

Abstract
Locomotion and respiration require motor axon connectivity and activation of the neuromuscular junction (NMJ). Through a forward genetic screen for muscle weakness, we recently reported an allele of ryanodine receptor type 1 (Ryr1AG). Here we reveal a role for functional RyR1 during acetylcholine receptor (AChR) cluster formation and embryonic synaptic transmission. Ryr1AG homozygous embryos are non-motile. Motor axons extend past AChR clusters and enlarged AChR clusters are found under fasciculated nerves. Using physiological and pharmacological methods, we show that contractility can be resumed through the masking of a potassium leak, and evoked vesicular release can be resumed via bypassing the defect in RyR1 induced calcium release. Moreover, we show the involvement of ryanodine receptors in presynaptic release at the NMJ. This data provides evidence of a role for RyR1 on both the pre- and postsynaptic sides of the NMJ.
Keywords: Diaphragm, Motoneuron, Acetylcholine, Excitation, contraction coupling
1. Introduction
The mechanisms that establish and maintain axon outgrowth and synaptic function during embryonic development are crucial for proper locomotion (Busetto et al., 2003; Hanson and Landmesser, 2004, 2006; Krull, 2010; Sanes and Lichtman, 1999, 2001) A key component of the locomotor circuit is the neuromuscular junction (NMJ), which is formed and refined by signaling between the motoneuron and the muscle (Sanes and Lichtman, 2001). Therefore, defects in pre- and postsynaptic signaling at the NMJ may have detrimental effects on neonatal survival.
Presynaptic vesicular release of acetylcholine (ACh) by motor nerve terminals leads to focal depolarization of the post-synaptic apparatus. A large action potential sweeps through the muscle fiber to induce global depolarization and muscle contraction. Contractions in muscle transpire through the opening of a muscle-specific voltage gated channel, dihydropyridine receptor (L-type calcium channel DHPR; Cacna1s, CaV1.1, (Franzini-Armstrong and Jorgensen, 1994)), which stimulates ryanodine receptor type 1 (RyR1) in the sarcoplasmic reticulum to flood the myoplasm with calcium. The increased calcium initiates contraction in the skeletal muscle (Numa et al., 1990). These events constitute excitation–contraction coupling (E–C coupling).
Transgenic studies have shown that skeletal muscle DHPR acts independently of E–C coupling to establish muscle prepatterning of AChR clusters, AChR cluster size, the frequency of ACh dependent depolarization, and expression of MuSK (Chen et al., 2011). Moreover, RyR1 is required for establishing muscle prepatterning of AChR clusters and proper axon outgrowth (Hanson and Niswander, 2014). However, the requirement for functional RyR1-DHPR complex in determining AChR cluster size, the frequency of ACh-dependent depolarization, and expression of MuSK is less clear.
The family of ryanodine receptors is required for proper Ca2+ release from the sarcoplasmic reticulum in skeletal muscle (RyR1 and RyR3) and heart (RyR2) (Coussin et al., 2000; Giannini et al., 1995; Nakai et al., 1990; Otsu et al., 1990; Takeshima et al., 1989; Zorzato et al., 1990). Mice that lack either Ryr1 or Ryr2 die at birth (Takeshima et al., 1994, 1998), whereas mice that lack Ryr3 are viable (Takeshima et al., 1996), but have deficiencies in synaptic plasticity and locomotion (Futatsugi et al., 1999). Embryonic rat spinal cord and cultured rat motoneurons express RyR1, RyR2, and RyR3 (Dayanithi et al., 2006) and this family of intracellular receptors is required for Ca2+ release from the endoplasmic reticulum of neurons (Fill and Copello, 2002). Therefore, RyRs regulate muscle Ca2+ release and may be involved in release of Ca2+ in the presynaptic terminal that results in release of transmitter.
Here we explore the function of RyR1 as it pertains to mechanisms of motoneuron axon extension, AChR cluster distribution, and presynaptic transmitter release. Mouse embryos that are homozygous for a point mutation in Ryr1 (Ryr1AG) have defects in axon extension, abnormally narrow distribution of AChR clusters and increased AChR cluster size. The Ryr1AG/AG embryonic muscle has a greater change in internal potassium levels and muscle paralysis but, by inhibiting the potassium leak, the muscle can be stimulated to undergo contractions. In addition, we use this mouse model to uncover a pre-synaptic role of ryanodine receptors at the mouse neuromuscular junction. Overall, these data illustrate that RyRs are involved in vesicular release at the NMJ, RyR1 is required for proper release of transmitter at the neuromuscular junction, and postsynaptic RyR1 is required for the size and distribution of AChR clusters.
2. Material and methods
2.1 Forward genetic screen and mouse strains
ENU mutagenesis was performed as described (Kasarskis et al., 1998) on males of C57BL/6J background and then outcrossed onto 129S1/Svlmj background to score at embryonic day 18.5 for recessive mutations that affect embryonic locomotion. The forward genetic screen and identification of the Ryr1AG mutation is described in Hanson et al. (2015). This allele is named RyR1m1Nisw but here we will refer to it as Ryr1AG. Ryr1−/− is the dyspedic null mutation (Ryr1tm1TAlle allele generated by Richard Allen (Buck et al., 1997)). To examine axon guidance errors in limb muscles, Ryr1AG/+ mice were crossed with B6.Cg-Tg(Hlxb9-GFP)1Tmj/J (Jackson laboratory, Maine) stated as Hlxb9-GFP then outcrossed 6 generations in 129S1/Svlmj background. For the locomotion screen, E18.5 embryos were dissected from timed pregnant dams and placed in room temperature oxygenated mouse Tyrode’s solution. To induce limb movement, forelimb and hindlimb footpads were pinched with tweezers to induce paw retraction and cross-extensor reflexes. Touching the forceps to the dorsal rib cage scored s-shaped movements in axial muscles. Touching the forceps to the nose scored neck extension reflex. Touch assay was performed on each embryo in the litter and the genotype of all embryos was determined.
2.2 Calcium transient measurements
Primary myoblasts isolated from E18.5 embryonic muscle were differentiated and then loaded with the fluorescent Ca2+ indicator Fluo-3 (Life Technologies, Grand Island, NY) injected by whole-cell configuration at a final concentration of 200 μM. Following loading, Fluo-3 dye was allowed to diffuse within the cell interior for 5 min. The total change in fluorescence (ΔF/F) was determined from the change in peak fluorescence from initial baseline during stimulation, where F was the fluorescence immediately before the test pulse minus the measured average background (non–Fluo-3) fluorescence before dye injection. The average values of fluorescence change (ΔF) for each test potential (V) were fitted according to: (ΔF)=(ΔF)max/{1 +exp [(VF −V)/kF]}, where (ΔF)max =1093 au, VF = −1.4 mV and kF =5.5 mV. Fluorescence emission was measured by fluorometer (Biomedical Instrumentation Group, University of Pennsylvania, Philadelphia, PA). Kurt Beam and Roger Bannister provided Ryr1-YFP cDNA and performed these experiments.
2.3 [K+]i/PBFI measurements
For [K+]i measurements, E18.5 diaphragm muscle was mechanically scraped of membrane bordering the muscle and affixed to glass bottom culture dishes (MatTeK) with Vetbond (3 M, St. Paul, MN). The ratiometric cell-permeant potassium indicator PBFI–AM (5 μM; Life Technologies, Grand Island, NY) together with 0.2% Pluronic F-127 for enhanced dye loading was applied to the diaphragm muscle for 30 min in Ringers Solution (3 mM K+). After loading, muscles were washed for 30 min to remove excess dye and to allow deesterification of the AM dye. Data was collected from greater than 10 fibers per muscle with 4 muscles per condition. Muscles were analyzed at 33 °C using the 340/380 filters on Zeiss Axio Observer Z1 with Photometrics CoolSNAP HQ2 camera for wide-field imaging. The filtered 340 and 380 intensities were analysized per second and the ratiometric intensity for the fibers were plotted. To calibrate intracellular potassium concentrations, ratiometric PBFI intensities were fitted to the intensities at different K+ concentrations with the addition of 10 μM gramicidin in extracellular solutions and using the intensity of ~10 fibers of interest per muscle in >3 muscles from each condition. Calibration solutions were prepared with appropriate volumes of a high [K+] solution with potassium gluconate.
