

Summary
S-acylation, S-glutathionylation, S-nitrosylation, and S-sulfenylation are prominent, chemically distinct, modifications that regulate protein function, redox-sensing, and trafficking. Although the biological significance of these modifications is increasingly appreciated, their integration in the proteome remain unknown. Novel MS-based technologies identified 2,596 predominately unique sites in 1,319 mouse liver proteins under physiological conditions. Structural analysis localized the modifications in unique, evolutionary conserved protein segments, outside commonly annotated functional regions. Contrary to expectations, propensity for modification did not correlate with biophysical properties that regulate cysteine reactivity. However, the in vivo chemical reactivity is fine-tuned for specificity, demonstrated by the nominal complementation between the four modifications and quantitative proteomics that showed a reduction in S-nitrosylation is not correlated with increased S-glutathionylation. A comprehensive survey uncovered clustering of modifications within biologically related protein networks. The data provide the first evidence for the occurrence of distinct, endogenous protein networks that undergo redox signaling through specific cysteine modifications.
Graphical abstract
Introduction
The functional diversity of the amino acid cysteine in proteins is well appreciated by the identification and characterization of structural, metal binding and catalytic cysteine residues (Marino and Gladyshev, 2010; Pace and Weerapana, 2014; Schmidt, et al., 2006). Structural cysteine residues forming disulfides are essential for protein folding and for the structural integrity of proteins. Metal binding and metal coordination is indispensable for the formation of metalloproteins including iron-sulfur clusters in the mitochondrial respiratory chain (Pace and Weerapana, 2014). Cysteine residues also serve catalytic function in protein phosphatases, acyl transferases, oxidoreductases, and RING binding domain proteins such as E3 ligases (Karisch, et al., 2011; Passmore and Barford, 2004; Paulsen, et al., 2012). Another class of distinct cysteine residues that serve a regulatory role has also recently emerged as a widespread signaling mechanism. These regulatory cysteine residues undergo post translational modifications, principally S-acylation, S-glutathionylation, S-nitrosylation, and S-sulfenylation to influence protein function and location, afford allosteric regulation, and provide a mechanism for signal transduction akin to phosphorylation or lysine acetylation (Finkel, 2011; Groitl and Jakob, 2014; Sen, et al., 2012; Seth, et al., 2012; Smith and Marletta, 2012). To appreciate the global biological impact of these modifications and to improve our understanding of how signaling and protein regulation is achieved, proteomic studies have explored the landscapes of cysteine post translational modifications (Hamnell-Pamment, et al., 2005; Paulsen, et al., 2012). However, these proteomes were acquired one modification at a time and in different organs and cells; not permitting critical investigations into the interface, complementation and organization of these modifications at the proteome level. This is important, considering isolated instances of documented complementation and coordination between multiple modifications can regulate essential biological functions such as neurotransmission, redox-dependent signaling, and metabolism (Sen, et al., 2012). Therefore acquisition of endogenous site-specific proteomes of all four cysteine modifications simultaneously from the same organ under physiological conditions will enable comprehensive and global evaluation of complementation and coordination while providing a rich resource for appreciating cysteine modifications in biology.
Acquisition and systematic interrogation of these proteomes will also provide invaluable insights for the long-standing challenge relating to the biochemical and biophysical properties that guide the reactivity of cysteine residues. The presence of cysteine residues localized in microenvironments that readily enable deprotonation, forming thiolate anions, is considered a principal attribute for reactivity. Data from proteome wide studies of cysteine residues reactive to electrophiles indicated that thiolates represent a small fraction of hyper-reactive cysteine residues (Weerapana, et al., 2010). Moreover, thiolate anion is an imprecise distinguishing characteristic of catalytic activity, with a majority of catalytic residues having been identified as hyper-reactive as opposed to thiolate. This presents a biological conundrum since highly reactive residues are more likely to be covalently modified, however proteomes of cysteine post-translational modifications have localized these modifications primarily in non-catalytic regions. This dichotomy between the necessity for functional cysteine reactivity and propensity for covalent post translational modification is unresolved.
Herein, we have combined state-of-the-art chemical enrichment methodologies with high resolution mass spectrometry to identify specific cysteine residues sites that are endogenously modified by S-acylation, S-glutathionylation, S-nitrosylation, and S-sulfenylation under physiological conditions. Computational analysis of the data indicated that modified cysteine residues principally do not participate in disulfide bonds, do not coordinate metals or serve as catalytic residues and are localized in unique, regulatory regions of proteins conserved among related species. Although the propensity for modification did not correlate with biophysical properties relating to the intrinsic chemical reactivity of cysteine, there was minimal complementation among the four modifications. Notably, our findings indicated that the modifications were clustered in functionally distinct pathways some of which were predicted from existing knowledge and some which offer new insights into the regulation of functional pathways by selective post translational modification of regulatory cysteine modification.
Results
Acquisition and characterization of endogenous cysteine proteomes
To better understand the basis for the structural and functional diversity of protein cysteine residues in biology, we devised and implemented proteome-wide mass spectrometric technologies that enable site-specific identification of post-translationally modified cysteine residues from liver tissue under normal physiological conditions. The work-flow is depicted in Figure 1A and it consists of four steps: alkylation of reduced cysteine residues, selective reduction or labeling, chemical enrichment or chromatographic separation, and protease digestion followed by MS peptide analysis. The selectivity, accuracy and reproducibility of the methodologies have been reported previously and are reinforced by the use of knockout mice herein (Doulias, et al., 2010). In six wild type mouse liver, we mapped a total of 2,596 sites of cysteine post-translational modifications in 1,319 proteins. Specifically, we identified 686 S-nitrosylation (SNO) sites in 438 proteins, 883 S-glutathionylation (SSG) sites in 580 proteins, 585 S-acylation (SAc) sites in 428 proteins and 442 S-sulfenylation (SOH) sites in 392 proteins (Fig 1A and Tables S1–4). As an experimental reference, 7,682 mercury-reactive cysteine residues were mapped to 5,664 proteins in samples bypassing the thiol blocking step (Table S5). Considerable overlap exists between our experimentally derived reactive cysteine proteome and published proteomes (Huttlin, et al., 2010; Kislinger, et al., 2006; Kruger, et al., 2008). Including proteins from published sources, we generated a mouse liver reference proteome consisting of 9,378 proteins of which 9,185 (98%) contain at least one cysteine residue. The 1,319 endogenously modified proteins represent 14% of the cysteine containing proteins in the reference proteome (Fig 2A) and their abundance, molecular weight and total number of cysteine residues falls within the overall distribution of other cysteine-containing proteins in the reference proteome (Fig 1B–D).
