

Abstract
Cholestasis, including primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC), results from an impairment or disruption of bile production and causes intracellular retention of toxic bile constituents, including bile salts. If left untreated, cholestasis leads to liver fibrosis and cirrhosis, which eventually results in liver failure and the need for liver transplantation. Currently, the only therapeutic option available for these patients is ursodeoxycholic acid (UDCA), which slows the progression of PBC, particularly in stage I and II of the disease. However some patients have an incomplete response to UDCA therapy, while other more advanced cases often remain unresponsive. For PSC, UDCA therapy does not improve survival, and recommendations for its use remains controversial. These considerations emphasize the need for alternative therapies.
Hepatic transporters, located along basolateral (sinusoidal) and apical (canalicular) membranes of hepatocytes, are integral determinants of bile formation and secretion. Nuclear receptors are critically involved in the regulation of these hepatic transporters and are natural targets for therapy of cholestatic liver diseases. One of these nuclear receptors is peroxisome proliferator-activated receptor alpha (PPARα) which plays a central role in maintaining cholesterol, lipid and bile acid homeostasis by regulating genes responsible for bile acid synthesis, and transport in humans, including Cytochrome P450 (CYP) isoform 7A1 (CYP7A1), CYP27A1, CYP8B1, UGT1A1, 1A3, 1A4, 1A6, SULT2A1, MDR3, and ASBT. The expression of many of these genes is altered in cholestatic liver diseases but few have been extensively studied or had the mechanism of PPARα effect identified. In this review we examine what is known about these mechanisms and consider the rationale for the use of PPARα ligand therapy in various cholestatic liver disorders.
I. Introduction
Hepatic bile salt secretion and bile formation are essential functions of the mammalian liver. Cholestasis, a condition where the production of bile is impaired, results in intracellular retention of toxic bile constituents, including bile salts (1). Hepatocytes are polarized epithelial cells with basolateral (sinusoidal) and apical (canalicular) domains (2), where several liver-specific transporters enable vectorial movement of endogenous and exogenous compounds from blood into bile (3). Canalicular secretion of bile components represents the rate-limiting step in bile formation (2). Bile acids, glutathione conjugates, and xenobiotics are removed from hepatocytes and concentrated into the bile by canalicular efflux transporters in an ATP-dependent manner. Several transporters that are members of the ATP-binding cassette (ABC) superfamily are expressed along the canalicular membrane of hepatocytes, where they are involved in the secretion of bile constituents. Consequently, hepatic transporters play important roles in bile formation. The multidrug resistance protein 3 (MDR3/Mdr2 in rodent), encoded by the ABC protein subtype B4 (ABCB4) gene, is the major determinant of biliary phosphatidylcholine (PC) secretion (4), where it functions as a floppase and translocates PC from the inner to the outer leaflet of the canalicular membrane of hepatocytes for bile salt extraction (2). The bile salt export pump (BSEP/Bsep), encoded by the ABCB11 gene, and first identified as the sister of P-glycoprotein (5), is essential for biliary secretion of bile salts, while the heterodimer of ABCG5 and ABCG8 secretes cholesterol into bile (6). Substrates of these transporters (bile salts, PC and cholesterol) form mixed biliary micelles which reduce the biliary detergent activity of the bile salts and maintain cholesterol homeostasis (7). Indeed, mutations in these transporters result in several types of inherited disorders (8), known as progressive familial intrahepatic cholangitis (PFIC) type 1, 2, and 3. Mutations in MDR3 also cause several liver disorders in addition to PFIC3 ranging from gallstone formation, to drug induced liver disease, intrahepatic cholestasis of pregnancy and even some forms of progressive cholestatic liver disease in adults (9).
A growing body of literature supports the notion that nuclear receptors are critically involved in the regulation of hepatic transporters, findings that have prompted the investigation of nuclear receptors as therapeutic targets for cholestatic liver diseases. In particular, fenofibrate, the peroxisome proliferator-activated receptor-alpha (PPARα) agonist is increasingly used to treat patients with chronic cholestatic liver disease who are refractory to ursodeoxycholic acid (UDCA) monotherapy. In this review we examine the data supporting the claim of PPARα as an alternative pharmacological target for the treatment of cholestatic disorders and provide insight into the mechanism(s) by which PPARα agonists may improve cholestatic liver injury.
II. Peroxisome Proliferator-Activated Receptor (PPAR)
Discovered in 1990, PPARs are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily (10). Three distinct PPAR isoforms: α, β/δ, and γ, are encoded by distinct genes and show different distribution patterns. All PPARs share the typical structure of other members in this family (Figure 1). Upon ligand activation, PPARs regulate gene transcription by forming a heterodimer with the retinoid x receptor and bind to specific DNA-response elements containing direct repeats of the consensus sequence AGGTCA separated by one or two nucleotides located within the regulatory regions of target genes (11). Activation of PPAR by an appropriate ligand results in the recruitment of co-activators and the loss of co-repressors which remodel the chromatin and activate gene transcription (11).
Figure 1.
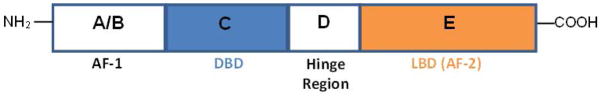
Typical structure of the functional domains of the human peroxisome proliferator-activated receptor (PPAR). A/B: Ligand-independent transactivation domain (AF-1), C: Ligand-dependent transactivation domain (AF-2), D: DNA binding domain (DBD), and E: Ligand binding domain (LBD).
Activation of PPARα, results in proliferation of peroxisomes in rodents but not humans (12). The molecular basis for this species difference is attributed to a murine-specific micro-RNA let-7c signaling cascade (13). PPARα is highly expressed in tissues including liver, muscle, kidney, and heart where it stimulates the β-oxidative degradation of fatty acids (22), and regulates the transcription of several genes involved in lipid metabolism (14). The phenotype of the fasted PPARα knock-out mouse is hypoglycaemia, hypoketonaemia, hypertriglyceridaemia and hepatic steatosis (15).
