

Abstract
Dengue is a mosquito-borne disease that affects millions of people worldwide yearly. Currently, there is no vaccine or specific treatment available. Further investigation on dengue pathogenesis is required to better understand the disease and to identify potential therapeutic targets. The chemokine system has been implicated in dengue pathogenesis, although the specific role of chemokines and their receptors remains elusive. Here we describe the role of the CC-chemokine receptor CCR5 in Dengue virus (DENV-2) infection. In vitro experiments showed that CCR5 is a host factor required for DENV-2 replication in human and mouse macrophages. DENV-2 infection induces the expression of CCR5 ligands. Incubation with an antagonist prevents CCR5 activation and reduces DENV-2 positive-stranded (+) RNA inside macrophages. Using an immunocompetent mouse model of DENV-2 infection we found that CCR5−/− mice were resistant to lethal infection, presenting at least 100-fold reduction of viral load in target organs and significant reduction in disease severity. This phenotype was reproduced in wild-type mice treated with CCR5-blocking compounds. Therefore, CCR5 is a host factor required for DENV-2 replication and disease development. Targeting CCR5 might represent a therapeutic strategy for dengue fever. These data bring new insights on the association between viral infections and the chemokine receptor CCR5.
Keywords: CC-chemokine receptor 5, chemokines, dengue virus, inflammation, viral replication
Introduction
Dengue is a major public-health concern in tropical and sub-tropical regions of the world. It is the most rapidly spreading mosquito-borne viral disease, with a 30-fold increase in global incidence over the past 50 years.1,2 Infection with any of the Dengue virus (DENV) serotypes can be asymptomatic, or can cause the classic dengue fever or evolve to severe disease, which leads to death if untreated.3–5 It is estimated that 390 million dengue infections occur each year, of which 96 million manifest as the fever.6 Besides its importance, disease pathogenesis is not fully understood, which renders treatment of dengue fever and severe dengue largely supportive. In addition, delay in the development of vaccines puts a serious burden on the health-care systems of low-income countries.7,8
Severe forms of dengue are characterized by an intense inflammatory response associated with haemorrhagic phenomena, including plasma leakage, thrombocytopenia, bleeding and hypovolaemic shock.9,10 The development of severe dengue involves a complex interplay between viral, host and environmental factors.11–13 DENV can interact and infect leucocytes, such as dendritic cells,14 monocytes and macrophages,15 resulting in cell activation and production of inflammatory mediators that shape both innate and acquired immune responses.
The C-C chemokine receptor 5 (CCR5) is involved in the recruitment of mononuclear leucocyte populations to tissues,16 among other functions.17,18 CCR5 was originally described by Gao et al. in 199319 as a receptor for the chemokines CCL3 and CCL5 and was later found to be also activated by CCL4.20 The discovery of CCR5 is concomitant with its association with HIV-1.20 Individuals homozygous for the CCR5Δ32 allele express a non-functional CCR5 that confers protection from HIV infection.21 This aspect of HIV biology resulted in the development of CCR5 inhibitors to treat HIV-seropositive patients.22–24 Among these inhibitors, the modified chemokine of regulated on activation normal T cell expressed and secreted (Met-RANTES) effectively blocks CCR5-mediated leucocyte recruitment and activation,25,26 and has been used in disease models with an inflammatory component, including arthritis,27 asthma26 and periodontitis.28
The association of CCR5 with HIV was the first to be described, followed by associations with different viral pathogens. CCR5 expression may be a risk factor for severe infections with flaviviruses, which share the same genus with DENV. Prevalence of the ccr5Δ32 allele is associated with severe meningoencephalitis in tick-borne encephalitis virus29 and symptomatic infection in West Nile encephalitis virus.30 In a mouse model of West Nile encephalitis virus infection, CCR5 expression is up-regulated in wild-type (WT) mice and CCR5-deficient mice are markedly susceptible to infection.31
The cytokine storm observed in patients with severe dengue fever includes increased levels of CC chemokines. All CCR5 ligands and CCR5 itself were demonstrated to be expressed in dengue patients32–34 and in different dengue infection models.35–38 Previous work from our group demonstrated that mice deficient for the CC chemokine receptors CCR2 and CCR4 have reduced disease severity after DENV-2 infection.35
The role of CCR5 in driving inflammatory diseases, together with clinical evidence on its role in flavivirus and dengue infection, places CCR5 and its ligands as interesting candidates for investigation. Here we demonstrate that CCR5 is a host factor required for DENV-2 replication and development of infection. DENV-2 poorly replicates in macrophages treated with CCR5-blocking compounds or deficient for this receptor. CCR5 deficiency or pharmacological blockade in vivo reduces DENV-2 load in tissues and prevents disease development. The role of CCR5 in dengue indicates clear differences in how flaviviruses exploit the chemokine system during infection, indicating that CCR5 is an important host factor for dengue development.
Materials and methods
Ethics statement
All experimental procedures were approved, and complied with the University of Minas Gerais's (UFMG) Committee for Ethics in Animal Experimentation (CETEA) regulations, under protocol number 113/2009. All efforts were made to minimize animal number and suffering during the experimental procedures.
Mice
Eight- to 12-week-old C57BL/6 (H-2Db) WT mice were acquired from Centro de Bioterismo (CEBIO) of UFMG (Belo Horizonte, Brazil). CCR5−/− mice, 8–12 weeks old, backcrossed at least 10 times in C57BL/6, were kindly provided by Dr Leda Q. Vieira and bred in the laboratory's animal facility. All animals were kept under controlled temperature (23°) with a strict 12-hr light/dark cycle, food and water available ad libitum under specific pathogen-free conditions.
Virus
Stocks of DENV-2 strain NGC were generated by clarified suspension of infected brains from a single passage in newborn mice and another passage in C6/36 mosquito cells. DENV-2 strain P23085 was obtained from the State Collection of Viruses, Moscow, Russia, and adapted as previously described.36 Virus adaptation was performed in a maximum containment biosafety level 3 laboratory of the SRC VB ‘Vector’, Koltsovo, Russia. Sequence of portions of the E and NS1 genes of the mouse-adapted virus was deposited at GenBank under the accession number AY927231. The identity of our DENV-2 P23085 strain was confirmed in vitro and in vivo.39 For the current set of experiments, virus stocks were generated by propagating the adapted virus stocks in LLC-MK2 cells [kidney, Rhesus monkey; American Type Culture Collection (ATCC), Manassas, VA] to a maximum of two passages. Cell culture media (Dulbecco's modified Eagle's medium; DMEM) containing DENV were harvested and stored at −80°, as well as the DENV-2 NGC stocks. Virus titres were determined by plaque assay in LLC-MK2 cells, expressed as plaque-forming units (PFU) per ml of suspension.
In vitro experimental infection
THP-1 cells (human leukaemic monocytes; ATCC) were activated by treatment with PMA at 0·5 μm for 3 hr at 37° in 5% CO2. All experiments used 1 × 106 THP-1 cells per well. Cells were incubated with or without Met-RANTES (Met-R, kindly donated by Merck-Serono, Zug, Switzerland) at a concentration of 1 μg/ml. Infections were performed with DENV-2 NGC at a multiplicity of infection (MOI) of 0·1. Cells were washed in RPMI-1640 between each step (plating, infection, treatment) and before harvesting for viral RNA. Samples were stored at −80° until processing.
For the viral entry experiment, the protocol described previously was followed.40 Briefly, cells were treated with or without Met-R and infected with DENV-2 NGC at an MOI of 1. After 2 hr, cells were kept at 4°, washed three times in PBS and incubated for 3 min in alkaline buffer to release bound viral particles. Cells were washed again in chilled PBS and harvested for RNA extraction.