2.4 ATP content
E18.5 limb skeletal muscles were assayed immediately after homogenization using buffer described in Hanson et al. (2015) to determine ATP content with a luciferin–luciferase based bioluminescence assay. The methodology of the ATP determination kit is provided in the experimental protocol by Molecular Probes (A-22066, Eugene, OR, USA). In order to determine ATP content, freshly extracted homogenate was added to a cuvette containing reaction buffer, D-luciferin, luciferase and DTT and placed in a Sirius luminometer v.2.2 (Berthold Detection Systems). Known concentrations of ATP standards were used to establish a standard curve.
2.5 Electrode recordings
Sharp electrode recordings were performed as described (Hanson et al., 2014; Plomp et al., 1992). E18.5 diaphragms were dissected and kept at room temperature in Normal Ringers solution. Diaphragm muscle fibers were impaled with a glass capillary microelectrode (30 MΩ resistance) made using a P-97 microelectrode puller (Sutter Instruments, Novata, CA) and filled with 3 M KCl (Brown et al., 2008). Evoked responses were elicited by a 0.2-ms maximal stimulus applied to the phrenic nerve using a suction electrode and pulse generator (STG 1002, ALA Scientific Instruments, Farmingdale, NY). Evoked and miniature endplate potentials (MEPP) were recorded using an Axoclamp-2A amplifier (Molecular Devices, Sunnyvale, CA) and DigiData 1322A (Molecular Devices). Muscle fiber membrane potentials were adjusted to Em = −70 mV under current clamp. Data was extracted and analyzed with Axoscope 10 software (Molecular Devices).
For EMG recordings and analysis of muscle contractions, explanted diaphragm muscle (Hanson and Niswander, 2014) was warmed to 30 °C and continually supplied with oxygenated Tyrode’s solution (3.0 mM KCl). EMG muscle recordings were made using fine-tip suction electrodes pulled from polyethylene tubing (PE-190; Clay Adams, NJ) and recorded via amplifiers (AI 401 amplifier connected with Digidata 1322a, Molecular Devices) directly on the computer with Axoscope 10 (Molecular Devices). Significance of data was evaluated by Student’s t-test, which determined the P value (Excel 2008). A P value below 0.05 was considered significant. E18.5 diaphragm preparations were treated with pharmacological reagents by addition of drugs to the bath at the indicated concentrations. For contractile studies, a stimulation electrode was placed onto the diaphragm. Evoked response was elicited by a 0.2-ms maximal stimulus compared to locomotor normal littermates within bath. A contractile movement was scored as a 0 if no contraction transpired after stimulation and 1 if contraction was recorded either through EMG recording or a visual confirmation. Drugs were bath applied for a minimum of 1 min before examination of contractility.
2.6 Electron microscopy
Diaphragm muscles of E18.5 embryos were fixed with 2.0% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) at 37 °C. The central region of muscle was dissected to focus on the predominant region of AChR clusters, followed by a secondary post-fixation in 2% osmium tetroxide and 1.5% potassium ferrocyanide in 0.1 M cacodylate (Sigma-Aldrich, St. Louis, MO) buffer for 1 h at room temperature. Samples were dehydrated through an ethanol series (50, 70, 90, 95 and 100% ETOH) and embedded in Epon/ Araldite resin polymerized overnight at 60 °C. Ultrathin sections were cut, placed on copper mesh grids and double contrasted with 2% aqueous uranyl acetate and Reynold’s lead citrate. Grids were imaged using a FEI Technai G2 BioTwin transmission electron microscope (FEI, Hillsboro, OR). Vesicle size and number were calculated using ImageJ.
2.7 In situ hybridization
Whole mount in situ hybridization was carried out as previously described (Lin et al., 2001). E15.5 embryos were dissected in oxygenated Tyrode’s Solution and pinned to a dish with a layer of Sylgard (184, Dow Corning) and the skin removed to expose the ribcage. A portion of the ribcage surrounding the diaphragm (last 5–8 caudal ribs) was removed from the embryo with small scissors and placed rostrally on another 100 mM Sylgard coated dish with the ribcage slightly stretched and pinned down using insect pins. Diaphragms were fixed in freshly prepared 4% PFA in 0.1 M phosphate buffer at 4 °C overnight and then dehydrated through a series of methanol solutions (25, 50, 75 and 100%). Hybridization was carried out at 70 °C overnight in hybridization buffer containing 50% formamide (Sigma), 1.3 × SSC, 50 μg/ml yeast tRNA, 0.2% Tween-20, and 1% Triton X-100 (Sigma), using digoxigenin-labeled Lrp4 or MuSK riboprobes. Diaphragms were washed with TBS containing 1% Tween-20, blocked with 5% goat serum in dilution buffer, and incubated with alkaline phosphatase conjugated anti-digoxigenin antibody (1:1000, Boehringer Mannheim GMBH, Indianapolis, IN) overnight at 4 °C, and hybridization signals were detected with a staining solution containing 100 mM Tris (pH 9.5), 0.4 mg/ml nitro blue tetrazolium chloride (NBT), 0.19 mg/ml 5-Bromo-4-chloro-3-indolyl phosphate (BCIP), 100 mM NaCl and 50 mM MgCl2 (Sigma).
2.8 Quantitative RT-PCR
RNA was extracted from spinal cord using Trizol® followed by DNaseI digestion and clean-up (Qiagen RNAeasy Minikit, Qiagen, Valencia, CA) and reverse transcribed using random hexamer primers and SuperScript III Reverse Transcriptase (Life Technologies) and amplified using TaqMan Universal PCR Master Mix (Life Technologies). Quantitative PCR was performed on a Roche LightCycler 480 Real-Time PCR System. Calculations were performed by a relative standard curve method. Probes for target genes were from TaqMan Assay-on-Demand kits (Life Technologies). Samples were adjusted for total RNA content by GAPDH in muscles of control embryos.
2.9 Immunohistochemistry
Diaphragms were dissected and pinned to sylgard dish in 4% paraformaldehyde for 10 min then permeabilized with 0.2% Triton/ PBS for 30 min. Non-specific reactivity was blocked with 2% BSA/ PBS for 2 h. When diaphragms were exposed to α-BTX conjugated Alexa 555 (1:500) alone, no permeabilization was performed to allow detection of AChRs at the membrane. Primary monoclonal antibody for all RyRs (34C, 1:20; Airey et al., 1990) was applied overnight at room temperature. Primary antibodies for neurofilament (1:100; Sigma) and synaptophysin (1:200; DSHB) were applied overnight at 4 °C. Secondary antibodies (Alexa Fluor 488-conjugated donkey anti-mouse, Alexa Fluor 568-conjugated goat anti-mouse IgG, 1:500, Life Technologies, #A11031) were applied for 1 h.
2.10 Confocal microscopy
Immunostained samples were examined by confocal laser scanning microscopy using a Zeiss LSM 510. For diaphragms, an area of 1 mm2 was selected from the field of view (×10). Alexa-Fluor 568 was excited at 543-nm by a HeNe laser line (1 mW maximum output, operated at 100%), directed via a 488/543 nm dual dichroic mirror. Emitted fluorescence was directed to a photomultiplier with a 560-nm long-pass filter. Confocal fluorescence intensity data were recorded as the average of four line scans per pixel and digitized at 8-bits, with photomultiplier gain adjusted such that maximum pixel intensities were <70% saturated.
