

Abstract
As ordered nanoscale architectures, viruses and virus-like particles (VLPs) remain unsurpassed by synthetic strategies to produce uniform and symmetric nanoparticles. Maintaining or mimicking the symmetry of pathogenic viruses, VLPs offer a ready platform for facilitating recognition, uptake, and processing by the immune system. An emerging understanding of how viruses interact with the immune system offers a means of precisely designing nanoparticles for biomedical use, both with respect to the structure of the particle as well as their ability to stimulate the immune system. Here we discuss recent advances by our group toward two parallel and complementary applications of VLPs, derived primarily from plants, bacteriophage, and nonviral sources, in biomedicine: diagnostic imaging and rational vaccine design. First we discuss advances in increasing VLP payloads of gadolinium magnetic resonance imaging (MRI) contrast agent as well as controlling the characteristics of individual gadolinium containing molecules to increase efficacy. In order to better understand the in vivo potential of VLP constructs, we then discuss the interface of protein-cages and the immune system beginning with the nonspecific innate immune system stimulation and continuing into the use of non-pathogenic VLPs as scaffolds for specific antigen presentation and control of the immune response.
INTRODUCTION
Typically the abilities of viruses to infiltrate, target, manipulate, and deliver cargo to their host are viewed as negative traits to be combatted. However, in the context of nanomedicine, these traits are hugely desirable. Engineering efforts aim to utilize the same characteristics that make viruses effect pathogens as tools for biological delivery and stimulation. As pathogens, viruses have driven immune system development making them optimum vehicles for interacting with the immune system.1,2 Noninfectious virus-like particles (VLPs) provide promising platforms for the development of biomimetic vessels for the delivery of therapeutic or diagnostic agents as well as precise stimulation of the immune system, because they provide the advantages of viruses without the risk of disease.
VLPs are protein-based, nanoscale, self-assembling, cage architectures. These particles can be derived not only from viral sources but also from nonviral cage forming systems. A key characteristic that separates VLPs from viruses themselves is that VLPs are either naturally noninfectious or have been rendered such. Virus-derived VLP’s can be made noninfectious either by removing genetic material and recovering only the structural capsid proteins or by expressing the structural proteins heterologously. Viruses are thought to be the most numerous biological entities on the planet.3 The library of useful VLPs is beyond extensive and encompasses both viral and nonviral particles from all domains of life. From an engineering perspective the ever-expanding list of well-characterized viruses provides multiple candidates for a specific nanoparticle application. Taking a limited subset of VLPs, this discussion will deal primarily with specific modification-tolerant VLPs derived from plant virus, bacteriophages, and nonviral sources.
Structurally, the hollow shell of these particles has evolved to carry and deliver a payload. When free of the natural nucleic acid contents, the VLP cage interior provides an appealing compartment for nonnative cargo encapsulation. The shell itself, comprised of repeated structural units of a limited number of distinct proteins, provides a platform for polyvalent conjugation either inside or outside the capsid; a single change being reflected at every symmetry-related site on the cage (Figure 1). Conjugation can be pursued through both synthetic and genetic modification of the VLP.
FIGURE 1.
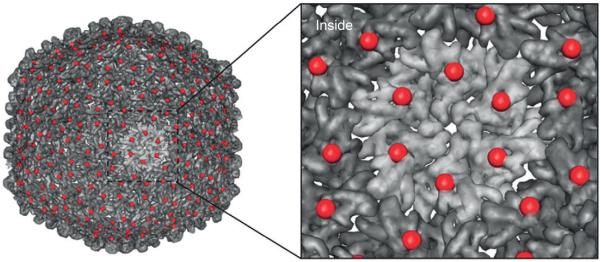
The symmetry of VLPs reflects a single change over the entire particle. The VLP from bacteriophage P22 is shown with a serine to cysteine point mutation at amino acid position 39, displayed as a red sphere. This single mutation provides a new site at each of the 420 coat proteins that make up the capsid. (Reprinted with permission from Ref 4. Copyright 2012 Nature Publishing Group)
Recent studies by our group and others have demonstrated that VLP’s imbue nonnative cargo molecules with the advantageous characteristics of a virus such as a mono-dispersed particle population, preferential cellular uptake, immune stimulation, and physical behavior of a nanoscale macromolecule. Here we begin with a discussion of the demonstrated potential of VLPs for the delivery of diagnostic imaging cargo molecules. The VLPs used in these systems are not invisible to the immune system. In order for VLP systems to be useful, the immune response needs to be characterized in detail. We continue our discussion focusing on work aimed at understanding this VLP interaction with the innate immune system and the potential for harnessing these immune interactions through engineered VLP vaccines. As will become evident, these immune interactions can be channeled and utilized for more efficacious technologies.
VLP’s AS CONTRAST AGENT DELIVERY SYSTEMS
Nanoparticle technologies have been widely adopted, in the medical and pharmaceutical sectors, as a solution to the problems of toxicity, bioavailability, solubility, controlled release, and cellular targeting.5–8 Many nanoparticles have biocompatible and bio-infiltration characteristics based largely on their size, symmetry, and surface chemistry. Presumably cargo molecules can assume these characteristics upon attachment to the nanoparticle. As natural nanoparticles, viruses have evolved primarily to deliver a payload of nucleic acid to a host organism and thus VLPs are particularly well suited to deliver cargos within the body. In this section, we discuss the utility of VLPs as sequestration and delivery vessels for inorganic magnetic resonance imaging (MRI) contrast agents as well as some of the challenges in moving these systems towards clinical use.
VLP MRI Contrast Agent Design
MRI is a versatile noninvasive tissue imaging technique. However, low sensitivity prevents the effective detection of low abundance tissue and hinders MRI use in early stage diagnostics. In an effort to resolve this problem, paramagnetic contrast agents are utilized to increase the difference in water-proton spin relaxation rates, referred to as relaxivity, between tissue types. The presence of these paramagnetic agents can be visualized as bright (T1) or dark (T2) contrast depending on the type of contrast agent used. Currently the most promising and clinically relevant paramagnetic contrast agents are based on gadolinium ions. However, in certain individuals the metal is toxic making sequestration and controlled bio-distribution important. Currently, gadolinium is clinically available in a variety of chelated forms. While these chelates significantly decrease the toxicity of gadolinium, the required clinical dosage is high, which has driven investigation of more efficient, targeted, contrast agents.