Figure 1. Acquisition and characterization of cysteine proteomes.

A. Schematic representation of the protocol utilizing selective reduction/reactivity coupled with enrichment methods for the detection of S-nitrosylation (SNO), S-glutathionylation (SSG), S-acylation (SAc), and S-sulfenylation (SOH) proteomes. The mercury reactive proteome (Hg-React) provides an experimental reference proteome used for biochemical and structural comparisons. Proteome counts for the identified peptides and the corresponding proteins are indicated in the table. B. Frequency distribution of protein abundance curated using PaxDB. All annotated liver proteins within PaxDB were utilized to generate the reference for examining cysteine post-translational modifications (PTMs). C. Distribution of protein molecular weights for cysteine PTMs compared to liver expressed reference proteins. D. Cysteine content normalized to length of proteins in the liver reference and individual modified cysteine proteomes. Proteome data is from 6 biological replicates.
Figure 2. Frequency and distribution of cysteine modifications.

A. Frequency of modified proteins within the liver proteome. The fraction of uniquely modified proteins correspond to the given annotated proteome, proteins that exhibit multiple modifications are clustered together. The combination of cysteine PTMs and Hg-react proteins represents the fraction of endogenous proteins that are experimentally detectable utilizing our methodology. B. Distribution of modified proteins among the four cysteine PTMs. C. Site specific frequency of cysteine modifications within the experimentally determined proteome. D. Site specific distribution of cysteine modifications. E. Overlap of identified S-glutathionylated sites in wild type and eNOS−/− liver tissue, sites in parentheses denote the number of sites that carry both SSG and SNO modifications. F. Label-free quantification of the 174 shared sites that are both S-glutathionylated and S-nitrosylated from wild type and eNOS−/−, fold-change is based on eNOS−/−, a positive value indicates greater abundance of peptide in the eNOS−/− tissue a negative value indicates greater abundance in wild type. Red lines are the cutoff for significantly changed peptides. G. Western blot of SOH in WT and eNOS−/− genotypes using antibody against dimedone (DMD) derivatized peptides.
The majority of modified cysteine residues were identified as having only a single post-translational modification. At the protein level, 937 proteins were distinguished by a single modification, representing 71% of all modified proteins identified. Moreover, only 8 of the 382 proteins carrying multiple modifications had cysteine residues with all four modifications (Fig 2B and Table S6). The largest overlap among the modifications was between SAc, SNO, and SSG, consisting of 118 proteins (9%). Among the redox-dependent modifications (SNO, SSG, SOH), the largest overlap was between SSG and SNO with 185 proteins (14%) indicating that protein selectivity exists for the majority of chemically reactive modifications. At the cysteine level, 1,653 sites were modified by only a single modification, representing 80% of the total modified residues and accounting for 19% of the total cysteine residues (Fig 2C). Only 5% of the cysteine residues detected carried multiple modifications. There were zero instances of a single cysteine residue modified by all four modifications (Fig 2D). SAc and SSG had the largest overlap with 262 (17%) shared cysteine residues. Among the redox-dependent modifications, the largest overlap was again between SNO and SSG with 171 (11%) cysteine residues identified in both proteomes. The aforementioned findings indicate that both selectivity (i.e., only one modification on a single cysteine residue within a protein) and to lesser extent complementation (i.e., more than one modification on protein cysteine residues) exists in native biological systems.
To assess the degree of overlap among different modifications, mice deficient in the endothelial isoform of nitric oxide synthase (eNOS−/−) were utilized. Since the largest overlap of non-enzymatic modifications is between SNO and SSG proteomes, and S-nitrosylation may in fact promote S-glutathionylation (Klatt, et al., 1999; Martínez-Ruiz and Lamas, 2007), we analyzed the SSG proteome in the eNOS−/− and WT liver (Fig 2E and Table S7). We have previously documented a 90% reduction in liver SNO sites in eNOS−/− compared to wild type (Doulias, et al., 2013). Between both genotypes, a total of 936 SSG sites were identified, with the majority of sites (905, 97%) being shared between the two genotypes. Of the 936 SSG sites identified, 177 sites correspond to sites of both S-glutathionylation and S-nitrosylation representing the fraction of cysteine residues that are potentially co-regulated. To assess whether changes in SSG correspond to the expected loss of SNO occupancy, a label free quantification approach was used on 174 shared sites between wild type and eNOS−/− (Fig 2F, Table S8). Using standard cutoffs for significance, only two peptides were increased in the eNOS−/− mice and two increased in wild type (Table S9). However, the four sites represent an extremely low percentage (0.4%) of the total SSG sites identified; indicating that complementation between modifications is a rare occurrence under normal physiological conditions. As a corresponding analysis, SOH modified proteins were visualized after dimedone derivatization via western blot (Fig 2G). Despite some non-specific binding of the antibody in negative controls treated with DTT during derivatization, overall protein SOH is unchanged in both WT and eNOS−/− genotypes. Moreover since neither the total number of SSG modified cysteine residues nor the magnitude of SSG or SOH modification significantly increased in the eNOS−/− due to the decrease in SNO, we infer that biological cysteine modifications occur in a specific manner and likely are not driven by cysteine reactivity.
However, from the 29 SSG containing peptides that were increased or detected exclusively in the wild type versus the eNOS−/− genotype, only three sites were also S-nitrosylated. One logical explanation for a peptide to have increased SSG in the wild type is that a SNO intermediate may promote the formation of SSG through an exchange reaction with GSH, as it has been shown in vitro (Klatt, et al., 1999). However, the data do not indicate that such chemical reactions are prominent in vivo, as only a very small subset of cysteine residues undergoes these types of reactions. Rather, our in vivo data argue for an intrinsic mechanism, possibly through a combination of biochemical and biological factors, which dictates specificity and selectivity among cysteine residues and the corresponding modifications.
