

Abstract
Membrane proteins mediate processes that are fundamental for the flourishing of biological cells. Membrane-embedded transporters move ions and larger solutes across membranes, receptors mediate communication between the cell and its environment and membrane-embedded enzymes catalyze chemical reactions. Understanding these mechanisms of action requires knowledge of how the proteins couple to their fluid, hydrated lipid membrane environment. We present here current studies in computational and experimental membrane protein biophysics, and show how they address outstanding challenges in understanding the complex environmental effects on the structure, function and dynamics of membrane proteins.
Keywords: membrane proteins, lipids, protein structure, protein function, protein dynamics, membrane-mediated interactions
Introduction
The lipid membranes surrounding cells and their various compartments host proteins that perform functions essential for both cell physiology and disease progression. Recent evidence indicates that lipid membranes can largely influence the local structure, dynamics, and even the activity of a membrane protein (Bondar et al., 2010; Bondar, del Val & White, 2009; Jardon-Valadez, Bondar & Tobias, 2010; Kalvodova et al., 2005; Urban & Wolfe, 2005). This discovery highlights the need to understand how membrane proteins operate in their physiological lipid environment, which can be very complex itself. For example, the outer membranes of Gram-negative bacteria are asymmetric in nature; the outer leaflet is composed of lipopolysaccharide, while the inner leaflet incorporates a mixture of approximately 25 phospholipid types.
The present perspective article arose from the workshop “Coupling between protein, water, and lipid dynamics in complex biological systems: Theory and Experiments”, organized within the framework of the Centre Européen de Calcul Atomique et Molecular (CECAM) in September 2013. The workshop aimed to place the most recent developments of computer simulation techniques into a biological context to identify future challenges for membrane protein computations, and to find unifying concepts in protein/lipid/water coupling. We review here selected aspects of membrane protein biophysics from both computational and experimental viewpoints. In what follows, we first introduce briefly the biomolecules that are specifically discussed in this perspective article, then provide details on the methodologies used to study them and finally discuss results.
Membrane proteins are recognized and inserted into the lipid bilayer by exquisite cellular machineries, such as the Sec protein translocase. A central component of the Sec protein translocase is the SecY/Sec61 protein translocon, which is thought to open a lateral gate and release transmembrane protein segments (du Plessis et al., 2009; Plath et al., 2004; Van den Berg et al., 2004). Opening of a lateral helical gate toward the lipid membrane is also observed in a different membrane protein, the GlpG rhomboid protease; in the case of GlpG, opening of a lateral gate allows docking of a transmembrane substrate (Baker et al., 2007).
Ion channels are fundamental molecular components of signaling in our nervous system, and have been the subject of major advances in structural determination over the last decade (see, for example (Brohawn, del Marmol & MacKinnon, 2012; Chen, Durr & Gouaux, 2014; Cuello et al., 2010; Gonzales, Kawate & Gouaux, 2009; Karakas & Furukawa, 2014; Kato et al., 2012; Lenaeus et al., 2014; Payandeh et al., 2012). The perspective of the community on the role played by lipids in channel modulation has recently started to shift: whereas earlier work only considered the membrane as an adaptable matrix for protein functioning, recent data suggest that lipid molecules play fundamental structural and functional roles in ion transport. For example, direct interactions of ligand-gated ion channels with cholesterol, which play a functional role, were observed (Hénin et al., 2014). Another challenging area in studying ion channel is how highly charged helical segments can transverse the cell membrane. For example, the S4 transmembrane helix of the voltage sensor domain (VSD) or channelrhodopsins contain highly charged helical segments, which, however, can be incorporated into the lipid membrane (Hessa, White & von Heijne, 2005) (Del Val et al., 2014), highlighting the need to understand how membrane protein segments partition into the lipid membrane.
Membrane proteins account for about two thirds of known druggable targets in the cell and about 50% of all known small molecule drugs bind to membrane proteins (Lappano & Maggiolini, 2011; Tautermann, 2014). In this regard, no discussion of biological membranes would be worth its salt without mentioning G protein-coupled receptors (GPCRs) and proteins related to amyloid diseases. For both GPCRs and amyloidogenic peptides, interactions with lipids are essential. GPCRs are sensitive to the hosting lipid environment (Brown, 1994; Goddard et al., 2013; Hille et al., 2014; Oates et al., 2012; Oates & Watts, 2011), and interactions between oligomers of amyloidogenic peptides and membranes or lipids appear central to the cellular toxicity of amyloid proteins (Tofoleanu & Buchete, 2012a; Walsh et al., 2002; Walsh & Selkoe, 2007).
Given the advances in computational methodologies and computer power, theoretical approaches are likely to become increasingly important in the study of membrane proteins and their reactions. Studying the potential energy landscape provides both conceptual and computational tools for understanding a wide range of observable properties in membrane protein science. In particular, we can exploit stationary points (minima and transition states) for structure prediction and analysis of global thermodynamic and kinetic properties. Upon passage through membrane pores, peptides undergo conformational transitions and sample intermediates that block the transmembrane current that would otherwise flow in an open pore under a potential drop. We consider here how these intermediate states can be considered “jammed” states, similar to the dynamical arrest of macroscopic granular matter and in macroscopic glasses. Finally, scaling up to mesoscale systems via coarse graining, coupled with three-dimensional membrane-coupled systems-level modeling, bioinformatics, and appropriate visualization techniques up to the cellular level will be required. New computational technologies will form the basis of our future understanding of integrated membrane structure and function.
MEMBRANE PROTEIN ASSEMBLY, INSERTION AND LIPID INTERACTIONS
Physical partitioning
Physical membrane protein partitioning properties directly determine membrane protein folding, stability, and function, and their understanding is vital for rational design of membrane-active peptides. However, whereas in the 1970s and 1980s the folding of proteins in membranes was considered in the context of the physical aqueous-membrane partitioning problem, it is now known that nascent transmembrane (TM) polypeptide segments are recognized and inserted into the lipid bilayer by cellular machineries such as the Sec protein translocase. The protein translocon (Sec61 in eukayotes, SecY in prokaryotes) is an essential component of the Sec protein secretion machinery in all organisms (for reviews see, e.g., (Driessen & Nouwen, 2008; Rapoport, 2007; White & von Heijne, 2008). The translocon mediates the insertion of membrane proteins into the lipid membrane according to recognition rules that correlate strongly with physical hydrophobicity scales that describe the free energy of insertion of TM helices from water (Wimley & White, 1996). However, the exact relationship between the physical and biological scales is unknown, because solubility problems limit our ability to measure the direct partitioning of hydrophobic peptides across lipid membranes experimentally.
Experimentally, sufficiently hydrophobic peptides tend to aggregate, while atomistic computer simulations at physiological temperatures cannot yet reach the long time scales required to capture partitioning. Elevating temperatures to accelerate simulation of the dynamics has been avoided, as this was thought to lead to rapid denaturing. However, it has been shown that model TM peptides (WALP) are exceptionally thermostable (Ulmschneider et al., 2010). At these temperatures, sampling is ~50–500 times faster than at room temperatures (Ulmschneider et al., 2011), sufficient to directly simulate spontaneous partitioning at atomic resolution. Elevated temperature simulation ensembles further allow the direct calculation of the insertion kinetics. High-temperature simulations of experimentally validated thermostable systems suggest a new avenue for systematic exploration of peptide partitioning properties.
Using the above high-temperature methodology, in recent microsecond molecular dynamics (MD) simulations monomeric polyleucine segments of different lengths were allowed to partition spontaneously into and out of lipid bilayers (Ulmschneider et al., 2011). This approach directly revealed all states populated at equilibrium and allowed the free energy of insertion to be directly obtained from the relative occupancy of these states. The partitioning free energy of the polyleucine segments thus determined was found to correlate strongly with values from translocon experiments. Arguably, the most interesting result of this study was the close correlation between the spontaneous surface-to-bilayer partitioning equilibrium and the translocon-mediated insertion scale. However, the results show that ~2 fewer leucines than predicted by the translocon scale are required for the peptides to insert on their own. The probable explanation for this is that two different partitioning processes are involved. In contrast to partitioning between bilayer interface and hydrocarbon core, the translocon crystal structure suggests that hydrophobic peptides are released laterally from the protein-conducting channel into the membrane. This means that the experiments measure the partitioning of peptides between translocon channel and bilayer, rather than between water-soluble and TM configurations. This is consistent with a translocon-to-bilayer equilibrium being at the heart of the translocon-mediated insertion probability. The close correlation of the simulation results with translocon experiment suggests that the surface-bound helical state of the peptides might be located in a region of hydrophobicity similar to that of the internal translocon pore. Thus, the equilibria of spontaneous partitioning and translocon-mediated insertion are likely independent, with no thermodynamic cycle connecting the two insertion paths. Both differ highly from the much larger free energy changes involved in non-equilibrium water-to-bilayer partitioning.
Dynamics of the translocon
The functioning of the translocon is associated with significant rearrangements of the protein, and crystal structures provide valuable snapshots of the translocon in different conformational states (Egea & Stroud, 2010; Tsukazaki et al., 2008; Van den Berg et al., 2004; Zimmer, Nam & Rapoport, 2008). Particularly important is the geometry of helices 2 and 7 of the translocon, because these two helices are thought to function as gates that open towards the lipid membrane to release hydrophobic peptides (du Plessis et al., 2009; Plath et al., 2004; Van den Berg et al., 2004). In the closed state of the translocon, helices 2 and 7 hydrogen bond to each other and to helix 3, which hydrogen bonds to four other segments of the translocon (Bondar et al., 2010). The translocon has numerous hydrogen bonds that interconnect its various structural elements, and the gate helices 2 and 7 are part of a hydrogen-bonded network that stabilizes the closed state of the translocon (Bondar et al., 2010).
The dynamics of the central hydrogen-bonded cluster of helices 2, 3 and 7 has some remarkable features that could not have been anticipated from the crystal structure: of the five hydrogen bonds of the cluster, three have a multi-state character, breaking and reforming on the picosecond-nanosecond timescale (Bondar & White, 2012). Importantly, in crystal structures that glimpse into open states of the translocon (Egea & Stroud, 2010; Tsukazaki et al., 2008; Zimmer et al., 2008) groups of the central hydrogen-bonded cluster have rearranged such that helix 7 appears to have broken away from hydrogen bonding with helices 2 and 3 (Bondar & White, 2012). The comparison of the hydrogen-bond analysis and of the crystal structure then suggests that hydrogen bonds with complex conformational modes are implicated in the control of the protein conformational state (Bondar & White, 2012).
Targeting cellular organelles by amphipathic helices
One crucial membrane property that has been suggested to favour translocon-assisted membrane protein folding is the extremely low cholesterol concentration of the endoplasmic reticulum (ER) of eukaryotic cells (Bretscher & Munro, 1993). The peculiar lipid composition of the ER, abundant in conical shaped lipids, such as diacylglycerols or those containing monounsaturated fatty acids (Bigay & Antonny, 2012), appears thus well adapted to decrease the energetic barriers for protein insertion thanks to its intrinsic plasticity. In contrast, late secretory pathway membranes (plasma membrane, late endosomes), are much stiffer, because of their enrichment in cholesterol and sphingolipids.
Remarkably, not only proteins that partition to the interior of the bilayer, but also those that partition to its surface, are sensitive to the lipid composition of the various membrane organelles, and can take advantage of the remarkable plasticity of early secretory pathway (ER, Golgi apparatus) membranes (Bigay & Antonny, 2012). The most common protein motifs able to recognize these specific membrane properties are amphipathic helices, short protein motifs that are usually unfolded in solution and that fold into an alpha helix upon binding with the membrane surface.
To accurately characterize how amphipathic helices can sense membrane properties that depend on lipid composition, all-atom MD simulations, in combination with a topographical method able to scan the bilayer surface at the lipid-water interface, have been used to show that bilayers containing a high percentage of conical lipids are also enriched in defects in lipid packing (Vamparys et al., 2013) and that these defects resembles those generated by curvature.
These defects can be recognized by amphipathic helices, such as the membrane-curvature sensing Amphipatic Lipid Packing Sensor (ALPS) motif: all-atom MD simulations show that the ALPS motifs partition into the bilayer through repetitive insertions of hydrophobic residues into pre-existing lipid packing defects and the partitioning is dramatically slowed down in the presence of tightly packed lipid bilayers (Vanni et al., 2013). These studies suggest that computer simulations are a powerful methodology to investigate, with atomic-level resolution, how membrane properties change as a function of lipid composition and how these properties can be sensed by protein motifs (Vanni et al., 2014).
PROTEOLYSIS IN THE LIPID MEMBRANE PLANE: RHOMBOID PROTEASES
The protein translocon discussed above uses a lateral gate to release transmembrane helices into the lipid bilayer. A lateral gate to the membrane is also used by rhomboid proteases to dock their transmembrane substrates. The tight communication between the lipids and the protease is highlighted by the observation from experiments that the proteolytic activity of the rhomboid proteases is drastically affected by the composition of the lipid bilayer – for example, the Escherichia coli GlpG rhomboid protease is active in phosphatodylethanolamine (PE) lipids, but not in phosphatodylcholine (PC) (Urban & Wolfe, 2005). This marked dependency of the proteolytic activity on lipids may be explained in part by the different hydrogen-bonding properties of different lipid head groups: MD simulations of GlpG in POPE vs. POPC lipid bilayers indicated that the lipid membrane composition affects not only the local hydrogen bonding between lipid head groups and protein surface amino acid residues, but it also influences hydrogen bonding between specific protein groups, and the orientation of the protease relative to the lipid membrane (Bondar et al., 2009). A key open question is how the transmembrane substrate is docked and cleaved by the protease. This question is addressed below from the viewpoint of X-ray crystallography, biochemistry and cell biology.
Rhomboids have low affinity for their transmembrane substrates
While rhomboid enzymes are the best understood intramembrane proteases structurally (Baker & Urban, 2012), the mechanistic details of their catalytic activity inside the membrane are still unclear. A new reconstitution system has been developed to study the kinetics of rhomboid proteolysis inside the membrane (Dickey et al., 2013). In this system prior hurdles were systematically overcome by substrate turnover and allowing the reaction to be initiated synchronously. Fluorescence of the released product provided a sensitive and quantitative means to measure initial rates within the first 12 minutes in real time. This is a new achievement, because current intramembrane protease assays are performed in detergent and over the course of hours. The resulting steady-state kinetics revealed rhomboid proteases have very low affinity for their substrates in the membrane, which was confirmed by measuring the Kd directly and validated by following cleavage in living cells. The reaction is also slow. These two features revealed a surprising similarity to DNA repair enzymes that monitor substrate dynamics. In a similar way, rhomboid proteases may target their substrates by identifying transmembrane segments that are able to unfold inside its active site. This property might also explain why no enzyme-substrate complex structure has been achieved despite nearly a decade of effort. Consequently, excellent opportunities exist for MD simulations to explore how substrates behave as they approach the protease, enter the active site, and are ultimately cleaved.
Dimer model for the full-length E. coli rhomboid intramembrane protease
To date the only crystal structures of rhomboid available are determined from Haemophilus influenzae (hiGlpG) (Lemieux et al., 2007), and the core E. coli GlpG membrane domain (Ben-Shem, Fass & Bibi, 2007; Brooks et al., 2011; Dickey et al., 2013; Lemieux et al., 2007; Vinothkumar, 2011; Vinothkumar et al., 2010; Wang & Ha, 2007; Wang et al., 2007; Wang, Zhang & Ha, 2006; Wu et al., 2006; Xue et al., 2012; Xue & Ha, 2012). Both are found in the same phylogenetic branch (Urban & Dickey, 2011), and not surprisingly, have similar three dimensional structures (Lemieux et al., 2007). Both consist of six TM domains that assemble to form an α-helical bundle. A catalytic Ser-His dyad buried 10–12 Å deep within the bilayer is located within this bundle. Between helices 1 and 2 is a loop, containing some invariant residues, that partially extends into the lipid bilayer.
The E. coli rhomboid GlpG represents a classical prokaryotic rhomboid that has a soluble N-terminal cytoplasmic domain in addition to the six transmembrane segments.. The physiological function for GlpG is unknown, and furthermore the role for this cytoplasmic domain in GlpG is unclear. No structural information exists for the full-length E. coli rhomboid. We have determined that the presence of the cytoplasmic domain does not affect the catalytic rate or specificity of GlpG (Lazareno-Saez et al., 2013). The crystal structure of the E. coli rhomboid cytoplasmic domain reveals a domain swapped dimer. This structure may be physiologically relevant as it is dimeric not only in solution but when in contact with the membrane domain (Arutyunova et al., 2014). The membrane domain of prokaryotic rhomboid is also dimeric independent of this cytoplasmic domain (Sampathkumar et al., 2012). We hypothesize that dimerization is a key step for regulation of intramembrane proteolysis.
Rhomboids are synthesized as active enzymes and are known to cleave substrates with minimal specificity. A mechanism must exist to achieve specificity for this enzyme. Movements in loop 5 and helix 5 around the buried active site are thought to provide access to the catalytic machinery (Baker et al., 2007; Brooks et al., 2011; Wang & Ha, 2007), and may provide a means for substrate specificity. Dimerization of the cytoplasmic domain would stabilize the dimer of the catalytic membrane domain, and may provide the structural changes necessary to promote conformational changes in the active site. Here we present a model where dimerization occurs at both the membrane domain and the cytoplasmic domain (Figure 1). The two domains are connected by a flexible linker. The dimerization interface of the membrane domain, here modeled to occur between the conserved loop 1, needs to be confirmed both experimentally and with molecular dynamics.
Fig 1.
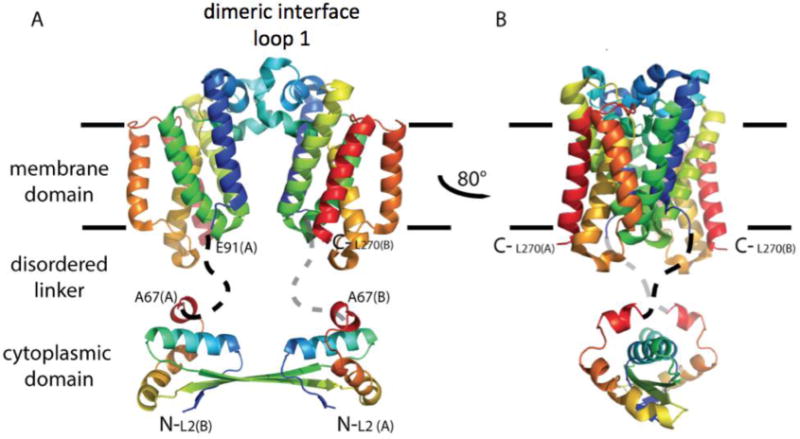
Hypothetical model for full-length E. coli GlpG dimerization. A cartoon illustration is shown in rainbow coloring with N-terminus in blue. The membrane domain of E. coli GlpG is dimeric. In this model, the dimer interface occurs through interaction of loop 1 that extends partially in the lipid bilayer (light blue). The cytoplasmic domain is separated from the membrane domain by a flexible linker. The structure of the cytoplasmic domain is from Ref. (Lazareno-Saez et al., 2013).