The current theoretical framework for paramagnetic relaxivity provides some guidance for developing materials with improved efficacy. From the Solomon–Bloembergen–Morgan (SBM) model for relaxivity there are three key parameters that have a large effect on the relaxation rate of water protons when associated with paramagnetic ion such as Gd3+ (Figure 2). These include the number of water molecules directly bound to the Gd ion (q), the residence time of the water molecule (τM) on the metal, and the rotational correlation time (τR).9 While increasing q or altering τM requires redesigning the metal chelator, the rotational correlation time, τR, can be readily modulated by attaching the metal chelate to a larger slowly tumbling macromolecule. Alternatively the overall efficacy of the contrast agent can be increased by selectively targeting the agent to a specific tissue type.
FIGURE 2.
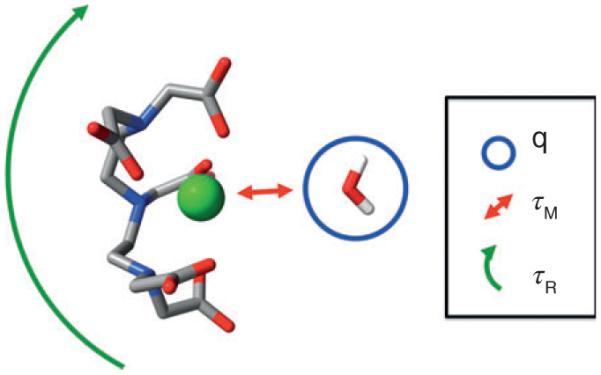
There are several targets for optimizing the relaxivity of a T1 contrast agent. DTPA-Gd is shown with a single water molecule in the binding position. Factors that affect relaxivity are the number of water molecules that can bind to the Gd ion (q), the exchange lifetime of the water molecule or molecules bound to the Gd (τM), and the rotational correlation time of the Gd complex (τR). The parameters q and τM are difficult to change for a given Gd-chelator complex leaving τR particle loading as optimization targets. (Reprinted with permission from Ref 9. Copyright 2009 American Chemical Society)
Nanoparticles in general have been pursued as means of sequestering gadolinium chelates and other contrast candidates in order to increase τR and provide a scaffold for the attachment of targeting moieties. Candidates have included but are not limited to liposomes,10 polymers, inorganic nanoparticles,11 and VLPs. VLP strategies, discussed here, have the advantage of genetic design and production, which affords precise spatial control and uniformity.
The VLP from Cowpea chlorotic mottle virus (CCMV) was first used to chelate Gd3+ via natural calcium binding sites at the quasi-threefold axes of the T = 3 icosahedral particle (CCMV-Gd).12 The CCMV-Gd system was found to increase the T1 and T2 relaxivity of bound gadolinium 10-fold and 19-fold respectively compared to free Gd ions. With ~140 Gd/particle, the per particle T1 relaxivity, which indicates the efficacy of a single particle reaching the target tissue, was 3.5 × 104 second−1 mM−1 compared to ~20 second−1 mM−1 for free Gd measured at the same frequency (Table 1).
TABLE 1.
The Strategies, Particle Loadings, and Effects on T1 Relaxivity (r1) of the VLP Systems Discussed in this Work are Tabulated
| VLP | Chelator | Attachment | Loading (Gd) | r1/ Gd (mM−1/second−1) |
r1/particle (mM−1/second−1) |
Ref. |
|---|---|---|---|---|---|---|
| GdCl3 | — | — | 1 | 20 | 20 | 12 |
| DTPA-Gd | DTPA | — | 1 | 4 | 4 | 4 |
| DOTA-Gd | DOTA | — | 1 | 5 | 5 | 13 |
| CCMV | Native Ca2+ site | Native | 140 | 202 | 28,280 | 12 |
| CCMV | Cal Domain | Genetic | 172 | 210 | 36,120 | 14 |
| CCMV | DOTA | NHS | 61 | 46 | 2,806 | 14 |
| MS2 | DTPA | SCN | 514 | 14 | 7,196 | 15 |
| CPMV | RNA & DOTA | RNA & Click | 268 | 15.5 | 4,154 | 16 |
| Qβ | DOTA | Click | 153 | 11.9 | 1,821 | 16 |
| TMV | DOTA | Click to interior | 3,417 | 10.7 | 36,562 | 13 |
| TMV | DOTA | Click to exterior | 1,712 | 18.4 | 31,501 | 13 |
| TMV sphere | DOTA | Click | 25,815 | 15.2 | 392,388 | 13 |
| sHSP-Branched polymer | DTPA | SCN | 159 | 19 | 3,021 | 9 |
| P22-AEMA | DTPA | SCN | 9,100 | 22 | 200,200 | 4 |
| Qβ-OEGMA | DOTA | Click | 610 | 11.6 | 7,092 | 17 |
All r1 measurements were taken between 60 and 64MHz Larmor frequency.
While the CCMV-Gd system exhibited impressive relaxivity enhancement, binding of Gd3+ to the native Ca2+ sites was too weak (Kd ~ 100 nM) to keep free Gd3+ below toxic levels.18 To remedy this, nonnative chelation sites were introduced to the CCMV capsid by both genetic and chemical conjugation.14 A nine-amino acid Ca binding sequence from calmodulin was fused to the N-terminus of the CCMV subunit (CCMV-Cal). Alternatively gadolinium-tetraazacyclododecane tetraacetic acid (Gd-DOTA), a clinically available contrast agent, was conjugated via reaction of an N-hydroxysuccinimide (NHS) ester to exposed lysine residues on the CCMV particle (CCMV-DOTA). Both constructs maintained a robust CCMV-VLP structure with Gd3+ loadings of 172 Gd/particle and 61 Gd/particle for CCMV-Cal and CCMV-DOTA, respectively. CCMV-Cal exhibited comparable ionic and per particle relaxivity to the native metal center CCMV-Gd. CCMV-DOTA exhibited slightly lower values for both T1 and T2 relaxivity values, likely due to differences in metal binding (and lower values of q) to these synthetic sites, but still markedly better than Gd3+ alone or Gd3+-DOTA. (Table 1)
The sheer number of viruses and protein-cages available in nature offers the opportunity to select an appropriate system tailored towards a specific application or to screen multiple systems for the same application. Anderson et al. coupled diethylene triamine pentaacetic acid-Gd (DTPA-Gd) to VLP from Bacteriophage MS2, via isothiocyanite coupling to native lysines, demonstrating maximum particle labeling of 514 Gd/VLP. This resulted in T1 relaxivities of 7.2 × 103 mM−1 second−1 per particle and 14 mM−1 second−1 per Gd ion.15 Utilizing intact infectious Cowpea Mosaic Virus (CPMV) and the VLP from Bacteriophage Qβ, Prasuhn et al. explored gadolinium attachment through native Lanthanide affinity to encapsulated RNA (CPMV) and directed bioconjugation of Gd-DOTA (CPMV and Qβ).16 A combined strategy of native Gd-RNA attachment and azide-alkyne 1,3-cycloaddition ‘click’ mediated attachment of Gd-DOTA resulted in 268 ± 30 Gd/capsid of CPMV. This particle exhibited 4150 mM−1 sec−1 per particle and 15.5 mM−1 sec−1 per Gd ion T1 relaxivity. With just bioconjugate attachment, Qβ was functionalized with 153 ± 15 Gd/capsid resulting in 1820 mM−1 sec−1 per particle and 11.9 mM−1 sec−1 per Gd ion T1 relaxivity.