Redox-modified cysteine residues occur outside common functional domains
To further explore possible underlying biochemical and biophysical factors that promote specificity of post-translational cysteine modifications, an in-depth structural and biochemical analyses of the proteomes was performed. We did not identify a specific primary sequence motif associated with the cysteine modifications, consistent with previous analyses of large data sets including those of reactive cysteine residues (defined by the reactivity towards iodoacetamide) (Marino and Gladyshev, 2010; Weerapana, et al., 2010). However, the frequency of certain amino acids within a thirteen amino acid window surrounding the modified cysteine residues revealed that cysteine, glutamine, serine, and tryptophan are generally under-represented and alanine, histidine, isoleucine, lysine, methionine and valine are generally over-represented amino acids (Fig S1). A notable exception relates to the over-representation of a second cysteine residue in the +3 position of SOH residues which prompted the interrogation of the data for common CXC and CXXC motifs (Fig 3A). Of the 427 uniquely mapped SOH cysteine residues, 18 (4.2%) are within CXC motifs and 61 (14.2%) are present in CXXC motifs. In contrast, the percentage of residues in a CXC or CXXC motif for SAc (0.8%, 0.4%), SNO (2.1%, 3.7%), and SSG (1.3%, 4.8%) are significantly lower than what would be expected based on the reference proteome. These findings correlate well with the higher frequency of unmodified cysteine residues proximal to S-sulfenylation sites in both three-dimensional structures and primary protein sequences (Salsbury, et al., 2008). Additionally, these data are in agreement with the established formation of stable sulfenic acid intermediates that can facilitate catalytic activity or catalytic regulation (Conway, et al., 2004).
Figure 3. Fraction of modified cysteine residues within functional cysteine groups.
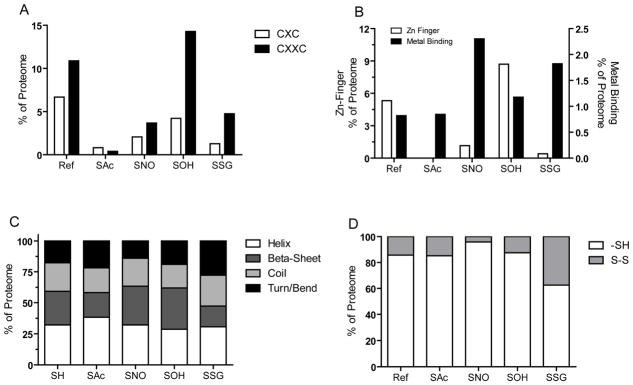
A. Distribution of reduced (SH) and modified cysteine residues within major structural domains. B. Fraction of cysteine residues within annotated CXC or CXXC domains. C. Distribution of cysteine residues annotated as zinc finger binding or metal binding. D. Fraction of cysteine residues within each proteome annotated within disulfide bonds.
Although not exclusive, a number of Zn-finger proteins contain CXXC domains and cysteine residues are commonly found in metal binding sites (Pace and Weerapana, 2014; Shi, et al., 2005). The proportion of proteomes that exhibit either Zn finger or other metal-binding properties were examined (Fig 3B). Consistent with the enrichment in CXXC domains, there is greater proportion of cysteine residues within Zn finger domains in the SOH proteome (8.5%) compared to the reference proteome (5.3%); while SAc (0%), SNO (1.2%) and SSG (0.4%) proteomes exhibited a significantly lower percentage than expected. Conversely, SNO (2.3%) or SSG (1.8%) residues were more likely to be within metal-binding regions compared to the reference (0.8%), SAc (0.8%), or SOH (1.2%) proteomes. The presence of metal ions in proximity to the modified cysteine is well known to modulate its reactivity or pKa (Stewart and Igumenova, 2012). However, in these cases it is difficult to discern whether the presence of the metal ion might promote or facilitate cysteine modification or whether it is a property of the individual cysteine, which facilitates metal ion coordination or redox-mediated covalent modification. Regardless, the overall percentage of modified cysteine residues within metal binding sites is low, indicating that cysteine modifications are less prone to directly disrupt catalytic or metal cofactor binding. Furthermore, searches for other functional motifs of classically redox-sensitive phosphatases ([IV]HCXGXGR[ST]G) and acyl transferases (C[X]5R) did not return any significant enrichment in any of the four modifications. The indication that cysteine modifications principally bind outside of commonly annotated functional regions of the proteins implies a greater potential for the existence of cysteine dependent redox regulatory domains in these proteins.
Beyond primary sequences and linear motifs, we also examined the influence of surrounding amino acids in the three-dimensional structure of proteins and their correlation with specific cysteine modifications (Fig 3C). Three-dimensional structures were available for 2,829 Hg-reactive sites and 111 SAc, 196 SNO, 70 SOH and 202 SSG sites. Cysteine residues that were not annotated as forming disulfide bonds or undergoing other modifications, and therefore presumed to exist predominately in the reduced state, were curated from our reference proteome and used as comparison group for the modified cysteine residues. Overall, these sites are distributed across the four structural motifs: helix (32.4%), beta-sheet (27.1%), coil (23.1%), and turn/bend (17.7%). Consistent with previously defined roles as an anchor for membrane proteins, SAc sites occurred in a higher proportion of helix domains compared to reduced cysteine residues (38.2% vs 32.4%). In contrast, sites of both SNO and SOH occurred with greater frequency in beta-sheet domains (31.3% and 33.3% respectively) and SSG in less structured turn/bend (27.8%) and coil (25%) domains, as compared to reduced cysteine residues. It is conceivable that steric hindrances are one limiting factor that selects for the size of modification and influences the domains within a protein where a modification can occur (Fig S2). The two smallest modifications, SNO and SOH, are estimated to have similar molecular volumes (Vw) of 108 Å and 94 Å respectively, thus it is not surprising that they share similarities in the distribution among structural motifs. If we consider S-palmitoylation, the addition of a 16:0 fatty acid and one of the most abundant S-acylation species, an estimated Vw of 570 Å is obtained compared to glutathione (Vw of 434 Å). However, the location of the sulfur at the center of GSH protruding roughly 2.8 Å from the carbon backbone may necessitate that GSH adopts a bent configuration to facilitate formation of a mixed-disulfide. In contrast, the majority of SAc modifying species are predominantly linear carbon chains with the thioester bond forming at one end. This highlights that acyl species, while slightly larger, adopt favorable linear conformations compared to GSH molecules, thereby allowing S-acylation to occur in more structured domains compared to S-glutathionylation, which occurs predominately in less structured flexible peptide domains.