Murine peritoneal macrophages were collected by peritoneal lavage in 0·34 m ice-cold sucrose, seeded in six-well plates (1 × 106 cells/well) and incubated at 37° in 5% CO2 for adherence. Then, cells were washed in DMEM and treated for 60 min with Met-R. After washing, cells were incubated with mouse-adapted DENV-2 P23085 at an MOI of 0·1 for 1 hr, washed twice and maintained in DMEM until sample collection. Cell culture supernatant and cellular extract samples were assessed for virus titres by plaque assay in LLC-MK2 cells.
Bone-marrow-derived macrophages and dendritic cells
Bone marrow was sterile harvested from femur and tibia of C57BL/6 mice. Cells were washed, counted and differentiated using two culture conditions. Bone-marrow-derived macrophages were cultured for 7 days in DMEM supplemented with 20% (v/v) horse serum and 30% L929 cell conditioned medium. Bone-marrow-derived dendritic cells were cultured in RPMI-1640 supplemented with 10% fetal calf serum (Hyclone), β-mercaptoethanol (50 nm; Invitrogen, Carlsbad, CA) and 5% J558L cells supernatant. At day 10, non-adherent cells were harvested. For infection, bone-marrow-derived macrophages and dendritic cells were plated at 1 × 106 cells/well and incubated with mouse-adapted DENV-2 P23085 at an MOI of 0·1 for 1 hr. Then, plate wells were washed twice and maintained in DMEM until sample collection at the desired time-points.
In vivo experimental infection
Mice were handled and kept in a biosafety level 2 laboratory animal facility. Mice were inoculated intraperitoneally with mouse-adapted DENV-2 P23085 (200 PFU) or mouse-adapted DENV-3 (1000 PFU, genotype I, accession number JN697379)41 diluted in 100 μl of endotoxin-free PBS. Lethality rates were observed every 12 hr until day 14 or 28 post-infection (p.i.) and disease parameters were evaluated at days 3, 5 and 7 p.i. Wild-type mice were treated with CCR5 antagonists Met-R or UK-484900, a Maraviroc analogue (a kind gift of Dr Stephen M. Shaw, Pfizer Global Research and Development, Sandwich, Kent, UK). Compounds were administered in a preventive schedule: 1 day before infection to day 6 p.i., or in a therapeutic schedule: 3 days after infection to day 6 p.i. Each animal received Met-R 0·5 mg/kg dissolved in 0·1% BSA weight/volume (w/v) in PBS or UK-484900 10 mg/kg dissolved in HCl 0·006 m in 0·9% NaCl solution. Both compounds were injected daily (subcutaneously). At day 7 p.i., mice were anaesthetized intraperitoneally with a ketamine (100 mg/kg)/xylazine (10 mg/kg) solution to recover blood samples and killed for spleen and liver collection. Samples were stocked at −80° before analysis.
Virus titration
Cell culture and tissue samples were assayed for viral load as previously described.42,43 Tissue samples were prepared as 10% w/v homogenates in DMEM. Samples were serially diluted, adsorbed in an LLC-MK2 monolayer for 1 hr and overlaid with Medium 199 (Gibco, Grand Island, NY) supplemented with 3% (v/v) fetal calf serum in 1·5% (w/v) carboxymethylcellulose, (Sigma, St Louis, MO). Cultures were fixed after 7 days and stained with crystal violet 1% (w/v) for counting of viral plaques and the results were expressed as PFU per 100 mg of tissue or per ml of blood/cell culture sample.
Quantitative RT-PCR
Total RNA was isolated from THP-1 cells using a QIAgen Viral RNA Isolation Kit and stocked at −80°. Real-time RT-PCR was performed on a StepOne sequence-detection system (Applied Biosystems, Foster City, CA) using SYBR Green PCR Master Mix (Applied Biosystems) after reverse transcription reaction of 2 μg of RNA using Moloney-murine leukaemia virus reverse transcriptase (Promega, Madison, WI). For DENV(+) strand detection, reverse transcription was carried out using the reverse primer (below) for the subsequent quantitative RT-PCR reaction, whereas the forward primer was used for reverse transcription to detect the DENV(−) strand. The relative level of DENV-2 RNA was determined by the ΔCt method, where Ct values for each sample were subtracted from Ct values from negative controls (2−ΔCt) and expressed as relative abundance of viral (+)-stranded RNA. The following primer pairs spanning the DENV NS4b gene were used: 5′-AAAGAATTCCCAGGACTTCAAGCAAAAGCA3′ (forward) and 5′-AAAGGATCCTCAGAGCATTACTTGTCCTAACTG-3′ (reverse).
Haematological parameters
Blood was obtained from the brachial plexus of anaesthetized mice in heparin-containing syringes and stocked before analysis. Platelets and leucocytes were quantified in an optical microscope using a Neubauer chamber and a haematocrit index was determined on centrifuged blood samples in heparinized glass capillaries. Results are presented as counts/mm3 per ml of blood (cells) or percentage (haematocrit).
Quantification of cytokine and chemokine concentrations
Concentrations of CCL5, CCL4, tumour necrosis factor-α and interferon-γ in culture supernatant or tissue samples were measured by ELISA using commercially available antibodies and according to manufacturer's procedures (R&D Systems, Minneapolis, MN). Samples were placed in duplicate and ELISA measurements for a given experiment were conducted in the same plate. Results are expressed as pg/ml, pg per 100 mg of tissue or fold increase in protein level in culture supernatant. The detection limit of the ELISA was in the range of 4–8 pg/ml. Graphs are representative of three independent experiments.
Determination of myeloperoxidase activity
For myeloperoxidase analysis, as an indirect index of neutrophil accumulation, tissue homogenates were prepared in 1 ml of PBS containing 0·5% hexadecyltrimethyl ammonium bromide and 5 mm EDTA using a Dispomix tissue homogenizer Medic Tools, Zug, Switzerland and the protocol was followed as already described.39 Results are expressed as arbitrary units (optical density at 492 nm) and were corrected for the activity of other peroxidases, which were not inhibited by 3-amino-1,2,4-triazole.
Histopathology
For histopathological analysis, a portion of liver was obtained from mice at day 7 p.i., immediately fixed in 4% buffered formaldehyde, processed and embedded in paraffin. Tissue sections (4 μm thick) were stained with haematoxylin and eosin and examined under light microscopy. Pictures were taken using an Olympus BX51 upright microscope (200× magnification; Olympus, Tokyo, Japan).
Immunofluorescence microscopy
PMA-activated THP-1 cells were seeded on gelatine-coated coverslips, with or without Met-R and infected with DENV-2 NGC at an MOI of 0·1, for 1 hr at 4°. Cells were washed and fixed with 4% (w/v) paraformaldehyde on ice. At room temperature, cells were permeabilized with 0·1% (v/v) Triton X-100 and stained with phycoerythrin-conjugated anti-CCR5 antibody (BD Biosciences, San Jose, CA) and/or anti-pan-DENV 2·5 μg/ml (anti-dengue virus 1 + 2 + 3 + 4 antibody; Abcam, Cambridge, UK) followed by Alexa Fluor 488-labelled secondary anti-mouse antibody (Molecular Probes, Eugene, OR). All experiments were performed in duplicate, together with control groups without primary antibodies. Coverslips were mounted in Mowiol 4-88 (Polysciences, Inc., Warrington, PA) and analysed using a C2 Eclipse Ti confocal microscope (Nikon, Tokyo, Japan). Images were analysed with Volocity 3D image analysis software (PerkinElmer Inc., Waltham, MA).