Exploring the upper end of the VLP size spectrum, Bruckman et al. made use of the rod-shaped tobacco mosaic virus (TMV).13 TMV is composed of 2130 coat protein subunits arranged in a 300 nm long right-handed helix providing vastly more labeling sites than the spherical viruses discussed above.19 Gd-DOTA azide was ‘clicked’ to the interior (iGd-TMV) or exterior (eGd-TMV) of the TMV capsid via terminal alkynes generated on either glutamate or tyrosine residues respectively. iGd-TMV was also thermally expanded to ~170 nM diameter spherical particles (iGd-SNP). All constructs exhibited impressive per particle loadings, 1,712 Gd/eGD-TMV particle, 3,417 Gd/iGd-TMV particle, and 25,815 Gd/iGd-SNP. Ionic relaxivities for all constructs fell in the expected range for attachment to a slow tumbling large particle but due to the high loading, per particle relaxivities reached 31,501 (eGd-TMV), 36,562 (iGd-TMV), and 392,388 (iGd-SNP) mM−1 second−1.13
Despite relaxivity improvements using VLPs as direct macromolecular scaffolds for Gd binding, VLPs are still largely comprised of unutilized interior space. Bioconjugate polymerization techniques offer a means of introducing additional binding sites, utilizing this empty space. To this end a branched ‘click’ copolymerization strategy was utilized through 3.5 generations to create a polymer-filled small heat-shock protein (sHSP) from Methanococcus jannaschii.9 sHSP is a relatively small (12 nm diameter 400 kDa) nonviral protein cage known to decrease heat-induced denaturation and aggregation of proteins.20 Internal polymerization resulted in the addition of 200 amine sites which when functionalized with DTPA-Gd resulted in 159 Gd3+ ions in the final construct. Ionic relaxivity for complexed Gd3+ was measured to be sixfold higher than DTPA-Gd alone but remained constant across increasing polymerization generations. The SBM model of relaxivity predicts that the measured tumbling rate of Gd3+ within the sHSP capsid (7.6 × 10−9 second) is within an expected optimum range and thus no increase in ionic relaxivity compared to previous macromolecular systems was expected. The main advantage to this approach was the dramatic increase in the per particle relaxivity which reached a maximum value of 4,200 second−1 mM−1 at 31 MHz for the fully polymerized construct.
With the goal of increasing the number of cargo sites per particle, larger capsids have the advantage of larger interior volume and the potential for even more chelation sites. One such cage is the VLP from bacteriophage P22, which has roughly 800 times the interior volume of sHSP. Stepwise co-polymerization of this large volume would be impractical. Instead a living polymerization strategy, atom transfer radical polymerization (ATRP), was employed (Figure 3). Polymerization was initiated at a cysteine site on the interior or the capsid using 2-bromoisobutyryl aminoethyl maleimide. Amine sites were introduced by polymerization with 2-aminoethyl methacrylate. The encapsulated polymer was subsequently labeled with DTPA-Gd.4 The final construct contained 9,100 ± 800 Gd per particle. As expected, individual ionic relaxivity rates remained within the characteristic range for large macromolecular Gd complexes. However, the per particle relaxivity for this construct reached values of 200,200 second−1 mM−1 (60 MHz), among the highest particle relaxivity observed to date.
FIGURE 3.

The interior space of the P22 VLP can be utilized by introducing a scaffold via atom-transfer radical polymerization. The radical initiator 2-bromoisobutyryl aminoeythl maleimide (1) was coupled to an internal cysteine of the P22 coat protein. Polymerization of the capsid interior with 2-aminoethyl methacrylate (AEMA) introduced as many as 9000 amine sites within the intra-capsid space. These sites could then be functionalized with Gd-DPTA-NCS resulting in high particle loading. (Reprinted with permission from Ref 4. Copyright 2012 Nature Publishing Group)
Similarly, Pokorski et al. utilized ATRP polymerization to create an exterior functionalized Qβ particle. Surface accessible amines were used to initiate the polymerization of either an azide or hydroxyl terminated oligo(ethylene glycol)-methacrylate monomer (OEGMA).17 This resulted in an ~10 nm thick polymer shell about the virus capsid to which Gd-DOTA with a reactive alkyne could be coupled via a ‘click’ reaction. Qβ particles with Gd-DOTA conjugated to the polymer matrix or directly to the initiation site amines on the capsid surface were compared with respect to loading and relaxivity. Addition of the polymer matrix increased the Gd particle loading (350 versus 610 Gd/particle) while ionic T1 relaxivity (60 MHz) remained unchanged, 11.6 second−1 mM−1 for the polymerized Qβ versus 10.7 second−1 mM−1 for the bare particle, leading to T1 relaxivities of 7092 second−1 mM−1 per particle for the polymerized sample. In addition, the polymer matrix was loaded with doxorubicin, which was shown to still be bioavailable, or a fluorescent dye demonstrating the modular versatility of a single VLP polymer system.
Polymerization can also drastically change the biological behavior of a VLP. Hovlid et al. examined internally ATRP polymerized Qβ VLPs with respect to the effect of polymerization on the capsid structure and cellular internalization.21 Cellular interactions were further facilitated in this case by the addition of a cyclic RGD integrin-targeting peptide, however, this was found to only partially improve cellular internalization. Cellular internalization was significantly affected by the positively charged interior polymer as shown from a sharp increase in nonendosomal entry of VLPs into the cell. This resulted in the efficient delivery of a model siRNA. The combination of cellular targeting, internal polymerization and payload delivery illustrates the real potential of VLPs as multifaceted engineering tools.
This progressive development of VLP-based contrast agents not only speaks to the ability of VLPs to effectively sequester specific cargo for delivery but also demonstrates the high tolerance of VLPs to chemical and genetic manipulation. Ongoing research is focused on the use of VLP systems with less toxic contrast agents such manganese22 and iron oxide23–25 and more efficient targeting of these agents. A specific example is the natural iron sequestration capability of ferritin which has been examined both as a T2 enhancement compound and for its behavior in noncontrasted MRI imaging within the body.26 While these metals lack the high base-line relaxivity of gadolinium, the appeal of nontoxic species coupled with the advantages of VLP-contrast agent complexes give these systems the potential to be more efficacious and safer than currently available gadolinium compounds.