ION CHANNELS
Studying ion channel function using fully atomistic molecular dynamics simulations
Ion channels catalyze rapid and selective transport of salt ions, such as Na+, K+, Cl− and Ca2+, across cell membranes to enable electrical and chemical activity in the body. They are associated with many neurological, muscular and cardiac disorders, which establishes them as important pharmacological targets (Clare, 2010).
The solution of the first high-resolution crystal structure in 1998 for the bacterial K+ channel, KcsA (Doyle et al., 1998) (see Figure 2a), opened the door to molecular-level insight into ion channel conduction. In the extracellular-facing pore segment, called the selectivity filter, the protein backbone forms a narrow, ion-sized pore, presenting a sequence of carbonyl cages to coordinate almost dehydrated K+ ions, and is responsible for ion discrimination. It has long been thought that this filter is inherently better at stabilizing K+ over Na+ ions (Neyton & Miller, 1988), suggesting selective conduction via a ‘thermodynamic’ mechanism of selective binding. Based on the KcsA structure, it was argued that this preference is due to a ‘snug fit’ of the K+ ion inside the selectivity filter, which is too big for a Na+ ion (Doyle et al., 1998). The high-resolution structure in itself does not, however, tell the full story. Proteins are often pictured as rigid entities, but they are in fact highly flexible, with even the most rigid protein backbones fluctuating (and able to deform) on the Å-scale, critical for ion channel conduction (Allen, Andersen & Roux, 2004; Noskov, Berneche & Roux, 2004). For the protein to prohibit ~0.4 Å adjustment of carbonyl groups to account for K+-Na+ size differences, despite the attractive interactions between ion and carbonyl groups, it would have to be as stiff as diamond (Allen et al., 2004). Clearly mechanisms of ion channel selectivity must emerge from models that incorporate the thermal fluctuations of the protein, from picosecond vibrations to structural isomerizations that span up towards the microsecond scale.
Fig 2.
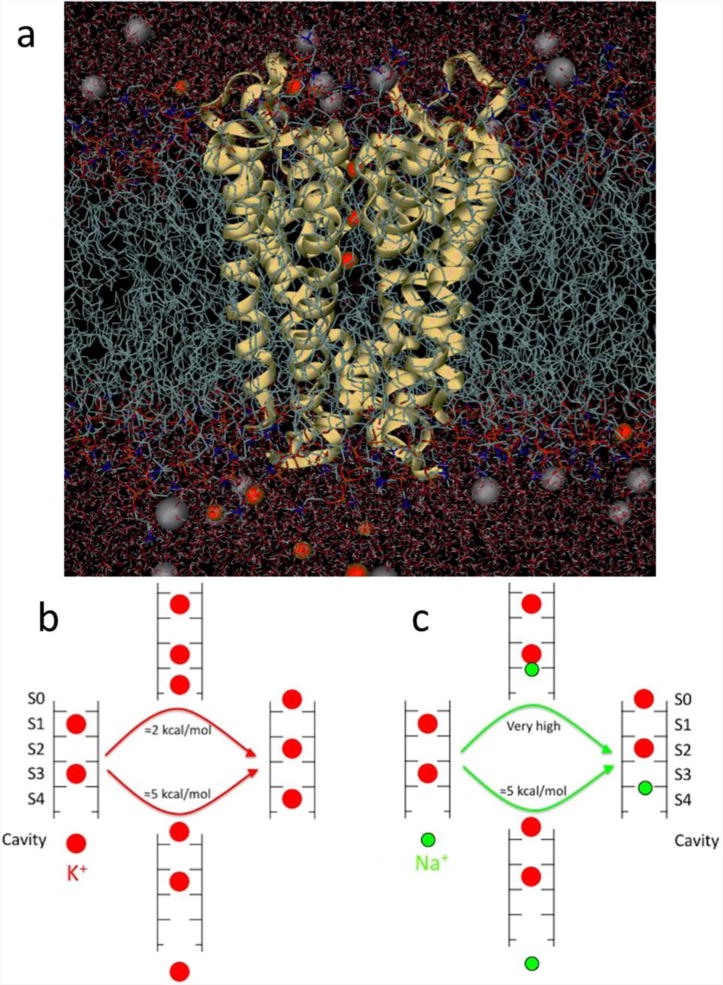
(a) Fully atomistic KcsA ion channel system (protein as yellow ribbons, lipid tails as chains, water as sticks and K+ and Cl− ions as red and grey balls, respectively). (b) Differing multi-ion barriers for K+ and Na+. The low barrier to K+ outward conduction results from the ability of a K+ ion (red) to bind to the entrance of the selectivity filter. (c) The shift of binding site for Na+ (green), eliminates this process and requires a substantial amount of energy. Based on (Nimigean & Allen, 2011).
Much progress has been made with MD simulation, based on this structure, to understand the mechanisms by which K+ channels achieve selective conduction (Andersen, 2011). Although there has been considerable attention given to the preference of carbonyl cage geometries for K+ ions, focusing on the field strength created by the ligands, the strain energies associated with ligand-ligand repulsion, and the role of thermal fluctuations (e.g. (Noskov et al., 2004), it has been suggested that the selective binding picture is oversimplified. Recent calculations have shown that this preference is smaller than previously thought, simply because the cage of carbonyl groups is the K+ binding site, but that the selectivity filter prefers to house the smaller Na+ ions at adjacent plane of carbonyl sites (such that focusing only on cage sites inherently biases calculations to favor K+). Instead, on average, there is only a small overall preference for K+ in the middle of the selectivity filter, and surprisingly, a preference for Na+ at the entrances of that region (Kim & Allen, 2011). It has been shown that selective permeation can instead arise from differences in the barriers for the concerted motions of multiple ions through the channel (Figure 2b and c; (Nimigean & Allen, 2011; Thompson et al., 2009). This ‘kinetic view’ of selective exclusion (Bezanilla & Armstrong, 1972) implies that one must elucidate the full free energy landscape governing the movements of all of the ions in the pore.
With this in mind, and armed with a new crystal structure for a bacterial Na+ channel (Payandeh et al., 2011), research has rapidly moved towards uncovering the contrasting mechanisms of selectivity in Na+ channels. Simulations have revealed a surprising degree of protein flexibility for this channel, especially in the important selectivity filter, and have shown that is essential to invest in sufficient sampling times, if one wants to obtain quantitative measures of function (Boiteux, Vorobyov & Allen, 2014a; Chakrabarti et al., 2013). Use of purpose-built computational architectures to simulate large membrane protein systems, for multiple microseconds, up to milliseconds, e.g. (Jensen et al., 2012), have indeed revealed protein conformational changes relevant to activity, e.g. (Boiteux et al., 2014a; Boiteux et al., 2014b).
Despite the ability to carry out long enough simulations to begin to observe function, we still seek quantiative and reproducible measures to understand mechanisms and compare to experiments. An obvious route is to carry out equilibrium sampling to produce a free energy surface, via its connection to the probability distribution, or the potential of mean force (PMF) along the reaction coordinate. The calculation is typically done using Umbrella Sampling (Roux, 1995), where a sequence of independent simulations, distributed along the coordinate, biased to ensure local sampling within every region of that coordinate, can then be recombined after simulation in an optimal way, such as via the Weighted Histogram Analysis Method, WHAM (Kumar et al., 1992). Interestingly, however, with the use of new computer architectures that allow multi-microsecond unbiased ion channel simulations, complete sampling of the multipleion mechanisms of conduction have been obtained without Umbrella Sampling (Boiteux et al., 2014a). Those same long simulations have also revealed conformational changes involving trans-membrane helix bending and gate collapses that relate to slow inactivation in Nav channels, despite these processes typically occurring on much longer timescales. They have also uncovered the interplay between the protein and the lipid membrane via fenestrations in the pore domain. Importantly, they have revealed the distribution of lipids and lipophilic drugs around the channel pore domain and fenestrations, as well as key details of the interactions responsible for drug binding and pathways (Boiteux et al., 2014a; Boiteux et al., 2014b). The field of biomolecular simulation is therefore beginning to uncover the quantitative mechanisms of ion channel function and modulation, enabling future developments in drug discovery.
Voltage Sensor Domain (VSD) Activation
The VSD is a transmembrane molecular device used by cells to transduce an electrical signal across the plasma membrane. Composed of a solvated four transmembrane helix bundle (Figure 3), this device can be coupled to an ion channel pore (voltage-gated cation channel – VGCC), to a phosphatase (voltage-sensing phosphatase-VSP) or can be a self-standing selective channel (proton voltage-gated channel – Hv). Amongst the four helices, S4 stands out with its large number (4 to 7) of positive charges (Arg and Lys) spread along its lumen facing side: as such, it constitutes the primary voltage-sensor (Palovcak et al., 2014). These charges are stabilized in a transmembrane position through their interaction, not only with the negative charges of S1–S3, but also with the negatively charged phosphate groups of the lipid membrane, creating a favorable solvated environment. When a hyperpolarizing electric field is applied, S4 is dragged downwards in a ratchet-like movement, as the salt bridge pairings between the S4 positive charges and their counterparts rearrange in a step-wise manner. Because of the hourglass volume accessible to water within the VSD, the electric field is focused over a narrow hydrophobic region (sometimes called the hydrophobic plug), made up by several hydrophobic residues, including a conserved PHE residue on S2 and ILE on S1. In VGCCs, an intracellular interfacial helix (the S4–S5 linker) couples the VSD to the pore helices (S5–S6) helping therefore to convert electrical energy of the membrane potential into the mechanical energy needed to open and close the S6 gate.
Fig 3.
The transmembrane Kv1.2 voltage-gated cation channel.
Kv1.2 is the first mammalian VGCC of which the open state crystal structure was revealed. Using an MD based protocol to trigger deactivation (closing) of the channel in a model POPC membrane the details of the S4 sensor downwards movement were revealed (Delemotte, Klein & Tarek, 2012; Delemotte et al., 2011; Tarek & Delemotte, 2013). Interestingly, several of the S4 positive charges are stabilized by their interaction with the lipid head groups, not only in the activated state, where the two top charges are bound to top leaflet phosphate groups, but also in the intermediate and resting states: in the middle intermediate state, the outermost charge is in contact with the top PO4-group and the innermost charge in contact with the inner leaflet PO4-groups and in the resting state, the innermost three charges interact with the inner leaflet phosphate group.
VGCC modulation by lipids
The perspective on how lipids modulate ion channels has recently started to change (Kasimova et al., 2014). Indeed, earlier work only considered the membrane as an adaptable matrix for protein functioning. Recent data, however, revealed that lipid molecules have fundamental structural and functional roles (Sands & Sansom, 2007; Schmidt, Jiang & MacKinnon, 2006; Treptow & Tarek, 2006). For example, the activation of the VSD is modified by the alteration of sphingomyelin head group of the outer membrane leaflet by sphingomyeliase (Combs et al., 2013; Ramu, Xu & Lu, 2006; Xu, Ramu & Lu, 2008). Such a mechanism is taken advantage of by the spider Loxoceles reclusa, which carries this enzyme in its venom and uses it to paralyze its prey. Another well-studied example is the specific role of PIP2, a signaling lipid of the membrane inner leaflet, which modulates several processes in the working cycle of VGCCs (Kruse, Hammond & Hille, 2012; Logothetis et al., 2010; Suh & Hille, 2008).
In principle, two hypotheses can be proposed to rationalize the macroscopic VGCC response at a molecular level: altering the lipid head groups may 1) modify the global properties of the membrane, and more specifically the electrostatic environment or 2) change the local interaction pattern between protein residues and their environment. Insights gained into this question are discussed using MD simulations.
1. Sphingomyelin alteration
Sphingomyelin is an abundant lipid (forms up to 15%) of the outer layer of mammalian cell membranes (Combs et al., 2013). Recently, Lu and co-workers have shown that enzymes present in the venom of Loxoceles reclusa target VGCCs, causing thereby paralysis (Combs et al., 2013; Ramu et al., 2006; Xu et al., 2008). The mode of action of these enzymes is very peculiar since they target the lipid matrix rather than the protein itself. Specifically, Smase D removes the sphygomyelin choline group leaving a negatively charged lipid (ceramide-1-phosphate) that promotes activation of the channel. Smase C, on the other hand, removes the entire sphygomyelin zwitterionic head group and leaves behind a polar molecule (ceramide). This favors the closed state of the channel.
The electrostatics of asymmetric bilayers in which the top leaflet is made of 100% of one of the three sphingomyelin derivatives (sphingomyelin itself, ceramide-1-phosphate and ceramide) and the bottom one of 100% phosphatidylcholine were studied. Interestingly, the contributions to the electrostatic potential profile along the normal to the bilayer from the individual components of the top layer (lipids, water and counterions) are system-dependent, i.e. the most polarizable elements counteract the rather static dipole potential from the lipid head groups. However, surprisingly, summing these different contributions leads to the same overall transmembrane potential of ~200 ± 50 mV. The Kv1.2 VSD (Delemotte et al., 2011; Long, Campbell & Mackinnon, 2005) in three different states (activated, intermediate and resting) in these three asymmetrical bilayers were then inserted. The gating charge, i.e. the quantity of charge transported by the VSD during activation was subsequently calculated (Delemotte et al., 2012; Treptow, Tarek & Klein, 2009). Through its energetic formulation, the gating charge is an indirect measurement of the rearrangement of the electric field acting on the protein upon external voltage application (Roux, 1997). Therefore, it can be computed for each transition from one state to another. In this particular case, the analyses have shown that the gating charges do not vary much with the environment, i.e. with the lipid composition. Taken together, these results indicate that the reshaping of the electric field is independent of the lipid composition and rule out any significant global effect of the electrostatic environment on the VSD functioning subsequent to modification of the lipid.
On the other hand, close inspection of the systems reveals lipid-dependent interactions with the S4 outermost positive residues, providing support to the local effect hypothesis. The presence of a phosphate in the sphingomyelin head group provides a stable binding site for the top two S4 arginines when the VSD is activated. In ceramide-1-phosphate, this interaction is expected to be more stabilizing due to the absence of a countercharge in the lipid head group. In ceramide, since there is no phosphate group, it was hypothesized that the interaction between the VSD and the lipids head groups is much weaker. Relaxation of the three systems using unbiased MD simulations exceeding the 100 ns time scale under large hyperpolarizing potentials were not enough to reveal any significant destabilization of the interactions between the VSD and the lipid head groups. Free energy calculations are now under way to fully characterize the interactions involved, and link the changes in activation kinetics to experimental observables.
2. PIP2 modification
Phosphatidylinositol-(4,5)-bisphosphate (PIP2), a high negatively-charged phospholipid found in the inner membrane leaflet, has been shown to modulate the function of voltage-gated potassium channels of the Shaker-family (Abderemane-Ali et al., 2012; Rodriguez-Menchaca et al., 2012). The application of PIP2 to these channels leads both to a gain-of-function, namely an increase of the ionic current, and to a loss-of-function manifested by the right shift of G/V and I/V curves, describing respectively the gating and ionic current voltage dependencies.
Because PIP2 is a rare component of the membrane (1% of the inner leaflet of most cells any global effect was ruled out. Rather, the focus was placed on the specific interactions between the channel and the lipids in a system in which the ring of phosphatidylcholine molecules of the inner leaflet embedding the channel was replaced by PIP2 lipids. From 100 ns MD simulations of the Kv1.2 channel embedded in such a model lipid bilayer, it was shown that PIP2 molecules interact electrostatically with positively charged residues of the channel in a state-dependent manner. In particular, PIP2 lipids were found to interact with two different sites of the channel that bear an excess amount of positive charges. The first site (I) is composed by the S4–S5 linker outermost residues (LYS312, ARG326). The other site (II) is located in the middle of the S4–S5 linker (LYS322) and is occupied by residues in a state-dependent manner. In the closed state, aside from LYS322, site II encloses the S4 lower basic residues ARG303, LYS306 and ARG309, consistent with PIP2 stabilizing the OFF position of the voltage sensor. In the open state, ARG419 of the S6 C-terminal are in close contact with the middle of the S4–S5 linker (witnessed in three subunits out of four) occupying therefore site II. This is consistent with PIP2 stabilizing the open gate conformation through a mechanism similar to the one described for Kir channels (Hansen, Tao & MacKinnon, 2011; Whorton & MacKinnon, 2011).
In the up state, all S4 basic residues are engaged in salt bridges with other residues of the VSD, or with the upper leaflet lipid head groups. Within the first ON to OFF transition, as S4 translate down, the lowest residue ARG309 comes in close contact with the lipids of the lower leaflet (Delemotte et al., 2011). Consistent with this, one expects that the presence of PIP2 in this lower leaflet would modify the kinetics associated with this early transition, i.e. leads to accelerated deactivation. Accordingly, the models seem to agree well with the electrophysiology data (Abderemane-Ali et al., 2012; Rodriguez-Menchaca et al., 2012) and provide an insight into the molecular mechanisms involved in the seemingly contradictory gain- and loss-of-function effects of intracellular PIP2.
This work is being currently extended to other channels of the VGKC family (Kv7 sub-family) that have been shown also to require PIP2 for proper function.
Drug design for ion channels
Ion channels are expressed ubiquitously in cells and are implicated in a variety of physiological processes. Thus, targeting them with small molecules that can act as drugs is particularly challenging (Bagal et al., 2013).
The use of computational methods can greatly enhance our understanding of drug-protein binding processes, provide estimates of drug binding affinities, and may ultimately contribute to the development of novel, potent protein inhibitors. Common methodologies used in estimating ligand-binding affinities include docking, continuum electrostatic methods such as the Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) techniques, or Free Energy Perturbation (FEP) calculations. FEP calculations are an accurate quantitative link between experimental and computational studies (Erion et al., 2007; Lamb & Jorgensen, 1997; Reddy et al., 2007; Reddy & Erion, 2001; Zwanzig, 1954).
Relative binding free energies are calculated using the Thermodynamic Cycle Perturbation method. Using this thermodynamic cycle, the relative change in the free energy of binding between two ligands A and B is calculated through nonphysical paths connecting a reference and a terminal state (Zwanzig, 1954). Using Cycle 1 (Figure 4), the difference in binding free energy between ligand A and ligand B can be calculated, while the relative solvation free energy difference between two ligands A and B can be calculated based on Cycle 2 (Figure 4)
Fig 4.
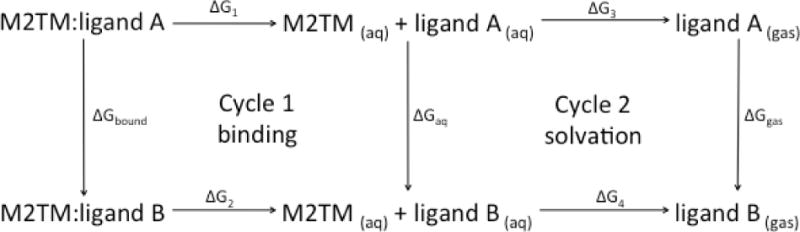
Thermodynamic cycle used to compute free energy differences for estimating the difference in the free energy of binding of a ligand to a protein (Cycle 1) or for the relative solvation free energy difference (Cycle 2).