2.11 Quantification
The ventral quadrant of the diaphragm shows a high degree of reproducibility in axon branching, AChR cluster number and distribution. Drawing a line between the most distal α-bungarotoxin positive AChR clusters and measuring the distance in ~10 μm intervals on each side of the central branch nerve in the ventral diaphragm quadrant quantified AChR cluster region. To determine the percent of the region occupied by AChR clusters, the distance to the distal edges of the diaphragm and the region of α-bungarotoxin labeled AChR clusters was measured in 100 μm increments. Percentage of AChR clusters was quantified using compressed z-stack images. Each condition was performed in at least 5 diaphragms for each condition. AChR cluster number was quantified on z-stack images of entire muscle by dividing the ventral quadrant into 0.1 mm bins from central branch in ventral quadrant outlined by purple boxes in Fig. 3A and B. The Zen 2009 software (Zeiss, Germany) was used to calculate the size of the AChR clusters.
Fig. 3.

Ryr1AG/AG muscle regains contractility with masked potassium leak. (A, B) Fluorescence imaging of PBFI at 340 nm in Ryr1+/+ (A) and Ryr1AG/AG (B) diaphragm in 3 mM Ringers solutions. Scale bar=50 μM. (C) Ratiometric potassium imaging obtained at 340 and 380 nm wavelengths provided a ratio of fluorescence in Ryr1+/+ (light gray), Ryr1AG/AG (dark gray) and Ryr1−/− (black) diaphragm muscle (normalized to Ryr1+/+). (D) ATP determination of E18.5 muscles. (E) Representation of the ratiometric imaging experimental paradigms for Ryr1+/+ (light gray), Ryr1AG/AG (dark gray) and Ryr1−/− (black) diaphragm muscle with bath applications of 3 mM KCl, 7 mM KCl, 0 mM KCl and 3 mM KCl with 2 μM glibenclamide in Ringers solutions. (F) Change of intracellular K+ fluorescence intensities in experimental conditions. (G) EMG recordings of diaphragm muscle from Ryr1+/+, Ryr1AG/AG, or Ryr1AG/AG treated with 6 mM KCl. (H) Analysis of contractility of E18.5 diaphragm muscle from Ryr1+/ +, Ryr1AG/AG, or RyR1−/− embryos. After equilibration, all samples were bathed first in 6 mM KCl, followed by wash out and then addition of one of the following conditions as indicated (3 and 6 mM KCl, 2 μM glibenclamide and 100 nM Bay K8644; n for experiments above bars).
2.12 Statistical analysis
The unpaired two-tailed Student’s t-test was used to examine statistical differences. Data in the manuscript are represented as mean ± SEM unless otherwise indicated. p<0.05 was considered significant.
2.13 Study approval
All experiments were conducted in accordance with the protocols described in the Guide for the Care and Use of Laboratory Animals (NIH, Revised 2011).
3. Results
3.1 RyR1AG protein is expressed but the mutation causes a non-motile embryonic phenotype
Following N-ethyl-N-nitrosourea (ENU) chemical mutagenesis and a multi-generation cross to identify recessive mutations, the Ryr1AG line was identified by muscle weakness (Hanson et al., 2015). The ENU-induced point mutation creates an E4242G change in exon 93, outside of the channel region of RyR1. To study the phenotype in mice homozygous for the Ryr1AG allele, Ryr1AG/+ mice were crossed. At embryonic day 18.5 (E18.5), Ryr1AG/AG homozygous mutant embryos displayed kyphosis (hunchback), carpoptosis (wrist drop), were non-motile during the locomotion screen (see methods) and died at birth (Fig. 1A). Gene identification was confirmed by a lack of complementation in trans-heterozygous embryos for our allele and RyR1 null allele and the Ryr1AG/AG phenotype was similar to RyR1−/− embryos (Fig. 1A). Next, Ryr1 RNA levels in the Ryr1AG/AG mouse embryonic spinal cord were determined at E14.5 and E18.5 and showed no significant difference compared to wildtype age matched embryos (n=4; Fig. S1). RyR1 is the predominant family member in muscle and using a pan RyR antibody we observed RyR immunoreactivity in puncta-like patterns in the tetrads of muscle fibers in both Ryr1AG/+ and Ryr1AG/AG muscle (Fig. 1B and C). In myotube cultures, locomotor normal and Ryr1AG/AG myotubes have detectable RyR protein expression (Fig. 1D and F), whereas no protein is detected in Ryr1 null myotubes (Fig. 1E and G is antibody control). These data indicate that the Ryr1AG allele has no apparent effect on Ryr mRNA or protein expression in the spinal cord or muscle.
Fig. 1.

ENU-induced mutation in Ryr1. (A) Complementation cross of Ryr1AG/+ with Ryr1−/ + results in trans-heterozygous embryos with the same gross phenotype as Ryr1AG/AG mutants and Ryr1−/− nulls. (B, C) Immunohistochemistry of muscle fibers labeled with pan-RyR antibody. (D–G) Punctate immunostaining was observed in normal (D) and Ryr1AG/AG (E) myotubes, but not in Ryr1−/− myotubes (F). Staining was absent when only the secondary antibody was present.
3.2 Defects in calcium dynamics in Ryr1AG/AG mice
RyR1 functions as a muscle isotype of ryanodine receptors that is mechanically activated by the dihydropyridine receptor to allow Ca2+ release from the sarcoplasmic reticulum (SR), thus evoking E–C coupling (Fill and Copello, 2002; Takeshima et al., 1994). To determine whether Ryr1AG mutant protein affects calcium release, myotube cultures were used, as RyR1 function has been well studied in this system (Bannister and Beam, 2009; Fill and Copello, 2002). Whole-cell clamping in combination with fluorescent Ca2+ indicator (Bannister et al., 2008) showed that, unlike normal myotubes, Ryr1AG/AG myotubes failed to release Ca2+ from the SR in response to change in membrane potential (Fig. 2A–C Ryr1+/ + light gray circles and Ryr1AG/AG dark gray circles). Moreover, Ryr1AG/AG myotubes failed to contract in response to electrical stimulation but electrically-evoked contractions could be rescued by expression of YFP-tagged wildtype RyR1 in Ryr1AG/AG myotubes (Fig. 2D), further evidence that the RyR1 mutation is causative of the phenotype. Addition of the RyR1 agonist, 4-chloro-m-cresol (4-CmC; 0.5 mM), produced substantial Ca2+ release in both normal and Ryr1AG/AG myotubes (peak ΔF/F of 1.4±0.1; n=5 vs. 1.2±0.1; n=3, respectively; p>0.05; Fig. 2E and F), compared to Ryr1−/− myotubes (ΔF/F 0.1±0.0; n=6; Fig. 2F). These results indicate that RyR1AG/AG protein is present in myotubes and can release SR Ca2+ stores with the help of an agonist to RyR. However, the rate of SR Ca2+ release in response to 4-CmC was much slower in Ryr1AG/AG myotubes than in normal myotubes (t1/2act =6.5±2.4 vs. 2.2±0.6 s, respectively; p<0.01; Fig. 2E). This suggests a functional disruption of the RyR1AG/AG channel is responsible for the lack of evoked release of calcium from internal stores.
Fig. 2.