PPAR is the molecular target for the fibrate hypolipidemic agents, including fenofibrate, gemfibrozil, and clofibrate, which are FDA-approved for the treatment of dyslipidemia, though each fibrate differs in its specificity for the different PPAR subtypes, α, β/δ, and γ (16), presented in Table 1. Another fibrate, bezafibrate, activates all three PPAR isoforms at equivalent molar concentrations, whereas fenofibrate exhibits approximately a 10-fold selectivity for PPAR-alpha vs. -gamma (16). Fenofibrate activates PPARα by binding to PPAR response elements (PPREs) in the upstream regions of target genes (17). The synthetic compound Wy-14,643 is also a PPARα ligand, often used in in vitro testing.
Table 1.
Potency of human PPAR agonists, modified from Wilson TM et al. (16).
| Ligand | Human receptor EC50 (μM) | ||
|---|---|---|---|
| PPARα | PPARβ/δ | PPARγ | |
| Wy-14,643 | 5 | 35 | 60 |
| Clofibratea | 55 | IA at 100 | ~500 |
| Fenofibrate | 30 | IA at 100 | 300 |
| Bezafibrate | 50 | 20 | 60 |
data is for the active metabolite, IA = inactive.
III. PPARα: Regulation of Bile Acid (BA) Synthesis
Ligand-activated PPARα contributes to a range of actions including cholesterol and BA homeostasis, summarized in Table 2. Also, fenofibrate significantly reduced serum levels of UDCA as well as toxic primary and secondary bile acids, including CDCA, deoxycholic acid, and lithocholic acid in non-cholestatic adult volunteers (18). PPARα primarily down-regulates BA synthesis through inhibition of the BA-synthesizing enzymes, cytochrome P450 cholesterol 7A1-hydroxylase (CYP7A1), the rate-limiting step in bile acid synthesis, and cytochrome sterol 27-hydroxylase (CYP27A1). Ciprofibrate and Wy-14,643 reduced CYP7A1 and CYP27A1 mRNA expression in parallel with reduction in their respective hydroxylase activities (19). Wy-14,643 suppresses Cyp7a1 gene promoter activity (28) via interruption of hepatocyte nuclear factor4-mediated activation of CYP7A1 (20), while ciprofibrate induces the promoter activity of human apical sodium-dependent bile salt transporter (ASBT) gene in Caco2 cells (30). In contrast, ligand-activated Pparα stimulates the expression and activity of sterol 12α-hydroxylase (CYP8B1), a hepatic microsomal enzyme that acts as a branch point in the bile acid synthetic pathway that determines the cholic acid (CA):chenodeoxycholic acid (CDCA) ratio (21). Treatment of wild-type animals with Wy-14,643 increased CA formation in a Pparα-dependent manner, that was blocked in Pparα-null mice (21). Additionally, recent findings indicate that PPARα also regulates BA detoxification via up-regulation of CYP3A4 (22), UGT2B4 (23), UGT1A1, 1A3, 1A4, 1A6 (24), SULT2A1 (25), and ASBT (26). Murine Pparα also plays a crucial role in BA synthesis, but reduces phospholipid concentration and mRNA expression of Abcb4, Abca1, Abcg5, and Abcg8 in Pparα-null mice. (27). These data suggest that PPARα activation decreases BA synthesis and bile salt secretion into bile. Furthermore, PPARα activators, i.e. fenofibrate, enhanced human hepatic glucuronidation activity and expression of the uridine 5′-diphospho-glucuronosyltransferase (UGT) family of enzymes, specifically UGT2B4 (23). SULT2A1 catalyzes the sulfation of BAs which facilitates the elimination of the toxic secondary bile acid lithocholic acid (28), thereby reducing cholestasis. Transcriptional regulation of SULT2A1 by PPARα occurs at PPREs located −5949 to −5929 bp upstream of the transcription start site (25). A thorough analysis of the UGT1 locus (24) revealed that PPARα positively regulates not only liver UGT1A1, but also 1A3, 1A4, and 1A6 isoforms. More recently, PPARα was shown to directly regulate CYP3A4 transcription as well as CYP1A1, 1A2, 2B6, and 2C8 (22). These findings introduce potential drug-drug interactions between PPARα agonists and CYP450 regulators.
Table 2.
Summary of key target genes involved in PPARα-induced beneficial effects in cholestatic disorders.
| Target gene(s) | Therapeutic effects | Ref. |
|---|---|---|
| Cyp7a1 and Cyp27a1 |
|
(19) |
| ASBT/SLC10A2 |
|
(26) |
| Cyp8b1 |
|
(21) |
| UGT2B4 |
|
(23) |
| UGT1A1, 1A3, 1A4 and 1A6 |
|
(24) |
| SULT2A1 |
|
(25) |
| Bile acid synthesizing and metabolizing enzymes, and transporting proteins: Cyp7a1, Cyp7b1, Cyp8b1, Cyp27a1, and Hsd3b7; Ntcp, Oatp1 and 4, Abcb4, Baat, Bacs |
|
(27) |
| MDR3/ABCB4 |
|
(37) |
| Mdr2/Abcb4 |
|
(39) |
| Mdr2 |
|
(40) |
| Primary and secondary BAs |
|
(18) |
CYP7A1: Cholesterol 7α-hydroxylase; CYP27A1: sterol 27-hydroxylase; ASBT/SLC10A2: Apical Sodium-dependent Bile Salt Transporter; Cyp8b1: Sterol 12α-hydroxylase ; UGT: UDP-glucuronosyltransferase; SULT2A1: Hydroxysteroid sulfotransferase enzyme; Hsd3b7:Hydroxyl-delta-5-steroid dehydrogenase; Ntcp: sodium-taurocholate cotransporting polypeptide; Oatp: organic anion transporting peptide; Baat/Slc27a5: bile acid-CoA synthetase; Bacs:bile acid-coA amino acid N-acetyltransferase; Mdr2: Multidrug resistance drug transporter 2; MDR3/ABCB4: Multidrug resistance transporter 3/ATP-binding cassette protein subfamily B4
IV. PPAR: Regulation of Bile Excretory Function
a. Efflux Transporter MDR3/Mdr2
MDR3 (ABCB4) is predominantly expressed in the liver (29) and localized to the canalicular membrane of hepatocytes, where it is the major determinant of biliary PC secretion (4). The functional importance of MDR3 was first demonstrated in Mdr2 knockout mice which lack biliary phospholipids and develop bile duct injury and progressive liver disease (30), closely resembling PSC in humans (31). We and others (7, 32) observed a linear relationship between biliary PC concentration and Mdr2 gene expression (Figure 2). Clinically, mutations and polymorphisms of MDR3 contribute to several types of cholestatic liver injury, including PFIC-3, intrahepatic cholestasis of pregnancy, low phospholipid cholelithiasis, drug-induced and idiopathic chronic cholestasis in adults (33). Thus, MDR3 represents an important pharmacological target.