Calculations of relative free energies of ligand binding in membrane proteins and in particular in ion channels using the FEP methodology combined with MD simulations (FEP/MD) have been performed only for a limited number of systems and ligand alchemical transformations (Gkeka et al., 2013; Gonzalez et al., 2011; Zhao, Caplan & Noskov, 2010). We review below the usage of FEP/MD calculations to design novel drug candidates for A/M2TM.
The A/M2TM protein is a tetrameric pore of a proton channel that is activated by low pH in the viral endosome. The main class of known A/M2TM blockers is adamantane derivatives, such as amantadine and rimantadine (Figure 5, left), that can block the A/M2TM pore via a pore blocking mechanism (Cady et al., 2010; Cady et al., 2011; Chuang et al., 2009; Gu et al., 2011; Rosenberg & Casarotto, 2010; Stouffer et al., 2008). Based on this pore blocking mechanism, docking calculations and FEP/MD calculations with a restrained A/M2TM protein system were performed and resulted in poor or low correlation with experimental data (R2 ≤ 0.20) (Gkeka et al., 2013). This low correlation was attributed to the limited flexibility of the protein in the system, as the use of a “frozen” environment introduces a dependency of the free energies of binding on the selection of the protein structure, as discussed elsewhere (Michel, Foloppe & Essex, 2010). An improved protocol, where protein flexibility of the system was taken into account by embedding the protein inside a fully hydrated DPPC lipid bilayer, yielded an excellent correlation with the available experimental data (R2 = 0.85, Figure 5, right), indicating that protein flexibility dramatically improves the accuracy of FEP calculations for this system. Comparison between the different MD trajectories indicated that protein flexibility greatly influences ligand location inside the pore, affecting their hydrogen bonding ability and overall interaction network. In addition, the calculated bundle tilt with respect to the membrane normal for the different ligands in the flexible protein system varied between 32° and 38° relative to the membrane plane in agreement with previous studies (Cady & Hong, 2008; Wang et al., 2001), while it was zero for the restrained system. This bundle tilt change reflects differences in tetramer flexibility. Though keeping the protein backbone rigid yields accurate results for certain binding pockets (Baggett et al., 2012), backbone flexibility appears essential in the case of A/M2TM.
Fig 5.
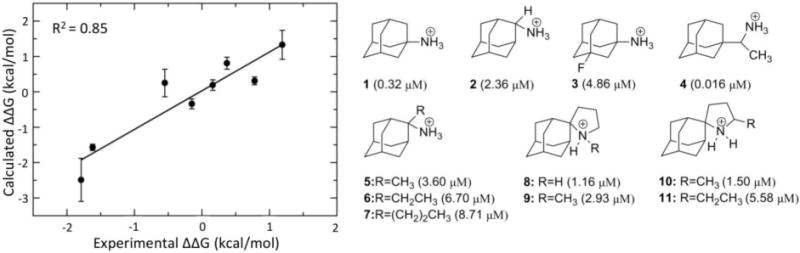
Structures of M2TM inhibitors and their binding constants. On the left side, note the excellent correlation between the FEP/MD calculations with a flexible protein and the experimental data. The image is adapted from (Gkeka et al., 2013).
In (Gkeka et al., 2013), ligand desolvation was used for the first time to provide an explanation for the binding preference of close aminoadamantine derivatives such as the 20-fold higher affinity of rimantadine 4 relative to amantadine 1 (Figure 5). It was shown that the desolvation free energy penalty is a major contributor to the A/M2TM binding affinity and should be considered for drug design purposes targeting membrane-associated systems. Decrease in the penalty of ligand desolvation facilitates its entrance into the binding site crevice and thus higher binding affinity can be obtained. However, a delicate balance is required between introducing hydrophobic elements on the ligands in order to decrease the ligand desolvation penalty and achieving optimal ligand-protein interactions (Gkeka et al., 2013).
Analysis of protein-ligand binding MD trajectories may reveal important interactions that affect the binding process. In the A/M2TM case, rimantadine 4 is implicated in favorable hydrophobic contacts due to its additional CHCH3 bridge with residues closer to the C-terminal of the A/M2TM pore, which are missing in amantadine 1. Also, hydrogen bond analysis showed that amantadine forms on average two hydrogen bonds with neighboring water molecules, while rimantadine forms three hydrogen bonds with water molecules almost throughout the simulation time. The increased hydrogen bonding ability of 4, the additional hydrophobic interactions as well as the higher desolvation free energy compared to 1 may well explain the higher binding affinity of rimantadine to the A/M2TM channel. Overall, based on similar analysis performed on compounds 1–11 (Figure 5), it was demonstrated that a delicate balance of intermolecular interactions, hydrogen bonding, and free energy of solvation is required to achieve enhanced ligand-protein binding in membrane protein systems (Gkeka et al., 2013).
Direct interactions of ligand-gated ion channels with cholesterol
Ligand-gated ion channels of the nicotinic acetylcholine receptor (nAChR) family have long been known to be extremely sensitive to their lipid environment, particularly cholesterol. The first evidence for molecular interactions between the nAChR and cholesterol is a functional requirement: the receptor is not functional when purified and reconstituted in phospholipid mixtures, unless cholesterol or chemically related compounds are added (Criado, Eibl & Barrantes, 1984; Dalziel, Rollins & McNamee, 1980; Kilian et al., 1980; Ochoa, Dalziel & McNamee, 1983). Although interactions of GABAA with cholesterol have received less attention, purification and reconstitution protocols for GABAA receptors either include cholesterol (Bristow & Martin, 1987; Dunn et al., 1989), or yield non-functional and unstable receptors (Bristow & Martin, 1987). Subsequent experiments depleted native membranes of cholesterol using liposomes (Lechleiter, Wells & Gruener, 1986; Leibel et al., 1987; Zabrecky & Raftery, 1985) or cyclodextrins (Baez-Pagan et al., 2008; Borroni et al., 2007; Santiago et al., 2001). An important result by Leibel et al. (Leibel et al., 1987) is that certain cholesterol molecules (about 36 molecules per receptor) remain in the system despite extensive depletion. Hence, this procedure may primarily modulate the cholesterol content of the bulk membrane. The theory that depletion/enrichment experiments access a different pool of cholesterol than reconstitution experiments is further supported by the opposing trend they yield: in depletion/enrichment experiments increased cholesterol content is associated with loss of nAChR function (Borroni et al., 2007; Santiago et al., 2001).
Functional modulation of GABAA receptors by cholesterol has been demonstrated by Sooksawate and Simmonds (Sooksawate & Simmonds, 2001a). In this system, both enrichment and depletion of cholesterol reduce the potency of the receptor, but while the enrichment effect can be reproduced using epicholesterol, introducing epocholesterol did not reverse the depletion effect. Thus, it was suggested that the depletion effect reflects direct interactions, whereas the enrichment effect is mediated by physical properties of the membrane.
Functional studies as summarized above are complemented by biophysical results demonstrating the existence of strong association between the nAChR and a pool of proximal cholesterol. The nAChR has a higher affinity for cholesterol than other lipids, as evidenced from early lipid monolayer experiments by Popot et al. (Popot et al., 1978). Electron paramagnetic resonance (EPR) measurements by Marsh et al. (Marsh & Barrantes, 1978; Marsh, Watts & Barrantes, 1981) using spin-labeled lipid probes found that some lipids are immobilized in the presence of the receptor – in particular, the steroid androstanol exhibits hindered rotation around its long axis. High affinity of the receptor for cholesterol or its analogues was further demonstrated by EPR (Ellena, Blazing & McNamee, 1983), photolabeling (Middlemas & Raftery, 1987), and fluorescence studies (Arias et al., 1990; Barrantes et al., 2000).
Cholesterol also has significant effects on the nAChR function at the cellular level, which have been reviewed recently in the case of the murine muscle-type nAChR (Barrantes, 2007). Here we focus on molecular-level interactions and their basis in the protein structures. For a review of cholesterol modulation of membrane receptors (nAChR and G protein-coupled receptors), see refs. (Barrantes, 2004; Burger, Gimpl & Fahrenholz, 2000).
Embedded cholesterol in a nicotinic receptor
Medium resolution cryo-electron microscopy images of the nAChR observed within a native lipid environment (Unwin, 2005) provided the earliest atomic-level insights into pLGIC structure (Figure 6, PDB ID:2BG9). Surprisingly, the structure indicated several significantly sized gaps in protein density within the transmembrane domain. Similar gaps have not been observed in more recent crystal structures of other (primarily prokaryotic) pLGICs.
Fig 6.
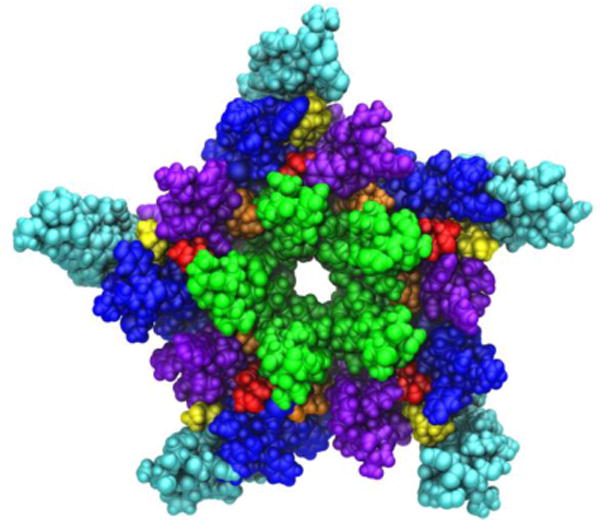
A view of the transmembrane domain of the nicotinic acetylcholine receptor from Torpedo marmorata, with proposed coordinates for embedded cholesterol molecules. Each subunit contains four membrane-spanning helices (M1 purple, M2 green, M3 blue, M4 cyan) and three bound cholesterol molecules (yellow, orange and red). The image is based on the crystal structure PDB ID: 2BG9 from Ref. (Unwin, 2005).
Using both automated and interactive docking, it was found that the gaps could contain cholesterol molecules embedded within the TM domain (Brannigan et al., 2008). Furthermore, the TM domain quickly collapsed to fill the gaps during MD simulations; such collapse was correlated with the loss of critical contacts in the interface between the transmembrane and extracellular ligand binding domains. Collapse and the subsequent decoupling effect were not observed when cholesterol was bound to internal sites revealed by docking, and the structure was most stable when all gaps were filled with cholesterol (15 molecules per pentameric assembly, see Figure 6). It was therefore proposed that the gaps in the structure (PDB ID:2BG9 (Unwin, 2005)) structure may represent regions filled with unresolved cholesterol molecules.
Specific binding of cholesterol to GABAA receptors
Despite the scarcity of direct information, a significant set of indirect evidence points to the intersubunit interfaces of the GABAA transmembrane domain as an interaction site for cholesterol. GABAA receptors are positively modulated by cholesterol derivatives known as neurosteroids (Belelli & Lambert, 2005). The interface between α and β subunits in the transmembrane domain of the GABAAR, in the same region as the ivermectin site in GluCl, has been identified through mutagenesis by (Hosie et al., 2006; Hosie, Wilkins & Smart, 2007) as the likely site of activation by the positively modulating neurosteroids.
Modulation by neurosteroids depends on cholesterol concentration (Sooksawate & Simmonds, 1998; Sooksawate & Simmonds, 2001b). Furthermore, the antagonistic effect upon cholesterol enrichment is selective of cholesterol versus epicholesterol (Sooksawate & Simmonds, 2001a), and may therefore be ascribed to specific cholesterol binding, possibly competitive binding to the intersubunit sites.
GABAA receptors are also known to be potentiated by ivermectin (Dawson et al., 2000; Krusek & Zemkova, 1994), a compound which is a potentiator and agonist of the glutamate-gated chloride channel (GluCl) of the nematode C. elegans. The X-ray structure of GluCl in complex with ivermectin (PDB ID:3RHW(Hibbs & Gouaux, 2011) indicates ivermectin bound to the five subunit interfaces of the transmembrane domain.
Hénin et al (Hénin et al., 2014) built homology models of a GABAA receptor type α1β1γ2 based on the crystal structure of GluCl, and investigated the possibility of cholesterol binding through three complementary approaches: alignment of cholesterol with crystallographic coordinates of ivermectin, automated docking with AutoDock, and explicit, atomistic MD simulations. Docking results confirmed the possibility of cholesterol binding to the five intersubunit clefts in a largely symmetric fashion, despite the asymmetric nature of this heteropentameric receptor (Hénin et al., 2014). The hydroxyl group of cholesterol mimics an ivermectin moiety in that it forms a hydrogen bond with a serine amino acid residue from the pore-forming helix M2. MD simulations indicated that cholesterol is stable within the cavities, but may reorient on a time scale of 100 ns or more. Two instances were observed of cholesterol unbinding, then spontaneously re-binding within the course of 200 ns trajectories, thus providing a picture of the spontaneous binding process in atomic detail (Figure 7) (Hénin et al., 2014).
Fig 7.
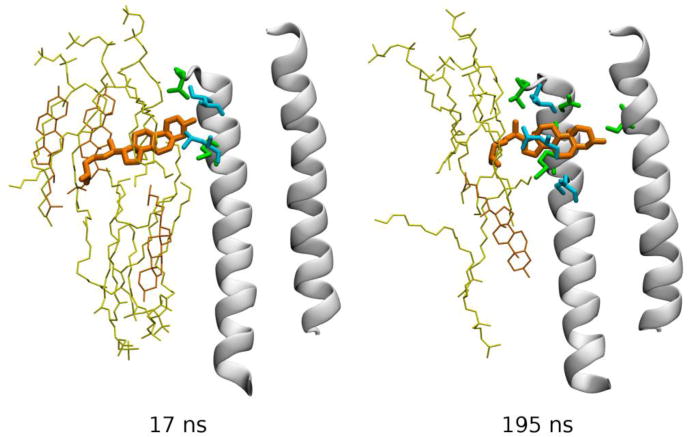
Spontaneous re-binding of cholesterol to an intersubunit cleft in the GABAA receptor. Two frames from a simulated MD trajectory are shown (Hénin et al., 2014). TM helices M2 (right) and M3 (left) forming the back of the pocket are shown as white cartoon, and cholesterol as orange bonds. Other specific groups are depicted as bonds using the following color code: yellow – phospholipid, orange – membrane cholesterol, green – protein amino acid residues from the back receptor subunit, and cyan – protein groups from the front receptor subunit. Data from (Hénin et al., 2014).
G-PROTEIN COUPLED RECEPTORS
Direct and indirect lipid effects modulate GPCR functionality
GPRCs are TM proteins that recognize and respond to a variety of molecules such as hormones and neurotransmitters, thus being involved in many aspects of cell physiology. The mechanisms by which GPCRs couple to their lipid membrane environment (Sadiq et al., 2013) remains elusive but, as discussed below, it appears to involve both direct lipid-GPCR interactions and indirect effects.
Direct lipid-GPCR interactions
One of the best-studied GPCRs in terms of its membrane environment is bovine rhodopsin. On the one hand, it has been experimentally demonstrated that a high content of polyunsaturated lipid chains destabilizes the native receptor state and enhances the kinetics of the photocycle (Mitchell, Niu & Litman, 2001). On the other hand, cholesterol seems to stabilize the native state, thus slowing down its kinetics (Niu, Mitchell & Litman, 2001). As reviewed in ref. (Sadiq et al., 2013), several computational works have investigated the structural basis of lipid coupling to rhodopsin at an all-atom level. Feller et al. observed a preference for unsaturated lipid chains of 1-stearoyl-2-docosahexaenoyl-sn-glyerco-3-phosphatidylcholine (SDPC) to interact with the protein, thereby excluding contacts with the saturated chains of SDPC (Feller, Gawrisch & Woolf, 2003). This finding was supported by a study of Pitman et al. (Pitman et al., 2005), who used a ternary membrane mixture of SDPC, 1-stearoyl-2-docosahexaenoyl phosphatidylethanolamine (SDPE) and cholesterol (2:2:1). Importantly, they found that the unsaturated docosahexaenoic acid (DHA) establishes contacts deep inside the protein, and hypothesized that this type of contacts could be responsible for receptor destabilization when placed into a DHA-rich environment (Grossfield, Feller & Pitman, 2006).
Recent X-ray crystal structures have captured direct lipid-receptor interaction also for other types of GPCRs. For example, a specific cholesterol binding site was observed in the case of the human beta2-adrenergic receptor (PDB ID:3D4S) (Hanson et al., 2008).. This specific sterol binding site is suggested to be important for enhanced stability of the beta2-adrenergic receptor (Hanson et al., 2008). A different cholesterol-binding site, in helix 6, was detected in the high-resolution structure of the adenosine 2A receptor (PDB ID:4EIY) (Liu et al., 2012). Remarkably, this site had been previously anticipated from all-atom MD simulation (Lee & Lyman, 2012), highlighting the predictive potential of computational approaches.
Indirect effects of lipids on GPCRs
Indirect effects of the lipid membrane are mainly mediated via bulk properties of the membrane such as membrane thickness and fluidity. Mondal et al (Mondal et al., 2011) showed that deformations around the rhodopsin receptor are radially asymmetric and involve local thickening and thinning. This asymmetric membrane adjustment allows partial alleviation of the hydrophobic mismatch between the protein and the membrane. In spite of the deformation of the surrounding lipid membrane, there remained a substantial residual hydrophobic exposure at specific regions of the protein (Mondal et al., 2011). Such residual exposure may contribute to the oligomerization of the receptors (Mondal et al., 2011).. The hydrophobic mismatch between the protein and the surrounding lipid bilayer could also lead to conformational adaptation of the protein. Such a conformational adaptation was observed in MD simulations of the metabotropic Glu2 GPCR (Bruno et al., 2012).
AMYLOIDS AND MEMBRANES
Membrane interactions in amyloid-related diseases
Amyloid-related diseases comprise a group of devastating human and animal illnesses. The most common among the human amyloid-related diseases are Alzheimer’s disease, Parkinson’s disease and type II diabetes mellitus. These diseases are characterized by abnormal protein aggregation into β-sheet rich molecular fibrils. Amyloid aggregation is a multi-step process. Initially, peptides that are natively or pathologically unfolded cluster together to form soluble bodies comprised of dimers, trimers or other low-order oligomers. Subsequently, they lead to the formation of higher order oligomers, filaments and fibrils. It is widely believed today that, with few exceptions, prefibrillar aggregates lead to cellular toxicity and mature fibrils are relatively harmless except of being a reservoir of monomers that are ready to form aggregates (Conway et al., 2000; Janson et al., 1996; Lambert et al., 1998).