Flow cytometry
Spleen samples were collected, homogenized in 40-μm sterile strainers and resuspended in PBS with 2% fetal calf serum and 2 mm EDTA. Red blood cells were removed with lysis buffer (Sigma-Aldrich, St Louis, MO). Cells were stained using monoclonal antibodies against mouse CD3 (peridinin chlorophyll protein-Cy5-conjugated), CD4 (Pacific Blue-conjugated), CD8 (allophycocyanin-Cy7-conjugated), CD69 (phycoerythrin-conjugated) and isotype controls. All antibodies were purchased from BD Biosciences. Limits for the quadrant markers were set based on negative populations and isotype controls. Cells were acquired (5 × 105 events) on a BD FACSCanto II (BD Biosciences) cytometer and analysed using the FlowJo 7.5.3 software (TreeStar Inc., Ashland, OR). The percentage of an analysed population in front of total acquired events was used in the construction of graphs.
Statistical analysis
Results are shown as means ± SEM. After checking for data normality, differences were compared using analysis of variance followed by a Student–Newman–Keuls post-hoc analysis. Differences between lethality curves were calculated using a Log-rank test (Graph Prism software 6.0). Results with P < 0·05 were considered significant. All data are representative of at least two experiments (n = 7 to n = 12 replicates or n = 8 to n = 14 mice).
Results
CCR5 is a host factor required for DENV-2 replication in macrophages
Mononuclear phagocytes are primary targets for DENV.15,44 These cells express the CCR5 receptor on their surface.45 We sought to investigate if CCR5 blockade would influence DENV infection by establishing an in vitro DENV-2 infection assay using the human monocyte-like cell lineage THP-1. Cells were stimulated with PMA for differentiation into adherent macrophages and then infected with DENV-2 strain NGC (Fig.1). We found that DENV-2 (+) RNA abundance was reduced in macrophages treated with the CCR5/CCR1 receptor antagonist Met-R before DENV-2 infection at 18, 24 and 48 hr p.i. when compared with non-treated infected macrophages (Fig.1a). DENV-2 infection of macrophages was accompanied by the production of CCR5 ligands. Indeed, CCL5 levels were increased in cell culture supernatants at early time-points (2 and 6 hr p.i.), followed by an increase in CCL4 levels from 12 to 48 hr p.i. when compared with non-infected controls (Fig.1b, c).
Figure 1.
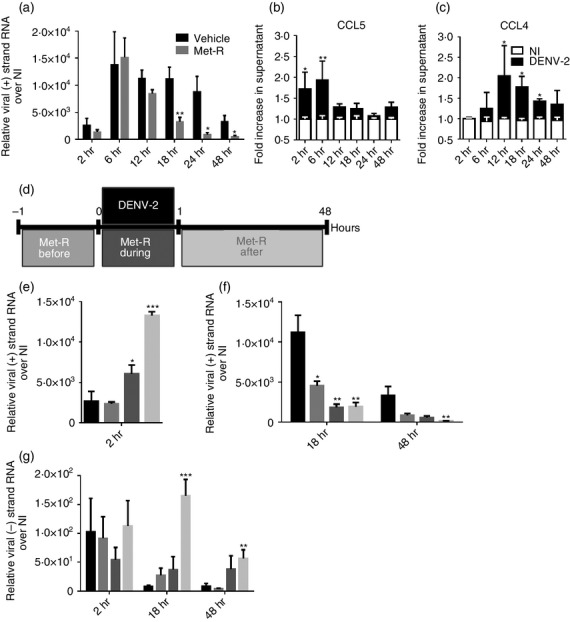
CC-chemokine receptor 5 (CCR5) blockade in vitro influences dengue virus 2 (DENV-2) infection of macrophages. (a) THP-1-derived macrophages were treated with or without Met-RANTES (Met-R), infected with DENV-2 NGC and harvested for viral (+) strand RNA at the defined time-points. (b) CCL5 and (c) CCL4 protein levels in culture supernatants of THP-1 derived macrophages after infection with DENV-2 NGC compared with levels in non-infected (NI) culture supernatants. (d) Experimental design and legend of results displayed in (e), (f) and (g), showing relative time and periods of Met-R treatment and DENV-2 infection in macrophage cultures. THP-1 macrophages were treated with Met-R and infected with DENV-2 NGC to evaluate influence in DENV-2 entry (e) and replication (f), as measured by (+) strand viral RNA inside cells or by (−) strand viral RNA (g). Data are representative of at least two independent experiments. *P < 0·05, **P < 0·01 or ***P < 0·001 when compared with the positive control.
To further study the effect of CCR5/CCR1 blockade on DENV-2 replication, THP-1-derived macrophages were incubated with Met-R before, during and after infection with DENV-2 (Fig.1d) and harvested for RNA extraction at 2, 18 and 48 hr p.i. (early, intermediate and late phases of infection, respectively). Met-R treatment before infection did not alter viral (+) RNA at 2 hr p.i. (Fig.1e), but reduced viral (+) RNA abundance at intermediate and late phases when compared with DENV-2-infected non-treated cells (Fig.1f). In contrast, Met-R addition to cell culture during or after infection could markedly increase the amount of viral (+) RNA inside macrophages at 2 hr p.i. (Fig.1e), but caused a greater reduction in viral (+) RNA levels at later phases of infection compared with positive or Met-R-treatment before infection control groups (Fig.1f). Macrophages that received Met-R after DENV-2 infection remained in contact with Met-R for the entire experiment, which may explain the greater efficiency of this strategy to reduce viral RNA levels inside infected macrophages. This suggests that exposure to Met-R would prevent CCR5 activation by chemokines, therefore decreasing macrophage permissiveness to DENV-2 replication.
Flavivirus replication requires the generation of viral negative strand (−) RNA, to act as substrate for the synthesis of novel (+) viral RNA molecules, which are used as templates for protein synthesis and viral progeny assembly.46,47 We complemented our experimental approach by assessing DENV-2 (−) RNA levels in macrophages treated with or without Met-R (Fig.1g). We observed that Met-R treatment had no effect on viral (−) RNA levels at 2 hr p.i. but increased the abundance of viral (−) RNA at 18 and 48 hr p.i. when compared with infected not-treated macrophages. Hence, the decrease in viral (+) RNA observed in Met-R-treated macrophages is associated with an increase in (−) RNA accumulation. Importantly, Met-R treatment after infection, which is most effective in reducing viral (+) RNA levels, markedly increased viral (−) RNA accumulation at intermediate and late phases of infection, indicating that CCR5 blockade interferes with DENV-2 RNA metabolism.
In summary, DENV-2 infection induces the expression of CCR5 ligands, whereas CCR5 blockade by Met-R prevents receptor activation. Moreover, CCR5 activation is necessary for macrophage permissiveness to DENV replication and largely influences viral RNA abundance in cells.
CCR5 is not a viral receptor but co-localizes with DENV-2 at the macrophage membrane
The ability of Met-R to increase viral (+) RNA abundance to high levels after 2 hr of infection led us to hypothesize that CCR5 could be involved in DENV-2 entry in macrophages. To directly assess the participation of CCR5 on viral entry, THP-1-derived macrophages were treated with or without Met-R, infected with DENV-2 and washed in an alkaline buffer to release viral particles not internalized during adsorption before RNA extraction (Fig.2a). Met-R presents partial agonist activity, which causes chemokine receptor internalization,48 leading to CCR5 depletion from macrophage surfaces before DENV-2 adsorption. We observed that CCR5 blockade and/or internalization by Met-R before DENV-2 infection did not alter the amount of viral (+) RNA inside macrophages. This indicates that CCR5 was not required for DENV entry into cells and therefore was not directly involved in DENV-2 entry. To further study these phenomena, THP-1-derived macrophages were infected with DENV-2 at 4°, followed by immediate fixation in formaldehyde, to allow for virus adsorption to macrophage surfaces but prevent virus internalization (Fig.2b). After staining for DENV-2 (green) and for CCR5 (red), we observed that DENV-2 and CCR5 fluorescence signals overlapped at the cell surface, yielding a yellow fluorescence signal, consistent with co-localization. Met-R treatment before infection prevented DENV-2/CCR5 fluorescence overlap (Fig.2c). Importantly, the intensity and coverage of the DENV-2 signal was unaltered by Met-R, which is in agreement with our data showing that viral entry occurs independently of CCR5 and with the current literature regarding DENV receptors.49–51 Also, Met-R treatment reduced the intensity and amount of CCR5 signal in the macrophage surface; which is consistent with the ability of Met-R to block and to cause receptor internalization, so reducing CCR5 availability at the surface.
