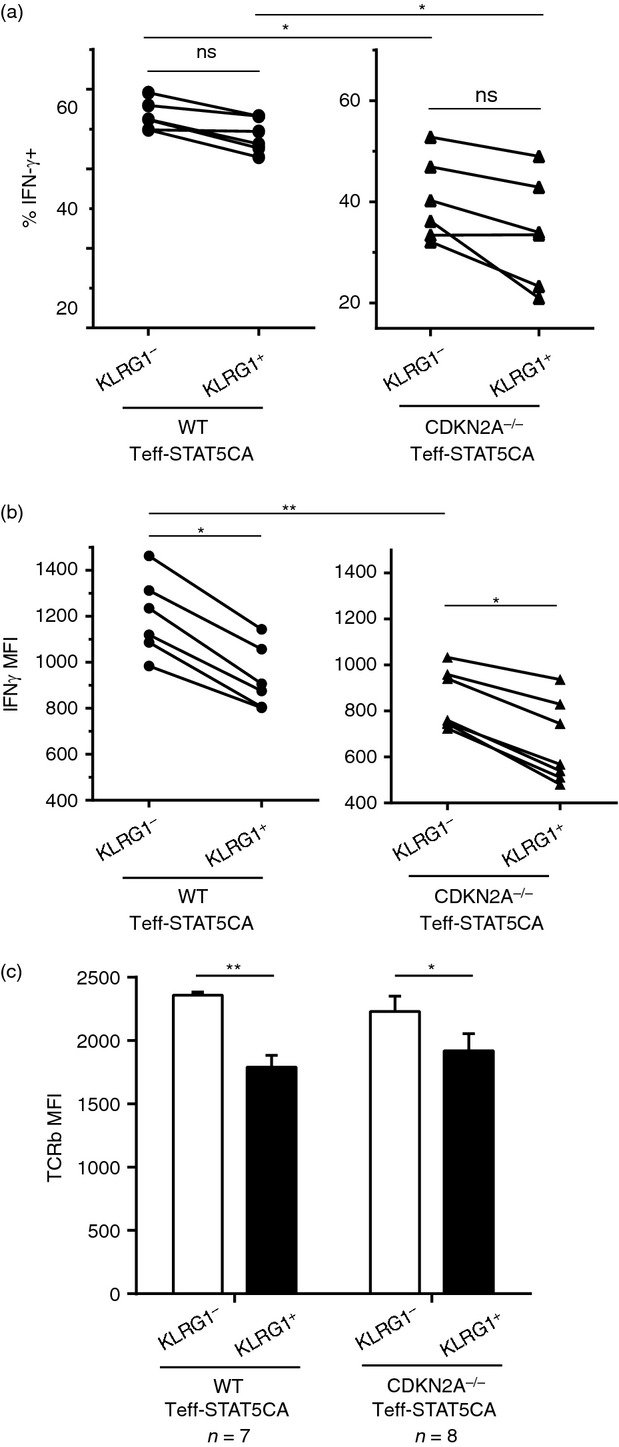
Comparison of interferon-γ (IFN-γ) production by KLRG1-positive (KLRG1+) T cells and their KLRG1− counterparts. (a, b) Cells were stimulated for 4 hr with anti-CD3 bound on a FcRγ+ tumour in the presence of monensin and stained for IFN-γ. For KLRG1hi and KLRG1lo T cell subpopulations, % IFN-γ-positive cells are shown in (a) and the MFI for IFN-γ staining is reported in (b). Data are representative of three independent experiments. (c) Surface staining for CD8, KLRG1 and TCR-β were performed. For KLRG1hi and KLRG1lo T subpopulations, the MFI for TCR-β staining is reported.
Altogether, these results suggest that STAT5CA expression allowed for long-term maintenance of CD8 effector memory T cells, primarily with a KLRG1lo phenotype. However, a proportion of these cells were committed to terminally differentiated KLRG1hi T cells, even upon genetic abrogation of CDKN2A expression.