Interactions between oligomers of amyloidogenic peptides and membranes or lipids appear to be one of the main reasons for the cellular toxicity of prefibrillar intermediates. For example, it has been shown that prefibrillar structures made by islet amyloid polypeptide (IAPP) monomers, whose aggregation is a pathological aspect of type 2 diabetes mellitus, can insert into membranes and lead to membrane damage (Engel, 2009). Interactions between amyloidogenic (poly)peptides and membranes, fatty-acids or other surfactants induce the formation of oligomers that appear to be involved in disease progression, through various mechanisms (see e.g., (Barghorn et al., 2005; Hasegawa et al., 2008; Vestergaard et al., 2013).
Challenges in experimental studies of protein oligomerization
Prefibrillar amyloidogenic structures are, by their nature, transient and dynamic, ranging in size from dimers to high order multimers and in structure from unordered, globular structures to filaments rich in β-sheet forming peptides. The monomers that aggregate typically lack ordered structures, and the native structures of disease-related amyloidogenic proteins such as amyloid-β (Aβ) or islet amyloid polypeptide are disordered or only partially structured (Danielsson et al., 2005; Vivekanandan et al., 2011).
The difficulty of studying amyloid aggregation experimentally is alleviated by the use of molecular simulations as an additional tool for the biophysical characterization of amyloid aggregates (Caflisch, 2006; Friedman, 2011a; Lemkul & Bevan, 2012; Shea & Urbanc, 2012). Simulations can shed light on times and length scales that may not be easily probed otherwise, providing insights into experimental findings (Engel, 2009) and leading to new testable hypotheses. Simulations suggest, for example, that amyloid fibril growth may be directional – e.g., for the HET-s prions, (Baiesi, Seno & Trovato, 2011; Friedman, 2013), and for Aβ1–40 (Buchete & Hummer, 2007; Friedman & Caflisch, 2014), but this has yet to be verified with experiments.
Coarse-grained simulations of amyloid aggregation in the presence of membranes and lipids
The sizes of many molecular oligomers, and the timescale for their formation are several orders of magnitude larger than those that can be followed by atomistic simulations. As a result, coarse-grained models are very common in the field (Derreumaux, 2013; Hung & Yarovsky, 2011; Liguori, Nerenberg & Head-Gordon, 2013; Seo et al., 2012). Fairly accurate coarsegrained force fields such as MARTINI (Marrink et al., 2007) or REACH (Moritsugu & Smith, 2007) are unfortunately not suitable to follow on amyloid aggregation where the conformational changes involve secondary structure. The approach used by Caflisch and co-workers has therefore been to develop phenomenological coarse-grained models for amyloid aggregation. In phenomenological models, a top-down approach is used to model a process. Instead of coarse-graining a collection of atoms to model membranes and peptides of different structures, lipids are represented by three beads having a single conformation (Friedman, Pellarin & Caflisch, 2009) and peptides by ten beads with two conformations: amyloid-prone β and amyloid-protected π (Pellarin & Caflisch, 2006). Such models are, in principle, simple and limited. Protein sequences, for example, are not modeled, and it is not possible to study features that are unique to a certain protein. Yet despite their simplicity, the models can be tailored to study biologically-relevant amyloid aggregation. The free energy difference between the β and π conformers can be tuned to represent peptides that form fibrils rapidly, more slowly, or not at all. Similarly, by changing the lipid’s head group size and the strength of the lipid/lipid interactions, the lipids can generate bilayered liposomes, micelles, worm-like structures and unordered aggregates, all of which are observed in experiments. The strength of the peptide-lipid interactions can be tuned, which can be used to emulate the effect of different types of biomolecular membranes (i.e., membranes whose interactions with peptides are weak, intermediate or strong).
The phenomenological coarse-grained model developed initially by Pellarin and Caflisch (Pellarin & Caflisch, 2006) and extended by Friedman et al. to include membranes (Friedman et al., 2009) have yield some interesting findings. Coarse-grained simulations were used to explain why growing filaments, but not mature fibrils, interfere with the membrane structure. An intriguing finding, that some membranes induce fibril degradation (Martins et al., 2008) that lead to the formation of smaller toxic aggregates (the fibrils themselves are not particularly toxic), was explained by molecular dynamics simulations (Friedman, Pellarin & Caflisch, 2010). Apparently, if the peptide/lipid interactions are very strong, this may lead to the disaggregation of fibrils and formation of oligomers. Finally, the same model has been used to simulate the formation of globulomers, i.e. of globular oligomers made of peptides and lipids (Friedman, 2011b). Their structures were shown to depend on the peptide-lipid ratio and the peptide’s amyloidogenicity.
A recurrent finding coming from these studies is that the amyloidogenicity of the peptides is important for the outcome of aggregation in the presence of lipids. For example, the kinetics of fibril formation is faster in the presence of membrane when highly amyloidogenic peptides are involved but slower when the peptides are non-amyloidogenic. The explanation was that unlike highly amyloidogenic peptides, peptides of low amyloidogenicity form fibrils only in solution. This has indeed been later observed experimentally with IAPP (high amyloidogenicity) and pro-IAPP (low amyloidogenicity) (Khemtemourian et al., 2009).
Probing the interactions of Alzheimer’s Aβ peptides with lipid membranes
As discussed above, the toxicity of Alzheimer’s disease-related amyloid β (Aβ) peptides depends on their ability to disrupt adjacent cellular membranes. Fibrillar and globular-like Aβ oligomers can cause both non-specific and specific ion leakage, either by membrane thinning or by forming pore-like trans-membrane structures that may conduct ions. Tofoleanu and Buchete, probed the molecular interactions between preformed fibrillar Aβ oligomers and lipid bilayers, in the presence of explicit water molecules, using all-atom MD simulations (see Figure 8) (Tofoleanu & Buchete, 2012b). These interactions play an important role in the stability and function of both Aβ fibrils, and of the contiguous cellular membrane (Tofoleanu & Buchete, 2012a; Tofoleanu & Buchete, 2012b). The MD simulations revealed the relative contributions of different structural elements to the dynamics and stability of Aβ protofilament segments near membranes, and the first steps in the mechanism of fibril–membrane interaction. The simulations identified the electrostatic attraction between the charged side chains of Aβ peptides and the lipid head groups as a major driving force. In addition, hydrogen bonds were observed between specific residues in the Aβ protofilaments and the lipid head groups. Taken as a whole, these interactions facilitate the permeation of the hydrophobic C-terminus amino acids of Aβ peptides through the lipid head group region, subsequently leading to further loss of the β-sheet-rich fibril structure, on one hand, and to local membrane-thinning effects, on the other hand (Tofoleanu & Buchete, 2012b). Recent atomistic MD simulations also show that the chemical composition of the lipid head groups can control in a specific manner both the type and magnitude of interactions between Aβ protofilaments and membranes (Tofoleanu, Brooks & Buchete, 2015)..
Fig 8.
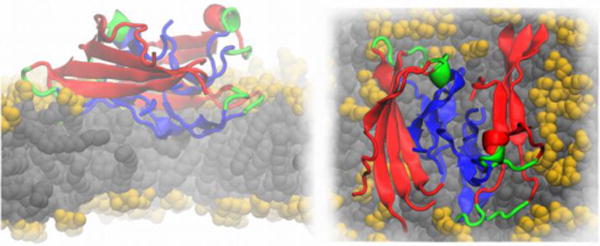
Cartoon representation of an Aβ protofilment segment (4 two-peptide layers) interacting with a lipid membrane, lateral (left) and top (right) views. The N-terminus is represented in red, the turn region in green and the C-terminus in blue. The lipid heavy atoms are represented as spheres, the headgroups are colored in yellow and the lipid tails in grey, respectively. The image is based on Ref. (Tofoleanu & Buchete, 2012b).
The findings summarized above advance our understanding of the detailed molecular mechanisms of Aβ-membrane interactions, and of their effects on both parties, and suggest a polymorphic structural character of amyloid ion channels embedded in lipid bilayers (Tofoleanu & Buchete, 2012a). Inter-peptide hydrogen bonds leading to the formation of long-range ordered β-strands could play a stabilizing role in the case of amyloid channel structures. However, these channels may also present a significant helical content in peptide regions (e.g., the N- and C-peptide termini) that are subject to direct interactions with lipids rather than with neighboring Aβ peptides. As currently there is no high-resolution structural information available on the amyloid channels, several models of Aβ pore-like structures traversing lipid bilayers were constructed, including helical, β-sheet and combinations of these two types of secondary structure (Tofoleanu & Buchete, 2012a). These results may guide new experiments that could test the assembly and structural features of membrane-formed amyloid channels.
COUPLING OF LIPID AND WATER DYNAMICS IN BACTERIAL MEMBRANES
Protein modulation by lipids also occurs in bacterial membranes. While bacteria are simple organisms, their cell envelopes are remarkably complex. The cell envelope of Gram-negative bacteria has a double membrane arrangement in contrast to the single membrane of Gram-positive bacteria. In both cases, the membranes provide a formidable barrier to permeation of solutes both into and out of the cell. The outer membranes of Gram-negative bacteria e.g E.coli and P. aeruginosa, are particularly complex. They are asymmetric in nature; the outer leaflet is composed of lipopolysaccharide (LPS) while the inner membrane is composed of a mixture of approximately 25 mixture of phospholipids, when all the different head group and tail combinations are included. This presents something of a challenge for MD simulations, which traditionally have treated bacterial membranes as a bilayer composed of one type of phospholipid (Bond, Derrick & Sansom, 2007; Khalid et al., 2006; Robertson & Tieleman, 2002). The reasons for the simplification of these membranes have largely been due to (i) simplicity of simulation setup and (ii) lack of parameters for LPS, (iii) the need for increased simulation lengths to allow for adequate sampling when considering lateral diffusion of multiple lipid species (ref: Disaccharides impact the lateral organization of lipid membranes (Moiset et al., 2014). While this usually an accurate representation of in vitro electrophysiology experiments, it clearly is far too simple to represent the heterogeneity of the in vivo environment.
Encouragingly, models of LPS for P. aeruginosa and E.coli have been reported in the literature in recent years. By pure serendipity models have been reported for each of the three most widely used families of force fields; Charmm, Amber and GROMOS thereby giving most membrane simulations groups an off-the-shelf solution (Lins & Straatsma, 2001; Piggot, Holdbrook & Khalid, 2011; Wu et al., 2013). The process of membrane electroporation has been studied via MD simulation (Delemotte & Tarek, 2012; Piggot et al., 2011; Tieleman, 2004).
Our recent simulation study has revealed the intricate interdependency of water and lipids in the process of electroporation (Piggot et al., 2011). The pore-forming process in a Gram-positive (S. aureus) and Gram-negative bacterium (E.coli) were compared. Our model of the outer membrane of E.coli comprised LPS molecules in the outer leaflet and a combination of phospholipids in the inner leaflet (phosphatidylethanolamine, phosphatidylglycerol and cardiolipin), whereas the model S. aureus membranes was modeled as a symmetric bilayer composed of lysyl-phosphatidylglycerol, phosphatidylglycerol and cardiolipin lipids. Briefly, our results showed two very different mechanisms of electroporation. In the symmetric membrane of S. aureus, the mechanism proceeds as follows: Initially, between 3–5 lipid headgroups move slightly towards the hydrophobic core of the bilayer. This is followed by water permeation into the core through the resulting ‘defect’ in the headgroup region of the bilayer. The molecules of water quickly extend through the bilayer core region and into the headgroups of the opposing leaflet, producing a continuous column or channel of water. This channel of water remained intact even when the magnitude of the external electric field was drastically reduced.
In contrast, in E.coli, the water channel was only observed to form, once a phospholipid had flip-flopped from the inner leaflet into the outer, LPS-containing leaflet. Furthermore, upon reduction of the applied electric field, the water was observed to exit the bilayer core, after which the headgroup region resealed, giving a defect-free bilayer. These results begin to reveal some of the molecular basis for the highly impermeable nature of Gram-negative bacteria. The LPS leaflet is tightly cross-linked by ions and extended LPS-LPS hydrogen bonds that the individual molecules are extremely immobile and therefore difficult to penetrate. Conversely, the phospholipid molecules in the inner leaflet diffuse an order of magnitude faster enabling them to expel water and quickly reseal if defects do form.
Atomistic MD simulations of bacterial outer membrane proteins embedded within realistic membrane models have also been reported recently, building upon the earlier work of Lins & Straatsma who simulated a homology model of OprF from P. aeruginosa in an asymmetric bilayer composed of LPS and phospholipids (Holdbrook et al., 2013; Piggot, Holdbrook & Khalid, 2013; Straatsma & Soares, 2009; Wu et al., 2014). More recently we have shown how the conformational behavior of the TonB dependent transporter, FecA from E. coli, and the autotransporter Hia from N. meningitides (see Figure 9) differ in phospholipid bilayers compared with more representative outer membrane models (Holdbrook et al., 2013; Piggot et al., 2013).
Fig 9.
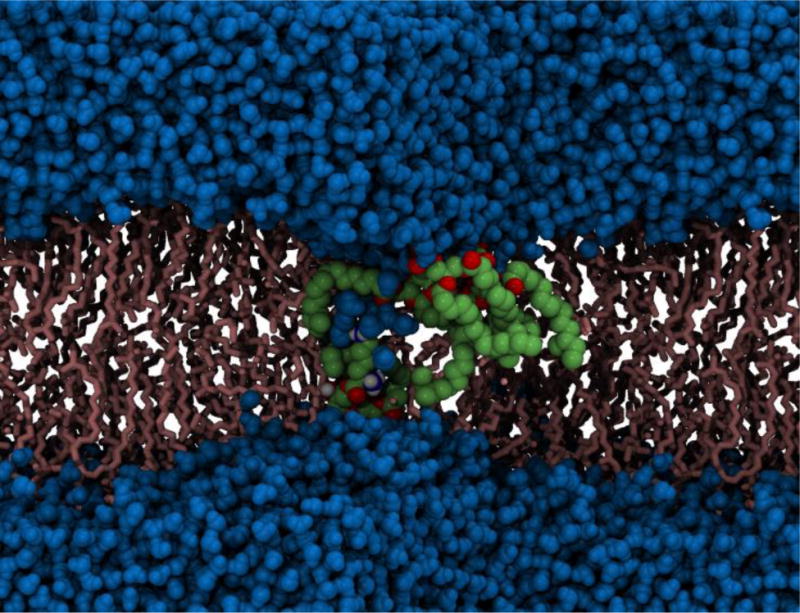
Electroporation in the cell membrane of the Gram-positive bacterium, S. aureus. Deformation of the lipid bilayer is clearly visible. Phospholipids that have been pulled in closer to the membrane core are shown in green and red, water molecules are shown in blue and the remaining lipid molecules are shown in pink (Piggot, Piñeiro & Khalid, 2012).
Given the recent availability of LPS models, the outlook for the future of bacterial membrane simulations is extremely promising. In addition to the complexity of the lipids, one of the next key steps will be to incorporate realistic copy numbers of the major proteins native to these membranes, to an extent this is already being addressed with coarse-grained MD simulations (Goose & Sansom, 2013). Thus, it is easy to envisage that simulations will play a major role in understanding the fine interplay between proteins, lipids and water in bacterial membranes.
NEW THEORETICAL CONCEPTS
Molecular transporters and transport processes – Membrane pores
Upon passage through membrane pores, peptides undergo conformational transitions and sample intermediates that block the transmembrane current (Dekker, 2013; Mohammad & Movileanu, 2008; Movileanu et al., 2000; Oukhaled et al., 2007; Pastoriza-Gallego et al., 2011; Payet et al., 2012; Stefureac et al., 2008) that would otherwise flow in an open pore under a potential drop. These intermediate states can be considered jammed states. On the other hand, the phenomenology of jamming has been widely studied, separately in the context of dynamical arrest of macroscopic granular matter and in that of macroscopic glasses (Freedman et al., 2013; Head, 2009; Heussinger & Barrat, 2009; Olsson & Teitel, 2007; Silbert, 2010; Snoeijer et al., 2004; Vagberg et al., 2011). Jamming can be identified by signature characteristics in the force distributions, p(f) – the force profile of a jammed system will exhibit a peak in p(f) that deviates from the exponential decay in the forces of an unjammed state (Dijksman et al., 2011; Reis, Ingale & Shattuck, 2007; Snoeijer et al., 2004; Tighe et al., 2010; Tighe et al., 2005). Such jamming analyses have been applied to study protein folding in aqueous solution (Jose & Andricioaei, 2012). Accelerated MD simulations captured the atomistic details of these jammed states of a single unstructured peptide molecule, CAMA (Asandei et al., 2011; Mereuta et al., 2014; Mereuta et al., 2012), as it traverses the pore during a translocation event, jamming signatures of which were further compared with that of the same peptide when in free state without the physical constriction of the pore.
Translocation of the above mentioned peptide, C ecropin A – MA gainin(CAMA) with a sequence of 20 amino acids KWKLFKKIGIGKFLQSAKKF-NH2, through biological nano-pore α-HL (Song et al., 1996), has been studied using single molecule electrophysiology, inserted from the trans-end (Krasilnikov et al., 2000; Mereuta et al., 2014). Molecular dynamics simulations of the above process at complete atomistic resolution (Aksimentiev, 2010; Aksimentiev & Schulten, 2005; Butler, Gundlach & Troll, 2006; Butler, Gundlach & Troll, 2007; Kantor & Kardar, 2004; Tian & Andricioaei, 2005), was successful in capturing the sub-states, which induce partial or complete blockage of the pore, reflected as spikes in the current blockage signature.
The peptide in its free state assumes a heterogeneous kinked shape due to the presence of two alternate glycines (G) at positions 9 and 11 separated by one Isoleucine (I) at position 10, showing force distribution profile most reminiscent of an un-jammed peptide. When the peptide is unfolded within the pore constriction (observed through biased MD simulations) and translocated through the beta-barrel of α-HL in a linear fashion it exhibits characteristics akin to a less jammed state whereas the signatures point towards a jammed peptide when CAMA traverses the pore in a wound, condensed state. Representative snapshots of the conformational ensemble assumed at each state are depicted with their respective non-bonded force profiles in Figure 10. All three states exhibit jamming signatures, as marked by the pronounced peak above the mean force, though this peak is shifted more to the right the more unjammed the peptide, e.g. the free CAMA, which is shifted farthest. Thus, it may be surmised that the position of the peaks depends upon the relative degree of jamming between the configurations. It is noteworthy that the simulation for the free CAMA was carried out at room temperature where jamming is shown, from our previous study, to be extant in peptides. A simulation at a much higher temperature will most likely wash out the jamming traits in the force profile, which may be the focus of investigation of a future study.
Fig 10.