Ryr1AG/AG show defects in calcium dynamics. (A, B) Representative recordings of myoplasmic Ca2+ transients elicited by 50-ms depolarizations from −50 mV to the indicated test potentials for locomotor normal myotubes (A; Ryr1+/ +) and Ryr1AG/AG myotubes (B). (C) Comparison of ΔF−V relationships for normal myotubes (light gray circles; n=4), and Ryr1AG/AG myotubes (dark gray circles; n=7). (D) Fraction of myotubes that contract in response to a 100 V, 10 ms electrical stimulus for normal myotubes (light gray bar), Ryr1AG/AG myotubes (dark gray bar, 0%), and Ryr1AG/AG myotubes transfected with RyR1-YFP (white bar). (E) Representative changes in Fluo-3 AM fluorescence in response to application of 4-chloro-m-cresol (4-CmC; 0.5 mM) elicited in a normal myotube (left) and Ryr1AG/AG myotube (right). Arrows indicate time of 4-CmC applications. (F) Half-rise times of Ca2+ transients in response to 4-CmC for normal (light gray bar), and Ryr1AG/AG (dark gray bar) myotubes compared to Ryr1−/− (black bar). Error bars represent ± SEM. Asterisks indicate a significant difference (p=0.006; t-test). For each group in (D) and (F), the total number of myotubes tested is indicated above each bar.
3.3 Inhibition of potassium leak in Ryr1AG/AG muscles
Our recent studies of adult Ryr1AG/+ mice demonstrated a myopathy with core-like structures and mechanistically this was related to a potassium leak (Hanson et al., 2015). The K+ leak could be masked either by raising external K+ or by inhibiting KATP channel outward current, which rescued the myopathy in Ryr1AG/+ muscles. To determine whether a similar K+ leak underlies the non-motile phenotype in Ryr1AG/AG embryos, we first assessed K+ homeostasis in Ryr1AG/AG diaphragms using the potassium indicator PBFI–AM (see Section 2) in combination with ratiometric potassium imaging of muscle fibers. Experiments using wildtype diaphragms in normal Ringers solution (3 mM K+) showed that intracellular K+ concentration was 144±7 mM (n=33 fibers from 4 diaphragms; Fig. 3A and C). Ryr1−/− null diaphragms exhibited intracellular concentration similar to wild-type diaphragms (139±11 mM; n=30 fibers from 4 diaphragms, Fig. 3C). However, Ryr1AG/AG diaphragms showed lower intracellular K+ concentrations of 85±15 mM (n=41 fibers from 5 diaphragms; p<0.001; Fig. 3B and C), suggestive of a potassium leak.
In adult Ryr1AG/+ muscles, the mutant RyR1 inhibits Ca2+ release, which results in decreased ATP content and dysregulation of genes and proteins involved in potassium homeostasis (Hanson et al., 2015). ATP content in Ryr1AG/AG diaphragm muscle was significantly decreased compared to wildtype (21±4 nmol/mg Ryr1AG/AG muscles versus 35±6 in wildtype; n=4 per mouse line; Fig. 3D). Ryr1−/− diaphragm muscles had ATP content (31±4 nmol/mg Ryr1−/− muscle; n=4) similar to wildtype (Fig. 3D), suggesting that Ca2+ internal release persist in Ryr1−/− muscle. This data suggests that RyR1AG/AG protein affects ATP levels which may suggest that potassium homeostasis is also affected in these diaphragms. As in Ryr1AG/+ soleus muscles (Hanson et al., 2015), Ryr1AG/AG diaphragm fibers showed a greater change in K+ fluorescent ratio intensity over the course of testing suggesting more fluctuations in K+ ion permeability compared to wildtype diaphragms (Fig. 3E and F; n=41 fibers in 5 Ryr1AG/AG diaphragms and n=33 fibers in 4 wildtype diaphragms and n=30 fibers in Ryr1−/− diaphragms). The K+ leak could be amplified in all muscles by depleting the muscle of extra-cellular K+, however the change in PBFI intensity was significantly greater in Ryr1AG/AG diaphragms compared to wildtype and Ryr1−/− diaphragms (Fig. 3E and F). The addition of 6 mM KCl or glibenclamide (2 μM) inhibited K+ loss and significantly increased intracellular K+ intensity of the muscle fibers of Ryr1AG/AG diaphragms compared to wildtype and Ryr1−/− diaphragms (Fig. 3E and F). These data suggest that Ryr1AG/AG diaphragms have decreased intracellular K+, decreased ATP content and greater fluctuations in internalized K+.
3.4 Resumption of contractility in Ryr1AG/AG muscles
The inhibition of the K+ leak by 6 mM KCl might increase intracellular K+ and this may be sufficient to initiate a contraction. Using electromyography recordings (EMG) we examined muscle contractions in whole diaphragm muscle in vitro upon direct muscle stimulation. In 3 mM KCl Ringers solution, stimulation of diaphragms initiated whole muscle contractions in wildtype diaphragms but not in Ryr1AG/AG or Ryr1−/− diaphragms (Fig. 3G and H). Increasing KCl from 3 mM to 6 mM together with electrical stimulation of the diaphragm induced bursts of electrical activity in the EMG recordings and whole muscle contractions in wildtype and Ryr1AG/AG muscle but not Ryr1 null muscle (Fig. 3G and H). The potassium leak in adult Ryr1AG/+ muscle can be masked through inhibiting KATP channels with glibenclamide (2 μM; Hanson et al., 2015). The addition of glibenclamide to the bath induced contractions in Ryr1AG/AG but not Ryr1−/− muscle (Fig. 3H). These data provide in vitro evidence that homozygous RyR1AG channels have the ability to be functional active, but the channel might be forced into an inactive state due to a potassium leak.
If the RyR1AG channel is in an inactive state due to defects in membrane excitability, the pharmacological activation of the voltage-dependent DHPR might force RyR1AG into an active state, allowing release of stored Ca2+ to instigate a contraction. To examine the ability of the DHPR channel to engage excitation–contraction coupling, Bay K8644 (100 nM), an agonist to the channel portion of DHPR, was applied to Ryr1AG/AG diaphragm muscle. Indeed, Bay K8644 addition could induce muscle contractions, whereas no contractions occurred in Ryr1 null diaphragm muscle (Fig. 3H). This indicates that orthograde DHPR-RyR1 signaling can be elicited in Ryr1AG/AG muscle.
3.5 RyR1 signaling participates in axon extension and postsynaptic AChR cluster distribution
Two mouse models that disrupt E–C coupling due to lack of evoked calcium release from internal stores-targeted deletion of β1 subunit of DHPR (Cacnb1null) (Chen et al., 2011) and deletion of Stac3 (Nelson et al., 2013), which disrupts the voltage activation of DHPR and affects RyR1 calcium release, show increased axon branching and extension and broad disorganization of AChR clusters throughout the muscle. In contrast, loss of RyR1 function causes regionalization of AChR clusters around large nerve bundles (Hanson and Niswander, 2014). To determine the effect of the Ryr1AG/AG allele, we examined AChR cluster distribution and axon extension in E18.5 Ryr1AG/AG muscle. In normal E18.5 diaphragm, postsynaptic AChR clusters are centralized and distributed under the branched axons (Fig. 4A and inset shown in Fig. 4C; labeled with α-bungarotoxin [αBTX, red], neurofilament and synaptophysin [green]). In Ryr1AG/AG mutant diaphragm, motor axons were present and there were similar numbers of AChR clusters in Ryr1AG/AG compared to RyR1AG/+ (Fig. 4A, B and E). However, Ryr1AG/AG diaphragms exhibited a narrowed region of AChR cluster distribution showing an ~50% reduction in width in the region of the AChR clusters compared to normal littermates (Fig. 4C, D and F). Furthermore, the axons extended past these AChR clusters resulting in longer axons in Ryr1AG/AG compared to Ryr1AG/+ diaphragms (Fig. 4D and G). In other muscles of Ryr1AG/AG embryos, axons extended past narrowed AChR clusters (Fig. S2 shows gluteus maximus and vastus lateralis muscles). In Ryr1 null diaphragms, axons also extended past the AChR clusters and the AChR cluster region was narrowed (Fig. 4E–G). Similar to RyR1−/− E15.5 diaphragms that lack phrenic nerves (Hanson and Niswander, 2014), MuSK and Lrp4, proteins important in postsynaptic AChR formation, axon outgrowth and synapse stability, show narrowed RNA expression patterns at E14.5 and E18.5 in Ryr1AG/AG diaphragms compared to normal (Fig. 4H–O). This is in contrast to Cacnb1null embryos in which Musk is over-expressed throughout the diaphragm and AChR clusters are distributed throughout the diaphragm (Chen et al., 2011). This data suggests significant functional differences resulting from mutations to either the DHPR or RyR1.