Cellular Targeting of VLP Particles
Future improvements in VLP-based MRI agents are likely to focus first on the ability to increase the number and type of cargo molecules incorporated per particle and second on the ability to effectively target those particles to a specific tissue or cell type in vivo. The typical size range of VLPs (20–200 nm) predisposes them to travel into lymphatic circulation and promotes uptake by nonspecific antigen presenting cells (APC) such as dendritic cells (DC) and macrophages (Mac).27 With little modification some VLPs are already targeted to specific cell types and organs such as lymphoid tissues. Pathogen-derived VLPs can retain receptor specific binding proteins. Examples include the VLPs derived from paramyxoviruses and HIV, which naturally target and facilitate uptake similar to the infectious parent pathogens.28 Certain nonhost associated VLPs have very specific binding partners in organisms to which they are not normally infectious. CPMV naturally targets and binds to the cellular surface protein vimentin facilitating cellular uptake in cell culture as well as mouse and rat models.29–31 The Canine parvovirus (CPV) VLP naturally not only binds to canine transferrin receptors but also binds to homologous human transferrin receptors which are overexpressed in certain types of cancer cells.32
In the case of other nonhost associated VLPs, the exterior surface of capsids provides a ready platform for the display of directing molecules. As with the capsid interiors, targeting and masking moieties can be introduced either genetically or through synthetic bio-conjugation. One specific strategy within the VLP targeting effort has used peptides, containing the RGD motif, derived from phage display against rapidly dividing cancer cells expressing integrins αvβ3 and αvβ5. To adapt this strategy for VLP targeting, the RGD-4C sequence was fused to either sHSP or human H-chain ferritin (Fn-RGD).24,33,34 Both of these cages successfully targeted melanoma cells in vitro. In the case of sHSP, alternative specificity could be designed through the conjugation of anti-CD4 antibodies to the cage exterior, which directed these cages to CD4+ lymphocytes as confirmed by FACS analysis.33
For applications in diagnostic imaging, the targeting potential of Fn-RGD was also assessed in vivo. In addition to overexpression in tumor vasculature, integrin αvβ3 is upregulated in Mac localized to atherosclerotic lesions.35 The near infrared fluorescence intensity at lesion sites of Fn-RGD conjugated to Cy5.5 dye was compared nontargeted particles.34 Signal was increased twofold at the lesion sites for Fn-RGD compared to nontargeted particles. While this increase is significant, this work also demonstrates one of the hurdles of biological targeting not limited to nanoparticle systems. Only a fraction of targeted particles make it to the intended site with significant accumulations in the lungs, spleen, liver, and kidneys regardless of if the particle is targeted.34 While this type of targeting can help improve accumulation at intended sites it does not prevent off-target effects nor, in the case of therapeutic delivery, does it necessarily ensure enough accumulation to significantly change downstream biological effects.
This contrast of programed targeting and selection of capsids with inherent targeting highlights the range of strategies available in VLP engineering. The modularity of VLP selection, cargo loading, and targeting strategies allows for multiple combinations in the design of functional diagnostic VLP nanoparticles.
IMMUNE RESPONSES TO VLPs
While VLPs exhibit impressive tolerance to manipulation and functionalization, effective use of these systems for biomedical applications will hinge on the ability to characterize, predict, and control the in vivo behavior of these particles. As stated above, the size of VLPs predisposes them to immune-circulation and uptake. In addition VLPs are protein-based technologies derived from a variety of biological sources. This immediately raises concerns about the potential for harmful immune responses to the VLP itself as well as to contaminant molecules from VLP production such as lipopolysaccharide, which is ubiquitous in many bacterial VLP production systems. In response to these concerns much focus has been placed on masking immune response and creating biologically invisible particles through PEGylation and alternative surface functionalization.
An alternative strategy is to characterize and utilize the in vivo behavior of VLPs as yet another engineering tool for the design of biomedically useful nanoparticles. In this section we discuss potentially beneficial interactions of VLP systems both specifically and nonspecifically with the immune system leading to better particle design and more complete understanding of viral immunity.
VLPs and the Innate Immune System
VLPs from pathogenic viruses have served as polyvalent antigens in prophylactic vaccines including current vaccines to hepatitis B virus (HBV), human papilloma virus (HPV), and others.28,36 Advantageous as low risk alternatives to attenuated virus vaccines, VLP vaccines maintain authentic presentation of conformational antigens by preserving the native structure, including symmetry and polyvalency, of the infectious virus.37 However, the usefulness of VLPs as stimulators of the immune system extends well beyond their ability to elicit strong adaptive immune responses. An emerging picture of VLPs shows that they are potent stimulators of the innate immune system as well as coordinators of the innate–adaptive interface. This makes them potentially useful as probes for elucidating immunological interactions as well as platforms for the design of specific immune stimulatory particles.
Emerging work continues to suggest that certain innate reactions to VLP particles are independent of the primary sequence of the VLP proteins or commonly associated pathogen associated molecular patterns (PAMPs). In viral immunology, much focus has been dedicated to the recognition of nucleic acid species and other PAMPs by the innate immune system through toll-like receptors, complement systems and other receptors in the stimulation of innate immune cascades.38–40 While identified PAMPs undoubtedly serve as powerful, specific, innate immune signals, the extent of the nonspecific contribution of the virus structure has been largely overlooked. VLP studies isolate the effects of the protein shell from those of nucleic acid and other PAMPs allowing for focus on nonspecific structural means of recognition.
VLP Induction of Secondary Lymphoid Tissue
The lung is of particular interest, as a common pathogen entry route, in the investigation of VLP innate immune interactions. Many pathogens infect the lung mucosal epithelium directly or enter other host systems though the mucosal interface.41 This delicate environment is particularly sensitive to inflammatory immune responses. Because of its prevalence as a pathogen entry point, the lung is also a target in vaccination efforts. Induction of immunity in the mucosal network is often efficacious toward preventing initial pathogen entry through the induction of IgA.41
Constant exposure of the lung environment to pathogens requires efficient and controlled immune regulation and activation. The presence of secondary lymphoid tissue in the form of bronchial associated lymphoid tissue (BALT) regulates the rapid recognition and response to lung pathogens. This tissue is composed of a mixture of APC and adaptive immune cells arranged similar to other secondary lymphoid tissues. This includes formation of B-cell follicles surrounded by a T-cell zone, high endothelial venules, and DC.42 These structures are positioned to maximize exposure to incoming pathogens, often adjacent to major airways and at airway bifurcations (Figure 4). In some mammals, humans and mice in particular, BALT is not constitutively maintained and instead is induced upon pathogen infection.42 Increasing evidence implicates innate immune cells, in particular DC, as the activators of this induced BALT or iBALT. iBALT has been shown to be key in influenza immunity where it localizes many immunological functions commonly associated with the spleen and constitutive secondary lymphoid tissues.42,44
FIGURE 4.