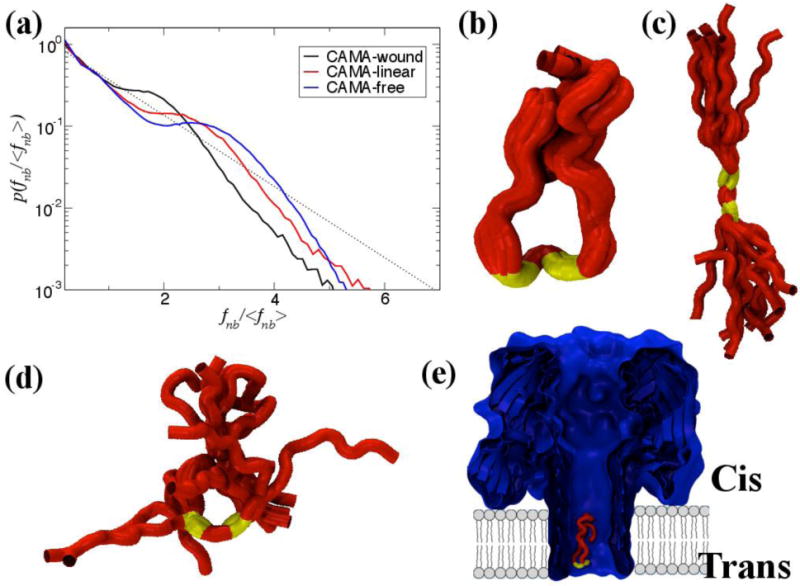
The non-bonded, normalized force distributions for three different states of CAMA, ranging from jammed (wound) to marginally jammed to relatively un-jammed (free). All three exhibit jamming signatures, namely a pronounced peak about the exponential line (dashed), though this is shifted to higher values the more apparently unjammed the peptide. Additionally, the tails of those that are less jammed are closer to the exponential line. Conformational ensembles of CAMA P6 in (b) wound state (c) linear and (d) free state. (e) Translocation of peptide CAMA – P6 through lumen of α-hemolysin. Panels b–e are adapted from Ref. (Mereuta et al., 2014).
The Energy Landscapes Framework
Studying the potential energy landscape of membrane proteins provides both conceptual and computational tools for understanding a wide range of observable properties in molecular science (Wales, 2003; Wales, 2010; Wales, 2005; Wales & Bogdan, 2006). In particular, we can exploit stationary points (minima and transition states) for structure prediction and analysis of global thermodynamic and kinetic properties.
Basin-hopping global optimization (Wales & Doye, 1997) represents a powerful tool for structure prediction, while basin-sampling/parallel tempering (Bogdan, Wales & Calvo, 2006; Wales, 2013) and discrete path sampling (Wales, 2002; Wales, 2006; Wales & Dewsbury, 2004) enable us to address broken ergodicity and rare event dynamics. Basin-hopping and basin-sampling require only local minimization; discrete path sampling involves location of transition states between local minima. This coarse-graining in terms of stationary points is founded upon efficient geometry optimization procedures; the corresponding software and energy landscape databases are available at URL http://www-wales.ch.cam.ac.uk.
Applications have been presented ranging from prediction and interpretation of high resolution spectra for small water clusters, (Wales & Walsh, 1997; Walsh & Wales, 1996) folding and misfolding of biomolecules, (Evans & Wales, 2003; Mortenson, Evans & Wales, 2002; Strodel, Whittleston & Wales, 2007) to the analysis of structural glass-formers (de Souza & Wales, 2008; Wales & Doye, 2003) and condensed soft matter systems (Chakrabarti & Wales, 2009; Fejer & Wales, 2007). Some recent results for small oligomers of the Aβ1–42 peptide are illustrated in Figure 11 (Strodel et al., 2010).
Fig 11.
Predicted structures for Aβ1–42 monomer (transmembrane and surface structures, left), dimer (middle) and octamer (right) in contact with an implicit membrane. The molecular graphics images are adapted from Ref. (Strodel et al., 2010).
The structures illustrated in Figure 11 were obtained using CHARMM19 (Neria, Fischer & Karplus, 1996) with the implicit membrane potential IMM1 (Lazaridis, 2003). A basin-hopping/parallel tempering scheme with exchanges between basin-hopping runs at different temperatures was used, (Strodel et al., 2010) together with intra-and intermolecular coordinate moves for the peptides. The most favorable monomer transmembrane structure has residues 17 to 42 inserted in the membrane. The most stable octamer structures can be viewed as displaced tetramers composed of two or three β-sheets (Strodel et al., 2010).
Coarse-grained modeling has been used to explain the underlying principles that lead to the emergence of shells, tubes, helices and spirals (Chakrabarti & Wales, 2009; Fejer & Wales, 2007). Recently we have discovered design principles that connect the morphology of amyloid fibers to seedpods and macroscopic helices formed from elliptical magnets (Figure 12) (Forman et al., 2013). Experimentally, the introduction of a cytochrome domain into an amyloid fiber permits dynamic adjustment of the fiber morphology via heme binding (Forman et al., 2012). The interlocking of fiber filaments introduces systematic kinking, (Forman et al., 2012) and a transition from a twisted ribbon to a spiral ribbon morphology was also observed (Baldwin et al., 2006). By constructing a rigid link between two anisotropic interacting units we have now shown that the precise helical morphology of aggregates formed from such a composite building block primarily depends on the internal geometry, rather than the nature of the interaction or the anisotropy of the interacting units. This framework can be viewed as a discrete version of the bilayered frustration principle, which drives the morphological transitions of the Bauhinia seedpod.
Fig 12.

Left: introduction of a cytochrome domain into an amyloid fibre can change the morphology from twisted to spiral ribbons and induce systematic kinking. Centre: rigid building blocks consisting of two ellipsoids can reproduce these structures. Right: the structure depends mostly on the internal geometry of the building blocks. The molecular graphics images are based on Ref. (Forman et al., 2013).
Biomolecular Visualization
Biomolecular visualization has played an important role since the beginning of computer-assisted biophysical and biochemical analysis. In the following, we present some visualization techniques that bear a great potential for the analysis of structure, dynamics and function of membrane proteins. Some of these techniques have been specifically developed for membrane proteins and others were designed with different molecular structures in mind. Nevertheless, we believe that all of them are readily applicable to support the analysis of membrane substructures, single membrane proteins or even whole membranes.
Molecular conformations
During the past two decades methods have been developed to identify metastable conformations of biomolecules and to reveal the dynamics of conformations (Schütte & Sarich, 2013). Here, a conformation is considered as a connected subset in state space. In order to better understand molecular systems, it is useful to depict molecular conformations, making the differences between them clearly visible. In references (Rheingans & Joshi, 1999; Schmidt-Ehrenberg, Baum & Hege, 2002), an approach has been proposed to represent sets of molecular configurations as spatial densities; these can be depicted using iso-surfacing or volume rendering; and with color-coding, different parts of the molecules can be distinguished or distinct conformations can be rendered in superposition (Figure 13).
Fig 13.
Direct volume rendering of the BSI molecule 2-(biphenyl-4-sulfonyl)-1,2,3,4-tetrahydro-isoquinoline-3-carboxylic acid based on 241.760 molecular configurations in 192 metastable sets, comprising 12 chemically different groups that are distinguished by color. On the left the spatial alignment is based on a fixed substructure, on the right on Procrustes analysis.
Molecular configurations that can be transformed into each other by a rigid transformation are considered physically equivalent. Therefore, configurations are usually defined without reference to an external coordinate system. For a graphical representation, however, they must be placed in a given external coordinate system. While this is no problem when displaying a single molecular system, especially when one can rotate and move it interactively, the optimal alignment for sets of molecular systems is less clear; to create informative depictions of such sets, the molecules must be aligned relative to each other in a specific way. For illustration, in Figure 14 four different types of alignments of a set of molecules are shown using the simple molecule pentane. For sufficiently similar geometries, generalized partial Procrustes analysis provides an objective function (sum of squared distances between corresponding atoms, or, equivalently sum of atom position variances) and an alignment algorithm. If groups of substantially different forms are to be compared, however a more sophisticated alignment strategy is necessary. In Ref. (Schmidt-Ehrenberg, 2008) it is proposed to perform a hierarchical decomposition of the molecule into maximally stiff and less stiff structures and to apply an alignment strategy adapted to this decomposition. The resulting visualization provides a clear impression of the stiff structure, while the rest of the molecule, exhibiting interesting form differences between the metastable sets, are depicted fuzzy.
Fig 14.
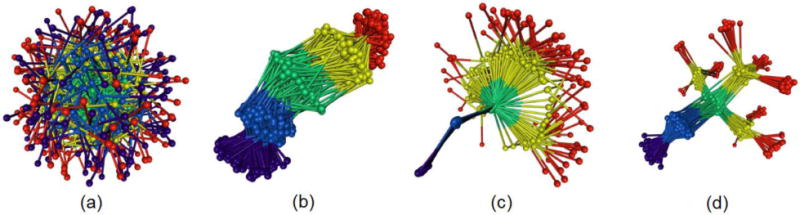
Illustration of the alignment problem by aligning 150 forms of Pentane by minimizing (a) the distances between the centers of gravity of each form, furthermore, the distances between corresponding atoms summed over (b) all atoms, (c) three selected atoms, and (d) atoms in a hierarchy of sets, defined by stiff substructures and an interactively selected anchor structure.
Extraction and visualization of molecular paths
Before molecular interactions can take place, the interacting molecules must come close enough to each other. For molecules like nanotransporters, which are often complex and endowed with specific binding sites, this is of particular interest. Identification of ligand binding sites via molecular dynamics simulations, however, is too expensive. In Ref. (Lindow, Baum & Hege, 2011) it was suggested to restrict the search space by first computing the paths that are geometrically possible. These paths can be found by computing the 3D Voronoi diagram of the atom spheres and selecting a subset of the Voronoi edges (i.e. all points that locally are furthest away from the van der Waals spheres of the molecule). Utilizing computer graphics techniques like deferred shading, ray-casting and screen space ambient occlusion, and by placing light sources along the paths, one obtains very informative depictions (Figure 15). All of these techniques are directly applicable to, for example, ion channels. In contrast to static channels, which can be observed in a single time step of a molecular dynamics trajectory, the evolution of molecular cavities needs to be analyzed over time to identify dynamic channels. This aspect is addressed below.
Fig 15.
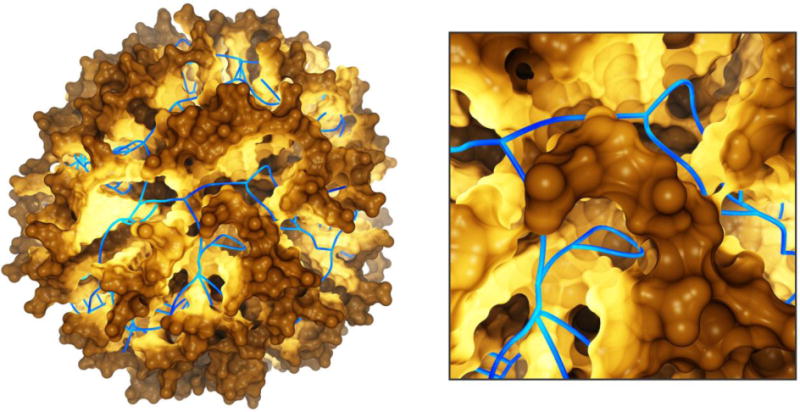
Geometric molecular paths (blue) in a nano-transporter, computed by the Voronoi diagram of the atom spheres and combined with a path-dependent illumination of the molecular surface. Based on Ref. (Lindow et al., 2011).
Interactive exploration of cavity dynamics in proteins
The internal cavities of proteins are dynamic structures and their dynamics is associated with conformational changes. In Refs. (Lindow et al., 2012a; Lindow et al., 2013), a tool was presented that allows rapid identification of internal cavities and an interactive assessment of their time-dependent shapes. Given a MD trajectory, in a pre-processing step the structure of the cavities is computed from the Voronoi diagram of the van der Waals spheres. Then the user can interactively select, visualize and analyze the time-dependent cavity components and the dynamic molecular paths with a user-defined minimum constriction size. The cavity dynamics can be depicted in a single image that shows the cavity surface colored according to its occurrence in time. As an example, in Figure 16 the dynamic structure of internal cavities in the bacteriorhodopsin proton pump is depicted and in Figure 17 the related splits and merges of the cavities as well as their position along a user-defined axis through the proton pump are shown. An alternative technique, based on object-space ambient occlusion for the detection of cavities, channels and pockets has been proposed in Refs. (Krone et al., 2014; Krone et al., 2013).
Fig 16.
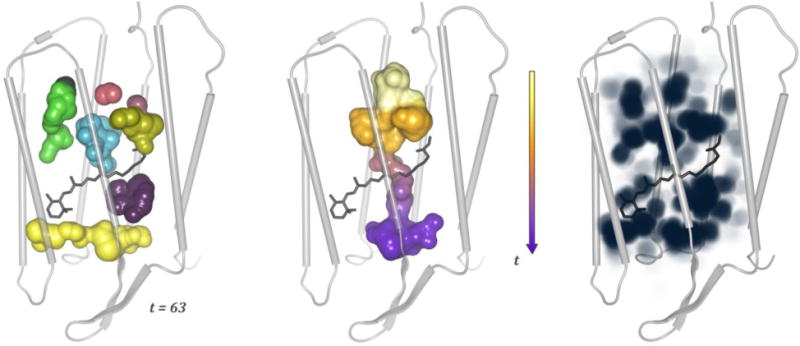
Cavity dynamics in a bacteriorhodopsin monomer. From left to right: the cavities traced at time step t = 63, a channel created by the dynamics from t = 50 till t = 64, and the overall spatial cavity probability (depicted with maximum intensity projection). The images are based on Refs. (Lindow et al., 2012a; Lindow et al., 2013)
Fig 17.
Temporal development of cavities in a bacteriorhodopsin monomer (for the simulation and the time steps also used in Figure 16); upper diagram: topology graph showing splits and merges of cavities; lower diagram: penetration graph depicting the cavities’ location along a user-defined axis. The image is based on Refs. (Lindow et al., 2012a; Lindow et al., 2013)
Multi-scale visualization of biological structures
Properties of inorganic and organic materials can often be understood by analyzing them simultaneously on different scales, e.g. on the atomic and nanoscopic scale. This raises the problem of rendering images with millions to billions of atoms. In Ref. (Lindow, Baum & Hege, 2012b), a simple yet efficient rendering method was proposed for visualizing interactively large sets of atomic data, bridging 5 orders of magnitude in length scale. The method exploits the recurrence of molecular structures and the fact that these are typically rendered opaquely, such that only a fraction of the atoms is visible. The method has been demonstrated by visualizing large molecular structures that where fitted to subcellular objects which had been imaged with electron tomography. On a commodity PC, 150 million atoms could be visualized with interactive speed; and for scenes with up to 10 billion atoms, still rates of 3 frames per second could still be achieved. An example is shown in Figure 18, depicting microtubules reconstructed from electron tomography data with fitted molecular structure. This method was further optimized in Ref. (Falk, Krone & Ertl, 2013) and extended to render triangular objects, such that arbitrary molecular models can be displayed.
Fig 18.
Visualization of microtubules (reconstructed from electron tomography data) containing about 10 billion atoms; rendered on commodity PC with subsecond framerate. The image is based on ref. (Lindow et al., 2012b).
Since membranes also consist of a large number of recurring molecules, the described methods should be directly applicable to data from membrane simulations. A great challenge here is to visualize membrane dynamics at an interactive speed.
CONCLUSIONS
The above considerations of recent progress in membrane protein structural biology present a unifying theme: even in atomic-detail considerations one cannot escape at least elements of a holistic approach. The protein, lipid and solvent components of a biological membrane system have been shown to be highly interdependent, such that consideration, say, of a simulation model of a protein in a single model phospholipid, such as DOPC, is becoming rather anachronistic. Increased computer power, improved computational methods and experimental advances in techniques such as, for example, the crystallography of large complexes are revealing the workings of integrated membrane systems of hundreds of thousands of atoms. Undoubtedly in the future it will be necessary to integrate these approaches still further, with “systems-level” imaging and simulation. Possibly only in the context of non-equilibrium cellular fluxes will the mechanics of membrane function be able to be fully comprehended.
Acknowledgments
ZC, ANB, JCS would like to acknowledge funding from Centre Européen de Calcul Atomique et Moléculaire (CECAM) to host the Workshop “Coupling between protein, water, and lipid dynamics in complex biological systems: Theory and Experiments” that took place in September 2013, Lausanne, Switzerland. JTD, IA, and MR used the computational resources of the Modeling Facility of the Department of Chemistry, University of California Irvine funded by NSF Grant CHE-0840513 for this work. A-NB is supported in part by the Marie Curie International Reintegration Award IRG-26920. TWA was supported by ARC DP120103548, NSF MCB1052477, DE Shaw Anton (PSCA00061P; NRBSC, through NIH RC2GM093307), VLSCI (VR0200) and NCI (dd7). BA and SV acknowledge the support by ERC advanced grant #268888. ZC and PG would like to acknowledge Reference Framework (NSRF) 2011 – 2013, National Action “Cooperation”, under grant entitled “Magnetic Nanoparticles for targeted MRI therapy (NANOTHER)”, with code “11ΣYN-1-1799”. The programme is cofunded by the European Regional Development Fund and national resources. Part of the calculations presented herein were performed using resources of the LinkSCEEM-2 project, funded by the EC under FP7 through Capacities Research Infrastructure, INFRA-2010-1.2.3 Virtual Research Communities, Combination of Collaborative Project and Coordination and Support Actions (CP-CSA) under grant agreement no RI-261600. GB is supported in part by NSF grant MCB1330728 from the National Science Foundation, and grant PO1GM55876-14A1 from the National Institutes of Health. LD receives funding from EU FP7 (PIOF-GA-2012-329534). LD and MLK used the computational resources of Temple University, supported by the National Science Foundation through major research instrumentation grant number CNS-09-58854. FT and NVB are grateful for financial support from the Irish Research Council, and for using the computational facilities of the Biowulf Linux cluster at the National Institutes of Health, USA and the Irish Centre for High-End Computing (ICHEC). JS acknowledges support from the Instituto de Salud Carlos III FEDER (CP12/03139) and the GLISTEN European Research Network.