Figure 2.
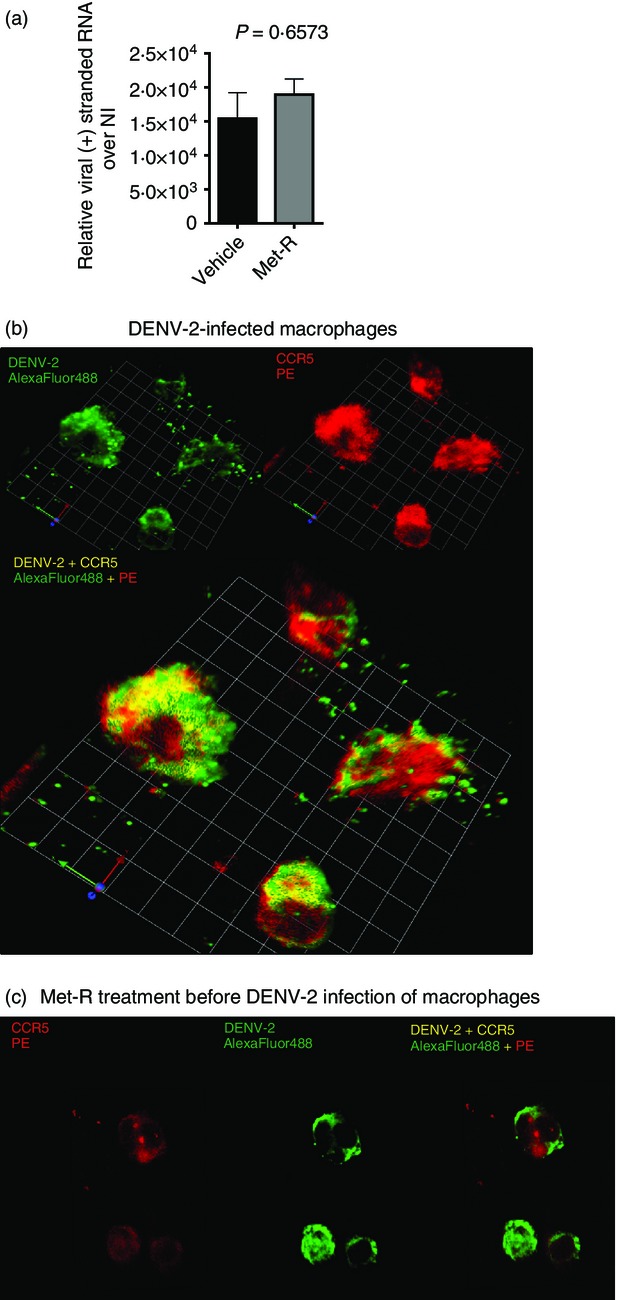
CC-chemokine receptor 5 (CCR5) is not a viral receptor but co-localizes with dengue virus 2 (DENV-2) at the macrophage membrane. (a) THP-1-derived macrophages were treated with or without Met-RANTES (Met-R), infected with DENV-2 NGC and washed in a high-salt alkaline buffer to remove membrane-bound virus. DENV-2 virions that were able to enter macrophages were assessed by quantitative RT-PCR. (b) THP-1 cells were infected at 4°C and stained immediately after virus adsorption for CCR5 (red) and DENV-2 (green) presence at the cell membrane. Sites of co-localization are shown in yellow. Images taken at 400× magnification. (c) THP-1 cells treated with Met-R before infection do not present co-localization of CCR5 and DENV-2. Images taken at 200× magnification. Graphs and panels are representative of two independent experiments.
CCR5 is required for DENV-2 replication in murine macrophages
To evaluate the in vivo role of CCR5 in experimental dengue infection in mice, we first aimed to reproduce our findings in vitro using primary murine macrophages and a mouse-adapted DENV-2 strain. Naive peritoneal macrophages from WT mice were treated in vitro with Met-R and infected with DENV-2 P23085 to evaluate viral replication kinetics (Fig.3a). DENV-2 P23085 was able to replicate in murine macrophages, reaching a peak of replication in culture at 72 hr p.i. Met-R treatment led to a decrease in DENV-2 titres throughout all evaluated time-points. This result corroborates the ability of Met-R to impair DENV-2 replication in vitro in different host cells. Also, DENV-2 infection of murine bone-marrow-derived macrophages induced the expression of CCL5 at 24 and 48 hr p.i. (Fig.3b), indicating that DENV-2 infection causes CCR5 ligand production in murine mononuclear phagocytes.
Figure 3.
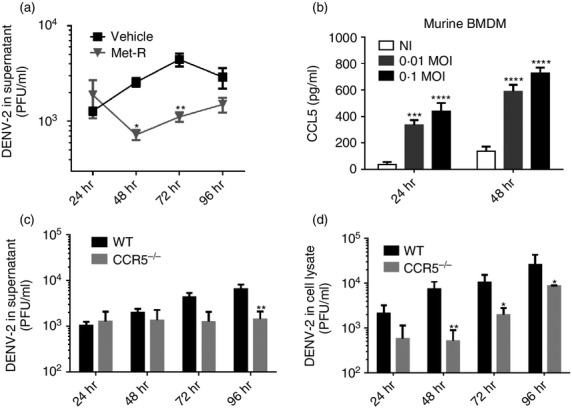
CC-chemokine receptor (CCR5) is required for dengue virus 2 (DENV-2) replication in murine macrophages. (a) Murine peritoneal macrophages were treated with or without Met-RANTES (Met-R) and infected with the mouse-adapted DENV-2 P23085 strain at a multiplicity of infection (MOI) of 0·1. Viral replication was measured up to 96 hr post-infection (p.i.) in the culture supernatant by plaque assays. (b) Wild-type (WT) murine bone marrow-derived macrophages (BMDM) infected with 0·1 or 0·01 MOI of DENV-2 P23085 express and secrete CCL5 in the culture supernatant, measured at 24 and 48 hr p.i. by ELISA. (c, d) Murine peritoneal macrophages from WT or CCR5−/− mice were infected with DENV-2 P23085 and assessed for infective DENV-2 in the culture supernatant (c) and inside cells (d) up to 96 hr p.i. by plaque assay. Data are representative of three independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001 or ****P < 0·0001 when compared with positive controls.
Next, we evaluated whether DENV-2 replication would be impaired in the absence of CCR5 using primary macrophages collected from CCR5−/− mice. Naive peritoneal macrophages from WT and CCR5−/− mice were infected with DENV-2 P23085 and assessed for the production of infective virus in the supernatant and inside cells (Fig.3c, d). We found that DENV-2 P23085 infection of WT macrophages led to increasing virus titres between 24 and 96 hr, both in culture supernatant (Fig.3c) and inside macrophages (Fig.3d). In contrast, CCR5 deficiency led to steady DENV titres in culture supernatant and reduced PFU levels inside macrophages, suggesting that DENV-2 replication is impaired in CCR5−/− macrophages. At 48 hr p.i., virus titres were 10-fold lower in the CCR5−/− group compared with WT (Fig.3d).
These results indicate that pharmacological blockade of CCR5 also reduces DENV-2 replication in murine macrophages. Moreover, the DENV-2 replication impairment observed in CCR5−/− macrophages provides evidence for the involvement of CCR5 receptor in DENV-2 replication in macrophages.