Figure 2. Biliary phosphatidylcholine (PC) is a function of Abcb4 gene expression.

Bile was collected from age-matched mice that express the Abcb4 gene (wild-type, WT), heterozygous expression (+/−), and homozygous deletion (−/−). Biliary PC concentration was determined using a colorimetric assay. Data are expressed as the mean ± SD (n=3). **p<0.01 vs. WT.
The observation that bezafibrate improved serum alkaline phosphatase (ALP) in patients treated for hyperlipidemia (34) introduced the possibility that fibrates could be beneficial for patients with cholestatic liver disease. Since then, case reports and pilot studies demonstrated the efficacy of bezafibrate and fenofibrate, reviewed in (35), in reducing biomarkers of cholestasis and liver function abnormalities in patients with PBC or PSC experiencing an incomplete response to UDCA monotherapy (13–15 mg/kg/day). However, most studies test bezafibrate, a non-specific PPAR agonist not FDA-approved nor clinically available in the United States, compared to fenofibrate, a specific FDA approved PPARα agonist. Despite in vitro and animal findings, and clinical studies showing efficacy, the mechanism(s) by which fibrates reduce biochemical markers of cholestasis remains unclear. Bezafibrate has been reported to act as a dual PPARα and -γ and pregnane X receptor (PXR) agonist (36), albeit at super-physiological doses. We examined the mechanism by which fenofibrate modulates MDR3 expression and found that activation of PPARα by fenofibrate directly up-regulates human MDR3 expression by binding to specific PPREs located on the gene’s promoter (37). In silico analysis of the 5′-upstream region of human MDR3 gene identified 21 putative PPREs. Reporter gene assays identified three novel PPREs located 6–10 kb upstream of the transcription start site of the MDR3 promoter. Further, fenofibrate increased the canalicular excretion of fluorescently labeled PC when incubated with isolated rat hepatocytes grown in collagen sandwich cultures (37). This is the first evidence that the human MDR3 gene is directly trans-activated by PPARα and strongly suggests that PPARα up-regulates the expression of MDR3 and facilitates hepatic export of phospholipids. Together, these findings provide a molecular and physiologic mechanism by which fenofibrate may improve symptoms and liver function in patients with chronic cholestatic liver disease.
Others have reported that bezafibrate increases MDR3 localization and NBC-PC within pseudo-canaliculi of HepG2 cells although the specificity for canalicular secretion of PC was not clear since bezafibrate also increased mRNA expression of MRP2 and BSEP (38). In contrast, when primary rat hepatocytes were cultured in a collagen sandwich and treated with fenofibrate there was a 2-fold increase in fluorescent intensity of NBD-PC secreted into the canalicular lumen closely mimicking the in vivo situation (37). These results strongly suggest that fenofibrate improves biochemical markers of cholestasis by enhancing biliary excretion of PC.
Results vary on the effects of fibrates on Mdr2 mRNA expression, i.e. ciprofibrate and Wy-14,643 treatment up-regulate Mdr2 mRNA expression in primary hepatocytes and liver tissue of wild-type but not Pparα-null mice, yet, wild-type mice displayed an increase in bile flow in spite of reduced biliary phospholipid and bile salt concentrations (39). Others (40) reported that a 0.5% fenofibrate diet did not increase biliary phospholipid output in wild-type mice, despite increased Mdr2 mRNA levels, while clofibrate and ciprofibrate increased biliary phospholipid levels in wild-type animals, but not in Pparα-null mice. Interestingly, the effects of clofibrate and ciprofibrate on phospholipid output occurred independently of changes in bile acid output and fibrate treatment did not alter the BA pool composition (40). However, in silico analysis of the mouse Abcb4 promoter reveals 25 Ppres located within the 10 kb upstream region (37). In particular, three Ppres in the 6–10 kb upstream region were located very close to the three PPREs described in the human ABCB4 promoter (37). These findings corroborate the conclusion that /Pparα/PPARα directly regulates Abcb4/ABCB4 expression in both rodents and humans. Opposing effects of fibrates may be the result of species-specificity or the variable affinity of fibrates for PPARα/Pparα, as previously noted (19). While these studies suggest a direct role for MDR3/Mdr2 by PPARα activation, additional effects of PPARα activation, via independent yet additive pathways, i.e. lipid metabolism, CYP450-, and UGT-mediated drug metabolism and BA synthesis, likely contribute to the benefit of fenofibrate therapy observed in patients with chronic cholestatic liver disease.