CDKN2A−/− STAT5CA-expressing CD8 T cells showed a compensatory CDKN2B expression correlated with induction of senescence-associated transcripts
We next determined whether CDKN2A deficient or WT Teff-STAT5CA differ in their transcriptome. As previously reported for WT Teff-STAT5CA,20 we here performed these analyses on CDKN2A−/− STAT5CA-expressing T cells that were adoptively transferred in congenic hosts and recovered from recipients' lymph nodes and spleen 1 month later. The transcriptomic analyses showed that both CDKN2A-deficient and WT T cells acquired an effector phenotype including a similar up-regulation of transcripts encoding granzymes a and b, perforin, ifng, fas ligand (all being absent from Table1 that reports differentially expressed genes). Comparing adoptively transferred CDKN2A−/− Teff-STAT5CA and WT Teff-STAT5CA, 580 genes were differentially expressed with increased expression (present in Table1) in CDKN2A−/− Teff-STAT5CA of Egr2 and cd244 transcripts previously reported in anergic T cells33 and in tumour-specific exhausted T cells,34 respectively. Importantly, we noticed a significant up-regulation of CDKN2B transcripts (coding for p15INK4B) in CDKN2A−/− Teff-STAT5CA (Table1). This observation was confirmed by quantitative RT-PCR on mRNA extracted from sorted KLRG1hi CDKN2A−/− Teff-STAT5CA (Fig.6a). Noticeably, quantification of the p19ARF transcript revealed its expression in WT Teff-STAT5CA whether KLRG1lo or KLRG1hi (Fig.6b). Given the genetic ablation of exons E2 and E3 in CDKN2A−/− mice, both p16INK4A and p19ARF expression was abrogated in CDKN2A−/− Teff-STAT5CA (Fig.6b,c). Therefore, activation at the CDKN2B locus in CDKN2A−/− Teff-STAT5CA may compensate for defective p16INK4A expression/activity.
Table 1.
Transcripts differentially expressed in CDKN2A−/− versus wild-type (WT) STAT5-expressing effector T cells (Teff-STAT5CA) both being recovered at late time-points after their adoptive transfer in congeneic recipients
| Gene symbol | Fold change CDKN2A−/− Teff-STAT5CA versus wild-type Teff-STAT5CA | P value CDKN2A−/− Teff-STAT5CA versus wild-type Teff-STAT5CA |
|---|---|---|
| Egr2 | 4·066 | 3·52E-06 |
| Rora | 3·358 | 5·03E-09 |
| Nr4a2 | 2·880 | 3·10E-04 |
| Nr4a3 | 2·680 | 3·76E-04 |
| Cx3cr1 | 2·403 | 1·93E-03 |
| Lmna | 2·381 | 1·25E-06 |
| Nr4a1 | 2·380 | 7·67E-03 |
| Rgs16 | 2·123 | 1·25E-05 |
| Zeb2 | 2·119 | 7·09E-04 |
| Cd244 | 2·056 | 1·40E-05 |
| Egr1 | 1·978 | 3·71E-02 |
| Klrg1 | 1·932 | 6·51E-02 |
| Gzmk | 1·678 | 2·87E-02 |
| Cdkn2b | 1·531 | 2·96E-05 |
Transcriptomic analyses also pointed to increased Lmna transcripts, encoding Lamin A/C, in CDKN2A−/− Teff-STAT5CA. Measure of Lamin A/C protein expression in WT and CDKN2A−/− Teff-STAT5CA confirmed its higher level in KLRG1hi CDKN2A−/− cells (Fig.6d).