CCR5−/− mice are protected against lethal challenge by DENV
We next performed in vivo experiments to investigate whether CCR5 deficiency or blockade would impair DENV-2 infection and disease development. Mice infected with the mouse-adapted DENV-2 P23085 strain manifest a disease sharing several features with human clinical disease.35,39,52,53 Both WT and CCR5−/− mice were infected with a semi-lethal inoculum of DENV-2 P23085, or a mouse-adapted DENV-3 strain,41 and monitored daily for survival rates (Fig.4). WT mice succumbed to infection between day 6 and day 8 p.i. with either DENV-2 P23085 (Fig.4a) or mouse-adapted DENV-3 (Fig.4b), with an average 30% survival. Surprisingly, CCR5−/− mice were protected from infection with both viruses, presenting no lethality up to 28 days (Fig.4 and see Supplementary material, Fig. S1). Hence, CCR5 deficiency confers protection to experimental dengue infection in vivo using two mouse-adapted strains of different serotypes.
Figure 4.
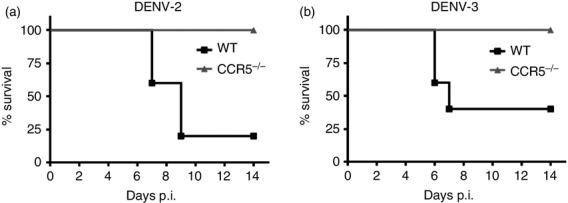
CC-chemokine receptor 5-deficient (CCR5−/−) mice are protected against lethal challenge by dengue virus (DENV). Wild-type (WT) and CCR5−/− mice were inoculated intraperitoneally with 200 plaque-forming units (PFU) of mouse-adapted DENV-2 P23085 (a) or 1000 PFU of mouse-adapted DENV-3 (b) diluted in 100 μl PBS and observed daily for 14 days post-infection (p.i.). Results are expressed as percentage of survival in each group and are representative of three different experiments. P < 0·01 and P < 0·05 for (a) and (b), respectively, compared with infected WT controls.
CCR5 deficiency prevents DENV-2 viral replication and disease development in vivo
To investigate if CCR5 deficiency affects disease development, WT and CCR5−/− mice were infected with DENV-2 P23085 and spleen, liver and blood samples were collected for viral load and immunopathology assessment (Fig.5). DENV-2 titres were detectable in tissues of WT mice as early as day 3 p.i., but not in tissues of CCR5−/− mice (Fig.5a). At day 5 p.i., DENV-2 infection was established in both groups, although viral load in the CCR5−/− group was reduced 10-fold in the spleen, slightly reduced in the liver and undetectable in the blood when compared with the WT group. At the peak of infection on day 7, viral load was undetectable in spleen and blood of CCR5−/− mice but was greatly increased in WT mice. Similarly, CCR5−/− mice presented a 100-fold reduction in DENV-2 load in the liver when compared with the high viral load observed in WT mice. This indicates that DENV-2 replication is impaired in vivo in the absence of CCR5.
Figure 5.

CC-chemokine receptor 5 (CCR5) deficiency prevents dengue virus 2 (DENV-2) -induced disease and reduces viral load in tissues. Wild-type (WT) and CCR5 −/− mice were inoculated with 200 plaque-forming units (PFU) of DENV-2 P23085 and killed at days 3, 5 and 7 post-infection (p.i.) for sample collection. (a) Viral loads in spleen, liver and blood were measured by plaque assay. (b) Platelet count and haematocrit, as indicators of haematological alteration, were measured at days 5 and 7 p.i., together with (c) interferon-γ (IFN-γ) and tumour necrosis factor-α (TNF-α) levels in spleen homogenates. (d) Neutrophil accumulation in the spleen (left) and liver (right) as indicated by myeloperoxidase (MPO) activity. (e) DENV-2 induction of CCR5 ligands CCL3 and CCL5 in the spleen and liver at day 7 p.i. as evaluated by ELISA. (f) Haematoxylin & eosin stained liver sections of experimental groups at day 7 p.i. (NI, non-infected). Data are representative of two independent experiments. *P < 0·05, **P < 0·01, when compared with positive controls, except for (e) where compared with negative controls. ND: not detectable.
The aforementioned reduction in viral load was accompanied by a significant reduction in disease parameters in CCR5−/− mice. At day 5 p.i., WT mice already presented discrete changes in haemoconcentration and platelet counts, which preceded the development of severe haematological alterations at day 7 p.i. (Fig.5b). These haematological changes were not observed in CCR5−/− mice at any time-point evaluated (Fig.5b). Also, WT mice presented high levels of interferon-γ and tumour necrosis factor-α in the spleen at day 7 p.i. (Fig.5c), whereas CCR5−/− mice presented reduced or non-detectable levels of these cytokines. Neutrophil accumulation in the spleen and liver, as measured by myeloperoxidase activity, was significantly reduced in CCR5−/− when compared with WT tissues (Fig.5d). Importantly, DENV-2 infection induced the expression of CCR5 ligands in the spleen and liver of infected WT mice (Fig.5e). Seven days after infection, CCL3 levels were increased in both spleen and liver when compared with the non-infected control group. CCL5 levels were increased in the liver when compared with the non-infected group, but no differences were observed in the spleen, because of high basal levels of CCL5 in this organ. The levels of these chemokines in DENV-2-infected CCR5−/− mice were similar to non-infected controls. Histological analysis of the hepatic parenchyma in infected WT mice revealed signs of hepatocyte degeneration, necrosis, loss of organ architecture and an extensive mononuclear cell recruitment, which were not observed in infected CCR5−/− or non-infected liver sections (Fig.5f).
Together, these data indicate that CCR5−/− mice are protected from DENV-2 P23085 infection and associated inflammatory responses. Although both groups were infected by DENV-2 P23085, the reduced DENV-2 load in CCR5−/− mice tissues during the infection suggests that CCR5 is a host factor required for virus replication in vivo.
Pre-treatment with CCR5 antagonists protects mice from DENV-2 infection
To validate our findings on the protection observed in CCR5−/− mice, WT mice were treated with the CCR5/CCR1 antagonist Met-R in a preventive (1 day before infection) or therapeutic (3 days post-infection) schedule before infection with DENV-2 P23085. We examined tissues and blood for disease parameters at day 7 p.i. and found that only the preventive treatment with Met-R was effective in protecting WT mice against experimental dengue infection (Fig.6). Preventive treatment (pre-treatment) reduced viral load at least 10-fold in infected organs and abolished viraemia in infected WT mice (Fig.6a), similar to the findings in CCR5−/− infected mice. Treatment after infection (post-treatment) was unable to reduce viral replication in tissues and blood compared with the DENV-2-infected control group (Fig.6a).
Figure 6.

Treatment with CC-chemokine receptor 5 (CCR5) antagonists before but not after infection resembles the phenotype of CCR5-deficient mice. Wild-type (WT) mice were treated daily with 10 μg of Met-RANTES 1 day before infection until day 6 p.i. (Pre-Treatment) or on day 3 until day 6 p.i. (Post-Treatment). Mice were inoculated with 200 plaque-forming units (PFU) of DENV-2 P23085 and killed at day 7 p.i. for sample collection. (a) Viral loads in spleen, liver and blood were measured by plaque assays. (b) Haematocrit and platelet count, as indicators of haematological alteration. (c) Interferon-γ (IFN-γ) and tumour necrosis factor-α (TNF-α) levels in spleen homogenates and (d) neutrophil accumulation in the spleen (left) and liver (right) as indicated by myeloperoxidase (MPO) activity. (e) DENV-2 induction of CCR5 ligands CCL3, CCL4 and CCL5 in the spleen as evaluated by ELISA. (f) Survival rates of DENV-2 P23085-infected WT mice treated with the CCR5 antagonist UK-484900 before (Pre-Treatment) and after (Post-Treatment) infection. Data are representative of two independent experiments. *P < 0·05, ***P < 0·001 when compared to positive controls.