Structural cysteine residues that form disulfides are essential for protein folding and account for the majority of established functional roles for cysteine residues within the proteome (Hansen, et al., 2009). Subsets of disulfides, described as allosteric in order to differentiate them from structural, can also alter function through conformational changes in the protein (Schmidt, et al., 2006). We parsed the proteomes for modified cysteine residues that may also form disulfides. Using structures that exhibit at least 80% homology with 100% sequence similarity within five amino acids of the cysteine of interest, on average, cysteine modifications are less likely (15.6% versus 28.7% random control) to form disulfide, as identified by C(α) distance of 8Å and S-S distance of 2.5Å. Based on the structural analysis of potential disulfide formation, 73.5% are SSG, 25% are SAc, 10% are SOH, and 7% are SNO modified. Available crystal structures limit the depth of analysis; therefore an additional examination of predicted disulfides was performed based on Uniprot annotations. A total of 109,960 cysteine residues were examined in the reference, 237 in SAc, 434 in SNO, 425 in SOH, and 494 in SSG (Fig 3D). The largest proportion of annotated disulfides 185 (37.4%) occurs within the SSG proteome, compared to 14.3% in the reference. The SNO proteome exhibited a relative underrepresentation of annotated disulfides (4.1%), while SAc (14.8%) and SOH (12.5%) are no different from the reference. Overall, the data indicate that the majority of modified cysteine residues identified in this study do not conform to the three major biological functions of cysteine residues (catalytic, metal binding, structural) and thus, can be distinctly considered as candidates for redox-dependent regulation, analogous to role of serine for phosphorylation or lysine for acetylation.
Functional cysteine residues, primarily structural and catalytic, are well conserved across highly divergent organisms (Marino and Gladyshev, 2010). We applied a previously published methodology for high-throughput analysis of amino acid conservation in proteins across different databases (Marino and Gladyshev, 2010), populated by increasingly diverging sets of organisms (Table 1). We used the UniProtKB/Swiss-Prot database as a reference, which was further divided in subsets consisting of only rodents, only mammals, or only vertebrates. By using the rodent database we found little difference between conservation values of modified cysteine residues (average conservation, 74%) and other cysteine sites (76%) in the same proteins. Similar results were obtained with the relatively divergent mammalian reference database (74% for PTM, 78% for controls). However, if a broader set of organisms was chosen (e.g., vertebrate database), representing a considerably more divergent set of homologs, the difference in conservation between PTM and control sites becomes larger (65% PTM, 73% controls). Finally, if the database is extended to contain all organisms (the full UniProtKB/Swiss-Prot dataset), then the difference increases even further (63% PTM, 76% controls). Overall, the difference in conservation increases with the degree of divergence between organisms. An important note is that internal controls are characterized by a higher frequency of disulfides and metal binding cysteine residues which are known to be highly conserved among divergent organisms. This contributes to the high conservation rates found for controls across all databases. Compared to other cysteine residues, modified sites are well conserved only in closely related organisms, but the similarity is lost at higher evolutionary distances. This indicates that cysteine modifications could be thought of as recently evolved functional sites. If we consider the modified cysteine residues as active points in the redox signaling network, then these results suggest that redox regulatory systems represent relatively “recent designs” within closely related phyla.
Table 1.
Cysteine PTM Conservation
| Databasea | PTM Cysb | Control Cysb | Difference |
|---|---|---|---|
| Rodent Only | 74.0 | 76.4 | 2.4 |
| Mammal Only | 74.5 | 78.0 | 3.5 |
| Vertebrate Only | 65.4 | 72.9 | 7.5 |
| UniProtKB/Swiss-Prot | 63.1 | 76.4 | 13.3 |
Indicates organism search database ordered by least to most divergent
Data presented as % conservation derived from the number of times each Cys site is present in BLASTP alignments for a given protein within the given database.
Biochemical properties of cysteine modifications
It is conceivable that underlying biochemical properties of the modified cysteine residues may promote modification and enable selective modification. Therefore we determined the Kyte-Doolittle hydropathy index of the peptides within a window of thirteen amino acids around the modified cysteine residue. In general, cysteine residues skew towards marginally hydrophilic regions of the protein, with few differences between modifications. The 7,579 experimentally mapped cysteine residues occurred in a relative normal distribution averaging only slightly hydrophilic with an index value of −0.16. Similarly, the averages of both SAc (−0.13) and SOH (−0.13) values did not significantly differ from the reference proteome. In contrast, the regions of the protein where SSG modifications occur are on average significantly more hydrophilic (−0.32, p<0.001) than cysteine residues in the reference proteome. Additionally, regions in which SNO modifications occur average significantly lower hydrophilicity (−0.04, p<0.001) compared to the reference (Fig S3). To account for the immense variability in the types and functions of the modified proteins, we analyzed the deviation of the hydropathy of the modified cysteine residues using other intra-protein cysteine residues as a reference (Fig 4A). Due to the high proportion of uniquely modified cysteine residues, unsupervised hierarchical clustering occurred within each of the modifications. This analysis mirrored the averages indicating that the majority of modified cysteine residues are more hydrophilic than other cysteine residues within the same protein. Smaller clusters of proteins exhibited modified residues that deviate toward greater hydrophobicity compared to other cysteine residues within the same protein. Due to the relative low abundance of proteins that exhibit multiple modifications, instances of differences in hydropathy between different modifications within the same protein is not sufficient to derive biological significance.
Figure 4. Biochemical properties of modified cysteine residues.
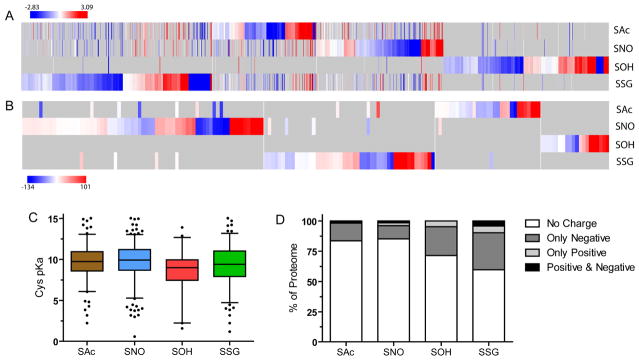
Heatmap resulting from the unsupervised hierarchical clustering of the deviation of hydropathy (A) and solvent accessibility (B) of modified cysteine residues compared to intra-protein unmodified cysteine residues. The x-axis represents all proteins which could be calculated, grey represents missing values resulting from uniquely modified proteins. C. Frequency distribution of pKa values that are able to be calculated using PROPKa. D. Charged amino acid frequency among cysteine proteomes within 10 Å of the modified cysteine.