Cd244 transcripts were also up-regulated in CDKN2A−/− compared with WT Teff-STAT5CA (Table1). CD244 (2B4) is a receptor expressed on NK cells as well as on a subset of memory CD8 T cells. In particular, high level expression of 2B4 and other inhibitory receptors (including KLRG1) was reported on exhausted CD8 T cells emerging in situations of chronic viral infections, such as lymphocytic choriomeningitis virus, human immunodeficiency virus, hepatitis C virus and hepatitis B virus (reviewed in ref. 35). Although the proportion of CD244+ cells within the KLRG1lo and KLRG1hi populations was similar for CDKN2A−/− and WT Teff-STAT5CA (Fig.6e), the level of surface expression of CD244 was increased in the KLRG1hi subpopulation of CDKN2A−/− compared with WT Teff-STAT5CA (Fig.6f).
Gene set enrichment analyses (GSEA) highlighted transcripts involved in mitochondrial composition and activity, RNA and DNA metabolic processes and transcription, as well as inflammatory responses among genes differentially expressed and down-modulated in the CDKN2A−/− compared with WT Teff-STAT5CA (see Supplementary material, Fig. S2). By measuring the mitochondrial mass by cytometry, we further showed that the down-regulation of transcripts encoding mitochondrial components was associated with a lower mitochondrial content in CDKN2A−/− compared with WT Teff-STAT5CA (Fig.6g). This observation is in agreement with previous reports on defective mitochondrial function in human senescent T cells.36 However, we found that this characteristic was not correlated with the KLRG1 phenotype (not shown).
Discussion
We previously reported the capacity of adoptively transferred STAT5CA-expressing tumour-specific CD8 T cells to infiltrate autochthonous melanomas and remain functional in the immunosuppressive environment of those tumours, thereby inducing efficient melanoma regression.17 We here provide evidence that CD8 Teff-STAT5CA remain sensitive to TCR-controlled regulation in vivo, as manifested by (i) their return to a resting stage after a number of cell divisions; and (ii) their dependence on ‘tonic’ TCR engagement for long-term maintenance (proliferation/survival).
Long-term expression of STAT5CA in CD8 T cells induced moderate STAT5a mRNA (2·5-fold over naive T cells20) and protein content (Fig.2e, lower panel). We correlated better in vivo survival for Teff-STAT5CA with moderate but sustained increased sensitivity of STAT5CA to signalling by means of γc cytokine receptors, in particular IL-15. Indeed the expression of the single mutated S710F STAT5a (STAT5S710F) that was shown to be independent of WT STAT5 protein for DNA binding,26 induced higher levels of both p-Y694- and total STAT5 in CD8 T cells (see Supplementary material, Fig. S3), as compared with Teff-STAT5CA, but failed to promote T cell engraftment upon adoptive transfer (data not shown). The proliferative advantage conferred by moderate p-Y694-STAT5 in CD8 T cells is reminiscent of a similar observation reported for human CD34+ progenitor cells.37 Using (STAT5F/F × Rosa-CreERT2) mice (in which both Stat5a and Stat5b loci are floxed), we tested whether the pairing with the quantitatively limiting WT endogenous STAT5 proteins was required to maintain an increased, but controlled, activation of the STAT5 mediated signalling. However, these experiments (not shown) were not conclusive because of an incomplete Cre-mediated deletion. Additionally, heterodimerization of STAT5CA with other STAT proteins might occur as previously described in NK cells.38