Met-R pre-treated mice also had less haemoconcentration and thrombocytopenia (Fig.6b), reduced levels of interferon-γ and tumour necrosis factor-α in the spleen (Fig.6c) and less neutrophil accumulation in the spleen and liver (Fig.6d) at day 7 p.i. when compared with infected WT mice. Liver histology indicated that pre-treatment with Met-R prevented hepatocyte degeneration and necrosis and preserved organ architecture (Fig.5f). Post-treatment with Met-R prevented thrombocytopenia (Fig.6b), but could not prevent haemoconcentration, cytokine production or neutrophil accumulation in organs (Fig.6c, d), which were similar to levels observed in non-treated WT infected mice. Interestingly, Met-R pre-treatment did not alter the levels of CCL3 observed in the spleen upon DENV-2 infection, but led to an increase in CCL4 and CCL5 levels in this organ (Fig.6e). Met-R post-treatment had no effect on the expression levels of any chemokine compared with control groups.
Finally, we tested a small molecule antagonist of CCR5, UK-484900, an analogue of Maraviroc which interacts with CCR5 differently from the polypeptide Met-R (Fig.6f). Mice pre-treated with UK-484900 were protected from lethal infection with DENV-2 P23085, presenting survival rates up to 70%, in contrast to the 20% survival observed in infected control groups. Treatment after infection (post-treatment) with UK-484900 could not protect mice from DENV-2-induced lethality. Hence, pharmacological blockade of CCR5 can prevent lethality and disease through experimental DENV-2 infection only when applied before infection, then mimicking CCR5 genetic ablation as observed when using CCR5−/− mice.
Discussion
We describe a hitherto unknown association between the chemokine receptor CCR5 and DENV, in which the virus requires receptor activation during infection to sustain replication and develop disease. These findings relate to the well-established association of CCR5 and flavivirus infections in an unexpected way: as a host factor that contributes to viral replication, rather than as a key molecule driving protective immune responses such as observed in West Nile virus, tick-borne encephalitis virus and Japanese encephalitis virus infections.29,31,54 However, diseases caused by these viruses are characterized by encephalitis, in contrast to the haemorrhagic fever and shock caused by DENV, with rare exceptions.55 Hence, these diseases must have pathogenic mechanisms relying on different roles for CCR5 and its ligands during infection.
A recent study discussed the possible association of CCR5Δ32 allele frequency and primary acute dengue disease.56 The authors found no association between CCR5Δ32 allele frequency and acute dengue, probably because of the small number of samples analysed in the study. Sierra et al.34 suggest that increased expression of CCR5 in peripheral blood mononuclear cells from primary-infected dengue patients would lead to a more efficient T-cell effector response in secondary DENV infection. Oliveira-Pinto et al.33 showed that during acute dengue infection, CCR5 expression on both CD4+ and CD8+ T-cell populations is markedly increased. In our in vivo model, the percentage of activated (CD69-expressing) CD4+ and CD8+ T cells is increased in the spleen of DENV-2-infected CCR5−/− mice, most notably CD8+ T cells (see Supplementary material, Fig. S2). At least in this experimental model, CCR5 does not seem to be essential for lymphocyte migration/activation in target organs. It is possible that due to its protective phenotype, CCR5−/− mice might present reduced lymphopenia,57 making their T-cell response more effective and sustained.
It remains elusive whether CCR5 blockade would impair DENV-2 replication in cell types other than macrophages. Our data showed that BMDC also express and secrete a CCR5 ligand upon DENV-2 infection (see Supplementary material, Fig. S3). Myeloid dendritic cells are known to support DENV-2 infection and express CCR5, which might require CCR5 activation to become permissive to DENV-2 infection. In contrast, in vitro experiments performed with the hepatocarcinoma cell line HepG2 showed that treatment with Met-R or CCL5 had no effect on DENV-2 replication (see Supplementary material, Fig. S4). Therefore, it is likely that DENV-2 replication in hepatocytes does not require CCR5 activation, which is in line with the recovery of higher virus titres from the liver of CCR5−/− infected mice, in comparison to the spleen from these animals (Fig.5a).
Although well described for HIV infection, molecular mechanisms suggesting the utilization of CCR5 by DENV (and other flavivirus) for effective viral replication in the host cells have not been described. Our data indicate that DENV-2 and CCR5 co-localize at the macrophage membrane but CCR5 is not required for viral entry, although Met-R treatment increases DENV (+) RNA inside macrophages if included during or immediately after DENV adsorption (Fig.1d). We suggest that this increase in DENV-2 RNA is an indirect consequence of Met-R-induced CCR5 internalization, because DENV receptors at the macrophage surface (e.g. CLEC-5A, mannose receptor) and CCR5 are anchored in membrane lipid rafts and can be internalized by clathrin-dependent endocytosis.48,58,59 Met-R addition after adsorption would induce internalization of CCR5 when most virions are bound to macrophage viral receptors at the cell membrane, therefore dragging DENV-2 virions together with Met-R/CCR5 complexes to the interior of macrophages. Le Sommer et al.40 showed that G protein-coupled receptor kinase 2 (GRK2) promotes flaviviridae entry and replication in host cells, where GRK2 participates in multiple steps of flavivirus life cycle and viral RNA synthesis. GRK2 is highly expressed in leucocytes, including monocytes,23 and is known to participate in CCR5 signalling events after ligand activation.60 Hence, it is likely that CCR5 and GRK2 roles in flavivirus RNA synthesis are associated, given that blockade of these molecules ultimately results in impaired viral replication. Although we do not propose a molecular mechanism to explain the dependency of CCR5 for DENV-2 productive infection in host cells, the involvement of signalling pathways (i.e. Erk1/2, GRK2) should be further explored.
Our initial in vitro data using Met-R treatment could not exclude a role for CCR1, which is also expressed in macrophages,25 but it is unlikely that this receptor participates in DENV replication. CCL4 expression was concomitant to the reduction in viral RNA abundance in Met-R-treated macrophages (Fig.1a, b), suggesting that CCL4 could be involved in chemokine receptor-mediated cell permissiveness to DENV-2 infection. CCL4 does not bind CCR1 and Met-R possess higher affinity for CCR5.61 More importantly, we previously described the role of CCR1 in experimental dengue infection in vivo, where no differences were observed in the viral load recovered from WT and CCR1−/− mouse tissues.35
It must be mentioned that delayed treatment with Met-R was ineffective in our experimental system, indicating that such strategies may be useful only if given very early in the course of infection. Our mouse model of DENV-2 infection resembles an acute and severe disease in immunocompetent animals and is based on a mouse-adapted DENV-2 strain that is highly virulent to the murine host.53 We believe that such a degree of viral adaptation is helpful to identify host molecules involved in DENV-2 infection, but poses several limitations, and so requires testing in other DENV infection models to fully assess the beneficial effects of CCR5 blockade and mechanism of action. In contrast to intracellular host or viral molecules, CCR5 and its chemokine ligands can be easily targeted, given that a small-molecule CCR5 antagonist developed against HIV is available (Maraviroc, Pfizer). Also, there are ongoing clinical trials for other CCR5 antagonists and CCL5-blocking molecules, which will increase the possibilities of pharmacological intervention in the chemokine system in the context of dengue.62
In summary, we provide experimental data highlighting the role of CCR5 in contributing to DENV-2 replication and disease development in vitro and in vivo. We believe that further studies aiming to decipher the molecular and cellular mechanisms underlying these findings must be conducted so as to envisage prevention and treatment using safe CCR5 antagonists in areas where dengue is endemic.
Acknowledgments
We thank Ilma Marçal and Gilvânia Ferreira da Silva Santos (ICB/UFMG) for providing technical assistance. This work was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq/Brazil) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG/Brazil). The work was done under the auspices of the programme INCT em Dengue (CNPq, Brazil).