It has been proposed that accessibility and cysteine reactivity are the two most influential factors for thiol redox reactions (Marino and Gladyshev, 2012). The cellular redox environment is primarily reducing, but smaller localized environments exist which can promote oxidation. A number of proteins exhibit ‘oxidant sensing’ functions presumably through highly reactive cysteine residues (Brandes, et al., 2009; Groitl and Jakob, 2014). We analyzed the solvent accessibility of the modified cysteine residues and found no difference between modified cysteine residues and the unmodified reference. Due to limitations in the quality and availability of structures, only 276 modified cysteine residues, compared to 1,744 unmodified residues, could be analyzed. Across all modifications, an average of 13.7% occurred in surface regions (defined as a RSA value above 50), compared to 10% in the reference. The SOH proteome exhibited the greatest percentage of cysteine residues localized in surface exposed regions (29%), although it also exhibited the lowest number of residues (21) that could be analyzed. We also examined deviation in solvent accessibility using intra-protein control cysteine residues (Fig 4B). Overall, the modified cysteine residues exhibited a distribution relatively equal to the unmodified cysteine residues within the same protein, only slightly skewing towards being higher solvent accessibility. Since a number of modified cysteine residues exhibit similar accessibility yet do not share modifications, our data argues that solvent exposure is not sufficient to induce the formation of a modification, further suggesting that these modifications demonstrate true, functionally targeted, redox regulatory elements that are both selective and specific.
The reactivity of a protein cysteine residue is in part controlled by intrinsic properties of the sulfur (i.e. pKa) and the amino acids that make up the microenvironment of the cysteine residue (Britto, et al., 2002). The majority of cysteine residues are protonated at physiological pH although deprotonation to form thiolate anions can occur rendering these residues more reactive (Denu and Tanner, 1998). It has been suggested that some redox-responsive proteins contain a stabilized thiolate anion due to lowering of the cysteine pKa (Lim, et al., 2012; Wood, et al., 2003). Additionally, it has been proposed that thiolate anion formation is sufficient to induce the formation of covalent modifications (Britto, et al., 2002). We investigated the predicted pKa distribution of modified cysteine residues. Compared to the average pKa of 9.7 for reference cysteine residues, modified residues did not significantly differ with an average pKa of 9.5 (Fig 4C), although there is the distinct possibility that local pKa of the cysteine can be affected by polar residues, causing a deviation between predicted and experimentally derived pKa. We investigated the distribution of charged amino acids vicinal to the modified cysteine to examine the contribution of various amino acids towards cysteine reactivity (Fig 4D). Greater than 50% of all four proteomes exhibited uncharged amino acids within 10 Å of the modified cysteine. Smaller subsets of cases which exhibit negatively charged amino acids near the modified cysteine occur in the SOH and SSG proteomes. Interestingly, basic amino acids that are more likely to lower the thiol pKa were minimally represented within 10 Å of the modified cysteine residues. Considering the low proportion of basic amino acids, our data indicates that thiolate anion formation may not be the primary determinant for the formation of cysteine modifications. Conversely, the relative higher proportion of negatively charged residues near SOH and SSG sites may act to stabilize the modifications rather than directly affecting cysteine reactivity. Collectively, these data indicate that commonly considered measures of general cysteine reactivity (accessibility and pKa/charge state) are not necessarily the sole factor to determine cysteine reactivity and cysteine modification state in vivo.
Biological clustering of modified cysteine residues
Specific biochemical properties of modified cysteine residues, and by extension cysteine reactivity, do not appear sufficient to distinguish the occurrence or type of modification on a given cysteine residue. Therefore we explored whether specific modifications can be attributed to biological locales or functions. Uniquely modified proteins were labeled with all gene ontology (GO) terms by starting with term associations provided by Uniprot, and adding each term’s ancestor associations using “is a” and “part of” relationships. GO terms were evaluated in each of the three GO categories: cellular location, biological process, and molecular function. Subsets of terms were selected as features that could be used to classify modified proteins. These GO terms had: (i) at least one modification with 10% of proteins associated with the term, (ii) at least one modification with more than 20% of proteins not associated with the term, and (iii) a significant association with modification status, as indicated by a Benjamini-Hochberg adjusted Chi-square p-value < 0.001. As an evaluation of how well these GO terms were associated with different modifications, we constructed a random forest classifier to predict modifications. Given the redundancy of GO term associations, we first clustered the distinguishing GO terms into seven groups using k-means clustering. Each GO term was represented by a feature vector holding the fraction of annotated proteins for each modification. The vector was normalized by the sum of each annotated fraction for a term. The result was seven distinct clusters of GO terms which included both cellular localization and biological/molecular processes. A heat-map of the relative distribution of proteins represented in each GO term, clustered within one of the seven significantly distinguishing clusters is depicted in Fig 5A. We used each cluster as features in a random forest classifier, and scored an average accuracy of 48% across 10 fold cross validation, which is better than the accuracy of 37% obtained by always predicting the most frequent modification.
Figure 5. Biological clustering of modified proteins.
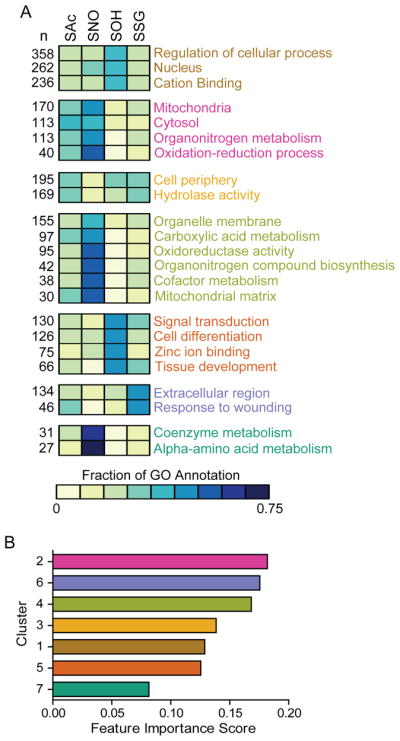
A. Clustering of aggregate GO terms of cellular localization and biological/molecular processes. The heatmap represents the relative proportion of proteins within each of the designated GO term, with the total protein number indicates to the left. Cluster number ranging from 1–7 assigned from top to bottom with text color differentiating the assigned clusters. Scale bar represents the relative proportion of proteins assigned within the GO term assigned to any one modification. B. Ranking of cluster importance score based on random forest rank tests of the clusters accuracy in distinguishing modifications from each other, bar coloring corresponds to cluster color text in A.