V. Additional Effects of PPARα
a. Efflux Transporters Mrp3 and Mrp4
Hepatic transporters have crucial roles in (ATP-dependent) BA and substrate efflux into the systemic circulation. MRP4/Mrp4, a member of the multidrug resistance-associated gene family, expressed on the basolateral membrane of hepatocytes undergoes an adaptive up-regulation in response to cholestatic injury or BA feeding (41). Mrp4-null mice show greater liver injury following BDL significant reductions in serum BA levels, and increased Mrp3 protein expression. MRP3 is also up-regulated during human cholestasis (42). Up-regulation of these basolateral export pumps presumably function as an adaptive compensatory mechanism to minimize the damaging cellular effects of cholestasis. These findings have led to the idea that pharmacological up-regulation of efflux transporters on the basolateral membrane, particularly MRP3 and MRP4, might be of therapeutic benefit. Indeed treatment with the PPARα agonist clofibrate increases Mrp3 and Mrp4 mRNA and protein expression (43). Whether human MRP3/4expression is affected by fibrates remains to be determined. However, bezafibrate, a dual PPAR and PXR agonist, increased mRNA expression of NTCP, CYP3A4, MDR1, MDR3, and MRP2, while down-regulating expression of CYP7A1 and CYP27A1 in human hepatoma cells (36). Up-regulation of these transporters may provide additional benefit in reducing cholestatic liver injury.
b. Regulation of the Pro-inflammatory Response
PPARα also interferes with pro-inflammatory transcription factor pathways by trans-repression (21). PPARα negatively interferes with transcription factor signaling, including nuclear factor-κB (NF-κB) (43), activator protein 1, signal transducers and activators of transcription. PPARα activation by fibrates and Wy-14,643 induced human mRNA and protein expression of the inhibitory protein IκBα in a PPARα-dependent manner, thereby reducing p65-mediated gene activation of the pro-inflammatory cytokine Il-1β (43). A significant increase in Tnfα mRNA expression was observed in Pparα-null mice (27). Human endothelial cells stimulated with cytokines and treated with fenofibrate or Wy-14,643, showed a significant reduction in vascular cell adhesion molecular-1 (VCAM-1) gene expression accompanied by reduced VCAM-1 promoter activity via inhibition of NF-κB translocation, thereby reducing leukocyte adhesion and inhibition of subsequent trans-endothelial migration (44). Whether fibrates exert their beneficial effect on cholestatic liver function by also regulating anti-inflammatory pathways seems likely but remains to be determined.
c. Crosstalk with Nuclear Farnesoid X Receptor (FXR)
Alternative pathways of human MDR3 regulation have also been reported. FXR response elements exist in the proximal region of the human MDR3 promoter and MDR3 mRNA levels increase following treatment with CDCA (45). Despite these findings, FXR may not be a direct activator of Abcb4 regulation since mice with a homozygous deletion of the Fxr gene still respond to a CA-enriched diet with an increase in Mdr2 mRNA (46). Thus, FXR-mediated effects may occur indirectly via PPARα activation. Combination treatment with CDCA and GW7647, a PPARα agonist, enhance PPARα activation compared to either agent alone (47). In contrast, treatment of primary human hepatocytes with fenofibrate increases MDR3 mRNA and protein expression greater than CDCA but together are not synergistic (37). Interestingly, many of the actions reported by PPARα, i.e. up-regulation of UGT2B4 and CYP7A1, are also described for FXR (48).
VI. Clinical Experience with Fibrates
Fibrates reduce serum triglyceride levels by at least 30% with a modest increase in high-density lipoprotein (HDL) in patients with hypertriglyceridemia (49). These effects are mediated in the liver through increased lipoprotein lipolysis and fatty acid uptake, decreased triglyceride production, and increased low-density lipoprotein (LDL) removal and reverse cholesterol transport. The effect of fibrates on lipoproteins is in large part triggered by fibrate activation of PPARα, which binds to a PPRE in the promoter of the lipoprotein lipase gene in humans (50).
Human studies in cholestasis have not been conducted with clofibrate, a drug which is no longer in clinical use. Gemfibrozil, though clinically used for treatment of hypertriglyceridemia, has also not been studied in human cholestatic disorders. Once early pharmacology studies observed that bezafibrate reduced serum liver-specific ALP and γ-glutamyl transferase levels in humans (51), Iwasiki et al. (52), demonstrated a 62.5% reduction in serum ALP for patients with PBC and persistent cholestasis despite UDCA. Since then, various studies confirmed this observation in PBC (53–66), and also reported ALP reductions in three patients with PSC (57). A prospective, randomized, multicenter study from Japan demonstrated improvement in cholestasis both with bezafibrate monotherapy and combination therapy with UDCA (55). Adverse effects included three instances of elevated serum creatinine kinase and one case of self-limited myalgia. A recent study in a Western patient population (60) further demonstrated the role of bezafibrate as an effective adjuvant therapy for pre-cirrhotic PBC patients treated with UDCA who have persistent cholestasis. In this trial there were isolated instances of mild symptoms, of nausea and heartburn, resulting in a 13% rate of adverse effects, but no reports of myalgias or rhabdomyolysis (60). However, bezafibrate is not clinically available in the United States.
Fenofibrate has been increasingly evaluated for cholestatic liver diseases. An initial report (67) showed similar reductions in cholestasis as seen with bezafibrate for PBC. Subsequent studies evaluated fenofibrate as adjunct to UDCA for treatment of refractory cholestasis in PBC, reviewed in (35). One study reported adverse effects consisting of limited heartburn, nausea, arthralgias, and transient aminotransferase elevations to 2–5x upper limit of normal (68). Two others reported mild, transient pruritus and liver enzyme elevations (67, 69). One trial compared fenofibrate to bezafibrate and reported a similar effectiveness in ALP reduction (75), although fenofibrate was superior in reducing serum LDL and uric acid levels. Of note, in a large randomized study for cardiovascular prevention in type 2 diabetes, the median plasma creatinine was reversibly increased by 12% in the fenofibrate arm (76). The potential nephrotoxic effect of fenofibrate remains controversial with multiple studies supporting (70, 71) and refuting (72, 73) these findings. One potential mechanism for acute kidney injury may be PPARα-induced blockade of prostaglandin-mediated renal vasodilation, resulting in vasoconstriction (74). Monitoring serum creatinine is recommended prior to initiation of a fibrate and throughout therapy with dose reduction if there is renal impairment (75).
a. Potential Clinical Drug-drug Interactions Involving Fibrates
Most drug-drug interactions involving fibrates are attributed to either competition for protein binding, primarily to albumin, or CYP3A4 metabolism (76). Clinically important potential drug interactions involving fenofibrate are listed in Table 3. All fibrates, but particularly gemfibrozil, displace warfarin from albumin and prolong the prothrombin time. Therefore, warfarin doses should be reduced by at least 30% and INR levels must be frequently monitored (76). Competition for the CYP3A4 enzyme may result in increased sensitivity to statin toxicity, particularly with hydrophobic drugs such as lovastatin when taken with gemfibrozil (77). However, a recent comprehensive review of fibrate toxicity in the cardiology literature suggests that with monitoring it is safe to co-administer statins and fibrates in the clinical setting of refractory hyperlipidemia (75). This has relevance to emerging evidence to support the role of FXR agonist obeticholic acid (OCA) for refractory cholestasis in PBC (78, 79), since OCA increases serum LDL levels consistent with its mechanism of action (80). No studies to date have evaluated fibrates in conjunction with FXR agonists for the treatment of cholestasis.