Glossary
- ATCC
American Type Culture Collection
- CCR
CC-chemokine receptor
- DENV
Dengue virus
- DMEM
Dulbecco's modified Eagle's medium
- MOI
multiplicity of infection
- p.i.
post-infection
- RANTES
regulated on activation normal T cell expressed and secreted
- THP-1
human monocytic leukaemia cell line
- WT
wild-type
- w/v
weight/volume
Author's contribution
REM, RG and MMT conceived the study. GBM, DGS, MLN and MMT provided tools and laboratory infrastructure. REM, RG, JDS, RF, ALQ, DC, CCP, PEM and CTF performed experiments and analysed data. REM, RG, DGS and MMT wrote the manuscript.
Disclosures
The authors declare that they have no financial or commercial conflicts of interest.
Supporting Information
Figure S1. CC-chemokine receptor 5 deficient (CCR5-/-) mice do not present delayed mortality after semi-lethal dengue virus 2 (DENV-2) infection. Wild-type (WT) and CCR5−/− mice were inoculated intraperitoneally with 200 plaque forming units of DENV-2 P23085 and observed daily for 28 days post-infection (p.i).
Figure S2. CC-chemokine receptor 5 deficient mice (CCR5-/-) develop an increased T lymphocyte response to dengue virus 2 (DENV-2) infection.
Figure S3. Dengue virus 2 (DENV-2) induces the expression of CC-chemokine receptor 5 (CCL5) in murine bone marrow dendritic cells (BMDCs).
Figure S4. Treatment with CC-chemokine receptor 5 (CCR5) antagonist or ligand does not alter dengue virus type 2 (DENV-2) replication in HepG2 cells.
References
- WHO Global strategy for dengue prevention and control 2012–2020 Publications W. 2012. In:, ed. WHO website ( www.who.int.
- Marques RE, Guabiraba R, Cisalpino D, Teixeira MM, Souza DG. Dengue. Colloquium Series on Integrated Systems Physiology: From Molecule to Function. 2014;6:1–104. [Google Scholar]
- Guzman MG, Kouri G. Dengue: an update. Lancet Infect Dis. 2002;2:33–42. doi: 10.1016/s1473-3099(01)00171-2. [DOI] [PubMed] [Google Scholar]
- Kalayanarooj S, Vaughn DW, Nimmannitya S, et al. Early clinical and laboratory indicators of acute dengue illness. J Infect Dis. 1997;176:313–21. doi: 10.1086/514047. [DOI] [PubMed] [Google Scholar]
- Who T. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control. Geneva: WHO Library; 2009. [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, et al. The global distribution and burden of dengue. Nature. 2013;496:504–7. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RV, Vaughn DW. Dengue: an escalating problem. BMJ. 2002;324:1563–6. doi: 10.1136/bmj.324.7353.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suaya JA, Shepard DS, Siqueira JB, et al. Cost of dengue cases in eight countries in the Americas and Asia: a prospective study. Am J Trop Med Hyg. 2009;80:846–55. [PubMed] [Google Scholar]
- Gubler DJ. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. 1998;11:480–96. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman AL, Ennis FA. Immunopathogenesis of Dengue hemorrhagic fever. Virology. 1999;257:1–6. doi: 10.1006/viro.1999.9656. [DOI] [PubMed] [Google Scholar]
- Guzman MG, Kouri G. Dengue haemorrhagic fever integral hypothesis: confirming observations, 1987–2007. Trans R Soc Trop Med Hyg. 2008;102:522–3. doi: 10.1016/j.trstmh.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Whitehorn J, Simmons CP. The pathogenesis of dengue. Vaccine. 2011;29:7221–8. doi: 10.1016/j.vaccine.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Halstead SB. Dengue. Lancet. 2007;370:1644–52. doi: 10.1016/S0140-6736(07)61687-0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Madoz JR, Bernal-Rubio D, Kaminski D, Boyd K, Fernandez-Sesma A. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J Virol. 2010;84:4845–50. doi: 10.1128/JVI.02514-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Wang SY. Activation of terminally differentiated human monocytes/macrophages by dengue virus: productive infection, hierarchical production of innate cytokines and chemokines, and the synergistic effect of lipopolysaccharide. J Virol. 2002;76:9877–87. doi: 10.1128/JVI.76.19.9877-9887.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M. Chemokine receptor CCR5: insights into structure, function, and regulation. Cell Signal. 2004;16:1201–10. doi: 10.1016/j.cellsig.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Barcelos LS, Coelho AM, Russo RC, et al. Role of the chemokines CCL3/MIP-1α and CCL5/RANTES in sponge-induced inflammatory angiogenesis in mice. Microvasc Res. 2009;78:148–54. doi: 10.1016/j.mvr.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Weiss ID, Shoham H, Wald O, et al. Ccr5 deficiency regulates the proliferation and trafficking of natural killer cells under physiological conditions. Cytokine. 2011;54:249–57. doi: 10.1016/j.cyto.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Gao JL, Kuhns DB, Tiffany HL, et al. Structure and functional expression of the human macrophage inflammatory protein 1α/RANTES receptor. J Exp Med. 1993;177:1421–7. doi: 10.1084/jem.177.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–8. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply exposed individuals to HIV-1 infection. Cell. 1996;86:367–77. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Telenti A. Safety concerns about CCR5 as an antiviral target. Curr Opin HIV AIDS. 2009;4:131–5. doi: 10.1097/COH.0b013e3283223d76. [DOI] [PubMed] [Google Scholar]
- Olbrich H, Proudfoot AE, Oppermann M. Chemokine-induced phosphorylation of CC chemokine receptor 5 (CCR5) J Leukoc Biol. 1999;65:281–5. doi: 10.1002/jlb.65.3.281. [DOI] [PubMed] [Google Scholar]
- Fatkenheuer G, Pozniak AL, Johnson MA, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11:1170–2. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Power CA, Hoogewerf AJ, et al. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem. 1996;271:2599–603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- Wells TN, Proudfoot AE, Power CA. Chemokine receptors and their role in leukocyte activation. Immunol Lett. 1999;65:35–40. doi: 10.1016/s0165-2478(98)00121-7. [DOI] [PubMed] [Google Scholar]
- Shahrara S, Proudfoot AE, Woods JM, et al. Amelioration of rat adjuvant-induced arthritis by Met-RANTES. Arthritis Rheum. 2005;52:1907–19. doi: 10.1002/art.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repeke CE, Ferreira SB, Jr, Vieira AE, et al. Dose–response met-RANTES treatment of experimental periodontitis: a narrow edge between the disease severity attenuation and infection control. PLoS One. 2011;6:e22526. doi: 10.1371/journal.pone.0022526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindberg E, Mickiene A, Ax C, Akerlind B, Vene S, Lindquist L, Lundkvist A, Svensson L. A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J Infect Dis. 2008;197:266–9. doi: 10.1086/524709. [DOI] [PubMed] [Google Scholar]