We used a random forest to rank the importance of each cluster in the correct assignment of modification labels according to its mean decrease impurity (Fig 5B). The importance measure represents the decrease in accuracy when the protein assignments for a cluster’s terms have been permuted to erase the association between modification and term. Based on these analyses, cluster 4 which predominately includes small molecule and nucleotide metabolic processes is a rather effective distinguishing classifier for SNO proteins and against SOH proteins. Combined with the abundance of SNO proteins within cluster 2, which is highly populated with mitochondria localization and oxidoreductase activities, and this fits well with the known cardio-protective effects of S-nitrosylation during ischemia and the role of S-nitrosylation in fatty acid β-oxidation (Chouchani, et al., 2013; Doulias, et al., 2013).
Other modifications, such as S-sulfenylation, were highly represented in clusters 5 and 6 which incorporate the higher proportion of SOH sites in zinc binding and CXXC domain proteins. SSG proteins were primarily represented by annotations in the response to wounding. Overall, SAc proteins were the least specific among each of the cysteine modifications examined, potentially due to the variety of acyl species which can be formed and the multiple different interactors of acyl-transferases. As would be expected based on the well-characterized function as a membrane anchor, S-acylated proteins did correlate well with proteins localized to organelle membranes and the cell periphery. Collectively our data reveal the presence of biologically-related protein networks containing discriminating, conserved cysteine residues among related species that undergo distinct modifications.
Discussion
Herein, we mapped 2,596 sites of four chemically distinct cysteine modifications, S-nitrosylation, S-glutathionylation, S-acylation and S-sulfenylation in wild type mouse livers under normal physiological conditions. The four different modifications target discrete protein populations and cysteine residues with minimal complementation between the modifications. Specifically, the data revealed that 80% of the modified residues carried a single modification, while at the protein level 71% of proteins exhibited only a single modification. This remarkable selectivity is maintained even under conditions of decreased occupancy of S-nitrosylation. In the eNOS knockout mice, which have decreased SNO site occupancy the data showed that S-glutathionylation does not significantly deviate in site identity or abundance compared to wild type. While some of the chemical reactions that give rise to these cysteine modifications in vitro are known, the in vivo chemical events, including those that may account for the selectivity of the different modifications are less defined. In part, selectivity can be derived by protein-protein interactions as it has been shown for prototypical redox-sensing proteins such as thioredoxin and glutathione-S-transferase, which facilitate the direct transfer of nitric oxide and glutathione equivalents respectively to target proteins (Benhar, et al., 2010; Mitchell and Marletta, 2005; Townsend, et al., 2009). There is also evidence that certain modifications (i.e. SNO or SOH) can be reduced by cellular glutathione resulting in a displacement of the nitroso or hydroxyl groups resulting in protein S-glutathionylation (Dalle-Donne, et al., 2007; Smith and Marletta, 2012). Furthermore, the general consensus to date is that under normal physiological conditions, the modifications are dictated by the reactivity of the cysteine residues and the microenvironment surrounding the modified cysteine residue. However extensive biochemical interrogation of the modified cysteine residues did not reveal a single overarching biochemical property of the modified cysteine residues that will dictate selectivity. The commonly considered properties that discriminate reactive thiols (i.e. solvent accessibility, pKa, and deprotonation) from unreactive cysteine residues cannot be used to access the propensity for modification. Moreover, recent evidence of S-glutathionylation in Titin indicates that conformational changes in the protein allow a previously buried cysteine to become exposed and modified (Alegre-Cebollada, et al., 2014). The principle of redox modifications depending on conformation presents an additional layer of complexity that could only be identified on a protein-by-protein basis. This introduces a concept wherein cysteine modifications may not only be dependent on the redox environment but also on temporal changes in protein biochemistry. Considering the specificity between modifications, it is possible that the propensity for thiol modification lies in the combination of cysteine and modifying agent reactivity, somewhat analogous to a ‘lock and key’ type mechanism. Regardless, it is becoming increasingly apparent that finding a single rule or property for cysteine post-translational modification on a proteome wide scale may not be possible.
The comprehensive mapping of the endogenous cysteine modifications it also provides a rich, validated, resource for understanding the landscape of thiol modifications in vivo, and may guide future studies since non-catalytic cysteine residues are often used as an effective strategy for drug design and targeting (Hagel, et al., 2011). The modifications were principally localized in protein segments outside of commonly annotated functional regions (disulfide-forming, metal binding and catalytic) and thus, can be distinctly considered as regulatory residues. This new class of regulatory cysteine residues is conserved among closely related species to the same degree as other functional cysteine classes. The unbiased, bioinformatics analysis of the proteomes also revealed several structural findings consistent with previous analyses of individual modifications. The localization of SOH modifications in CXC and CXXC motifs correlated well with the overabundance of zinc finger proteins in the SOH proteome and their well-known CXXC active site motif. Furthermore, the greater proportion of SAc modifications found in helical domains is consistent with the higher frequency of S-palmitoylation localization at the N-termini, where helical conformations are also common (Linder and Deschenes, 2003). Finally, the fact that SSG modifications had the highest disulfide annotation is unsurprising since the chemical distinction between inter/intra protein disulfides and mixed disulfide is largely non-existent. Thus it is expected that S-glutathionylated cysteine residues share similar properties characteristic of cysteine residues that form disulfides.
Overall, the data identified subsets of cysteine residues in the proteome that are functionally related and selectively modified supporting the emerging concept for the presence of regulatory cysteine residues wherein post-translational modifications trigger, harmonize and expand physiological signaling and protein function.