Fig. 4.

Narrowed AChR cluster distribution and increased motor axon outgrowth in Ryr1AG/AGdiaphragms. (A, B) E18.5 whole diaphragms show clustered AChRs that form neuromuscular junctions visualized with α–bungarotoxin (red) and axons with synaptophysin and neurofilament (green; scale bar in A, B=1 mm). (C–E) Left ventral quadrants of diaphragms (scale bar=200 μm). (C) In Ryr1AG/+ diaphragms, AChR clusters extend along the axon branches. (D, E) In Ryr1AG/+ and Ryr1−/− diaphragms, AChR clusters surround the fasciculated nerve but do not extend along the axon branch. (F–G) Quantification in the ventral quadrant (outlined by purple boxes in A, B) of AChR cluster regionalization (F) and axon length (G) (n=5 for each). H–O, At 14.5 (H–K, scale bar=100 μm) and at E18.5 (L–O, scale bar=200 μm), MuSK and Lrp4 (yellow dashes) are expressed in the central region of Ryr1AG/+ and Ryr1AG/AG diaphragms.
3.6 RyR1 restricts size of AChR clusters
Recently, our examination of E18.5 Ryr1−/− diaphragms provided evidence that the majority of AChR clusters do not distribute with the overextended axons (Hanson and Niswander, 2014). At E18.5, individual synapses appeared to be larger in Ryr1AG/AG diaphragm muscle than in the age-matched control embryos (Fig. 5A, B and D), while Ryr1−/− AChR clusters varied in size and shape with nonfluorescent regions within the middle and to the sides of the disk (Fig. 5C and D). Measurements of individual AChR clusters showed an ~50% increase in size of AChR clusters in Ryr1AG/AG muscles compared with normal controls (Fig. 5D). This suggests that functional RyR1 channels in muscle or nerve may regulate AChR cluster size and distribution during NMJ development.
Fig. 5.

RYR1 controls AChR cluster size. (A–C) AChR clusters of Ryr1AG/+, Ryr1AG/AG, and Ryr1−/− endplates (scale bar=20 μm). (D) Quantification in the ventral quadrant of the diaphragm of AChR cluster size.
3.7 Enhanced spontaneous transmitter release on Ryr1AG/AG muscle
In order to examine the presynaptic side of the NMJ, an electron microscopy analyzes was performed on E18.5 mouse diaphragm muscle/phrenic nerve region. In Ryr1AG/AG diaphragms (n=15), the presynaptic nerve terminals contained increased numbers of vesicles compared to Ryr1AG/+ AChR clusters (265±38 in Ryr1AG/AG compared to 165±25 in Ryr1AG/+ per terminal; n=20 sections used per genetic background) with decreased size (32 nm2±11 in Ryr1AG/AG (n=621) compared to 52±18 in Ryr1AG/+ (n=605)), while the basal lamina was well defined (Fig. 6A–D). In order to determine whether the NMJ in the Ryr1AG/AG embryos were functional, electrophysiological analysis was performed on E18.5 diaphragm muscle (Fig. 6E–I). Spontaneous neuromuscular synaptic activity, as measured by miniature endplate potential (mEPP) frequency in the absence of stimulation (events per minute), was increased more than 80-fold in Ryr1AG/AG muscles (69.11±16.79, n=21 cells, n=6 embryos, 8915 total events in 129 min) compared with controls (0.79±0.21, n=25 cells, n=5 embryos, 113 total events in 143 min; p<0.005; Fig. 6. E, F and H). In RyR1−/− diaphragm muscle mEPP were modestly increased compared to controls (5.92±2.21, n=18 cells, n=5 embryos, 959 total events in 162 min; Fig. 6G and H). Examination of the magnitude of mEPP show variability in the amplitudes in Ryr1AG/AG muscles, but no significant increase in size (2.38±0.81 mV, n=21 cells and 6 embryos) compared with controls (1.97±0.22 mV, n=25 cells and 5 embryos; p<0.05; Fig. 5I) or Ryr1−/− muscle (2.08±0.62 mV, n=19 cells and 4 embryos; p<0.05).
Fig. 6.
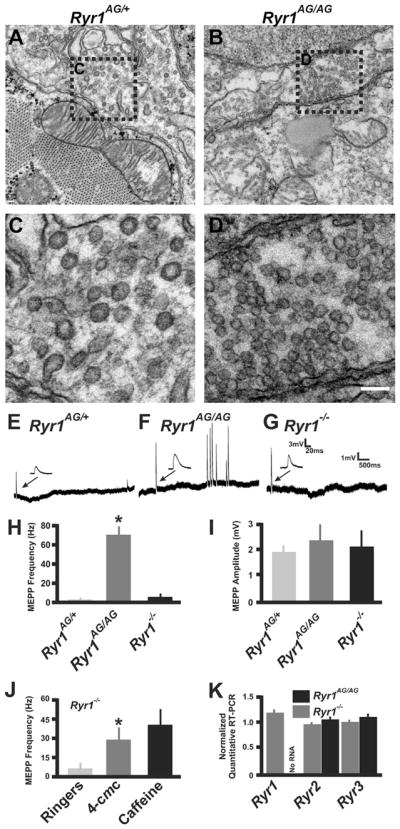
Ryr1AG/AG embryos show increased synaptic vesicles and miniature endplate potentials. (A–D) Transmission EM of E18.5 phrenic nerve terminals and synaptic vesicles in Ryr1AG/AG (A, C) compared to Ryr1AG/+ (B, D). A, B Scale bar=500 nm. (C and D) magnified view of boxed regions in A, B. (E–G) Recordings of spontaneous events demonstrate increased frequency of mEPPs in Ryr1AG/AG (F) compared to Ryr1AG/+ (E) and Ryr1−/− (G). (H) Quantification of mEPP frequency. (I) Quantification of mEPP amplitude. (J) Examination of mEPPs per minute after application of either 500 μM 4-CmC or 1 μM caffeine in Ryr1−/− diaphragm. (K) Quantitative RT-PCR of Ryr1, Ryr2 and Ryr3 from the ventral spinal cord of Ryr1+/+, Ryr1AG/AG, Ryr1−/− embryos (n=4 each) and the data normalized to the level of expression in wild type.