Cell proliferation and senescence are tightly regulated through the CDKN2A locus in both non-lymphoid and lymphoid lineages.8 Interestingly, the self-renewal capacity of haematopoietic stem cells induced by STAT5CA was correlated with an increased expression of Bmi-1,39 a repressor that abrogates both transcription at the p16INK4A locus and associated senescence.5 An accumulation of p16INK4A expressing T cells was also observed in aged humans and silencing of p16INK4A in mouse T cells abolished age-related functional decline of the T cell responses.29
We here report that STAT5CA expression in CD8 Teff cells does not abrogate the emergence of a KLRG1-positive T cell population with up-regulated transcription at the CDKN2A locus (see Supplementary material, Fig. S4). The accumulation of a KLRG1hi fraction in the Teff-STAT5CA population, which was not observed for untransduced Teff cell, may result from (i) an increased rate of KLRG1hi cells generation as a result of a more sustained antigen-driven proliferation, as suggested by the prolonged BrdU incorporation in STAT5CA-expressing T cells as compared with unmanipulated counterparts; (ii) a STAT5-driven prolonged maintenance of the KLRG1hi cells; or (iii) a direct effect of active STAT5 on transcription of the KLRG1 and of the p16INK4A encoding genes. The latter possibility appears unlikely as IL-2 signalling was found to maintain expression of Bmi-1. It is more likely, in line with the second possibility, that the increased sensitivity of STAT5CA-expressing Teff cells to IL-15 allows for maintenance of the KLRG1hi cells within that population. This is consistent with a previous report on the dependence of virus-induced KLRG1hi CD8 Teff cells on IL-15 for survival.2
Several groups have reported a correlation between the absence of CD4 help and the expression of T-Bet and KLRG1 in CD8 effector memory T cells,2,40 suggesting that the level of T-Bet not only controls CD8 effector functions (cytolysis, IFN-γ) but also, directly or indirectly, controls the susceptibility of CD8 effector memory T cells to terminal differentiation or senescence. However, we have shown that STAT5CA regulates T-Bet transcription20 and we did not observe statistically significant differences in T-Bet expression between KLRG1lo and KLRG1hi Teff-STAT5CA (not shown).
CDKN2A-encoded Alternate open Reading Frame (ARF) proteins (p14ARF in human or p19ARF in mouse) also play a major role in senescence by trapping the Mdm2 ubiquitin ligase to the nucleolus and therefore preventing p53 ubiquitination and degradation.41 In WT Teff-STAT5CA, p19ARF transcripts were detected in both KLRG1lo and KLRG1hi cell subpopulations, suggesting that differentiation between these two STAT5CA-expressing subpopulations is controlled mainly by p16INK4A, independently of p19ARF.
When evaluating the effect of the combined genetic deficiency in the senescence-promoting CDKN2A locus with STAT5CA expression in CD8 Teff cells, we made the counterintuitive observation that the KLRG1hi CD8 Teff-STAT5CA CDKN2A−/− accumulated to a greater extent than their WT counterparts. Interestingly, the CDKN2A−/− T cells had up-regulated CDKN2B transcripts (see Supplementary material, Fig. S4). Compensation for CDKN2A deficiency by increased CDKN2B expression was reported in CDKN2A−/− mouse embryonic fibroblasts exposed to stress42 and might therefore be a general feature independent of the cell lineage. It is not clear, however, whether the increased accumulation of KLRG1hi CD8 Teff cell results from more intense cell proliferation or from a survival advantage for CDKN2A−/− T cells. Both processes are eventually influenced by the p19ARF-mediated control of a p53-dependent pathway of apoptosis.
Comparison of the transcriptomes of CDKN2A−/− and WT Teff-STAT5CA highlighted, in addition to the increased expression of CDKN2B transcripts, that of other genes relevant to the control of senescence induction such as Lmna, cd244 and klrg1. In contrast, among down-regulated genes those encoding mitochondrial components were prominent.