References
- Abderemane-Ali F, Es-Salah-Lamoureux Z, Delemotte L, Kasimova MA, Labro AJ, Snyders DJ, Fedida D, Tarek M, Baro I, Loussouarn G. Dual effect of phosphatidylinositol (4,5)-bisphosphate PIP(2) on Shaker K(+) [corrected] channels. J Biol Chem. 2012;287:36158–67. doi: 10.1074/jbc.M112.382085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksimentiev A. Deciphering ionic current signatures of DNA transport through a nanopore. Nanoscale. 2010;2:468–483. doi: 10.1039/b9nr00275h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksimentiev A, Schulten K. Imaging alpha-Hemolysin with Molecular Dynamics: Ionic Conductance, Osmotic Permeability, and the Electrostatic Potential Map. Biophysical Journal. 2005;88:3745–3761. doi: 10.1529/biophysj.104.058727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TW, Andersen OS, Roux B. On the importance of atomic fluctuations, protein flexibility, and solvent in ion permeation. J Gen Physiol. 2004;124:679–90. doi: 10.1085/jgp.200409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OS. Perspectives on: Ion selectivity. J Gen Physiol. 2011;137:393–5. doi: 10.1085/jgp.201110651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias HR, Sankaram MB, Marsh D, Barrantes FJ. Effect of local anaesthetics on steroid-nicotinic acetylcholine receptor interactions in native membranes of Torpedo marmorata electric organ. Biochim Biophys Acta. 1990;1027:287–94. doi: 10.1016/0005-2736(90)90320-n. [DOI] [PubMed] [Google Scholar]
- Arutyunova E, Panwar P, Skiba PM, Gale N, Mak MW, Lemieux MJ. Allosteric regulation of rhomboid intramembrane proteolysis. EMBO J. 2014;33:1869–81. doi: 10.15252/embj.201488149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asandei A, Apetrei A, Park Y, Hahm K-S, Luchian T. Investigation of Single-Molecule Kinetics Mediated by Weak Hydrogen Bonds within a Biological Nanopore. Langmuir. 2011;27:19–24. doi: 10.1021/la104264f. [DOI] [PubMed] [Google Scholar]
- Baez-Pagan CA, Martinez-Ortiz Y, Otero-Cruz JD, Salgado-Villanueva IK, Velazquez G, Ortiz-Acevedo A, Quesada O, Silva WI, Lasalde-Dominicci JA. Potential role of caveolin-1-positive domains in the regulation of the acetylcholine receptor’s activatable pool: implications in the pathogenesis of a novel congenital myasthenic syndrome. Channels. 2008;2:180–90. doi: 10.4161/chan.2.3.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, Sidders B, Skerratt SE, Stevens EB, Storer RI, Swain NA. Ion channels as therapeutic targets: a drug discovery perspective. J Med Chem. 2013;56:593–624. doi: 10.1021/jm3011433. [DOI] [PubMed] [Google Scholar]
- Baggett AW, Cournia Z, Han MS, Patargias G, Glass AC, Liu SY, Nolen BJ. Structural characterization and computer-aided optimization of a small-molecule inhibitor of the Arp2/3 complex, a key regulator of the actin cytoskeleton. ChemMedChem. 2012;7:1286–94. doi: 10.1002/cmdc.201200104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baiesi M, Seno F, Trovato A. Fibril elongation mechanisms of HET-s prion-forming domain: topological evidence for growth polarity. Proteins. 2011;79:3067–3081. doi: 10.1002/prot.23133. [DOI] [PubMed] [Google Scholar]
- Baker RP, Urban S. Architectural and thermodynamic principles underlying intramembrane protease function. Nat Chem Biol. 2012;8:759–68. doi: 10.1038/nchembio.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci U S A. 2007;104:8257–62. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AJ, Bader R, Christodoulou J, MacPhee CE, Dobson CM, Barker PD. Cytochrome display on amyloid fibrils. J Am Chem Soc. 2006;128:2162–3. doi: 10.1021/ja0565673. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner R, Harlan J, Barlow E, Ebert U, Hillen H. Globular amyloid beta-peptide oligomer-a homogenous and stable neuropathological protein in A lzheimer’s disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- Barrantes FJ. Structural basis for lipid modulation of nicotinic acetylcholine receptor function. Brain Res Brain Res Rev. 2004;47:71–95. doi: 10.1016/j.brainresrev.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Barrantes FJ. Cholesterol effects on nicotinic acetylcholine receptor. J Neurochem. 2007;103(Suppl 1):72–80. doi: 10.1111/j.1471-4159.2007.04719.x. [DOI] [PubMed] [Google Scholar]
- Barrantes FJ, Antollini SS, Blanton MP, Prieto M. Topography of nicotinic acetylcholine receptor membrane-embedded domains. J Biol Chem. 2000;275:37333–9. doi: 10.1074/jbc.M005246200. [DOI] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat Rev Neurosci. 2005;6:565–75. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci U S A. 2007;104:462–6. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Negative conductance caused by entry of sodium and cesium ions into the potassium channels of squid axons. J Gen Physiol. 1972;60:588–608. doi: 10.1085/jgp.60.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigay J, Antonny B. Curvature, lipid packing, and electrostatics of membrane organelles: defining cellular territories in determining specificity. Dev Cell. 2012;23:886–95. doi: 10.1016/j.devcel.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Bogdan TV, Wales DJ, Calvo F. Equilibrium thermodynamics from basin-sampling. J Chem Phys. 2006;124:044102. doi: 10.1063/1.2148958. [DOI] [PubMed] [Google Scholar]
- Boiteux C, Vorobyov I, Allen TW. Ion conduction and conformational flexibility of a bacterial voltage-gated sodium channel. Proc Natl Acad Sci U S A. 2014a;111:3454–9. doi: 10.1073/pnas.1320907111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiteux C, Vorobyov I, French RJ, French C, Yarov-Yarovoy V, Allen TW. Local anesthetic and antiepileptic drug access and binding to a bacterial voltage-gated sodium channel. Proc Natl Acad Sci U S A. 2014b;111:13057–62. doi: 10.1073/pnas.1408710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond PJ, Derrick JP, Sansom MS. Membrane simulations of OpcA: gating in the loops? Biophys J. 2007;92:L23–5. doi: 10.1529/biophysj.106.097311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar AN, del Val C, Freites JA, Tobias DJ, White SH. Dynamics of SecY translocons with translocation-defective mutations. Structure. 2010;18:847–57. doi: 10.1016/j.str.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar AN, del Val C, White SH. Rhomboid protease dynamics and lipid interactions. Structure. 2009;17:395–405. doi: 10.1016/j.str.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar AN, White SH. Hydrogen bond dynamics in membrane protein function. Biochim Biophys Acta. 2012;1818:942–50. doi: 10.1016/j.bbamem.2011.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni V, Baier CJ, Lang T, Bonini I, White MM, Garbus I, Barrantes FJ. Cholesterol depletion activates rapid internalization of submicron-sized acetylcholine receptor domains at the cell membrane. Mol Membr Biol. 2007;24:1–15. doi: 10.1080/09687860600903387. [DOI] [PubMed] [Google Scholar]
- Brannigan G, Hénin J, Law R, Eckenhoff R, Klein ML. Embedded cholesterol in the nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A. 2008;105:14418–23. doi: 10.1073/pnas.0803029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher MS, Munro S. Cholesterol and the Golgi apparatus. Science. 1993;261:1280–1. doi: 10.1126/science.8362242. [DOI] [PubMed] [Google Scholar]
- Bristow DR, Martin IL. Solubilisation of the gamma-aminobutyric acid/benzodiazepine receptor from rat cerebellum: optimal preservation of the modulatory responses by natural brain lipids. J Neurochem. 1987;49:1386–93. doi: 10.1111/j.1471-4159.1987.tb01004.x. [DOI] [PubMed] [Google Scholar]
- Brohawn SG, del Marmol J, MacKinnon R. Crystal structure of the human K2P TRAAK, a lipid-and mechano-sensitive K+ ion channel. Science. 2012;335:436–41. doi: 10.1126/science.1213808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Lazareno-Saez C, Lamoureux JS, Mak MW, Lemieux MJ. Insights into substrate gating in H. influenzae rhomboid. J Mol Biol. 2011;407:687–97. doi: 10.1016/j.jmb.2011.01.046. [DOI] [PubMed] [Google Scholar]
- Brown MF. Modulation of rhodopsin function by properties of the membrane bilayer. Chem Phys Lipids. 1994;73:159–80. doi: 10.1016/0009-3084(94)90180-5. [DOI] [PubMed] [Google Scholar]
- Bruno A, Costantino G, de Fabritiis G, Pastor M, Selent J. Membrane-sensitive conformational states of helix 8 in the metabotropic Glu2 receptor, a class C GPCR. PLoS One. 2012;7:e42023. doi: 10.1371/journal.pone.0042023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchete NV, Hummer G. Structure and dynamics of parallel beta-sheets, hydrophobic core, and loops in Alzheimer’s A beta fibrils. Biophysical Journal. 2007;92:3032–3039. doi: 10.1529/biophysj.106.100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger K, Gimpl G, Fahrenholz F. Regulation of receptor function by cholesterol. Cell Mol Life Sci. 2000;57:1577–92. doi: 10.1007/PL00000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler T, Gundlach J, Troll M. Determination of RNA Orientation during Translocation through a Biological Nanopore. Biophysical Journal. 2006;90:190–199. doi: 10.1529/biophysj.105.068957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler T, Gundlach J, Troll M. Ionic current blockades from DNA and RNA molecules in the alpha-hemolysin nanopore. Biophysical journal. 2007;93:3229–3240. doi: 10.1529/biophysj.107.107003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–U127. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. Specific Binding of Adamantane Drugs and Direction of Their Polar Amines in the Pore of the Influenza M2 Transmembrane Domain in Lipid Bilayers and Dodecylphosphocholine Micelles Determined by NMR Spectroscopy. Journal of the American Chemical Society. 2011;133:4274–4284. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caflisch A. Computational models for the prediction of polypeptide aggregation propensity. Curr Opin Chem Biol. 2006;10:437–444. doi: 10.1016/j.cbpa.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Chakrabarti D, Wales DJ. Simulations of rigid bodies in an angle-axis framework. Phys Chem Chem Phys. 2009;11:1970–6. doi: 10.1039/b818054g. [DOI] [PubMed] [Google Scholar]
- Chakrabarti N, Ing C, Payandeh J, Zheng N, Catterall WA, Pomes R. Catalysis of Na+ permeation in the bacterial sodium channel Na(V)Ab. Proc Natl Acad Sci U S A. 2013;110:11331–6. doi: 10.1073/pnas.1309452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Durr KL, Gouaux E. X-ray structures of AMPA receptor-cone snail toxin complexes illuminate activation mechanism. Science. 2014;345:1021–6. doi: 10.1126/science.1258409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang G-Y, Kozakov D, Brenke R, Beglov D, Guarnieri F, Vajda S. Binding Hot Spots and Amantadine Orientation in the Influenza A Virus M2 Proton Channel. Biophysical Journal. 2009;97:2846–2853. doi: 10.1016/j.bpj.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clare JJ. Targeting ion channels for drug discovery. Discov Med. 2010;9:253–60. [PubMed] [Google Scholar]
- Combs DJ, Shin HG, Xu Y, Ramu Y, Lu Z. Tuning voltage-gated channel activity and cellular excitability with a sphingomyelinase. J Gen Physiol. 2013;142:367–80. doi: 10.1085/jgp.201310986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway K, Lee S, Rochet J, Ding T, Williamson R, Lansbury P. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset P arkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criado M, Eibl H, Barrantes FJ. Functional properties of the acetylcholine receptor incorporated in model lipid membranes. Differential effects of chain length and head group of phospholipids on receptor affinity states and receptor-mediated ion translocation. J Biol Chem. 1984;259:9188–98. [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K(+) channels. Nature. 2010;466:203–8. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel AW, Rollins ES, McNamee MG. The effect of cholesterol on agonist-induced flux in reconstituted acetylcholine receptor vesicles. FEBS Lett. 1980;122:193–6. doi: 10.1016/0014-5793(80)80435-2. [DOI] [PubMed] [Google Scholar]
- Danielsson J, Jarvet J, Damberg P, Graslund A. The Alzheimer beta-peptide shows temperature-dependent transitions between left-handed 3-helix, beta-strand and random coil secondary structures. FEBS J. 2005;272:3938–49. doi: 10.1111/j.1742-4658.2005.04812.x. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Wafford KA, Smith A, Marshall GR, Bayley PJ, Schaeffer JM, Meinke PT, McKernan RM. Anticonvulsant and adverse effects of avermectin analogs in mice are mediated through the gamma-aminobutyric acid(A) receptor. J Pharmacol Exp Ther. 2000;295:1051–60. [PubMed] [Google Scholar]
- de Souza VK, Wales DJ. Energy landscapes for diffusion: analysis of cage-breaking processes. J Chem Phys. 2008;129:164507. doi: 10.1063/1.2992128. [DOI] [PubMed] [Google Scholar]
- Dekker C. Solid-state nanopores. Nat Nano. 2013;2:209–215. doi: 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- Del Val C, Royuela-Flor J, Milenkovic S, Bondar AN. Biochim Biophys Acta, 2013. Elsevier B.V; Netherlands: 2014. Channelrhodopsins: a bioinformatics perspective; pp. 643–55. [DOI] [PubMed] [Google Scholar]
- Delemotte L, Klein ML, Tarek M. Molecular dynamics simulations of voltage-gated cation channels: insights on voltage-sensor domain function and modulation. Front Pharmacol. 2012;3:97. doi: 10.3389/fphar.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delemotte L, Tarek M. Molecular dynamics simulations of lipid membrane electroporation. J Membr Biol. 2012;245:531–43. doi: 10.1007/s00232-012-9434-6. [DOI] [PubMed] [Google Scholar]
- Delemotte L, Tarek M, Klein ML, Amaral C, Treptow W. Intermediate states of the Kv1.2 voltage sensor from atomistic molecular dynamics simulations. Proc Natl Acad Sci U S A. 2011;108:6109–14. doi: 10.1073/pnas.1102724108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derreumaux P. Coarse-grained models for protein folding and aggregation. Methods Mol Biol. 2013;924:585–600. doi: 10.1007/978-1-62703-017-5_22. [DOI] [PubMed] [Google Scholar]
- Dickey SW, Baker RP, Cho S, Urban S. Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity. Cell. 2013;155:1270–81. doi: 10.1016/j.cell.2013.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijksman JA, Wortel GH, van Dellen LTH, Dauchot O, van Hecke M. Jamming, Yielding, and Rheology of Weakly Vibrated Granular Media. Phys Rev Lett. 2011;107:108303–108307. doi: 10.1103/PhysRevLett.107.108303. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Driessen AJ, Nouwen N. Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem. 2008;77:643–67. doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]
- du Plessis DJ, Berrelkamp G, Nouwen N, Driessen AJ. The lateral gate of SecYEG opens during protein translocation. J Biol Chem. 2009;284:15805–14. doi: 10.1074/jbc.M901855200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn SM, Martin CR, Agey MW, Miyazaki R. Functional reconstitution of the bovine brain GABAA receptor from solubilized components. Biochemistry. 1989;28:2545–51. doi: 10.1021/bi00432a030. [DOI] [PubMed] [Google Scholar]
- Egea PF, Stroud RM. Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proc Natl Acad Sci U S A. 2010;107:17182–7. doi: 10.1073/pnas.1012556107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellena JF, Blazing MA, McNamee MG. Lipid-protein interactions in reconstituted membranes containing acetylcholine receptor. Biochemistry. 1983;22:5523–35. doi: 10.1021/bi00293a012. [DOI] [PubMed] [Google Scholar]
- Engel M. Membrane permeabilization by Islet Amyloid Polypeptide. Chem Phys Lipids. 2009;160:1–10. doi: 10.1016/j.chemphyslip.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Erion MD, Dang Q, Reddy MR, Kasibhatla SR, Huang J, Lipscomb WN, van Poelje PD. Structure-guided design of AMP mimics that inhibit fructose-1,6-bisphosphatase with high affinity and specificity. Journal of the American Chemical Society. 2007;129:15480–15490. doi: 10.1021/ja074869u. [DOI] [PubMed] [Google Scholar]
- Evans DA, Wales DJ. The free energy landscape and dynamics of met-enkephalin. The Journal of Chemical Physics. 2003;119:9947–9955. [Google Scholar]
- Falk M, Krone M, Ertl T. Atomistic visualization of mesoscopic whole-cell simulations using ray-casted instancing. Comput Graph Forum. 2013;32 [Google Scholar]
- Fejer SN, Wales DJ. Helix self-assembly from anisotropic molecules. Phys Rev Lett. 2007;99:086106. doi: 10.1103/PhysRevLett.99.086106. [DOI] [PubMed] [Google Scholar]
- Feller SE, Gawrisch K, Woolf TB. Rhodopsin exhibits a preference for solvation by polyunsaturated docosohexaenoic acid. J Am Chem Soc. 2003;125:4434–5. doi: 10.1021/ja0345874. [DOI] [PubMed] [Google Scholar]
- Forman CJ, Fejer SN, Chakrabarti D, Barker PD, Wales DJ. Local frustration determines molecular and macroscopic helix structures. J Phys Chem B. 2013;117:7918–28. doi: 10.1021/jp4040503. [DOI] [PubMed] [Google Scholar]
- Forman CJ, Nickson AA, Anthony-Cahill SJ, Baldwin AJ, Kaggwa G, Feber U, Sheikh K, Jarvis SP, Barker PD. The morphology of decorated amyloid fibers is controlled by the conformation and position of the displayed protein. ACS Nano. 2012;6:1332–46. doi: 10.1021/nn204140a. [DOI] [PubMed] [Google Scholar]
- Freedman KJ, Haq SR, Edel JB, Jemth P, Kim MJ. Single molecule unfolding and stretching of protein domains inside a solid-state nanopore by electric field. Sci Rep. 2013;3:1638. doi: 10.1038/srep01638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R. Aggregation of amyloids in a cellular context: modelling and experiment. Biochem J. 2011a;438:415–26. doi: 10.1042/BJ20110369. [DOI] [PubMed] [Google Scholar]
- Friedman R, Caflisch A. Wild type and mutants of the HET-s(218–289) prion show different flexibility at fibrillar ends: A simulation study. Proteins. 2014;82:399–404. doi: 10.1002/prot.24402. [DOI] [PubMed] [Google Scholar]
- Friedman R, Pellarin R, Caflisch A. Amyloid aggregation on lipid bilayers and its impact on membrane permeability. J Mol Biol. 2009;387:407–415. doi: 10.1016/j.jmb.2008.12.036. [DOI] [PubMed] [Google Scholar]
- Friedman R, Pellarin R, Caflisch A. Soluble Protofibrils as Metastable Intermediates in Simulations of Amyloid Fibril Degradation Induced by Lipid Vesicles. J Phys Chem Lett. 2010;1:471–474. [Google Scholar]
- Friedman RaCA. Surfactant effects on amyloid aggregation kinetics. J Mol Biol. 2011b;414:303–312. doi: 10.1016/j.jmb.2011.10.011. [DOI] [PubMed] [Google Scholar]
- Friedman RaCA. Wild type and mutants of the HET-s(218–289) prion show different flexibility at fibrillar ends: A simulation study. Proteins. 2013 doi: 10.1002/prot.24402. In Press: 0. [DOI] [PubMed] [Google Scholar]