Table 3.
Summary of potential clinical drug-drug interactions involving fenofibrate (82).
| Drug | Concern | Recommendation |
|---|---|---|
| Warfarin |
|
|
| Calcineurin Inhibitors (CI), i.e. cyclosporine, tacrolimus |
|
|
| HMG-CoA reductase inhibitors, i.e. statins |
|
|
| Colchicine |
|
|
Fenofibrate can increase the lithogenicity of bile (81) and thus patients with pre-existing gallbladder disease are advised against its use (82). However, as opposed to clofibrate and gemfibrozil, there is no evidence that fenofibrate therapy induces formation of new gallstones (81, 82). Interestingly, the Physicians’ Desk Reference specifically mentions PBC as a contraindication to use of fenofibrate (82). This appears to be a reference to much older literature in which clofibrate exacerbated hypercholesterolemia in patients with biliary cirrhosis (83, 84). Similar findings have not been described in more recent clinical studies of bezafibrate or fenofibrate. Published data on fibrate use in refractory cholestasis has thus far been limited mostly to pre-cirrhotic patients without overt jaundice. Also, no formal pharmacokinetic studies have been performed with these fibrates in patients with hepatic impairment. Therefore, further information is needed on the metabolism and safety of fibrates in advanced liver disease due to cholestasis before recommendations could be made in this specific setting.
VII. Conclusions
This review summarizes the current literature highlighting the beneficial actions of PPARα in the context of chronic cholestatic liver disease in adults. A summary of these effects is illustrated in Figure 3. It is likely that fenofibrate ameliorates cholestatic liver disease through its transcriptional activation of MDR3, in addition to the other reported beneficial actions of PPARα activation in liver. Altogether, these findings strongly support the use of PPARα agonists such as fenofibrate, as therapeutic alternatives for adult patients with cholestatic liver diseases, specifically those with an incomplete therapeutic response to UDCA.
Figure 3.

Proposed pathway of fenofibrate-mediated reduction of cholestasis via PPARα in the liver.
Acknowledgments
This manuscript has been supported in part by USPHS DK 025636.
References
- 1.Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology. 2010;139:1481–1496. doi: 10.1053/j.gastro.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. 2003;83:633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 3.Nathanson MH, Boyer JL. Mechanisms and regulation of bile secretion. Hepatology. 1991;14:551–566. [PubMed] [Google Scholar]
- 4.Oude Elferink RP, Ottenhoff R, van Wijland M, Smit JJ, Schinkel AH, Groen AK. Regulation of biliary lipid secretion by mdr2 P-glycoprotein in the mouse. J Clin Invest. 1995;95:31–38. doi: 10.1172/JCI117658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Childs S, Yeh RL, Georges E, Ling V. Identification of a sister gene to P-glycoprotein. Cancer Res. 1995;55:2029–2034. [PubMed] [Google Scholar]
- 6.Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110:671–680. doi: 10.1172/JCI16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elferink RP, Tytgat GN, Groen AK. Hepatic canalicular membrane 1: The role of mdr2 P-glycoprotein in hepatobiliary lipid transport. FASEB J. 1997;11:19–28. doi: 10.1096/fasebj.11.1.9034162. [DOI] [PubMed] [Google Scholar]
- 8.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 9.Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. 30:134–146. doi: 10.1055/s-0030-1253223. [DOI] [PubMed] [Google Scholar]
- 10.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 11.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez FJ, Shah YM. PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246:2–8. doi: 10.1016/j.tox.2007.09.030. [DOI] [PubMed] [Google Scholar]
- 13.Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol Cell Biol. 2007;27:4238–4247. doi: 10.1128/MCB.00317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duval C, Fruchart JC, Staels B. PPAR alpha, fibrates, lipid metabolism and inflammation. Arch Mal Coeur Vaiss. 2004;97:665–672. [PubMed] [Google Scholar]
- 15.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 17.Issemann I, Green S. Cloning of novel members of the steroid hormone receptor superfamily. J Steroid Biochem Mol Biol. 1991;40:263–269. doi: 10.1016/0960-0760(91)90191-7. [DOI] [PubMed] [Google Scholar]
- 18.Trottier J, Caron P, Straka RJ, Barbier O. Profile of serum bile acids in noncholestatic volunteers: gender-related differences in response to fenofibrate. Clin Pharmacol Ther. 2011;90:279–286. doi: 10.1038/clpt.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Post SM, Duez H, Gervois PP, Staels B, Kuipers F, Princen HM. Fibrates suppress bile acid synthesis via peroxisome proliferator-activated receptor-alpha-mediated downregulation of cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase expression. Arterioscler Thromb Vasc Biol. 2001;21:1840–1845. doi: 10.1161/hq1101.098228. [DOI] [PubMed] [Google Scholar]
- 20.Marrapodi M, Chiang JY. Peroxisome proliferator-activated receptor alpha (PPARalpha) and agonist inhibit cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription. J Lipid Res. 2000;41:514–520. [PubMed] [Google Scholar]