The Lmna gene encodes A-type lamins, producing lamins A and C and two variant isoforms through alternative splicing. Most laminopathies are associated with premature aging. Lamins function as nucleoplasmic scaffolds for nuclear chromatin and consequently DNA replication and transcription are affected by modifications of the nuclear lamina.43 Therefore, changes in regulation of Lamin A, DNA transcription and RNA metabolism (see Supplementary material, Fig. S2), in CDKN2A−/− Teff-STAT5CA argues for a senescent rather than a transformed phenotype. A more complete understanding of the mechanisms linking Lamin A and senescence is needed. Interestingly, mitochondrial dysfunction was recently reported in the premature aging disorder associated to laminopathies.44
Dampened recall responses of KLRG1hi CDKN2A−/− Teff-STAT5CA (Fig.5b) may result from their increased levels of expression of inhibitory receptors KLRG1 and CD244. Signalling pathways triggered by the latter have not yet been clearly defined, however. The question of the role of KLRG1 surface expression as a receptor inhibiting TCR-mediated signalling is still debated.9,45 It was reported, in particular, that KLRG1-mediated inhibition under physiological conditions could be observed only in human, but not in mouse lymphocytes.9,46 This was based on the observations by Pircher and colleagues that KLRG1-deficient mice produced normal virus-induced CD8 T cell differentiation and function in vivo9 and that human and mouse KLRG1 presented with different oligomeric states in biochemical analyses.46 More recently, however, the same group reported that mouse KLRG1 inhibitory activity was dampened by its association with the transferrin receptor, but was effective on resting T cells with low transferrin receptor expression.11 In humans, the recent report that KLRG1 blockade on CD4 T cells in hepatitis C virus infection partially restores their proliferative potential concomitant with decreased p16INKA expression45 requires confirmation in different conditions of senescence induction in subpopulations of T cells. In the present study, no evidence is presented for a role of KLRG1 in the lower response of the KLRG1hi compared with KLRG1-negative Teff-STAT5CA. Additionally, our observation of a reduced level of TCR surface expression on the KLRG1hi population may also contribute to the lower recall responses of that population (Fig.5c). Our results are in line with the observed hypo-responsiveness of late-stage TCR-stimulated human T cells with a KLRG1hi phenotype.47
With respect to the differences observed in expression of transcripts for mitochondrial components between CDKN2A−/− and WT Teff-STAT5CA, evidence exists for a regulation of survival of memory CD8 T cells by their mitochondrial respiratory capacity.48 In humans, a subset of effector memory CD8 T cells present features of proliferative senescence with poor mitochondrial function, yet are efficient at producing IFN-γ.36 Further studies are required to decipher how terminal differentiation/senescence manifests itself in T cells.
Loss of p16INK4A/p14ARF is a genetic alteration found in 50% of human melanomas and germline mutations of the CDKN2A gene are found in melanoma-prone families.30 In the latter patients, tumour-specific autologous T cells also bear the genetic alterations. Therefore, use of these T cells in adoptive therapy requires caution, as deletion of a tumour suppressor gene might impact their long-term behaviour. Altogether, our results suggest that T cells from patients, even those bearing genetic alteration at the CDKN2A locus, can be manipulated to express an active form of STAT5 for their use in immunotherapy of solid tumours, as these T cells maintained a senescence programme that may limit the risk of transformation. Additionally, mature T cells appear to be difficult to transform49,50 and no transformation of STAT5CA-transduced CD8 T cells was observed in this study and in a previous study.51 Similarly, somatic ablation of p16INK4A in mature T cells did not lead to neoplasia in mice.29
In summary (see Supplementary material, Fig. S4), our study demonstrates that STAT5CA-expressing CD8 T cells that present increased expansion, survival and effector functions17 nevertheless maintain TCR- and cytokine-mediated control of in vivo expansion. A fraction of those cells acquires some features of senescence with expression of KLRG1 and p16Ink4a transcripts. Importantly, genetic deficiency in the CDKN2A locus does not impair the characteristics of STAT5CA-expressing CD8 T cells and maintains their capacity to acquire senescent features, a property that may be associated with up-regulation of CDKN2B transcripts in the absence of the CDKN2A gene products.
Acknowledgments
We acknowledge Pascal Barbry and Chimene Moreihlon from the Plate-Forme Transcriptome, Nice-Sophia Antipolis, France. We thank the CIML bioinformatics platform (headed by Sebastien Jagger) and personnel funded by Canceropole PACA (Amira Djebbari) for their help in analysing transcriptomic data. We are grateful to Jonathan Irish (Stanford University) and Chris Coveney (Cytobank) for an introduction to Cytobank analyses and discussions. We also thank Sandrine Henri, Paulo Viera and Jim Disanto for providing mice, the CIML imaging and animal facilities personnel for assistance and Lee Leserman for editing the manuscript.