CACNB1/DHPR (Chen et al., 2011) and STAC3 (Nelson et al., 2013) increase the incidence of mEPPs in age-matched diaphragm muscle, suggesting that signaling in muscle affects the frequency of spontaneous ACh release onto the post-synaptic endplate. To determine whether ryanodine receptors in the nerve regulate mEPP frequency, mEPPs were examined in E18.5 Ryr1−/− diaphragms. The application of 4-CmC (0.5 mM; to activate endogenous presynaptic RyR2 without activating pre- and post-synaptic RyR3 (Choisy et al., 1999; Fessenden et al., 2000)) to the bath increased the incidence of mEPPs in Ryr1−/− muscle (29.87±10.29, n=20 cells, n=5 embryos, 3734 total events in 125 min; Fig. 6J). Bath application of caffeine (10 mM; agonist for all three RyRs) showed an even greater incidence of mEPPS in Ryr1−/− muscle, suggesting that both RyR2 and RyR3 mediated Ca2+ release can affect mEPP frequency (43.76±12.77, n=19 cells, n=5 embryos, 5252 total events in 120 min; Fig. 6J). Moreover, these data suggest that RyR2 channels are in the pre-synaptic apparatus and involved in NMJ transmission. To determine whether genetic alteration of RyR1 function can change the amount of RNA produced from the three Ryr family member genes, we performed quantitative RT PCR on the ventral spinal cords of Ryr1−/−, Ryr1AG/AG and Ryr1+/+ E18.5 embryos. Under these conditions, no significant differences were found in the level of Ryr2 or Ryr3 RNA (Fig. 6K), suggesting the RyR2 effect on mEPP frequency is not due to an alteration in Ryr2 transcript in the spinal cord.
3.8 Lack of evoked endplate potentials in Ryr1AG/AG diaphragms
To further examine the motor axon electrophysiology of the NMJ, evoked endplate potentials (eEPPs) were induced through phrenic nerve stimulation. Amplitudes of eEPPs were similar between control (33.25±5.25 mV, n=23 cells and 4 embryos; Fig. 7A) and Ryr1−/− muscles (26.75±9.22 mV, n=27 cells and 4 embryos; Fig. 7C) in response to electrical stimulation of the nerve. Notably, in Ryr1AG/AG diaphragm muscle, stimulation did not induce eEPPs (n=35 cells and 5 embryos; Fig. 7B compared to locomotor normal littermate in Fig. 7A). The lack of evoked release in Ryr1AG/AG could be due to a presynaptic defect and, if so, this would suggest a role for presynaptic RyR1 at the neuromuscular junction. The non-functioning neuronal RyR1 might compromise the production or availability of components required for vesicular release. To test this, 10 mM Ca2+ was bath applied and this was capable of restoring evoked potentials in Ryr1AG/AG diaphragm muscles (Fig. 8A; compared to Fig. 7B without additional Ca2+). The initial application of 10 mM Ca2+ to the bath induced asynchronous release of transmitter which suppressed the high number of mEPPs produced in control Ca2+ solutions. This data suggests that the components for evoked release are present and able to function. This data suggest that the Ryr1AG/AG mutation might affect the availability of necessary Ca2+ to support evoked ACh release onto the muscle. To increase cytosolic Ca2+ levels in Ryr1AG/AG preparation, treatment with 4-CmC (500 μM) and phrenic nerve stimulation produced eEPPs in the muscle and increased the number of mEPPs (Fig. 8D compared to 7B, arrowheads highlight mEPPs in Fig. 8). 4-CmC induced evoked response in Ryr1AG/AG muscle immediately after application suggesting that presynaptic RyR (RyR1 and/or RyR2) are involved in evoked release of ACh. Moreover, depleting internally stored Ca2+ with bath application of thapsigargin (1 μM) temporarily restored eEPPs in Ryr1AG/AG diaphragms (Fig. 8F), suggesting the evoked release involvement of ER-like membranes on the presynaptic side of the NMJ. These data suggests that functional neuronal ryanodine receptor channels can be involved in the action potential-mediated release and spontaneous release of synaptic vesicles at the NMJ.
Fig. 7.

Lack of evoked endplate potentials in RyR1AG/AG muscle. Sharp electrode recordings of responses from diaphragm muscle following a 0.2 ms suprathreshold stimulus to the phrenic nerve in normal Ringer’s solution. (A–C) Representative recording of evoked response in Ryr1AG/+ (A) and Ryr1−/−(C) compared to complete lack of evoked response in Ryr1AG/AG (B). (D) Quantification of evoked end-plate potentials from Ryr1AG/+, Ryr1AG/AG and Ryr1−/− diaphragms.
Fig. 8.
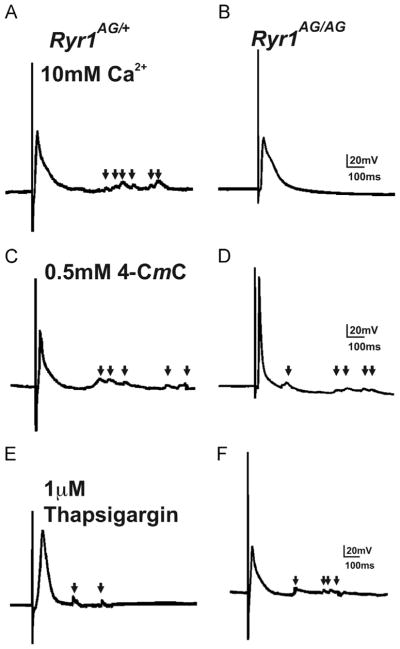
Rescue of evoked EPP in Ryr1AG/AG muscle. Resumption of evoked EPPs in Ryr1AG/AG by application of 10 mM Ca2+ (B), 500 μM 4-CmC (D), or 1 μM thapsigargin (F) compared to Ryr1AG/+ (A, C and E). Arrows represent mEPPs.
4. Discussion
Here we present homozygous embryonic data from a myopathic RyR1AG mouse model that demonstrates structural and functional defects of the muscle and nerve. Of the structural defects, we first substantiate that axons extend beyond the AChR cluster region and AChR clusters become more centralized in Ryr1AG and Ryr1−/− muscle. Second, we show that AChR clusters in dysfunctional Ryr1AG muscle are consistently larger than AChR clusters in wildtype and Ryr1−/− null muscle. Of the functional defects, we first describe that the homozygous RyR1AG protein is present but dysfunctional in the muscle. Second, we provide evidence of a K+ leak in Ryr1AG/AG diaphragms, and that masking of the K+ leak induces muscle contractions in otherwise paralyzed muscle. Third, we demonstrate that unlike wildtype and Ryr1−/− NMJs, NMJs in Ryr1AG/AG lack release of Ca2+ in the presynaptic terminal that results in release of transmitter. However, supplementation by external Ca2+ can re-initiate evoked release. Lastly, we show that ryanodine receptors are involved in vesicular release of ACh at the NMJ. These data suggests that RyR1 contributes to structural and functional events at the neuromuscular interface beyond the well-studied role in SR Ca2+ release to induce muscle contractility.
The inhibition of RyR1-dependent SR Ca2+ release may underscore the majority of structural changes in Ryr1AG/AG muscle. In Cacna1smdg/mdg, Cacnb1 null and Ryr1 null diaphragms, similarities exist in extensive axon overgrowth and AChR cluster size (Fig. 4D compared with work by Powell and work by Chen et al. (2011), Powell et al. (1984)). Our data on Ryr1AG/AG muscle and on muscle lacking Ryr1 allows us to suggest that restriction of AChR cluster size might be dependent on RyR1 downstream signaling. However, dissimilarity exists in AChR cluster distribution between DHPR mouse models and RyR1 mouse models, AChR cluster distribution is expanded in diaphragms that lack DHPR subunits while restricted in models with abnormal RyR1 function (Fig. 4A; Chen et al., 2011; Hanson and Niswander, 2014; Powell et al., 1984). Therefore, our data and work by Chen et al. (2011) suggest an additional role for DHPR and RyR1 function beyond muscle contractility and SR Ca2+ release, and might play roles in axon extension, AChR distribution and AChR cluster size.