iBALT readily forms in the murine lung after i.n. administration of sHSP. (a) iBALT structures,indicated by arrows, emerge adjacent to airways and blood vessels after five administrations of sHSP compared to a PBS control (b). Stained fluorescence microscopy reveals that, compared to a control (c), iBALT structures contain (d) CD4+ T-cells, B220+ B cells, and (e) CD21+ follicular DC. (Reprinted with permission from Ref 43. Copyright 2009 Public Library of Science (PLoS))
A difficultly in studying iBALT arises from the inflamed, infection-associated environment under which iBALT naturally occurs. To circumvent this, noninfectious VLPs can be used as dummy viruses. Administration of the nonviral sHSP VLP induced iBALT formation without negative inflammatory effects commonly associated with pulmonary pathogenesis.43 Pulmonary administration of sHSP (5 × 100 μg sHSP) in murine subjects led to the formation of iBALT with similar structure and content to those induced by influenza infection and constitutive BALT in other species (Figure 4).
When mice, pre-inoculated with sHSP, were challenged with lethal doses of multiple serotypes of influenza as well as murine-adapted SARS and pneumovirus they exhibited almost complete resistance to infection including minimal weight loss and viral burden. This nonspecific protection extended to pulmonary bacterial challenge as demonstrated by lethal challenge with Coxiella burnetii, Francisella tularemia, and Yesinia pestis. While sHSP treated mice demonstrated less weight loss due to bacterial infection they failed to show an advantage toward pathogen clearance. This protection was shown to be highly dependent on the iBALT structures as knock-out mice for lymphotoxin-α, a key regulator of iBALT formation, showed minimal protection while mice lacking other secondary lymphoid organs showed similar protection to WT mice.
A secondary effect of VLP induced, prophylactic iBALT formation is that inflammation due to subsequent challenge is curtailed. In the above study, this is shown by the reduced levels of the damage markers serum albumin and lactate dehydrogenase. This suggests a mechanism for protection centered on cellular recruitment, but without the inflammation seen in pathogenic infections. This balance may be a consequence of the absence of potent PAMPs in the VLPs, though Escherichia coli expressed VLPs contain some level of LPS, unless explicitly removed.
This innate response to VLPs is not limited to the M. jannaschii sHSP. Administration of the bacteriophage P22 VLP elicited similar iBALT formation and immunity to subsequent influenza challenge.44 While both sHSP and P22 are symmetric protein cage architectures, the type of symmetry, size, and primary structure of these VLPs are quite different, suggesting that iBALT formation occurs due to a nonspecific mechanism of particle or polyvalent symmetry recognition. One major contributor to VLP recognition, and that of nanoparticles in general, is the size of the particles. The size range of many VLPs, 20–200 nm, predisposes them for cellular uptake as well as ready entrance into lymphatic circulation where they are collected in lymph nodes rich in DC and Mac.27 In addition, the repeated symmetry of VLP particles may contribute to immune recognition in a nonspecific fashion. Any non-specific associations with cellular surface components or with serum corona proteins are likely to reflect the underlying symmetry or repeated structure and encourage engulfment due to the spherical shape of the capsids. The attachment of nonspecific corona proteins could potentially translate the repeated symmetry or shape of a particle into a specific signal for uptake or immune stimulation.45 Further work is needed to elucidate the mechanism of nonspecific VLP innate recognition. This characteristic makes these particles exceptionally attractive platforms for the rapid transport and processing of guest antigens displayed on the interior or exterior of the capsid.
Designed VLP Vaccines
VLPs have long been known to stimulate the adaptive immune system in ways similar to their parent infectious viruses. Initially this effect was recognized through B-cell mediated adaptive immunity, to a particular pathogenic virus, gained through exposure to the corresponding noninfectious VLP. The polyvalent nature of VLP antigens leads to a high frequency of B-cell receptor (BCR) crosslinking and a stronger B-cell stimulation as well the potential for broader spectrum protection.46,47
While direct use of a VLP from the target pathogen is effective in some specific cases as an alternative to traditionally produced vaccines, many pathogen-derived VLPs remain difficult to purify and stabilize.48 In addition, VLPs derived directly from pathogenic viral precursors lack the ability to selectively present only specific antigenic sequences, which could focus the immune response to conserved antigenic regions. To circumvent these difficulties, nonhost associated VLPs can be used to design particles that can be selectively engineered to encapsulate and or present specific antigens and adjuvants. These systems have the potential to produce designed vaccines with a higher tendency to generate memory responses and broader immunity than traditionally produced vaccines or target pathogen VLPs.
Toward this goal, the potent humoral antigen Ovalbumin (OVA) was conjugated through exposed lysine residues to engineered cysteine residues on the surface of sHSP with an NHS-maleimide bifunctional PEG crosslinker.49 Compared to OVA alone, intranasal administration of the sHSP-OVA induced antibody formation 9 days earlier. Capitalizing on the nonspecific VLP stimulation of iBALT, the P22-VLP was administered prior to sHSP-OVA and further accelerated the antibody response. Priming with P22 also accelerated isotype switching to IgA and IgG-1. This accelerated behavior suggests that BCR crosslinking can be maintained for nonnative VLP-displayed antigens. The VLPs in this case are thought to be acting both as polyvalent scaffolds and as adjuvants through demonstrated preference for APC uptake and the recruitment of innate and adaptive cells.