Glossary
- BrdU
bromodeoxyuridine
- FMO
fluorescence minus one
- GFP
green fluorescent protein
- IFN
interferon
- IL-2
interleukin-2
- KLRG1
killer-cell lectin like receptor G1
- MFI
mean of fluorescence intensity
- NK
natural killer
- STAT5
signal transducer and activator of transcription 5
- TCR
T-cell receptor
- Teff cells
effector T cells
- WT
wild-type
Author contribution
MGr performed and analysed the functional assays and performed all the sample preparations for transcriptomic analyses; NAA validated transcript expression with the help of MGi and AM; RR performed biochemical analyses. NAA and MGr performed the phospho-flow analyses with the help of GF and JN. The STAT5S710F construct and (STAT5F/F × Rosa-CreERT2) mice were provided by JG who also made suggestions on experimental designs. AMSV participated in the design of experiments and writing the paper. NAA designed the study, analysed data and wrote the manuscript.
Funding
This work was supported by funding from INSERM and CNRS and by grants from the ‘Association pour la Recherche sur le Cancer’ (ARC: NAA and Fondation ARC: AMSV) and the Institut National du Cancer (INCA) (AMSV). MGr was the recipient of a doctoral fellowship from ARC.
Disclosures
The authors declare that they have no conflict of interest.
Supporting Information
Figure S1.Teff-STAT5CA mediate specific and inducible recall responses in vitro and in vivo.
Figure S2. Representative gene set enrichment analysis (GSEA) of CDKN2A-/-Teff-STAT5CA vs wt Teff-STAT5CA gene signatures.
Figure S3. TCRP1A CD8 T cells were stimulated by peptide P1A and transduced to express STAT5CA (Teff-STAT5CA) or STAT5S710F(Teff-STAT5S710F).
Figure S4. Model for STAT5CA-driven differentiation of CD8 effector memory T cells.
References
- Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–95. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo AN, Weyand CM, Goronzy JJ. T-cell senescence: a culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med. 2004;10:119–24. doi: 10.1016/j.molmed.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Buttitta LA, Edgar BA. Mechanisms controlling cell cycle exit upon terminal differentiation. Curr Opin Cell Biol. 2007;19:697–704. doi: 10.1016/j.ceb.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffner M, Fearon DT. Loss of T cell receptor-induced Bmi-1 in the KLRG1+ senescent CD8+ T lymphocyte. Proc Natl Acad Sci USA. 2007;104:13414–9. doi: 10.1073/pnas.0706040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–40. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessmer MS, Fugere C, Stevenaert F, Naidenko OV, Chong HJ, Leclercq G, Brossay L. KLRG1 binds cadherins and preferentially associates with SHIP-1. Int Immunol. 2007;19:391–400. doi: 10.1093/intimm/dxm004. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Grundemann C, Schwartzkopff S, Koschella M, Schweier O, Peters C, Voehringer D, Pircher H. The NK receptor KLRG1 is dispensable for virus-induced NK and CD8+ T-cell differentiation and function in vivo. Eur J Immunol. 2010;40:1303–14. doi: 10.1002/eji.200939771. [DOI] [PubMed] [Google Scholar]
- Henson SM, Franzese O, Macaulay R, Libri V, et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood. 2009;113:6619–28. doi: 10.1182/blood-2009-01-199588. [DOI] [PubMed] [Google Scholar]
- Schweier O, Hofmann M, Pircher H. KLRG1 activity is regulated by association with the transferrin receptor. Eur J Immunol. 2014;44:1851–6. doi: 10.1002/eji.201344234. [DOI] [PubMed] [Google Scholar]
- Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo CJ, Fearon DT. T-bet-mediated differentiation of the activated CD8+ T cell. Eur J Immunol. 2011;41:60–6. doi: 10.1002/eji.201040873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, Lefrançois L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci USA. 2010;107:193–8. doi: 10.1073/pnas.0909945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441:890–3. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdeil G, Puthier D, Nguyen C, Schmitt-Verhulst AM, Auphan-Anezin N. STAT5-mediated signals sustain a TCR-initiated gene expression program toward differentiation of CD8 T cell effectors. J Immunol. 2006;176:4834–42. doi: 10.4049/jimmunol.176.8.4834. [DOI] [PubMed] [Google Scholar]
- Grange M, Buferne M, Verdeil G, Leserman L, Schmitt-Verhulst AM, Auphan-Anezin N. Activated STAT5 promotes long-lived cytotoxic CD8+ T cells that induce regression of autochthonous melanoma. Cancer Res. 2012;72:76–87. doi: 10.1158/0008-5472.CAN-11-2187. [DOI] [PubMed] [Google Scholar]
- Huijbers IJ, Krimpenfort P, Chomez P, van der Valk MA, Song JY, Inderberg-Suso EM, Schmitt-Verhulst AM, Berns A, Van den Eynde BJ. An inducible mouse model of melanoma expressing a defined tumor antigen. Cancer Res. 2006;66:3278–86. doi: 10.1158/0008-5472.CAN-05-3216. [DOI] [PubMed] [Google Scholar]
- Soudja SM, Wehbe M, Mas A, Chasson L, de Tenbossche CP, Huijbers I, van den Eynde B, Schmitt-Verhulst AM. Tumor-initiated inflammation overrides protective adaptive immunity in an induced melanoma model in mice. Cancer Res. 2010;70:3515–25. doi: 10.1158/0008-5472.CAN-09-4354. [DOI] [PubMed] [Google Scholar]
- Grange M, Verdeil G, Arnoux F, Griffon A, Spicuglia S, Maurizio J, Buferne M, Schmitt-Verhulst AM, Auphan-Anezin N. Active STAT5 regulates T-bet and eomesodermin expression in CD8 T cells and imprints a T-bet-dependent Tc1 program with repressed IL-6/TGF-β1 signaling. J Immunol. 2013;191:3712–24. doi: 10.4049/jimmunol.1300319. [DOI] [PubMed] [Google Scholar]
- Buferne M, Chasson L, Grange M, et al. IFNg producing CD8 T cells modified to resist major immune checkpoints induce regression of MHC class I-deficient melanomas. Oncoimmunology. 2015;4:e974959. doi: 10.4161/2162402X.2014.974959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanker A, Verdeil G, Buferne M, et al. CD8 T cell help for innate antitumor immunity. J Immunol. 2007;179:6651–62. doi: 10.4049/jimmunol.179.10.6651. [DOI] [PubMed] [Google Scholar]
- Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–62. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- Liao NS, Bix M, Zijlstra M, Jaenisch R, Raulet D. MHC class I deficiency: susceptibility to natural killer (NK) cells and impaired NK activity. Science. 1991;253:199–202. doi: 10.1126/science.1853205. [DOI] [PubMed] [Google Scholar]
- Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, Miyajima A, Kitamura T. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998;18:3871–9. doi: 10.1128/mcb.18.7.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld JW, Grebien F, Kerenyi MA, et al. The different functions of Stat5 and chromatin alteration through Stat5 proteins. Front Biosci. 2008;13:6237–54. doi: 10.2741/3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. 2002;195:1523–32. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voehringer D, Blaser C, Brawand P, Raulet DH, Hanke T, Pircher H. Viral infections induce abundant numbers of senescent CD8 T cells. J Immunol. 2001;167:4838–43. doi: 10.4049/jimmunol.167.9.4838. [DOI] [PubMed] [Google Scholar]
- Liu Y, Johnson SM, Fedoriw Y, Rogers AB, Yuan H, Krishnamurthy J, Sharpless NE. Expression of p16(INK4a) prevents cancer and promotes aging in lymphocytes. Blood. 2011;117:3257–67. doi: 10.1182/blood-2010-09-304402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AM, Chan M, Harland M, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007;44:99–106. doi: 10.1136/jmg.2006.043802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TW, Morre M, Kaech SM. Expression of IL-7 receptor α is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc Natl Acad Sci USA. 2007;104:11730–5. doi: 10.1073/pnas.0705007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged Interleukin-2Rα expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- Baitsch L, Baumgaertner P, Devevre E, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner SN, Kumar V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front Immunol. 