- 21.Hunt MC, Yang YZ, Eggertsen G, Carneheim CM, Gafvels M, Einarsson C, Alexson SE. The peroxisome proliferator-activated receptor alpha (PPARalpha) regulates bile acid biosynthesis. J Biol Chem. 2000;275:28947–28953. doi: 10.1074/jbc.M002782200. [DOI] [PubMed] [Google Scholar]
- 22.Thomas M, Burk O, Klumpp B, Kandel BA, Damm G, Weiss TS, Klein K, et al. Direct transcriptional regulation of human hepatic cytochrome P450 3A4 (CYP3A4) by peroxisome proliferator-activated receptor alpha (PPARalpha) Mol Pharmacol. 2013;83:709–718. doi: 10.1124/mol.112.082503. [DOI] [PubMed] [Google Scholar]
- 23.Barbier O, Duran-Sandoval D, Pineda-Torra I, Kosykh V, Fruchart JC, Staels B. Peroxisome proliferator-activated receptor alpha induces hepatic expression of the human bile acid glucuronidating UDP-glucuronosyltransferase 2B4 enzyme. J Biol Chem. 2003;278:32852–32860. doi: 10.1074/jbc.M305361200. [DOI] [PubMed] [Google Scholar]
- 24.Senekeo-Effenberger K, Chen S, Brace-Sinnokrak E, Bonzo JA, Yueh MF, Argikar U, Kaeding J, et al. Expression of the human UGT1 locus in transgenic mice by 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (WY-14643) and implications on drug metabolism through peroxisome proliferator-activated receptor alpha activation. Drug Metab Dispos. 2007;35:419–427. doi: 10.1124/dmd.106.013243. [DOI] [PubMed] [Google Scholar]
- 25.Fang HL, Strom SC, Cai H, Falany CN, Kocarek TA, Runge-Morris M. Regulation of human hepatic hydroxysteroid sulfotransferase gene expression by the peroxisome proliferator-activated receptor alpha transcription factor. Mol Pharmacol. 2005;67:1257–1267. doi: 10.1124/mol.104.005389. [DOI] [PubMed] [Google Scholar]
- 26.Jung D, Fried M, Kullak-Ublick GA. Human apical sodium-dependent bile salt transporter gene (SLC10A2) is regulated by the peroxisome proliferator-activated receptor alpha. J Biol Chem. 2002;277:30559–30566. doi: 10.1074/jbc.M203511200. [DOI] [PubMed] [Google Scholar]
- 27.Li F, Patterson AD, Krausz KW, Tanaka N, Gonzalez FJ. Metabolomics reveals an essential role for peroxisome proliferator-activated receptor alpha in bile acid homeostasis. J Lipid Res. 2012;53:1625–1635. doi: 10.1194/jlr.M027433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitada H, Miyata M, Nakamura T, Tozawa A, Honma W, Shimada M, Nagata K, et al. Protective role of hydroxysteroid sulfotransferase in lithocholic acid-induced liver toxicity. J Biol Chem. 2003;278:17838–17844. doi: 10.1074/jbc.M210634200. [DOI] [PubMed] [Google Scholar]
- 29.Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet. 1995;29:607–649. doi: 10.1146/annurev.ge.29.120195.003135. [DOI] [PubMed] [Google Scholar]
- 30.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, Mol CA, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 31.Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Weiglein AH, Lammert F, Marschall HU, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123:1238–1251. doi: 10.1053/gast.2002.35948. [DOI] [PubMed] [Google Scholar]
- 32.Cai SY, Mennone A, Soroka CJ, Boyer JL. Altered expression and function of canalicular transporters during early development of cholestatic liver injury in Abcb4-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2014;306:G670–676. doi: 10.1152/ajpgi.00334.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trauner M, Fickert P, Halilbasic E, Moustafa T. Lessons from the toxic bile concept for the pathogenesis and treatment of cholestatic liver diseases. Wien Med Wochenschr. 2008;158:542–548. doi: 10.1007/s10354-008-0592-1. [DOI] [PubMed] [Google Scholar]
- 34.Day AP, Feher MD, Chopra R, Mayne PD. The effect of bezafibrate treatment on serum alkaline phosphatase isoenzyme activities. Metabolism. 1993;42:839–842. doi: 10.1016/0026-0495(93)90056-t. [DOI] [PubMed] [Google Scholar]
- 35.Ghonem NS, Boyer JL. Fibrates as adjuvant therapy for chronic cholestatic liver disease - it’s time has come. Hepatology. 2012 doi: 10.1002/hep.26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Honda A, Ikegami T, Nakamuta M, Miyazaki T, Iwamoto J, Hirayama T, Saito Y, et al. Anticholestatic effects of bezafibrate in patients with primary biliary cirrhosis treated with ursodeoxycholic acid. Hepatology. 2013;57:1931–1941. doi: 10.1002/hep.26018. [DOI] [PubMed] [Google Scholar]
- 37.Ghonem NS, Ananthanarayanan M, Soroka CJ, Boyer JL. Peroxisome proliferator-activated receptor alpha activates human multidrug resistance transporter 3/ATP-binding cassette protein subfamily B4 transcription and increases rat biliary phosphatidylcholine secretion. Hepatology. 2014;59:1030–1042. doi: 10.1002/hep.26894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shoda J, Inada Y, Tsuji A, Kusama H, Ueda T, Ikegami T, Suzuki H, et al. Bezafibrate stimulates canalicular localization of NBD-labeled PC in HepG2 cells by PPARalpha-mediated redistribution of ABCB4. J Lipid Res. 2004;45:1813–1825. doi: 10.1194/jlr.M400132-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Kok T, Bloks VW, Wolters H, Havinga R, Jansen PL, Staels B, Kuipers F. Peroxisome proliferator-activated receptor alpha (PPARalpha)-mediated regulation of multidrug resistance 2 (Mdr2) expression and function in mice. Biochem J. 2003;369:539–547. doi: 10.1042/BJ20020981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chianale J, Vollrath V, Wielandt AM, Amigo L, Rigotti A, Nervi F, Gonzalez S, et al. Fibrates induce mdr2 gene expression and biliary phospholipid secretion in the mouse. Biochem J. 1996;314(Pt 3):781–786. doi: 10.1042/bj3140781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mennone A, Soroka CJ, Cai SY, Harry K, Adachi M, Hagey L, Schuetz JD, et al. Mrp4−/− mice have an impaired cytoprotective response in obstructive cholestasis. Hepatology. 2006;43:1013–1021. doi: 10.1002/hep.21158. [DOI] [PubMed] [Google Scholar]