In Ryr1AG/AG muscles, we show that axons extend beyond the centralized AChR clusters and beyond Musk and Lrp4 expression regions. In addition, Musk and Lrp4 expression regions are narrowed and similar in extent as the centralized AChR clusters. Motor axons require proper MuSK signaling for correct placement (Kim and Burden, 2008). Moreover, Lrp4 is the receptor for agrin and component of MuSK signaling and is required for AChR cluster formation (Kim et al., 2008; Lin et al., 2001; Weatherbee et al., 2006; Yang et al., 2001). Recently, RyR1 signaling was shown to be involved in proper prepatterning of AChR clusters and axon extension (Hanson and Niswander, 2014). Therefore, our data suggest that muscle RyR1 may help the MuSK and Lrp4 signaling complex to halt axon extension. Since Cacnb1 null (DHPR β1 null) mice show an increase in Musk expression (Chen et al., 2011), which contrasts with our data in Ryr1AG/AG muscles, we can conclude that muscle RyR1 may have a distinct function in axon extension separate from the role of the DHPR–RyR1 complex in SR Ca2+ release.
Ryanodine receptors may also have a distinct role in release of Ca2+ in the presynaptic terminal that results in release of transmitter at the presynaptic NMJ. Synaptic vesicles are increased at Ryr1AG/AG presynaptic nerve terminals and our pharmacological studies suggest that functional RyR1 and RyR2 are involved in release of Ca2+ that results in release of ACh at the NMJ and thus may also be involved in the Ca2+ dependent release of other factors required during NMJ development. Interactions between RyR1 and Snapin, a protein involved in peptide secretion, enhances RyR1 channel activation (Zissimopoulos et al., 2006) and stimulates peptide secretion in non-neuronal cells (Kinoshita et al., 2013). RyR channels and the vesicular release protein, SNAP25 share an overlapping binding site on Snapin, suggesting a direct link between RyR and vesicular release (Zissimopoulos et al., 2006). In Drosophila, dense core vesicles (DCV) are activated through CaMKII and can be modulated by ryanodine receptors (Levitan, 2008; Shakiryanova et al., 2007). In vertebrates, golgi-like apparatus release DCVs that contain agrin (Magill-Solc and McMahan, 1988). Therefore, DCV might utilize RyR dependent stores of Ca2+ to evoke release of agrin onto the muscle. In addition, RyR might be involved in ACh and agrin release during the presynaptic instigation of the evoked endplate potentials, two molecules that are involved in neuronal synapse elimination and stabilization (Lin et al., 2001; Misgeld et al., 2005; Reist et al., 1992; Rimer, 2010; Wang et al., 2014; Yang et al., 2001).
Even though synapses are formed in Ryr1AG/AG diaphragms, action potential evoked SR Ca2+ release and muscle contractility are abolished. Muscle contractility can be rescued by masking the potassium leak from the muscle. A possible explanation for the rescue might be that signaling between the voltage sensitive DHPR channels and RyR1 channels is disrupted. DHPR channels and RyR1 channels interact through orthograde and retrograde signaling (Andronache et al., 2009; Bannister and Beam, 2009). Orthograde signaling from DHPR opens the RyR1 channel in the SR, while retrograde signaling from RyR1 controls the gating of DHPR (Bannister and Beam, 2009). The DHPR agonist Bay K8644 was able to induce contractions in Ryr1AG/AG diaphragms suggesting that orthograde signaling is unaffected. In addition, 4-CmC, the agonist of RyR1, can induce calcium release in homozygous Ryr1AG/AG myotubes. Therefore, Ryr1AG/AG channels have the ability to become activated but cannot activate under normal physiological conditions. In the future, experiments can be performed to examine the retrograde communication from Ryr1AG/AG to DHPR in a manner similar to Bannister and Beam (2009). Ryanodine receptors are expressed in excitable cells of vertebrates (Giannini et al., 1995). Therefore, ryanodine receptors may have functions beyond the release of internal Ca2+ stores and might contribute to the formation and maintenance of synaptic structures throughout the nervous system. Further study of RyR1 models might help to establish appropriate therapies for patients with myopathies.
Supplementary Material
Acknowledgments
The authors would like to thank Roger Bannister and Kurt Beam for generating the myotube data in Figs. 1 and 2; Danielle Marck, Bleu Knight, and Lori Bulwith for technical assistance; Dorothy Dill for TEM assistance; Rosa Moreno for critical comments on the manuscript; Steve Burden and Scott Weatherbee for probes to MuSK and Lrp4 respectively. Imaging experiments were performed in the University of Colorado Anschutz Medical Campus Advance Light Microscopy Core supported in part by Rocky Mountain Neurological Disorders Core Grant number P30NS048154 and by NIH/NCRR Colorado CTSI Grant number UL1 RR025780. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
This work was supported by Department of Pediatrics and Neuroscience Program NS 48154. MGH was supported by postdoctoral fellowships from Muscular Dystrophy Association MDA69338 and NIH F32 NS059267.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.ydbio.2015.05.018
Footnotes
Disclosures
The authors have declared that no conflict of interest exists.
References
- Airey JA, Beck CF, Murakami K, Tanksley SJ, Deerinck TJ, Ellisman MH, Sutko JL. Identification and localization of two triad junctional foot protein isoforms in mature avian fast twitch skeletal muscle. J Biol Chem. 1990;265:14187–14194. [PubMed] [Google Scholar]
- Andronache Z, Hamilton SL, Dirksen RT, Melzer W. A retrograde signal from RyR1 alters DHP receptor inactivation and limits window Ca2+ release in muscle fibers of Y522S RyR1 knock-in mice. Proc Natl Acad Sci USA. 2009;106:4531–4536. doi: 10.1073/pnas.0812661106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister RA, Beam KG. Ryanodine modification of RyR1 retrogradely affects L-type Ca(2+) channel gating in skeletal muscle. J Muscle Res Cell Motil. 2009;30:217–223. doi: 10.1007/s10974-009-9190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister RA, Colecraft HM, Beam KG. Rem inhibits skeletal muscle EC coupling by reducing the number of functional L-type Ca2+ channels. Biophys J. 2008;94:2631–2638. doi: 10.1529/biophysj.107.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Johnson BE, Goodman MB. Making patch-pipettes and sharp electrodes with a programmable puller. J Vis Exp. 2008 Oct 8;(20) doi: 10.3791/939. pii: 939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck ED, Nguyen HT, Pessah IN, Allen PD. Dyspedic mouse skeletal muscle expresses major elements of the triadic junction but lacks detectable ryanodine receptor protein and function. J Biol Chem. 1997;272:7360–7367. doi: 10.1074/jbc.272.11.7360. [DOI] [PubMed] [Google Scholar]
- Busetto G, Buffelli M, Cangiano L, Cangiano A. Effects of evoked and spontaneous motoneuronal firing on synapse competition and elimination in skeletal muscle. J Neurocytol. 2003;32:795–802. doi: 10.1023/B:NEUR.0000020624.48032.ed. [DOI] [PubMed] [Google Scholar]
- Chen F, Liu Y, Sugiura Y, Allen PD, Gregg RG, Lin W. Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat Neurosci. 2011;14:570–577. doi: 10.1038/nn.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choisy S, Huchet-Cadiou C, Leoty C. Sarcoplasmic reticulum Ca(2+) release by 4-chloro-m-cresol (4-CmC) in intact and chemically skinned ferret cardiac ventricular fibers. J Pharmacol Exp Ther. 1999;290:578–586. [PubMed] [Google Scholar]
- Coussin F, Macrez N, Morel JL, Mironneau J. Requirement of ryanodine receptor subtypes 1 and 2 for Ca(2+)-induced Ca(2+) release in vascular myocytes. J Biol Chem. 2000;275:9596–9603. doi: 10.1074/jbc.275.13.9596. [DOI] [PubMed] [Google Scholar]