Experimental Procedures
Tissue Collection
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Children’s Hospital of Philadelphia. Wild-type C57BL/6J and Nos3tm1Unc (eNOS−/−) animals were obtained from Jackson Laboratories. Mice were anesthetized using CO2 and exsanguinated via cervical dislocation.
Preparation of modified cysteine proteomes
Two methods were used for the detection of in vivo cysteine redox modifications, selective reduction coupled with phenylmercury enrichment or chemical derivatization coupled with 2D separation.
Analysis of S-nitrosylation proteome follows essentially as previously described (Doulias, et al., 2013).
S-glutathionylation proteome analysis follows a similar procedure as S-nitrosylation with the main exception being that endogenous cysteine bound nitric oxide is removed using UV prior to alkylation with n-ethylmalemide (Sigma). SSG proteins are selectively reduced using recombinant Glutaredoxin 1 (Cayman) and enriched using phenylmercury resin.
S-acylation proteome analysis follows the same steps as that of the SSG proteome except proteins are selectively reduced with 800 mM hydroxylamine (Sigma-Aldrich) for 60 min at 50°C(Martin, et al., 2012).
Since S-sulfenylated proteomes necessitate a chemical derivatization, mercury enrichment is not employed; instead proteins are separated in gels and fractionated. 700 μg of proteins were treated with 25 mM dimedone (DMD) for 2h at RT with constant shaking as previously described (Martínez-Acedo, et al., 2014). The protein samples were separated by concentrating SDS-PAGE. The unique gel band obtained for each proteome was visualized by Coomassie brilliant blue G-250 (Amresco), and then cut into small gel pieces, treated with 10 mM DTT and 50 mM NEM at pH 8.0 prior to tryptic digestion. The resulting peptide pool was desalted onto Oasis C18 HLB extraction cartridges (Waters) and dried down. Peptide were suspended in focusing buffer (5% glycerol and 2% IPG buffer pH 4–7 (GE Healthcare)), loaded onto 24-well, 24 cm-long ImmobilineDryStrip, pH 4–7 (GE Healthcare) and separated by isoelectric focusing on a 3100 OFFgel fractionator (Agilent). The recovered fractions were acidified and the peptides were desalted using OMIX C18 tips (Varian, Inc). After elution, the peptides were dried-down prior to reverse phase-high performance liquid chromatography (RP-HPLC)-LIT analysis. Visualization of protein sulfenylation utilized the same dimedone derivatization as above followed by reducing SDS-PAGE. The blot was incubated with anti-cysteine sulfenic acid antibody raised against dimedone derivatized peptide (Millipore).
The generation of the experimental proteome, termed mercury reactive (Hg-React) consisted of loading un-alkylated protein suspension on the mercury column. Processing follows the same enrichment, wash, elution, and lyophilization methods as the other three proteomes that utilize the resin enrichment. After lyophilization, the resulting peptides are resupended in 200 μL 0.1% formic acid followed by the addition of 200 μL 2% acetonitrile and 5 mM ammonium formate, pH 10. The volume is then reduced to 10 μL via Speedvac. The resulting peptide solution was then fractionated using alkaline-reverse phase fractionation on a Waters NanoAcquity UPLC instrument with a Agilent Zorbax-300Extended C18 3.5μM 100 × 0.3mm Column (Mertins, et al., 2013). Flow rate was set at 6 μl/min for 50 minutes with a 20 min gradient. Mobile phase A consisted of 5 mM NH4CO2, pH 10 and 2% acetonitrile. Mobile phase B consisted of 5 mM NH4CO2, pH 10 and 90% acetonitrile. Peptide elution was monitored at 280 nm wavelength. A total of 10 fractions were generated and combined non-continuously into 5 final fractions which were analyzed by LC-MS/MS.
More detailed methodology, including mass spectrometry, peptide identification and bioinformatic analysis parameters can be found in the supplementary experimental methods section.
Supplementary Material
Significance.
Redox signaling mediated by cysteine post-translational modifications represent a recent area of intense interest with wide reaching implications for multiple scientific disciplines ranging from physiology to cardiovascular and neurological diseases. These diverse protein cysteine modifications represent newly realized regulatory mechanisms that influence protein function and location, afford allosteric regulation, and provide a mechanism of signal transduction akin to phosphorylation or acetylation. To enable investigations into the interface, complementation and organization of cysteine modifications at the proteome level we mapped the sites of S-acylation, S-glutathionylation, S-nitrosylation, and S-sulfenylation. The assembled proteomes represent a unique resource and the first comprehensive identification of endogenous cysteine residues in proteins that are modified by these four chemically distinct modifications under normal physiological conditions. Analysis of the acquired proteomes offered the following significant advances: 1) discovered that redox dependent post-translational modifications occur in biologically unique protein networks containing differentiating, conserved cysteine residues that possess capacity for redox regulation. 2) The redox-dependent modification of cysteine residues are surprisingly selective and minimal complementation exists in the proteome. 3) Through comprehensive bioinformatic interrogation, we established that modifications are not localized in common cysteine functional domains, rather they exist in unique segments of proteins that may represent recent evolutionary designs to accommodate specific redox-sensing, signaling and functional regulation of proteins. Collectively these findings are on the forefront for understanding the basic tenets of redox-dependent regulatory cysteine residues at the proteome level and their biological role in normal signaling and disease pathways and provide a rich resource for future work on these pathways.
Highlights.
Post-translational modification of cysteine is selective and specific in vivo.
Minimal crosstalk exists between different modifying agents.
The propensity for thiol modification is not due solely to chemical reactivity.
Acknowledgments
We thank Lynn Spruce, Palaniappan Sevugan Chetty, and Dr. Steve Seeholzer from the Children’s Hospital of Philadelphia Protein and Proteomics Core for assistance with sample analysis. This work was supported by National Institute of Health grants HL54926 (HI), GM102187 (KC), CA174986 (KC), GM065204 (VNG), and by a TUBITAK grant 113Z524 (SMM).