- 42.Chai J, He Y, Cai SY, Jiang Z, Wang H, Li Q, Chen L, et al. Elevated hepatic multidrug resistance-associated protein 3/ATP-binding cassette subfamily C 3 expression in human obstructive cholestasis is mediated through tumor necrosis factor alpha and c-Jun NH2-terminal kinase/stress-activated protein kinase-signaling pathway. Hepatology. 2012;55:1485–1494. doi: 10.1002/hep.24801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delerive P, Gervois P, Fruchart JC, Staels B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem. 2000;275:36703–36707. doi: 10.1074/jbc.M004045200. [DOI] [PubMed] [Google Scholar]
- 44.Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99:3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang L, Zhao A, Lew JL, Zhang T, Hrywna Y, Thompson JR, de Pedro N, et al. Farnesoid X receptor activates transcription of the phospholipid pump MDR3. J Biol Chem. 2003;278:51085–51090. doi: 10.1074/jbc.M308321200. [DOI] [PubMed] [Google Scholar]
- 46.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 47.Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17:259–272. doi: 10.1210/me.2002-0120. [DOI] [PubMed] [Google Scholar]
- 48.Barbier O, Torra IP, Sirvent A, Claudel T, Blanquart C, Duran-Sandoval D, Kuipers F, et al. FXR induces the UGT2B4 enzyme in hepatocytes: a potential mechanism of negative feedback control of FXR activity. Gastroenterology. 2003;124:1926–1940. doi: 10.1016/s0016-5085(03)00388-3. [DOI] [PubMed] [Google Scholar]
- 49.Fruchart JC, Brewer HB, Jr, Leitersdorf E. Consensus for the use of fibrates in the treatment of dyslipoproteinemia and coronary heart disease. Fibrate Consensus Group. Am J Cardiol. 1998;81:912–917. doi: 10.1016/s0002-9149(98)00010-1. [DOI] [PubMed] [Google Scholar]
- 50.Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res. 1996;37:907–925. [PubMed] [Google Scholar]
- 51.Monk JP, Todd PA. Bezafibrate. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in hyperlipidaemia. Drugs. 1987;33:539–576. doi: 10.2165/00003495-198733060-00002. [DOI] [PubMed] [Google Scholar]
- 52.Iwasaki STK, Ueta H, Aono R, Ono M, Saibara T. Benzafibrate may have beneficial effect in pre-cirrhotic primary biliary cirrhosis. Hepatol Res. 1999;16:12–18. [Google Scholar]
- 53.Akbar SM, Furukawa S, Nakanishi S, Abe M, Horiike N, Onji M. Therapeutic efficacy of decreased nitrite production by bezafibrate in patients with primary biliary cirrhosis. J Gastroenterol. 2005;40:157–163. doi: 10.1007/s00535-004-1518-3. [DOI] [PubMed] [Google Scholar]
- 54.Hazzan R, Tur-Kaspa R. Bezafibrate treatment of primary biliary cirrhosis following incomplete response to ursodeoxycholic acid. J Clin Gastroenterol. 2010;44:371–373. doi: 10.1097/MCG.0b013e3181c115b3. [DOI] [PubMed] [Google Scholar]
- 55.Iwasaki S, Ohira H, Nishiguchi S, Zeniya M, Kaneko S, Onji M, Ishibashi H, et al. The efficacy of ursodeoxycholic acid and bezafibrate combination therapy for primary biliary cirrhosis: A prospective, multicenter study. Hepatol Res. 2008;38:557–564. doi: 10.1111/j.1872-034X.2007.00305.x. [DOI] [PubMed] [Google Scholar]
- 56.Kanda T, Yokosuka O, Imazeki F, Saisho H. Bezafibrate treatment: a new medical approach for PBC patients? J Gastroenterol. 2003;38:573–578. doi: 10.1007/s00535-002-1102-7. [DOI] [PubMed] [Google Scholar]
- 57.Kita R, Takamatsu S, Kimura T, Kokuryu H, Osaki Y, Tomono N. Bezafibrate may attenuate biliary damage associated with chronic liver diseases accompanied by high serum biliary enzyme levels. J Gastroenterol. 2006;41:686–692. doi: 10.1007/s00535-006-1831-0. [DOI] [PubMed] [Google Scholar]
- 58.Kurihara T, Maeda A, Shigemoto M, Yamashita K, Hashimoto E. Investigation into the efficacy of bezafibrate against primary biliary cirrhosis, with histological references from cases receiving long term monotherapy. Am J Gastroenterol. 2002;97:212–214. doi: 10.1111/j.1572-0241.2002.05413.x. [DOI] [PubMed] [Google Scholar]
- 59.Kurihara T, Niimi A, Maeda A, Shigemoto M, Yamashita K. Bezafibrate in the treatment of primary biliary cirrhosis: comparison with ursodeoxycholic acid. Am J Gastroenterol. 2000;95:2990–2992. doi: 10.1111/j.1572-0241.2000.03220.x. [DOI] [PubMed] [Google Scholar]
- 60.Lens S, Leoz M, Nazal L, Bruguera M, Pares A. Bezafibrate normalizes alkaline phosphatase in primary biliary cirrhosis patients with incomplete response to ursodeoxycholic acid. Liver Int. 2014;34:197–203. doi: 10.1111/liv.12290. [DOI] [PubMed] [Google Scholar]