One reason for utilizing a nonhost associated VLP for humoral antigen presentation is to control the serotypic specificity of a vaccine. Influenza serves as a model pandemic virus with a complex multispecies host system and high degree of variability that can lead to the sudden emergence of highly infectious serotypes with widespread global impact. Current annual influenza vaccines are composed of the three serotypes anticipated to be most dangerous that year, two A strain and one B strain, referred to as a trivalent inactivated influenza vaccine (TIV).50 These vaccines take time to produce and are not able to confer immunity toward unanticipated seasonal strains. The need for seasonal vaccine production for influenza is largely due to the genetic drift facilitated variability seen in the highly antigenic hemagglutinin (HA) protein of influenza. This exterior, membrane-bound, trimeric spike protein facilitates fusion of the virus phospholipid layer with the target cell.51
Effective humoral immunity to influenza, both during infection and vaccination, is primarily directed at the immunodominant but highly variable epitopes of the HA head. Broad-spectrum antibodies to the conserved stalk region of the HA protein, with activity toward multiple serotypes of influenza, have been identified but B-cells carrying these antibodies are likely outcompeted by populations carrying antibodies to the immunodominant regions.52 Kanekiyo et al. demonstrated that the VLP-like protein cage of Helicobacter pylori ferritin (HpFn) can be used to display 1999 NC (H1N1) HA through genetic fusion of the HA gene to the HpFn subunit gene.53 Subsequent expression of the construct led to the formation of eight trimeric HA spikes at the natural threefold axes of the HpFn cage (Figure 5). Intramuscular administration of this designed vaccine resulted in comparable protection to a traditionally produced TIV containing the 1999 NC serotype. When the immune response was amplified through the use of the common adjuvant Ribi, the HpFn-HA construct exhibited 7.2-fold increase in neutralizing antibody titers compared to TIV immunized mice with Ribi. In addition, the breadth of immunity was expanded for the HpFn-HA compared to TIV largely due to the production of two classes of broadly neutralizing antibodies either to the HA-stalk region or a conserved receptor-binding site on the HA-head region. The reason for this improved immunization breadth has yet to be elucidated but may be due, in part, to the size of VLP used in this study. The small size of the HpFn VLP compared to the native influenza A membrane layer (8–12 nm versus 80–120 nm) will inevitably increase the angle of display between HA spikes, or other neighboring membrane proteins, increasing the exposure of conserved regions of the protein. Increased accessibility could lead to increased chance of binding to HA-stalk-specific, broadly neutralizing antibody precursor BCRs.
FIGURE 5.

Biomimetic display of influenza hemagglutinin (HA) on HpFn leads to improved protection compared to a traditional trivalent vaccine (TIV) in the presence of adjuvant (Ribi). (a) A transmission electron micrograph showing 1999 (NC) HA genetically fused to HpFn and displayed as the native trimer at the threefold axis of the VLP (HA-np). (b) HA-np provides greater protection than TIV when administered to ferrets challenged with 106.5 EID50 dose of 2007 Bris. Viral titers are reported as 50% tissue culture infectious dose (TCID50). (Reprinted with permission from Ref 53. Copyright 2013 Nature Publishing Group)
It has been shown that immunization efficacy of both VLP and traditional vaccines is highly dependent on the route of administration.54,55 With influenza, Ichinohe et al. compared intranasal versus subcutaneous immunization of traditional TIV (Japan 2005–2006), with no strain specificity, for emerging H5N1 (more commonly referred to as avian or bird flu). Subsequent challenge with various H5N1 serotypes demonstrated that intranasal administration of TIV protected against H5N1 through cross-reactive IgA and IgG while subcutaneous administration produced no detectable immunity.55 This preference for lung administration likely extends to VLP vaccines such as HpFn-HA. It may be possible to further amplify the protection of HpFn-HA by vaccinating intranasally.
As an alternative to primarily humoral immunity it has been demonstrated that, though noninfectious and thus extracellular, VLPs show a preference for cellular uptake and subsequent crosspresentation on MHC-1 compared to nonparticulate proteins.28,56 However, successful MHC-1 presentation does not necessitate a potent cytotoxic T-cell response and adjuvants such as anti-CD40 are necessary to ensure immune efficacy.57 Owing to precise spatial control within engineered VLP constructs, VLP vaccines also have the potential to further influence the pathway of antigen processing and the resultant immune response by shielding or readily presenting antigens. This type of control is highly desirable in many viral infections where certain antigens are more efficacious in stimulating effective CD4+ or CD8+ mediated immunity. Certain bacterial pathogens such as Y. pestis have been shown to require a balanced humoral and cellular response for effective immunity.58
Drawing again on influenza as a relevant model epidemic pathogen, a CD8+ directed VLP encapsulated antigen construct was created utilizing the VLP from bacteriophage P22. The assembly of the P22-VLP involves coexpression of a primary coat protein with an essential scaffolding protein. This scaffold protein can be truncated to an essential C-terminal domain which, when fused to a cargo gene product, can direct encapsulation of a protein cargo. In this study the P22-VLP encapsulation system was used to encapsulate influenza nucleoprotein (NP) which functions to coordinate the viral genome within the infectious influenza capsid and is conserved across multiple serotypes of influenza A.59
Intranasal administration of P22 containing the first 163 residues of the NP protein (P22-NP163), in high copy number, protected against subsequent challenge with 100× LD50 of PR8 (H1N1) and 50× LD50 of X31 (H3N2) compared to nonimmunized mice or those immunized with P22 alone.60 Immunized mice demonstrated an expected initial weight loss but subsequent recovery suggesting a primarily CD8+ mechanism of protection (Figure 6). This was further supported by NP-specific MHC-1 tetramer staining, the negation of protection by addition of CD8+ T-cell depleting IgG (TIB210) and the absence of neutralizing antibodies to PR8. Inefficacious NP-antibodies were detected in high titer after immunization with P22 encapsulated full-length NP but not NP163 suggesting that nonhost VLP associated antigens can readily undergo crosspresentation but subsequent effective antibody production is sequence dependent, possibly due simply to the surface exposure of an epitope in the native antigen.
FIGURE 6.

Biomimetic encapsulation of influenza nucleoprotein can elicit CD8+ mediated immunity. (a) Nucleoprotein (NP) was encapsulated in the P22 VLP (left) via genetic fusion to the scaffolding protein (SP) mimicking the natural position of the NP in Influenza an artist’s rendering of which is shown (right). (b) Mice vaccinated with NP163-P22 (the first third of the NP gene encapsulated in P22) showed improved survival after subsequent challenge with PR8 and X31 compared to the empty P22 or a PBS control. Protection could be negated by the addition of the CD8+ T-cell depleting IgG (TIB210). (c) The initial weight loss of all the immunized mice suggests that even the NP163-P22 mice initially are infected and that the mechanism of protection is not humoral. (Reprinted with permission from Ref 60. Copyright 2013 American Chemical Society)
Another application for use of VLPs in selective antigen processing and CD8+ activation is in the design of anti-cancer vaccines, which require a potent CTL response to be effective. Speiser and colleagues demonstrated that conjugation of a melanoma self antigen (melan-A) and a short immune-stimulatory nucleotide to the bacteriophage Qβ VLP results in an immune response, largely antibody driven, divided between specificity for Melan-A antigen and the VLP capsid.61,62 While the T-cell component of the Melan-A response was significant, further modification was necessary in the form of an accompanying adjuvant, either Incomplete Freund’s Adjuvant or Imiquimod, both of which are known to boost antigenicity. Tumorous tissue showed significant reduction in Melan-A expression after vaccination indicating biological activity and vaccine potential.63 Encapsulation of the Melan-A antigen could potentially further encourage an effective CTL response by avoiding any direct Melan-A specific B-Cell stimulation and necessitating intracellular processing before the antigen becomes exposed.