- Gkeka P, Eleftheratos S, Kolocouris A, Cournia Z. Free Energy Calculations Reveal the Origin of Binding Preference for Aminoadamantane Blockers of Influenza A/M2TM Pore. Journal of Chemical Theory and Computation. 2013;9:1272–1281. doi: 10.1021/ct300899n. [DOI] [PubMed] [Google Scholar]
- Goddard AD, Dijkman PM, Adamson RJ, Watts A. Methods Cell Biol. Elsevier Inc; United States: 2013. Lipid-dependent GPCR dimerization; pp. 341–57. copyright 2013. [DOI] [PubMed] [Google Scholar]
- Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature. 2009;460:599–604. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Murcia M, Benhamu B, Campillo M, Lopez-Rodriguez ML, Pardo L. The importance of solvation in the design of ligands targeting membrane proteins. Medchemcomm. 2011;2:160–164. [Google Scholar]
- Goose JE, Sansom MS. Reduced lateral mobility of lipids and proteins in crowded membranes. PLoS Comput Biol. 2013;9:e1003033. doi: 10.1371/journal.pcbi.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossfield A, Feller SE, Pitman MC. A role for direct interactions in the modulation of rhodopsin by omega-3 polyunsaturated lipids. Proc Natl Acad Sci U S A. 2006;103:4888–93. doi: 10.1073/pnas.0508352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu R-X, Liu LA, Wei D-Q, Du J-G, Liu L, Liu H. Free Energy Calculations on the Two Drug Binding Sites in the M2 Proton Channel. Journal of the American Chemical Society. 2011;133:10817–10825. doi: 10.1021/ja1114198. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 2011;477:495–8. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Tsutsumi-Yasuhara S, Ookoshi T, Ohhashi Y, Kimura H, Takahashi N, Yoshida H, Miyazaki R, Goto Y, Naiki H. Growth of beta(2)-microglobulin-related amyloid fibrils by non-esterified fatty acids at a neutral p H. Biochem J. 2008;416:307–315. doi: 10.1042/BJ20080543. [DOI] [PubMed] [Google Scholar]
- Head D. Critical Scaling and Aging in Cooling Systems Near the Jamming Transition. Phys Rev Lett. 2009;102:138001. doi: 10.1103/PhysRevLett.102.138001. [DOI] [PubMed] [Google Scholar]
- Hénin J, Salari R, Murlidaran S, Brannigan G. A predicted binding site for cholesterol on the GABAA receptor. Biophys J. 2014;106:1938–49. doi: 10.1016/j.bpj.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T, White SH, von Heijne G. Membrane insertion of a potassium-channel voltage sensor. Science. 2005;307:1427. doi: 10.1126/science.1109176. [DOI] [PubMed] [Google Scholar]
- Heussinger C, Barrat J-L. Jamming Transition as Probed by Quasistatic Shear Flow. Phys Rev Lett. 2009;102:218303. doi: 10.1103/PhysRevLett.102.218303. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B, Dickson EJ, Kruse M, Vivas O, Suh BC. Phosphoinositides regulate ion channels. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbalip.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdbrook DA, Piggot TJ, Sansom MS, Khalid S. Stability and membrane interactions of an autotransport protein: MD simulations of the Hia translocator domain in a complex membrane environment. Biochim Biophys Acta. 2013;1828:715–23. doi: 10.1016/j.bbamem.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–9. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, Smart TG. Neurosteroid binding sites on GABA(A) receptors. Pharmacol Ther. 2007;116:7–19. doi: 10.1016/j.pharmthera.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Hung A, Yarovsky I. Inhibition of peptide aggregation by lipids: insights from coarse-grained molecular simulations. J Mol Graph Model. 2011;29:597–607. doi: 10.1016/j.jmgm.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Janson J, Soeller W, Roche P, Nelson R, Torchia A, Kreutter D, Butler P. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci U S A. 1996;93:7283–7288. doi: 10.1073/pnas.93.14.7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardon-Valadez E, Bondar AN, Tobias DJ. Coupling of retinal, protein, and water dynamics in squid rhodopsin. Biophys J. 2010;99:2200–7. doi: 10.1016/j.bpj.2010.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MO, Jogini V, Borhani DW, Leffler AE, Dror RO, Shaw DE. Mechanism of voltage gating in potassium channels. Science. 2012;336:229–33. doi: 10.1126/science.1216533. [DOI] [PubMed] [Google Scholar]
- Jose PP, Andricioaei I. Similarities between protein folding and granular jamming. Nat Commun. 2012;3:1161. doi: 10.1038/ncomms2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem. 2005;280:36815–23. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- Kantor Y, Kardar M. Anomalous dynamics of forced translocation. Phys Rev E. 2004;69:021806–021818. doi: 10.1103/PhysRevE.69.021806. [DOI] [PubMed] [Google Scholar]
- Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–7. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasimova MA, Tarek M, Shaytan AK, Shaitan KV, Delemotte L. Biochim Biophys Acta, 2014. Elsevier B.V; Netherlands: 2014. Voltage-gated ion channel modulation by lipids: insights from molecular dynamics simulations; pp. 1322–31. [DOI] [PubMed] [Google Scholar]
- Kato HE, Zhang F, Yizhar O, Ramakrishnan C, Nishizawa T, Hirata K, Ito J, Aita Y, Tsukazaki T, Hayashi S, Hegemann P, Maturana AD, Ishitani R, Deisseroth K, Nureki O. Crystal structure of the channelrhodopsin light-gated cation channel. Nature. 2012;482:369–74. doi: 10.1038/nature10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid S, Bond PJ, Deol SS, Sansom MS. Modeling and simulations of a bacterial outer membrane protein: OprF from Pseudomonas aeruginosa. Proteins. 2006;63:6–15. doi: 10.1002/prot.20845. [DOI] [PubMed] [Google Scholar]
- Khemtemourian L, Lahoz Casarramona G, Suylen D, Hackeng T, Meeldijk J, de Kruijff B, Hoeppener J, Killian J. Impaired processing of human Pro-Islet Amyloid Polypeptide is not a causative factor for fibril formation or membrane damage in vitro. Biochemistry. 2009;48 doi: 10.1021/bi901076d. [DOI] [PubMed] [Google Scholar]
- Kilian PL, Dunlap CR, Mueller P, Schell MA, Huganir RL, Racker E. Reconstitution of acetylcholine receptor from Torpedo Californica with highly purified phospholipids: effect of alpha-tocopherol, phylloquinone, and other terpenoid quinones. Biochem Biophys Res Commun. 1980;93:409–14. doi: 10.1016/0006-291x(80)91092-x. [DOI] [PubMed] [Google Scholar]
- Kim I, Allen TW. On the selective ion binding hypothesis for potassium channels. Proc Natl Acad Sci U S A. 2011;108:17963–8. doi: 10.1073/pnas.1110735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikov OV, Merzlyak PG, Yuldasheva LN, Rodrigues CG, Bhakdi S, Valeva A. Electrophysiological evidence for heptameric stoichiometry of ion channels formed by Staphylococcus aureus alpha-toxin in planar lipid bilayers. Mol Microbiol. 2000;37:1372–8. doi: 10.1046/j.1365-2958.2000.02080.x. [DOI] [PubMed] [Google Scholar]
- Krone M, Kauker D, Reina G, Ertl T. Pacific Visualization Symposium (PacificVis), 2014 IEEE. IEEE; 2014. Visual analysis of dynamic protein cavities and binding sites; pp. 301–305. [Google Scholar]
- Krone M, Reina G, Schulz C, Kulschewski T, Pleiss J, Ertl T. Interactive extraction and tracking of biomolecular surface features. Comput Graph Forum. 2013;32:331–340. [Google Scholar]
- Kruse M, Hammond GR, Hille B. Regulation of voltage-gated potassium channels by PI(4,5)P2. J Gen Physiol. 2012;140:189–205. doi: 10.1085/jgp.201210806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusek J, Zemkova H. Effect of ivermectin on gamma-aminobutyric acid-induced chloride currents in mouse hippocampal embryonic neurones. Eur J Pharmacol. 1994;259:121–8. doi: 10.1016/0014-2999(94)90500-2. [DOI] [PubMed] [Google Scholar]
- Kumar S, Rosenberg J, Bouzida D, RH S, Kollman P. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method (pages 1011–1021) J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- Lamb ML, Jorgensen WL. Computational approaches to molecular recognition. Current Opinion in Chemical Biology. 1997;1:449–457. doi: 10.1016/s1367-5931(97)80038-5. [DOI] [PubMed] [Google Scholar]
- Lambert M, Barlow A, Chromy B, Edwards C, Freed R, Liosatos M, Morgan T, Rozovsky I, Trommer B, Viola K, Wals P, Zhang C, Finch C, Krafft G, Klein W. Diffusible, nonfibrillar ligands derived from A beta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov, England. 2011:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- Lazareno-Saez C, Arutyunova E, Coquelle N, Lemieux MJ. Domain swapping in the cytoplasmic domain of the Escherichia coli rhomboid protease. J Mol Biol. 2013;425:1127–42. doi: 10.1016/j.jmb.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins. 2003;52:176–92. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- Lechleiter J, Wells M, Gruener R. Halothane-induced changes in acetylcholine receptor channel kinetics are attenuated by cholesterol. Biochim Biophys Acta. 1986;856:640–5. doi: 10.1016/0005-2736(86)90159-8. [DOI] [PubMed] [Google Scholar]
- Lee JY, Lyman E. Predictions for cholesterol interaction sites on the A2A adenosine receptor. J Am Chem Soc. 2012;134:16512–5. doi: 10.1021/ja307532d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibel WS, Firestone LL, Legler DC, Braswell LM, Miller KW. Two pools of cholesterol in acetylcholine receptor-rich membranes from Torpedo. Biochim Biophys Acta. 1987;897:249–60. doi: 10.1016/0005-2736(87)90421-4. [DOI] [PubMed] [Google Scholar]
- Lemieux MJ, Fischer SJ, Cherney MM, Bateman KS, James MN. The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proc Natl Acad Sci U S A. 2007;104:750–4. doi: 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul J, Bevan D. The role of molecular simulations in the development of inhibitors of amyloid beta-peptide aggregation for the treatment of Alzheimer’s disease. ACS Chem Neurosci. 2012;3:845–856. doi: 10.1021/cn300091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaeus MJ, Burdette D, Wagner T, Focia PJ, Gross A. Structures of KcsA in complex with symmetrical quaternary ammonium compounds reveal a hydrophobic binding site. Biochemistry. 2014;53:5365–73. doi: 10.1021/bi500525s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori N, Nerenberg P, Head-Gordon T. Embedding Abeta42 in heterogeneous membranes depends on cholesterol asymmetries. Biophys J. 2013;105:899–910. doi: 10.1016/j.bpj.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindow N, Baum D, Bondar A, Hege HC. Dynamic channels in biomolecular systems: Path analysis and visualization. Biological Data Visualization (BioVis); 2012 IEEE Symposium on; IEEE; 2012a. pp. 99–106. [Google Scholar]
- Lindow N, Baum D, Bondar AN, Hege HC. Exploring cavity dynamics in biomolecular systems. BMC Bioinformatics. 2013;14(Suppl 19):S5. doi: 10.1186/1471-2105-14-S19-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindow N, Baum D, Hege H-C. Voronoi-based extraction and visualization of molecular paths. IEEE Trans Vis Comput Graph. 2011;17:2025–2034. doi: 10.1109/TVCG.2011.259. [DOI] [PubMed] [Google Scholar]
- Lindow N, Baum D, Hege H-C. Interactive rendering of material and biological structures on atomic and nanoscopic scale. Comput Graph Forum. 2012b;31:1045–1054. [Google Scholar]
- Lins RD, Straatsma TP. Computer simulation of the rough lipopolysaccharide membrane of Pseudomonas aeruginosa. Biophys J. 2001;81:1037–46. doi: 10.1016/S0006-3495(01)75761-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, AP IJ, Cherezov V, Stevens RC. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–6. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis DE, Petrou VI, Adney SK, Mahajan R. Channelopathies linked to plasma membrane phosphoinositides. Pflugers Arch. 2010;460:321–41. doi: 10.1007/s00424-010-0828-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Marrink S, Risselada H, Yefimov S, Tieleman D, de Vries A. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Marsh D, Barrantes FJ. Immobilized lipid in acetylcholine receptor-rich membranes from Torpedo marmorata. Proc Natl Acad Sci U S A. 1978;75:4329–33. doi: 10.1073/pnas.75.9.4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D, Watts A, Barrantes FJ. Phospholipid chain immkilization and steroid rotational immobilization in acetylcholine receptor-rich membranes from Torpedo marmorata. Biochim Biophys Acta. 1981;645:97–101. doi: 10.1016/0005-2736(81)90516-2. [DOI] [PubMed] [Google Scholar]
- Martins I, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, Van Gelder P, Hartmann D, D’Hooge R, De Strooper B, Schymkowitz J, Rousseau F. Lipids revert inert A beta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. Embo J. 2008;27:224–233. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereuta L, Roy M, Asandei A, Lee JK, Park Y, Andricioaei I, Luchian T. Slowing down single-molecule trafficking through a protein nanopore reveals intermediates for peptide translocation. Sci Rep. 2014;4:3885. doi: 10.1038/srep03885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereuta L, Schiopu I, Asandei A, Park Y, Hahm K-S, Luchian T. Protein Nanopore-Based, Single-Molecule Exploration of Copper Binding to an Antimicrobial-Derived, Histidine-Containing Chimera Peptide. Langmuir. 2012;28:17079–17091. doi: 10.1021/la303782d. [DOI] [PubMed] [Google Scholar]
- Michel J, Foloppe N, Essex JW. Rigorous Free Energy Calculations in Structure-Based Drug Design. Molecular Informatics. 2010;29:570–578. doi: 10.1002/minf.201000051. [DOI] [PubMed] [Google Scholar]
- Middlemas DS, Raftery MA. Identification of subunits of acetylcholine receptor that interact with a cholesterol photoaffinity probe. Biochemistry. 1987;26:1219–23. doi: 10.1021/bi00379a003. [DOI] [PubMed] [Google Scholar]
- Mitchell DC, Niu SL, Litman BJ. Optimization of receptor-G protein coupling by bilayer lipid composition I: kinetics of rhodopsin-transducin binding. J Biol Chem, United States. 2001:42801–6. doi: 10.1074/jbc.M105772200. [DOI] [PubMed] [Google Scholar]
- Mohammad M, Movileanu L. Excursion of a single polypeptide into a protein pore: simple physics, but complicated biology. European Biophysics Journal. 2008;37:913–925. doi: 10.1007/s00249-008-0309-9. [DOI] [PubMed] [Google Scholar]
- Moiset G, Lopez CA, Bartelds R, Syga L, Rijpkema E, Cukkemane A, Baldus M, Poolman B, Marrink SJ. Disaccharides impact the lateral organization of lipid membranes. J Am Chem Soc. 2014;136:16167–75. doi: 10.1021/ja505476c. [DOI] [PubMed] [Google Scholar]
- Mondal S, Khelashvili G, Shan J, Andersen OS, Weinstein H. Quantitative modeling of membrane deformations by multihelical membrane proteins: application to G-protein coupled receptors. Biophys J. 2011;101:2092–101. doi: 10.1016/j.bpj.2011.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritsugu K, Smith JC. Coarse-grained biomolecular simulation with REACH: realistic extension algorithm via covariance Hessian. Biophys J. 2007;93:3460–9. doi: 10.1529/biophysj.107.111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortenson PN, Evans DA, Wales DJ. Energy landscapes of model polyalanines. The Journal of Chemical Physics. 2002;117:1363–1376. [Google Scholar]
- Movileanu L, Howorka S, Braha O, Bayley H. Detecting protein analytes that modulate transmembrane movement of a polymer chain within a single protein pore. Nat Biotechnol. 2000;18:1091–5. doi: 10.1038/80295. [DOI] [PubMed] [Google Scholar]
- Neria E, Fischer S, Karplus M. Simulation of activation free energies in molecular systems. The Journal of Chemical Physics. 1996;105:1902–1921. [Google Scholar]
- Neyton J, Miller C. Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+ -activated K+ channel. J Gen Physiol. 1988;92:569–86. doi: 10.1085/jgp.92.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimigean CM, Allen TW. Origins of ion selectivity in potassium channels from the perspective of channel block. J Gen Physiol. 2011;137:405–13. doi: 10.1085/jgp.201010551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu SL, Mitchell DC, Litman BJ. Optimization of receptor-G protein coupling by bilayer lipid composition II: formation of metarhodopsin II-transducin complex. 2001 doi: 10.1074/jbc.M105778200. [DOI] [PubMed] [Google Scholar]
- Optimization of receptor-G protein coupling by bilayer lipid composition I: kinetics of rhodopsin-transducin binding. J Biol Chem, United States. :42807–11. doi: 10.1074/jbc.M105772200. [DOI] [PubMed] [Google Scholar]
- Noskov SY, Berneche S, Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–4. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- Oates J, Faust B, Attrill H, Harding P, Orwick M, Watts A. Biochim Biophys Acta, 2012. Elsevier B.V; Netherlands: 2012. The role of cholesterol on the activity and stability of neurotensin receptor 1; pp. 2228–33. [DOI] [PubMed] [Google Scholar]
- Oates J, Watts A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr Opin Struct Biol. 2011;21:802–7. doi: 10.1016/j.sbi.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Ochoa EL, Dalziel AW, McNamee MG. Reconstitution of acetylcholine receptor function in lipid vesicles of defined composition. Biochim Biophys Acta. 1983;727:151–62. doi: 10.1016/0005-2736(83)90379-6. [DOI] [PubMed] [Google Scholar]
- Olsson P, Teitel S. Critical scaling of shear viscosity at the jamming transition. Phys Rev Lett. 2007;99:178001. doi: 10.1103/PhysRevLett.99.178001. [DOI] [PubMed] [Google Scholar]
- Oukhaled G, Mathe J, Biance AL, Bacri L, Betton JM, Lairez D, Pelta J, Auvray L. Unfolding of proteins and long transient conformations detected by single nanopore recording. Phys Rev Lett. 2007;98:158101. doi: 10.1103/PhysRevLett.98.158101. [DOI] [PubMed] [Google Scholar]
- Palovcak E, Delemotte L, Klein ML, Carnevale V. Evolutionary imprint of activation: the design principles of VSDs. J Gen Physiol. 2014;143:145–56. doi: 10.1085/jgp.201311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastoriza-Gallego M, Rabah L, Gibrat G, Thiebot B, van der Goot F, Auvray L, Betton J-M, Pelta J. Dynamics of Unfolded Protein Transport through an Aerolysin Pore. Journal of the American Chemical Society. 2011;133:2923–2931. doi: 10.1021/ja1073245. [DOI] [PubMed] [Google Scholar]
- Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486:135–9. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–8. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payet L, Martinho M, Pastoriza-Gallego M, Betton J-M, Auvray L, Pelta J, Mathe J. Thermal Unfolding of Proteins Probed at the Single Molecule Level Using Nanopores. Analytical Chemistry. 2012;84:4071–4076. doi: 10.1021/ac300129e. [DOI] [PubMed] [Google Scholar]
- Pellarin R, Caflisch A. Interpreting the aggregation kinetics of amyloid peptides. J Mol Biol. 2006;360:882–892. doi: 10.1016/j.jmb.2006.05.033. [DOI] [PubMed] [Google Scholar]