- 61.Miyaguchi S, Ebinuma H, Imaeda H, Nitta Y, Watanabe T, Saito H, Ishii H. A novel treatment for refractory primary biliary cirrhosis? Hepatogastroenterology. 2000;47:1518–1521. [PubMed] [Google Scholar]
- 62.Nakai S, Masaki T, Kurokohchi K, Deguchi A, Nishioka M. Combination therapy of bezafibrate and ursodeoxycholic acid in primary biliary cirrhosis: a preliminary study. Am J Gastroenterol. 2000;95:326–327. doi: 10.1111/j.1572-0241.2000.01667.x. [DOI] [PubMed] [Google Scholar]
- 63.Ohmoto K, Mitsui Y, Yamamoto S. Effect of bezafibrate in primary biliary cirrhosis: a pilot study. Liver. 2001;21:223–224. doi: 10.1034/j.1600-0676.2001.021003223.x. [DOI] [PubMed] [Google Scholar]
- 64.Ohmoto K, Yoshioka N, Yamamoto S. Long-term effect of bezafibrate on parameters of hepatic fibrosis in primary biliary cirrhosis. J Gastroenterol. 2006;41:502–503. doi: 10.1007/s00535-006-1778-1. [DOI] [PubMed] [Google Scholar]
- 65.Takeuchi Y, Ikeda F, Fujioka S, Takaki T, Osawa T, Yasunaka T, Miyake Y, et al. Additive improvement induced by bezafibrate in patients with primary biliary cirrhosis showing refractory response to ursodeoxycholic acid. J Gastroenterol Hepatol. 2011;26:1395–1401. doi: 10.1111/j.1440-1746.2011.06737.x. [DOI] [PubMed] [Google Scholar]
- 66.Yano K, Kato H, Morita S, Takahara O, Ishibashi H, Furukawa R. Is bezafibrate histologically effective for primary biliary cirrhosis? Am J Gastroenterol. 2002;97:1075–1077. doi: 10.1111/j.1572-0241.2002.05645.x. [DOI] [PubMed] [Google Scholar]
- 67.Ohira H, Sato Y, Ueno T, Sata M. Fenofibrate treatment in patients with primary biliary cirrhosis. Am J Gastroenterol. 2002;97:2147–2149. doi: 10.1111/j.1572-0241.2002.05944.x. [DOI] [PubMed] [Google Scholar]
- 68.Levy C, Peter JA, Nelson DR, Keach J, Petz J, Cabrera R, Clark V, et al. Pilot study: fenofibrate for patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Aliment Pharmacol Ther. 2011;33:235–242. doi: 10.1111/j.1365-2036.2010.04512.x. [DOI] [PubMed] [Google Scholar]
- 69.Han XF, Wang QX, Liu Y, You ZR, Bian ZL, Qiu de K, Ma X. Efficacy of fenofibrate in Chinese patients with primary biliary cirrhosis partially responding to ursodeoxycholic acid therapy. J Dig Dis. 2012;13:219–224. doi: 10.1111/j.1751-2980.2012.00574.x. [DOI] [PubMed] [Google Scholar]
- 70.Attridge RL, Frei CR, Ryan L, Koeller J, Linn WD. Fenofibrate-associated nephrotoxicity: a review of current evidence. Am J Health Syst Pharm. 2013;70:1219–1225. doi: 10.2146/ajhp120131. [DOI] [PubMed] [Google Scholar]
- 71.Attridge RL, Linn WD, Ryan L, Koeller J, Frei CR. Evaluation of the incidence and risk factors for development of fenofibrate-associated nephrotoxicity. J Clin Lipidol. 2012;6:19–26. doi: 10.1016/j.jacl.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 72.Kostapanos MS, Florentin M, Elisaf MS. Fenofibrate and the kidney: an overview. Eur J Clin Invest. 2013;43:522–531. doi: 10.1111/eci.12068. [DOI] [PubMed] [Google Scholar]
- 73.Ting RD, Keech AC, Drury PL, Donoghoe MW, Hedley J, Jenkins AJ, Davis TM, et al. Benefits and safety of long-term fenofibrate therapy in people with type 2 diabetes and renal impairment: the FIELD Study. Diabetes Care. 2012;35:218–225. doi: 10.2337/dc11-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsimihodimos V, Miltiadous G, Bairaktari E, Elisaf M. Possible mechanisms of the fibrate-induced increase in serum creatinine. Clin Nephrol. 2002;57:407–408. [PubMed] [Google Scholar]
- 75.Davidson MH, Armani A, McKenney JM, Jacobson TA. Safety considerations with fibrate therapy. Am J Cardiol. 2007;99:3C–18C. doi: 10.1016/j.amjcard.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 76.Miller DB, Spence JD. Clinical pharmacokinetics of fibric acid derivatives (fibrates) Clin Pharmacokinet. 1998;34:155–162. doi: 10.2165/00003088-199834020-00003. [DOI] [PubMed] [Google Scholar]
- 77.Pierce LR, Wysowski DK, Gross TP. Myopathy and rhabdomyolysis associated with lovastatin-gemfibrozil combination therapy. JAMA. 1990;264:71–75. [PubMed] [Google Scholar]
- 78.Kowdley KVJD, Luketic V, Chapman R, Burroughs A, Hirschfield G, et al. An international study evaluating the farnesoid X receptor agonist obeticholic acid as monotherapy in PBC. J Hepatol. 2011;54:S13. [Google Scholar]
- 79.Silveira MG, Lindor KD. Obeticholic acid and budesonide for the treatment of primary biliary cirrhosis. Expert Opin Pharmacother. 2014;15:365–372. doi: 10.1517/14656566.2014.873404. [DOI] [PubMed] [Google Scholar]
- 80.Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574–582. e571. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 81.Blane GF. Comparative toxicity and safety profile of fenofibrate and other fibric acid derivatives. Am J Med. 1987;83(5B):26–36. doi: 10.1016/0002-9343(87)90868-0. [DOI] [PubMed] [Google Scholar]
- 82.Tricor. Physicians’ Desk Reference. 68. Montvale, NJ: PDR Network; 2014. pp. 585–90. [Google Scholar]
- 83.Clofibrate in biliary cirrhosis. Drug Ther Bull. 1970;8(3):12. [PubMed] [Google Scholar]
- 84.Summerfield JA, Elias E, Sherlock S. Effects of clofibrate in primary biliary cirrhosis hypercholesterolemia and gallstones. Gastroenterology. 1975;69:998–1000. [PubMed] [Google Scholar]