2012;3:377. doi: 10.3389/fimmu.2012.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson SM, Lanna A, Riddell NE, et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J Clin Invest. 2014;124:4004–16. doi: 10.1172/JCI75051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga AT, Vellenga E, Schuringa JJ. Maximal STAT5-induced proliferation and self-renewal at intermediate STAT5 activity levels. Mol Cell Biol. 2008;28:6668–80. doi: 10.1128/MCB.01025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallal LE, Biron CA. Changing partners at the dance: variations in STAT concentrations for shaping cytokine function and immune responses to viral infections. JAKSTAT. 2013;2:e23504. doi: 10.4161/jkst.23504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuringa JJ, Wu K, Morrone G, Moore MA. Enforced activation of STAT5A facilitates the generation of embryonic stem-derived hematopoietic stem cells that contribute to hematopoiesis in vivo. Stem Cells. 2004;22:1191–204. doi: 10.1634/stemcells.2004-0033. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, Shen H, Wherry EJ, Reiner SL. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med. 2007;204:2015–21. doi: 10.1084/jem.20070841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715–25. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, Berns A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007;448:943–6. doi: 10.1038/nature06084. [DOI] [PubMed] [Google Scholar]
- Andres V, Gonzalez JM. Role of A-type lamins in signaling, transcription, and chromatin organization. J Cell Biol. 2009;187:945–57. doi: 10.1083/jcb.200904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Torres J, Acin-Perez R, Cabezas-Sanchez P, et al. Identification of mitochondrial dysfunction in Hutchinson–Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J Proteomics. 2013;91C:466–77. doi: 10.1016/j.jprot.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Shi L, Wang JM, Ren JP, et al. KLRG1 impairs CD4+ T cell responses via p16ink4a and p27kip1 pathways: role in hepatitis B vaccine failure in individuals with hepatitis C virus infection. J Immunol. 2014;192:649–57. doi: 10.4049/jimmunol.1302069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M, Schweier O, Pircher H. Different inhibitory capacities of human and mouse KLRG1 are linked to distinct disulfide-mediated oligomerizations. Eur J Immunol. 2012;42:2484–90. doi: 10.1002/eji.201142357. [DOI] [PubMed] [Google Scholar]
- Rappl G, Riet T, Awerkiew S, Schmidt A, Hombach AA, Pfister H, Abken H. The CD3-ζ chimeric antigen receptor overcomes TCR Hypo-responsiveness of human terminal late-stage T cells. PLoS One. 2012;7:e30713. doi: 10.1371/journal.pone.0030713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newrzela S, Cornils K, Li Z, et al. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278–86. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- Scholler J, Brady TL, Binder-Scholl G, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, Liu Y, Kaech SM. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci USA. 2010;107:16601–6. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.Teff-STAT5CA mediate specific and inducible recall responses in vitro and in vivo.
Figure S2. Representative gene set enrichment analysis (GSEA) of CDKN2A-/-Teff-STAT5CA vs wt Teff-STAT5CA gene signatures.
Figure S3. TCRP1A CD8 T cells were stimulated by peptide P1A and transduced to express STAT5CA (Teff-STAT5CA) or STAT5S710F(Teff-STAT5S710F).
Figure S4. Model for STAT5CA-driven differentiation of CD8 effector memory T cells.