- Piggot T, Piñeiro A, Khalid S. Molecular Dynamics Simulations of Phosphatidylcholine Membranes: A Comparative Force Field Study. J Chem Theory Comput. 2012;8:4593–4609. doi: 10.1021/ct3003157. [DOI] [PubMed] [Google Scholar]
- Piggot TJ, Holdbrook DA, Khalid S. Electroporation of the E. coli and S. Aureus membranes: molecular dynamics simulations of complex bacterial membranes. J Phys Chem B. 2011;115:13381–8. doi: 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- Piggot TJ, Holdbrook DA, Khalid S. Conformational dynamics and membrane interactions of the E. coli outer membrane protein FecA: a molecular dynamics simulation study. Biochim Biophys Acta. 2013;1828:284–93. doi: 10.1016/j.bbamem.2012.08.021. [DOI] [PubMed] [Google Scholar]
- Pitman MC, Grossfield A, Suits F, Feller SE. Role of cholesterol and polyunsaturated chains in lipid-protein interactions: molecular dynamics simulation of rhodopsin in a realistic membrane environment. J Am Chem Soc. 2005;127:4576–7. doi: 10.1021/ja042715y. [DOI] [PubMed] [Google Scholar]
- Plath K, Wilkinson BM, Stirling CJ, Rapoport TA. Interactions between Sec complex and prepro-alpha-factor during posttranslational protein transport into the endoplasmic reticulum. Mol Biol Cell. 2004;15:1–10. doi: 10.1091/mbc.E03-06-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popot JL, Demel RA, Sobel A, Van Deenen LL, Changeux JP. Interaction of the acetylcholine (nicotinic) receptor protein from Torpedo marmorata electric organ with monolayers of pure lipids. Eur J Biochem. 1978;85:27–42. doi: 10.1111/j.1432-1033.1978.tb12209.x. [DOI] [PubMed] [Google Scholar]
- Ramu Y, Xu Y, Lu Z. Enzymatic activation of voltage-gated potassium channels. Nature. 2006;442:696–9. doi: 10.1038/nature04880. [DOI] [PubMed] [Google Scholar]
- Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–9. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- Reddy AS, Pati SP, Kumar PP, Pradeep HN, Sastry GN. Virtual screening in drug discovery – a computational perspective. Curr Protein Pept Sci. 2007;8:329–351. doi: 10.2174/138920307781369427. [DOI] [PubMed] [Google Scholar]
- Reddy MR, Erion MD. Calculation of relative binding free energy differences for fructose 1,6-bisphosphatase inhibitors using the thermodynamic cycle perturbation approach. Journal of the American Chemical Society. 2001;123:6246–6252. doi: 10.1021/ja0103288. [DOI] [PubMed] [Google Scholar]
- Reis P, Ingale R, Shattuck M. Forcing independent velocity distributions in an experimental granular fluid. Phys Rev E. 2007;75:051311–051325. doi: 10.1103/PhysRevE.75.051311. [DOI] [PubMed] [Google Scholar]
- Rheingans P, Joshi S. Data Visualization’99. Springer; 1999. Visualization of molecules with positional uncertainty; pp. 299–306. [Google Scholar]
- Robertson KM, Tieleman DP. Molecular basis of voltage gating of OmpF porin. Biochem Cell Biol. 2002;80:517–23. doi: 10.1139/o02-145. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Menchaca AA, Adney SK, Tang QY, Meng XY, Rosenhouse-Dantsker A, Cui M, Logothetis DE. PIP2 controls voltage-sensor movement and pore opening of Kv channels through the S4–S5 linker. Proc Natl Acad Sci U S A. 2012;109:E2399–408. doi: 10.1073/pnas.1207901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg MR, Casarotto MG. Coexistence of two adamantane binding sites in the influenza A M2 ion channel. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13866–13871. doi: 10.1073/pnas.1002051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B. The calculation of the potential of mean force using computer simulations. Comput Phys Commun. 1995;91:275–282. [Google Scholar]
- Roux B. Influence of the membrane potential on the free energy of an intrinsic protein. Biophys J. 1997;73:2980–9. doi: 10.1016/S0006-3495(97)78327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadiq SK, Guixa-Gonzalez R, Dainese E, Pastor M, De Fabritiis G, Selent J. Molecular modeling and simulation of membrane lipid-mediated effects on GPCRs. Curr Med Chem. 2013;20:22–38. [PubMed] [Google Scholar]
- Sampathkumar P, Mak MW, Fischer-Witholt SJ, Guigard E, Kay CM, Lemieux MJ. Oligomeric state study of prokaryotic rhomboid proteases. Biochim Biophys Acta. 2012;1818:3090–7. doi: 10.1016/j.bbamem.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Sands ZA, Sansom MS. How does a voltage sensor interact with a lipid bilayer? Simulations of a potassium channel domain. Structure. 2007;15:235–44. doi: 10.1016/j.str.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago J, Guzman GR, Rojas LV, Marti R, Asmar-Rovira GA, Santana LF, McNamee M, Lasalde-Dominicci JA. Probing the effects of membrane cholesterol in the Torpedo californica acetylcholine receptor and the novel lipid-exposed mutation alpha C418W in Xenopus oocytes. J Biol Chem. 2001;276:46523–32. doi: 10.1074/jbc.M104563200. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Jiang QX, MacKinnon R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 2006;444:775–9. doi: 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- Schmidt-Ehrenberg J. Analysis and Visualization of Molecular Conformations. Freie Universitat Berlin; Berlin: 2008. [Google Scholar]
- Schmidt-Ehrenberg J, Baum D, Hege H-C. Visualizing dynamic molecular conformations. Proc IEEE Visualization. 2002:235–242. [Google Scholar]
- Schütte C, Sarich M. Metastability and Markov State Models in Molecular Dynamics: Modeling, Analysis, Algorithmic Approaches. American Mathematical Society; 2013. [Google Scholar]
- Seo M, Rauscher S, Poms R, Tieleman D. Improving Internal Peptide Dynamics in the Coarse-Grained MARTINI Model: Toward Large-Scale Simulations of Amyloid- and Elastin-like Peptides. J Chem Theory Comput. 2012;8:1774–1785. doi: 10.1021/ct200876v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea J, Urbanc B. Insights into Abeta aggregation: a molecular dynamics perspective. Curr Top Med Chem. 2012;12:2596–2610. doi: 10.2174/1568026611212220012. [DOI] [PubMed] [Google Scholar]
- Silbert L. Jamming of frictional spheres and random loose packing. Soft Matter. 2010;6:2918–2924. [Google Scholar]
- Snoeijer JH, Vlugt TJH, Ellenbroek WG, van Hecke M, van Leeuwen JMJ. Ensemble theory for force networks in hyperstatic granular matter. Phys Rev E. 2004;70:061306–061322. doi: 10.1103/PhysRevE.70.061306. [DOI] [PubMed] [Google Scholar]
- Song L, Hobaugh M, Shustak C, Cheley S, Bayley H, Gouaux J. Structure of Staphylococcal alpha-Hemolysin, a Heptameric Transmembrane Pore. Science. 1996;274:1859–1865. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- Sooksawate T, Simmonds MA. Increased membrane cholesterol reduces the potentiation of GABA(A) currents by neurosteroids in dissociated hippocampal neurones. Neuropharmacology. 1998;37:1103–10. doi: 10.1016/s0028-3908(98)00113-0. [DOI] [PubMed] [Google Scholar]
- Sooksawate T, Simmonds MA. Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology. 2001a;40:178–84. doi: 10.1016/s0028-3908(00)00159-3. [DOI] [PubMed] [Google Scholar]
- Sooksawate T, Simmonds MA. Influence of membrane cholesterol on modulation of the GABA(A) receptor by neuroactive steroids and other potentiators. Br J Pharmacol. 2001b;134:1303–11. doi: 10.1038/sj.bjp.0704360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefureac R, Waldner L, Howard P, J L. Nanopore Analysis of a Small 86-Residue Protein. Small. 2008;4:59–63. doi: 10.1002/smll.200700402. [DOI] [PubMed] [Google Scholar]
- Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–U13. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straatsma TP, Soares TA. Characterization of the outer membrane protein OprF of Pseudomonas aeruginosa in a lipopolysaccharide membrane by computer simulation. Proteins. 2009;74:475–88. doi: 10.1002/prot.22165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strodel B, Lee JW, Whittleston CS, Wales DJ. Transmembrane structures for Alzheimer’s Abeta(1–42) oligomers. J Am Chem Soc. 2010;132:13300–12. doi: 10.1021/ja103725c. [DOI] [PubMed] [Google Scholar]
- Strodel B, Whittleston CS, Wales DJ. Thermodynamics and kinetics of aggregation for the GNNQQNY peptide. J Am Chem Soc. 2007;129:16005–14. doi: 10.1021/ja075346p. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–95. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarek M, Delemotte L. Omega currents in voltage-gated ion channels: what can we learn from uncovering the voltage-sensing mechanism using MD simulations? Acc Chem Res. 2013;46:2755–62. doi: 10.1021/ar300290u. [DOI] [PubMed] [Google Scholar]
- Tautermann CS. Bioorg Med Chem Lett, 2014 The Author. Elsevier Ltd.; England: 2014. GPCR structures in drug design, emerging opportunities with new structures; pp. 4073–9. [DOI] [PubMed] [Google Scholar]
- Thompson AN, Kim I, Panosian TD, Iverson TM, Allen TW, Nimigean CM. Mechanism of potassium-channel selectivity revealed by Na(+) and Li(+) binding sites within the KcsA pore. Nat Struct Mol Biol. 2009;16:1317–24. doi: 10.1038/nsmb.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian P, Andricioaei I. Repetitive Pulling Catalyzes Co-translocational Unfolding of Barnase During Import Through a Mitochondrial Pore. Journal of Molecular Biology. 2005;350:1017–1034. doi: 10.1016/j.jmb.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Tieleman DP. The molecular basis of electroporation. BMC Biochem. 2004;5:10. doi: 10.1186/1471-2091-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe B, Snoeijer J, Vlugt T, van Hecke M. The force network ensemble for granular packings. Soft Matter. 2010;6:2908–2917. [Google Scholar]
- Tighe B, Socolar J, Schaeffer D, Mitchener W, Huber M. Force distributions in a triangular lattice of rigid bars. Phys Rev E. 2005;72:031306–031316. doi: 10.1103/PhysRevE.72.031306. [DOI] [PubMed] [Google Scholar]
- Tofoleanu F, Brooks BR, Buchete NV. Modulation of Alzheimer’s abeta protofilament-membrane interactions by lipid headgroups. ACS Chem Neurosci. 2015;6:446–55. doi: 10.1021/cn500277f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofoleanu F, Buchete NV. Alzheimer Abeta peptide interactions with lipid membranes: fibrils, oligomers and polymorphic amyloid channels. Prion. 2012a;6:339–45. doi: 10.4161/pri.21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofoleanu F, Buchete NV. Molecular interactions of Alzheimer’s Abeta protofilaments with lipid membranes. J Mol Biol. 2012b;421:572–86. doi: 10.1016/j.jmb.2011.12.063. [DOI] [PubMed] [Google Scholar]
- Treptow W, Tarek M. Environment of the gating charges in the Kv1.2 Shaker potassium channel. Biophys J. 2006;90:L64–6. doi: 10.1529/biophysj.106.080754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treptow W, Tarek M, Klein ML. Initial response of the potassium channel voltage sensor to a transmembrane potential. J Am Chem Soc. 2009;131:2107–9. doi: 10.1021/ja807330g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukazaki T, Mori H, Fukai S, Ishitani R, Mori T, Dohmae N, Perederina A, Sugita Y, Vassylyev DG, Ito K, Nureki O. Conformational transition of Sec machinery inferred from bacterial SecYE structures. Nature. 2008;455:988–91. doi: 10.1038/nature07421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider JP, Smith JC, White SH, Ulmschneider MB. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J Am Chem Soc. 2011;133:15487–95. doi: 10.1021/ja204042f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider MB, Doux JP, Killian JA, Smith JC, Ulmschneider JP. Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J Am Chem Soc. 2010;132:3452–60. doi: 10.1021/ja909347x. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–89. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Urban S, Dickey SW. The rhomboid protease family: a decade of progress on function and mechanism. Genome Biol. 2011;12:231. doi: 10.1186/gb-2011-12-10-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Wolfe M. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci U S A. 2005;102:1883–8. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagberg D, Valdez-Balderas D, Moore MA, Olsson P, Teitel S. Finite-size scaling at the jamming transition: C orrections to scaling and the correlation-length critical exponent. Physical Review E. 2011;83:030303(R). doi: 10.1103/PhysRevE.83.030303. [DOI] [PubMed] [Google Scholar]
- Vamparys L, Gautier R, Vanni S, Bennett WF, Tieleman DP, Antonny B, Etchebest C, Fuchs PF. Conical lipids in flat bilayers induce packing defects similar to that induced by positive curvature. Biophys J. 2013;104:585–93. doi: 10.1016/j.bpj.2012.11.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- Vanni S, Hirose H, Barelli H, Antonny B, Gautier R. A sub-nanometre view of how membrane curvature and composition modulate lipid packing and protein recruitment. Nat Commun, England. 2014:4916. doi: 10.1038/ncomms5916. [DOI] [PubMed] [Google Scholar]
- Vanni S, Vamparys L, Gautier R, Drin G, Etchebest C, Fuchs PF, Antonny B. Amphipathic lipid packing sensor motifs: probing bilayer defects with hydrophobic residues. Biophys J. 2013;104:575–84. doi: 10.1016/j.bpj.2012.11.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard MC, Morita M, Hamada T, Takagi M. Membrane fusion and vesicular transformation induced by A lzheimer’s amyloid beta. Biochim Biophys Acta. 2013;1828:1314–1321. doi: 10.1016/j.bbamem.2013.01.015. [DOI] [PubMed] [Google Scholar]
- Vinothkumar KR. Structure of rhomboid protease in a lipid environment. J Mol Biol. 2011;407:232–47. doi: 10.1016/j.jmb.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar KR, Strisovsky K, Andreeva A, Christova Y, Verhelst S, Freeman M. The structural basis for catalysis and substrate specificity of a rhomboid protease. Embo J. 2010;29:3797–809. doi: 10.1038/emboj.2010.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem Biophys Res Commun. 2011;411:312–6. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales D. Discrete path sampling. Molecular Physics. 2002;100:3285–3305. [Google Scholar]
- Wales D. Energy Landscapes. Cambridge University Press; Cambridge: 2003. [Google Scholar]
- Wales D. Energy landscapes: calculating pathways and rates. International Reviews in Physical Chemistry. 2006;25:237–282. [Google Scholar]
- Wales D. Energy landscapes: some new horizons. Curr Opin Struct Biol. 2010;20:3–10. doi: 10.1016/j.sbi.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Wales D. Surveying a complex potential energy landscape: Overcoming broken ergodicity using basin-sampling. Chemical Physics Letters. 2013;584:1–9. [Google Scholar]
- Wales D, Doye J. Global Optimization by Basin-Hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms. The Journal of Physical Chemistry A. 1997;101:5111–5116. [Google Scholar]
- Wales D, Doye J. Stationary points and dynamics in high-dimensional systems. The Journal of Chemical Physics. 2003;119:12409–12416. [Google Scholar]
- Wales D, Walsh T. Theoretical Study of the Water Tetramer. J Chem Phys. 1997;106:7193–7207. [Google Scholar]
- Wales DJ. The energy landscape as a unifying theme in molecular science. Philos Trans A Math Phys Eng Sci. 2005;363:357–75. doi: 10.1098/rsta.2004.1497. discussion 375–7. [DOI] [PubMed] [Google Scholar]
- Wales DJ, Bogdan TV. Potential energy and free energy landscapes. J Phys Chem B. 2006;110:20765–76. doi: 10.1021/jp0680544. [DOI] [PubMed] [Google Scholar]
- Wales DJ, Dewsbury PE. Effect of salt bridges on the energy landscape of a model protein. J Chem Phys. 2004;121:10284–90. doi: 10.1063/1.1810471. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers-a decade of discovery. J Neurochem, England. 2007:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Walsh T, Wales D. Rearrangements of the water trimer. J Chem Soc Faraday T. 1996;92:2505–2517. [Google Scholar]
- Wang JF, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Protein Science. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ha Y. Open-cap conformation of intramembrane protease GlpG. Proc Natl Acad Sci U S A. 2007;104:2098–102. doi: 10.1073/pnas.0611080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Maegawa S, Akiyama Y, Ha Y. The role of L1 loop in the mechanism of rhomboid intramembrane protease GlpG. J Mol Biol. 2007;374:1104–13. doi: 10.1016/j.jmb.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–80. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- White SH, von Heijne G. How translocons select transmembrane helices. Annu Rev Biophys. 2008;37:23–42. doi: 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- Whorton MR, MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011;147:199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–8. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- Wu EL, Engstrom O, Jo S, Stuhlsatz D, Yeom MS, Klauda JB, Widmalm G, Im W. Molecular dynamics and NMR spectroscopy studies of E. coli lipopolysaccharide structure and dynamics. Biophys J. 2013;105:1444–55. doi: 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EL, Fleming PJ, Yeom MS, Widmalm G, Klauda JB, Fleming KG, Im W. E. coli Outer Membrane and Interactions with OmpLA. Biophys J. 2014;106:2493–502. doi: 10.1016/j.bpj.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13:1084–91. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ramu Y, Lu Z. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature. 2008;451:826–9. doi: 10.1038/nature06618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Chowdhury S, Liu X, Akiyama Y, Ellman J, Ha Y. Conformational change in rhomboid protease GlpG induced by inhibitor binding to its S’ subsites. Biochemistry. 2012;51:3723–31. doi: 10.1021/bi300368b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ha Y. Catalytic mechanism of rhomboid protease GlpG probed by 3,4-dichloroisocoumarin and diisopropyl fluorophosphonate. J Biol Chem. 2012;287:3099–107. doi: 10.1074/jbc.M111.310482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabrecky JR, Raftery MA. The role of lipids in the function of the acetylcholine receptor. J Recept Res. 1985;5:397–417. doi: 10.3109/10799898509041890. [DOI] [PubMed] [Google Scholar]
- Zhao C, Caplan DA, Noskov SY. Evaluations of the Absolute and Relative Free Energies for Antidepressant Binding to the Amino Acid Membrane Transporter LeuT with Free Energy Simulations. Journal of Chemical Theory and Computation. 2010;6:1900–1914. doi: 10.1021/ct9006597. [DOI] [PubMed] [Google Scholar]
- Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–43. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwanzig R. High-Temperature Equation of state by a perturbation method. 1. Nonpolar gases. 1954;22:1420–1426. [Google Scholar]