CONCLUSIONS
The results discussed here show the essential exchange between fundamental immunological understanding and VLP engineering efforts to create new immune complementary methods and diagnostic-imaging nanotechnology, combining some of nature’s best infiltrating structures with biomimetic design efforts. VLPs offer an immune complementary platform for the design of in vivo delivery systems. These systems serve as model biomimetic scaffolds, providing unmatched spatial and chemical control. Modularity afforded by the variety of engineering strategies and natural capsid candidates allows for the rational design of constructs for a biological need, taking advantage of each capsids unique characteristics.
In the case of MRI contrast agents, VLPs not only drastically improve the contrast on a per particle basis, through high metal contrast agent loading, but also fundamentally improve the relaxivity characteristics of each individual metal center. The demonstrated tolerance of VLPs to a wide range of bioconjugation, labeling, and polymerization techniques allows for the delivery a large spectrum of molecules in addition to MRI contrast agents. An advantage of cargo encapsulation is that the capsid surface can be used to further influence particle behavior through presentation of targeting domains or signal decoration. Despite impressive imaging potential, these particles are not immunologically inert even when masked. Thus actual application of VLP systems as contrast agents requires an intimate understanding of how the immune system responds to these constructs.
VLPs stimulate the innate immune system in a non-VLP-specific manner. This characteristic can be utilized to create nonspecific activators of immunity both as probes of viral recognition processes as well as adjuvants for the administration of vaccines. Combining this immune stimulation with the same resilience to modification demonstrated with MRI contrast agents, specific antigens can be conjugated to VLP capsids. VLPs carrying antigen through polyvalent presentation or multicopy encapsulation, have the potential to generate potent and directed immune responses.
The in vivo usage of VLPs in both of the discussed applications is not without obvious practical concerns. Biomimetic applications of viruses utilize the very structures that the immune system has evolved to effectively recognize and combat. The immune stimulatory nature of VLPs has been discussed as a weakness of VLP systems for delivery applications as development of protective adaptive immunity could cause rapid clearance and potential adverse side effects.64 In the case of diagnostic contrasts agents the major goal is to effectively image biological tissue without impacting or changing the system. For immune stimulatory and vaccine constructs repeated administration is often required to achieve an effective immune response. Subsequent administration of VLP constructs could suffer from inflammation, improved immune clearance, or at least variable biological response as the immune system develops adaptive memory to the viral shell. This was not reported to be problematic for the constructs described here but does not eliminate this problem as a real worry in the future VLP application.
While the immunogenic nature of VLP particles can be tempered and masked through bioconjugation,45 masking efforts will inevitably decrease some of the advantageous immune recognition processes that make these systems appealing as vaccine design systems and immune delivery systems. An alternative solution is afforded by the immense variety of available VLPs that can be engineered. The use of multiple VLP parent particles for subsequent administration toward a desired biological effect could avoid the risk of outright immune-rejection of a VLP construct due to adaptive memory. Regardless of the specific application, further understanding of how these particles look, behave and transform in biological systems is necessary to guide future engineering efforts.
Despite some reservations, the potential of VLPs as biomimetic nanoparticles in diagnostic, therapeutic, and prophylactic applications is enormous. In addition to the direct engineering of VLP systems, elucidation of immune recognition processes, especially those of the innate immune system, are invaluable for the future design of synthetic nanoparticle systems, vaccine strategies, and small molecule activities.
ACKNOWLEDGEMENT
This work was supported in part by grants from the NIH, NIAID R01-AI104905 and NIBIB R01-EB012027; B. Schwarz was supported by the DoD through the NDSEG program.
Footnotes
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1.Prugnolle F, Manica A, Charpentier M, Guégan JF, Guernier V, Balloux F. Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol. 2005;15:1022–1027. doi: 10.1016/j.cub.2005.04.050. [DOI] [PubMed] [Google Scholar]
- 2.Woolhouse MEJ, Webster JP, Domingo E, Charlesworth B, Levin BR. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat Genet. 2002;32:569–577. doi: 10.1038/ng1202-569. [DOI] [PubMed] [Google Scholar]
- 3.Suttle CA. Viruses in the sea. Nature. 2005;437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- 4.Lucon J, Qazi S, Uchida M, Bedwell GJ, LaFrance B, Prevelige PE, Jr, Douglas T. Use of the interior cavity of the P22 capsid for site-specific initiation of atom-transfer radical polymerization with high-density cargo loading. Nat Chem. 2012;4:781–788. doi: 10.1038/nchem.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel MV, Chen FJ. Solid carriers for improved delivery of active ingredients in pharmaceutical compositions. 2001 Available at: http://www.google.com/patents/US6248363. (Accessed June, 25 2014)
- 6.Faraji AH, Wipf P. Nanoparticles in cellular drug delivery. Bioorg Med Chem. 2009;17:2950–2962. doi: 10.1016/j.bmc.2009.02.043. [DOI] [PubMed] [Google Scholar]
- 7.Weissleder R, Kelly K, Sun EY, Shtatland T, Josephson L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotechnol. 2005;23:1418–1423. doi: 10.1038/nbt1159. [DOI] [PubMed] [Google Scholar]
- 8.Slowing II, Vivero-Escoto JL, Wu C-W, Lin VSY. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv Drug Deliv Rev. 2008;60:1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Liepold LO, Abedin MJ, Buckhouse ED, Frank JA, Young MJ, Douglas T. Supramolecular protein cage composite MR contrast agents with extremely efficient relaxivity properties. Nano Lett. 2009;9:4520–4526. doi: 10.1021/nl902884p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mulder WJM, Strijkers GJ, van Tilborg GAF, Griffioen AW, Nicolay K. Lipid-based nanoparticles for contrast-enhanced MRI and molecular imaging. NMR Biomed. 2006;19:142–164. doi: 10.1002/nbm.1011. [DOI] [PubMed] [Google Scholar]