Footnotes
Author Contributions
NG designed and performed the experiments, analyzed data and wrote the manuscript. PMA performed the experiment and analysis for the SOH proteome and edited the manuscript. KSC conceived and directed the analysis of the SOH proteome and edited the manuscript. PE, SMM, and VNG provided bioinformatic analysis and edited the manuscript. HI conceived and directed the project, and edited manuscript.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Warren CM, Solaro RJ, Linke WA, Fernández JM. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell. 2014;156:1235–1246. doi: 10.1016/j.cell.2014.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar M, Thompson JW, Moseley MA, Stamler JS. Identification of S-nitrosylated targets of thioredoxin using a quantitative proteomic approach. Biochemistry. 2010;49:6963–6969. doi: 10.1021/bi100619k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britto PJ, Knipling L, Wolff J. The local electrostatic environment determines cysteine reactivity of tubulin. J Biol Chem. 2002;277:29018–29027. doi: 10.1074/jbc.M204263200. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cochemé HM, Reinhold J, Lilley KS, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway ME, Poole LB, Hutson SM. Roles for cysteine residues in the regulatory CXXC motif of human mitochondrial branched chain aminotransferase enzyme. Biochemistry. 2004;43:7356–7364. doi: 10.1021/bi0498050. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. S-glutathionylation in protein redox regulation. Free Radic Biol Med. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- Doulias P-T, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010;107:16958–16963. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulias PT, Raju K, Greene JL, Tenopoulou M, Ischiropoulos H. Mass spectrometry-based identification of S-nitrosocysteine in vivo using organic mercury assisted enrichment. Methods. 2013;62:165–170. doi: 10.1016/j.ymeth.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Science signaling. 2013;6:rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groitl B, Jakob U. Thiol-based redox switches. Biochim Biophys Acta. 2014;1844:1335–1343. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagel M, Niu D, St Martin T, Sheets MP, Qiao L, Bernard H, Karp RM, Zhu Z, Labenski MT, Chaturvedi P, et al. Selective irreversible inhibition of a protease by targeting a noncatalytic cysteine. Nat Chem Biol. 2011;7:22–24. doi: 10.1038/nchembio.492. [DOI] [PubMed] [Google Scholar]
- Hamnell-Pamment Y, Lind C, Palmberg C, Bergman T, Cotgreave IA. Determination of site-specificity of S-glutathionylated cellular proteins. Biochem Biophys Res Commun. 2005;332:362–369. doi: 10.1016/j.bbrc.2005.04.130. [DOI] [PubMed] [Google Scholar]
- Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karisch R, Fernandez M, Taylor P, Virtanen C, St-Germain JR, Jin LL, Harris IS, Mori J, Mak TW, Senis YA, et al. Global proteomic assessment of the classical protein-tyrosine phosphatome and “Redoxome”. Cell. 2011;146:826–840. doi: 10.1016/j.cell.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- Klatt P, Molina EP, Lamas S. Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J Biol Chem. 1999;274:15857–15864. doi: 10.1074/jbc.274.22.15857. [DOI] [PubMed] [Google Scholar]
- Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fassler R, Mann M. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell. 2008;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- Lim JC, Gruschus JM, Kim G, Berlett BS, Tjandra N, Levine RL. A low pKa cysteine at the active site of mouse methionine sulfoxide reductase A. J Biol Chem. 2012;287:25596–25601. doi: 10.1074/jbc.M112.369116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Löwenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- Linder ME, Deschenes RJ. New insights into the mechanisms of protein palmitoylation. Biochemistry. 2003;42:4311–4320. doi: 10.1021/bi034159a. [DOI] [PubMed] [Google Scholar]
- Marino SM, Gladyshev VN. Analysis and functional prediction of reactive cysteine residues. J Biol Chem. 2012;287:4419–4425. doi: 10.1074/jbc.R111.275578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino SM, Gladyshev VN. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J Mol Biol. 2010;404:902–916. doi: 10.1016/j.jmb.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino SM, Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J Mol Biol. 2010;395:844–859. doi: 10.1016/j.jmb.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Wang C, Adibekian A, Tully SE, Cravatt BF. Global profiling of dynamic protein palmitoylation. Nat Methods. 2012;9:84–89. doi: 10.1038/nmeth.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Acedo P, Gupta V, Carroll KS. Proteomic analysis of peptides tagged with dimedone and related probes. J Mass Spectrom. 2014;49:257–265. doi: 10.1002/jms.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Ruiz A, Lamas S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: convergences and divergences. Cardiovasc Res. 2007;75:220–228. doi: 10.1016/j.cardiores.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- Pace NJ, Weerapana E. Zinc-binding cysteines: diverse functions and structural motifs. Biomolecules. 2014;4:419–434. doi: 10.3390/biom4020419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore LA, Barford D. Getting into position: the catalytic mechanisms of protein ubiquitylation. Biochem J. 2004;379:513–525. doi: 10.1042/BJ20040198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salsbury FR, Knutson ST, Poole LB, Fetrow JS. Functional site profiling and electrostatic analysis of cysteines modifiable to cysteine sulfenic acid. Protein Sci. 2008;17:299–312. doi: 10.1110/ps.073096508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B, Ho L, Hogg PJ. Allosteric disulfide bonds. Biochemistry. 2006;45:7429–7433. doi: 10.1021/bi0603064. [DOI] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, Snyder SH. Hydrogen Sulfide-Linked Sulfhydration of NF-kB Mediates Its Antiapoptotic Actions. Molecular Cell. 2012;45:13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth D, Hausladen A, Wang YJ, Stamler JS. Endogenous protein S-Nitrosylation in E. coli: regulation by OxyR. Science. 2012;336:470–473. doi: 10.1126/science.1215643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YY, Tang W, Hao SF, Wang CC. Contributions of cysteine residues in Zn2 to zinc fingers and thiol-disulfide oxidoreductase activities of chaperone Dna. J Biochemistry. 2005;44:1683–1689. doi: 10.1021/bi0480943. [DOI] [PubMed] [Google Scholar]
- Smith BC, Marletta MA. Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr Opin Chem Biol. 2012;16:498–506. doi: 10.1016/j.cbpa.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MD, Igumenova TI. Reactive cysteine in the structural Zn(2+) site of the C1B domain from PKCalpha. Biochemistry. 2012;51:7263–7277. doi: 10.1021/bi300750w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, Friedman DB, Ham AJ. Verification of automated peptide identifications from proteomic tandem mass spectra. Nat Protoc. 2006;1:2213–2222. doi: 10.1038/nprot.2006.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J Biol Chem. 2009;284:436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood ZA, Schröder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.