- Dayanithi G, Mechaly I, Viero C, Aptel H, Alphandery S, Puech S, Bancel F, Valmier J. Intracellular Ca2+ regulation in rat motoneurons during development. Cell Calcium. 2006;39:237–246. doi: 10.1016/j.ceca.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Fessenden JD, Wang Y, Moore RA, Chen SR, Allen PD, Pessah IN. Divergent functional properties of ryanodine receptor types 1 and 3 expressed in a myogenic cell line. Biophys J. 2000;79:2509–2525. doi: 10.1016/S0006-3495(00)76492-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Jorgensen AO. Structure and development of E–C coupling units in skeletal muscle. Annu Rev Physiol. 1994;56:509–534. doi: 10.1146/annurev.ph.56.030194.002453. [DOI] [PubMed] [Google Scholar]
- Futatsugi A, Kato K, Ogura H, Li ST, Nagata E, Kuwajima G, Tanaka K, Itohara S, Mikoshiba K. Facilitation of NMDAR-independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron. 1999;24:701–713. doi: 10.1016/s0896-6273(00)81123-x. [DOI] [PubMed] [Google Scholar]
- Giannini G, Conti A, Mammarella S, Scrobogna M, Sorrentino V. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J Cell Biol. 1995;128:893–904. doi: 10.1083/jcb.128.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Fregoso VL, Vrana JD, Tucker CL, Niswander LA. Peripheral nervous system defects in a mouse model for peroxisomal biogenesis disorders. Dev Biol. 2014;395:84–95. doi: 10.1016/j.ydbio.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Normal patterns of spontaneous activity are required for correct motor axon guidance and the expression of specific guidance molecules. Neuron. 2004;43:687–701. doi: 10.1016/j.neuron.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Increasing the frequency of spontaneous rhythmic activity disrupts pool-specific axon fasciculation and pathfinding of embryonic spinal motoneurons. J Neurosci. 2006;26:12769–12780. doi: 10.1523/JNEUROSCI.4170-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Niswander LA. An explant muscle model to examine the refinement of the synaptic landscape. J Neurosci Methods. 2014;238:95–104. doi: 10.1016/j.jneumeth.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Wilde JJ, Moreno RL, Minic AD, Niswander L. Potassium dependent rescue of a myopathy with core-like structures in mouse. Elife. 2015:4. doi: 10.7554/eLife.02923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasarskis A, Manova K, Anderson KV. A phenotype-based screen for embryonic lethal mutations in the mouse. Proc Natl Acad Sci USA. 1998;95:7485–7490. doi: 10.1073/pnas.95.13.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Burden SJ. MuSK controls where motor axons grow and form synapses. Nat Neurosci. 2008;11:19–27. doi: 10.1038/nn2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita SM, Kogure A, Taguchi S, Nolan GP. Snapin, positive regulator of stimulation- induced Ca(2)(+) release through RyR, is necessary for HIV-1 replication in T cells. PLoS One. 2013;8:e75297. doi: 10.1371/journal.pone.0075297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krull CE. Neural crest cells and motor axons in avians: common and distinct migratory molecules. Cell Adhes Migr. 2010;4:631–634. doi: 10.4161/cam.4.4.13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan ES. Signaling for vesicle mobilization and synaptic plasticity. Mol Neurobiol. 2008;37:39–43. doi: 10.1007/s12035-008-8014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- Magill-Solc C, McMahan UJ. Motor neurons contain agrin-like molecules. J Cell Biol. 1988;107:1825–1833. doi: 10.1083/jcb.107.5.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kummer TT, Lichtman JW, Sanes JR. Agrin promotes synaptic differentiation by counteracting an inhibitory effect of neurotransmitter. Proc Natl Acad Sci USA. 2005;102:11088–11093. doi: 10.1073/pnas.0504806102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Imagawa T, Hakamat Y, Shigekawa M, Takeshima H, Numa S. Primary structure and functional expression from cDNA of the cardiac ryanodine receptor/calcium release channel. FEBS Lett. 1990;271:169–177. doi: 10.1016/0014-5793(90)80399-4. [DOI] [PubMed] [Google Scholar]
- Nelson BR, Wu F, Liu Y, Anderson DM, McAnally J, Lin W, Cannon SC, Bassel-Duby R, Olson EN. Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc Natl Acad Sci USA. 2013;110:11881–11886. doi: 10.1073/pnas.1310571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numa S, Tanabe T, Takeshima H, Mikami A, Niidome T, Nishimura S, Adams BA, Beam KG. Molecular insights into excitation–contraction coupling. Cold Spring Harb Symp Quant Biol. 1990;55:1–7. doi: 10.1101/sqb.1990.055.01.003. [DOI] [PubMed] [Google Scholar]
- Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM, MacLennan DH. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem. 1990;265:13472–13483. [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol. 1992;458:487–499. doi: 10.1113/jphysiol.1992.sp019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JA, Rieger F, Blondet B, Dreyfus P, Pincon-Raymond M. Distribution and quantification of ACh receptors and innervation in diaphragm muscle of normal and mdg mouse embryos. Dev Biol. 1984;101:168–180. doi: 10.1016/0012-1606(84)90127-1. [DOI] [PubMed] [Google Scholar]
- Reist NE, Werle MJ, McMahan UJ. Agrin released by motor neurons induces the aggregation of acetylcholine receptors at neuromuscular junctions. Neuron. 1992;8:865–868. doi: 10.1016/0896-6273(92)90200-w. [DOI] [PubMed] [Google Scholar]
- Rimer M. Modulation of agrin-induced acetylcholine receptor clustering by extracellular signal-regulated kinases 1 and 2 in cultured myotubes. J Biol Chem. 2010;285:32370–32377. doi: 10.1074/jbc.M110.144774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Shakiryanova D, Klose MK, Zhou Y, Gu T, Deitcher DL, Atwood HL, Hewes RS, Levitan ES. Presynaptic ryanodine receptor-activated calmodulin kinase II increases vesicle mobility and potentiates neuropeptide release. J Neurosci. 2007;27:7799–7806. doi: 10.1523/JNEUROSCI.1879-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H, Iino M, Takekura H, Nishi M, Kuno J, Minowa O, Takano H, Noda T. Excitation–contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature. 1994;369:556–559. doi: 10.1038/369556a0. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Ikemoto T, Nishi M, Nishiyama N, Shimuta M, Sugitani Y, Kuno J, Saito I, Saito H, Endo M, Iino M, Noda T. Generation and characterization of mutant mice lacking ryanodine receptor type 3. J Biol Chem. 1996;271:19649–19652. doi: 10.1074/jbc.271.33.19649. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Komazaki S, Hirose K, Nishi M, Noda T, Iino M. Embryonic lethality and abnormal cardiac myocytes in mice lacking ryanodine receptor type 2. EMBO J. 1998;17:3309–3316. doi: 10.1093/emboj/17.12.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, Matsuo H, Ueda M, Hanaoka M, Hirose T, Numa S. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439–445. doi: 10.1038/339439a0. [DOI] [PubMed] [Google Scholar]
- Wang JY, Chen F, Fu XQ, Ding CS, Zhou L, Zhang XH, Luo ZG. Caspase-3 cleavage of dishevelled induces elimination of postsynaptic structures. Dev Cell. 2014;28:670–684. doi: 10.1016/j.devcel.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 2001;30:399–410. doi: 10.1016/s0896-6273(01)00287-2. [DOI] [PubMed] [Google Scholar]
- Zissimopoulos S, West DJ, Williams AJ, Lai FA. Ryanodine receptor interaction with the SNARE-associated protein snapin. J Cell Sci. 2006;119:2386–2397. doi: 10.1242/jcs.02936. [DOI] [PubMed] [Google Scholar]
- Zorzato F, Fujii J, Otsu K, Phillips M, Green NM, Lai FA, Meissner G, MacLennan DH. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1990;265:2244–2256. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.