- Glass WG, McDermott DH, Lim JK, et al. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. 2006;203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202:1087–98. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza FA, Cruz OG, Zagne SM, et al. Multiplex cytokine profile from dengue patients: MIP-1β and IFN-γ as predictive factors for severity. BMC Infect Dis. 2008;8:86. doi: 10.1186/1471-2334-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de-Oliveira-Pinto LM, Marinho CF, Povoa TF, et al. Regulation of inflammatory chemokine receptors on blood T cells associated to the circulating versus liver chemokines in dengue fever. PLoS One. 2012;7:e38527. doi: 10.1371/journal.pone.0038527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra B, Perez AB, Garcia G, Aguirre E, Alvarez M, Gonzalez D, Guzman MG. Role of CC chemokine receptor 1 and two of its ligands in human dengue infection. Three approaches under the Cuban situation. Microbes Infect. 2014;16:40–50. doi: 10.1016/j.micinf.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Guabiraba R, Marques RE, Besnard AG, Fagundes CT, Souza DG, Ryffel B, Teixeira MM. Role of the chemokine receptors CCR1, CCR2 and CCR4 in the pathogenesis of experimental dengue infection in mice. PLoS One. 2010;5:e15680. doi: 10.1371/journal.pone.0015680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atrasheuskaya A, Petzelbauer P, Fredeking TM, Ignatyev G. Anti-TNF antibody treatment reduces mortality in experimental dengue virus infection. FEMS Immunol Med Microbiol. 2003;35:33–42. doi: 10.1111/j.1574-695X.2003.tb00646.x. [DOI] [PubMed] [Google Scholar]
- St John AL, Rathore AP, Yap H, Ng ML, Metcalfe DD, Vasudevan SG, Abraham SN. Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc Natl Acad Sci U S A. 2011;108:9190–5. doi: 10.1073/pnas.1105079108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra B, Perez AB, Vogt K, et al. MCP-1 and MIP-1α expression in a model resembling early immune response to dengue. Cytokine. 2010;52:175–83. doi: 10.1016/j.cyto.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Guabiraba R, Besnard AG, Marques RE, et al. IL-22 modulates IL-17A production and controls inflammation and tissue damage in experimental dengue infection. Eur J Immunol. 2013;43:1529–44. doi: 10.1002/eji.201243229. [DOI] [PubMed] [Google Scholar]
- Le Sommer C, Barrows NJ, Bradrick SS, Pearson JL, Garcia-Blanco MA. G protein-coupled receptor kinase 2 promotes flaviviridae entry and replication. PLoS Negl Trop Dis. 2012;6:e1820. doi: 10.1371/journal.pntd.0001820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa VV, Fagundes CT, Valadao DF, et al. A model of DENV-3 infection that recapitulates severe disease and highlights the importance of IFN-γ in host resistance to infection. PLoS Negl Trop Dis. 2012;6:e1663. doi: 10.1371/journal.pntd.0001663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DG, Fagundes CT, Sousa LP, et al. Essential role of platelet-activating factor receptor in the pathogenesis of Dengue virus infection. Proc Natl Acad Sci U S A. 2009;106:14138–43. doi: 10.1073/pnas.0906467106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assuncao-Miranda I, Amaral FA, Bozza FA, et al. Contribution of macrophage migration inhibitory factor to the pathogenesis of dengue virus infection. FASEB J. 2010;24:218–28. doi: 10.1096/fj.09-139469. [DOI] [PubMed] [Google Scholar]
- Wu SJ, Grouard-Vogel G, Sun W, et al. Human skin Langerhans cells are targets of dengue virus infection. Nat Med. 2000;6:816–20. doi: 10.1038/77553. [DOI] [PubMed] [Google Scholar]
- Mueller A, Strange PG. The chemokine receptor, CCR5. Int J Biochem Cell Biol. 2004;36:35–8. doi: 10.1016/s1357-2725(03)00172-9. [DOI] [PubMed] [Google Scholar]
- Brinton MA. The molecular biology of West Nile Virus: a new invader of the western hemisphere. Annu Rev Microbiol. 2002;56:371–402. doi: 10.1146/annurev.micro.56.012302.160654. [DOI] [PubMed] [Google Scholar]
- Fernandez-Garcia MD, Mazzon M, Jacobs M, Amara A. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe. 2009;5:318–28. doi: 10.1016/j.chom.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Longden J, Cooke EL, Hill SJ. Effect of CCR5 receptor antagonists on endocytosis of the human CCR5 receptor in CHO-K1 cells. Br J Pharmacol. 2008;153:1513–27. doi: 10.1038/sj.bjp.0707691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JL, de Wet BJ, Martinez-Pomares L, Radcliffe CM, Dwek RA, Rudd PM, Gordon S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008;4:e17. doi: 10.1371/journal.ppat.0040017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassaneetrithep B, Burgess TH, Granelli-Piperno A, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. 2003;197:823–9. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ST, Lin YL, Huang MT, et al. CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008;453:672–6. doi: 10.1038/nature07013. [DOI] [PubMed] [Google Scholar]
- Renneson J, Guabiraba R, Maillet I, et al. A detrimental role for invariant natural killer T cells in the pathogenesis of experimental dengue virus infection. Am J Pathol. 2011;179:1872–83. doi: 10.1016/j.ajpath.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guabiraba R, Ryffel B. Dengue virus infection: current concepts in immune mechanisms and lessons from murine models. Immunology. 2014;141:143–56. doi: 10.1111/imm.12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larena M, Regner M, Lobigs M. The chemokine receptor CCR5, a therapeutic target for HIV/AIDS antagonists, is critical for recovery in a mouse model of Japanese encephalitis. PLoS One. 2012;7:e44834. doi: 10.1371/journal.pone.0044834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon T, Dung NM, Vaughn DW, et al. Neurological manifestations of dengue infection. Lancet. 2000;355:1053–9. doi: 10.1016/S0140-6736(00)02036-5. [DOI] [PubMed] [Google Scholar]
- Brestovac B, Halicki LA, Harris RP, Sampson I, Speers DJ, Mamotte C, Williams D. Primary acute dengue and the deletion in chemokine receptor 5 (CCR5δ32) Microbes Infect. 2014;16:518–21. doi: 10.1016/j.micinf.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Malavige GN, Ogg GS. T cell responses in dengue viral infections. J Clin Virol. 2013;58:605–11. doi: 10.1016/j.jcv.2013.10.023. [DOI] [PubMed] [Google Scholar]
- Acosta EG, Castilla V, Damonte EB. Differential requirements in endocytic trafficking for penetration of dengue virus. PLoS One. 2012;7:e44835. doi: 10.1371/journal.pone.0044835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiva K, Khiati A, Hery C, Salim H, Leclerc P, Horellou P, Tardieu M. CCR5-, DC-SIGN-dependent endocytosis and delayed reverse transcription after human immunodeficiency virus type 1 infection in human astrocytes. AIDS Res Hum Retroviruses. 2006;22:1152–61. doi: 10.1089/aid.2006.22.1152. [DOI] [PubMed] [Google Scholar]
- Aramori I, Ferguson SS, Bieniasz PD, Zhang J, Cullen B, Cullen MG. Molecular mechanism of desensitization of the chemokine receptor CCR-5: receptor signaling and internalization are dissociable from its role as an HIV-1 co-receptor. EMBO J. 1997;16:4606–16. doi: 10.1093/emboj/16.15.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot AE, Buser R, Borlat F, Alouani S, Soler D, Offord RE, et al. Amino-terminally modified RANTES analogues demonstrate differential effects on RANTES receptors. J Biol Chem. 1999;274:32478–85. doi: 10.1074/jbc.274.45.32478. [DOI] [PubMed] [Google Scholar]
- Marques RE, Guabiraba R, Russo RC, Teixeira MM. Targeting CCL5 in inflammation. Expert Opin Ther Targets. 2013;17:1439–60. doi: 10.1517/14728222.2013.837886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CC-chemokine receptor 5 deficient (CCR5-/-) mice do not present delayed mortality after semi-lethal dengue virus 2 (DENV-2) infection. Wild-type (WT) and CCR5−/− mice were inoculated intraperitoneally with 200 plaque forming units of DENV-2 P23085 and observed daily for 28 days post-infection (p.i).
Figure S2. CC-chemokine receptor 5 deficient mice (CCR5-/-) develop an increased T lymphocyte response to dengue virus 2 (DENV-2) infection.
Figure S3. Dengue virus 2 (DENV-2) induces the expression of CC-chemokine receptor 5 (CCL5) in murine bone marrow dendritic cells (BMDCs).
Figure S4. Treatment with CC-chemokine receptor 5 (CCR5) antagonist or ligand does not alter dengue virus type 2 (DENV-2) replication in HepG2 cells.