- 11.Ananta JS, Godin B, Sethi R, Moriggi L, Liu X, Serda RE, Krishnamurthy R, Muthupillai R, Bolskar RD, Helm L, et al. Geometrical confinement of gadolinium-based contrast agents in nanoporous particles enhances T1 contrast. Nat Nano. 2010;5:815–821. doi: 10.1038/nnano.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen M, Bulte JWM, Liepold L, Basu G, Zywicke HA, Frank JA, Young M, Douglas T. Paramagnetic viral nanoparticles as potential high-relaxivity magnetic resonance contrast agents. Magn Reson Med. 2005;54:807–812. doi: 10.1002/mrm.20614. [DOI] [PubMed] [Google Scholar]
- 13.Bruckman MA, Hern S, Jiang K, Flask CA, Yu X, Steinmetz NF. Tobacco mosaic virus rods and spheres as supramolecular high-relaxivity MRI contrast agents. J Mater Chem B. 2013;1:1482–1490. doi: 10.1039/C3TB00461A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liepold L, Anderson S, Willits D, Oltrogge L, Frank JA, Douglas T, Young M. Viral capsids as MRI contrast agents. Magn Reson Med. 2007;58:871–879. doi: 10.1002/mrm.21307. [DOI] [PubMed] [Google Scholar]
- 15.Anderson EA, Isaacman S, Peabody DS, Wang EY, Canary JW, Kirshenbaum K. Viral nanoparticles donning a paramagnetic coat: conjugation of MRI contrast agents to the MS2 capsid. Nano Lett. 2006;6:1160–1164. doi: 10.1021/nl060378g. [DOI] [PubMed] [Google Scholar]
- 16.Prasuhn JDE, Yeh RM, Obenaus A, Manchester M, Finn MG. Viral MRI contrast agents: coordination of Gd by native virions and attachment of Gd complexes by azide-alkyne cycloaddition. Chem Commun. 2007:1269–1271. doi: 10.1039/b615084e. doi: 10.1039/B615084E. [DOI] [PubMed] [Google Scholar]
- 17.Pokorski JK, Breitenkamp K, Liepold LO, Qazi S, Finn MG. Functional virus-based polymer–protein nanoparticles by atom transfer radical polymerization. J Am Chem Soc. 2011;133:9242–9245. doi: 10.1021/ja203286n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rocklage SM, Worah D, Kim SH. Metal ion release from paramagnetic chelates: what is tolerable? Magn Reson Med. 1991;22:216–221. doi: 10.1002/mrm.1910220211. discussion 229–232. [DOI] [PubMed] [Google Scholar]
- 19.Klug A. The tobacco mosaic virus particle: structure and assembly. Philos Trans R Soc Lond B Biol Sci. 1999;354:531–535. doi: 10.1098/rstb.1999.0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim R, Kim KK, Yokota H, Kim SH. Small heat shock protein of Methanococcus jannaschii, a hyperthermophile. Proc Natl Acad Sci U S A. 1998;95:9129–9133. doi: 10.1073/pnas.95.16.9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hovlid ML, Lau JL, Breitenkamp K, Higginson CJ, Laufer B, Manchester M, Finn MG. Encapsidated atom-transfer radical polymerization in Qβ virus-like nanoparticles. ACS Nano. 2014;8:8003–8014. doi: 10.1021/nn502043d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qazi S, Uchida M, Usselman R, Shearer R, Edwards E, Douglas T. Manganese (III) porphyrins complexed with P22 virus-like particles as T 1-enhanced contrast agents for magnetic resonance imaging. J Biol Inorg Chem. 2014;19:237–246. doi: 10.1007/s00775-013-1075-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terashima M, Uchida M, Kosuge H, Tsao PS, Young MJ, Conolly SM, Douglas T, McConnell MV. Human ferritin cages for imaging vascular macrophages. Biomaterials. 2011;32:1430–1437. doi: 10.1016/j.biomaterials.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchida M, Flenniken ML, Allen M, Willits DA, Crowley BE, Brumfield S, Willis AF, Jackiw L, Jutila M, Young MJ, et al. Targeting of cancer cells with ferrimagnetic ferritin cage nanoparticles. J Am Chem Soc. 2006;128:16626–16633. doi: 10.1021/ja0655690. [DOI] [PubMed] [Google Scholar]
- 25.Uchida M, Klem MT, Allen M, Suci P, Flenniken M, Gillitzer E, Varpness Z, Liepold LO, Young M, Douglas T. Biological containers: protein cages as multifunctional nanoplatforms. Adv Mater. 2007;19:1025–1042. [Google Scholar]
- 26.Vymazal J, Brooks RA, Zak O, McRill C, Shen C, Chiro GD. T1 and t2 of ferritin at different field strengths: effect on mri. Magn Reson Med. 1992;27:368–374. doi: 10.1002/mrm.1910270218. [DOI] [PubMed] [Google Scholar]
- 27.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38:1404–1413. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 28.Grgacic EVL, Anderson DA. Virus-like particles: Passport to immune recognition. Methods. 2006;40:60–65. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koudelka KJ, Rae CS, Gonzalez MJ, Manchester M. Interaction between a 54-kilodalton mammalian cell surface protein and cowpea mosaic virus. J Virol. 2007;81:1632–1640. doi: 10.1128/JVI.00960-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G, Manchester M. Endothelial targeting of cowpea mosaic virus (CPMV) via surface vimentin. PLoS Pathog. 2009;5:e1000417. doi: 10.1371/journal.ppat.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinmetz NF, Cho CF, Ablack A, Lewis JD, Manchester M. Cowpea mosaic virus nanoparticles target surface vimentin on cancer cells. Nanomedicine (Lond) 2011;6:351–364. doi: 10.2217/nnm.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh P, Destito G, Schneemann A, Manchester M. Canine parvovirus-like particles, a novel nanomaterial for tumor targeting. J Nanobiotechnol. 2006;4:1–11. doi: 10.1186/1477-3155-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flenniken ML, Willits DA, Harmsen AL, Liepold LO, Harmsen AG, Young MJ, Douglas T. Melanoma and lymphocyte cell-specific targeting incorporated into a heat shock protein cage architecture. Chem Biol. 2006;13:161–170. doi: 10.1016/j.chembiol.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Kitagawa T, Kosuge H, Uchida M, Dua MM, Iida Y, Dalman RL, Douglas T, McConnell MV. RGD-conjugated human ferritin nanoparticles for imaging vascular inflammation and angiogenesis in experimental carotid and aortic disease. Mol Imaging Biol. 2012;14:315–324. doi: 10.1007/s11307-011-0495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uchida M, Willits DA, Muller K, Willis AF, Jackiw L, Jutila M, Young MJ, Porter AE, Douglas T. Intracellular distribution of macrophage targeting ferritin–iron oxide nanocomposite. Adv Mater. 2009;21:458–462. [Google Scholar]
- 36.Garcea RL, Gissmann L. Virus-like particles as vaccines and vessels for the delivery of small molecules. Curr Opin Biotechnol. 2004;15:513–517. doi: 10.1016/j.copbio.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 37.Noad R, Roy P. Virus-like particles as immunogens. Trends Microbiol. 2003;11:438–444. doi: 10.1016/s0966-842x(03)00208-7. [DOI] [PubMed] [Google Scholar]
- 38.Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 39.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 40.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 41.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 42.Randall TD. Chapter 7—Bronchus-associated lymphoid tissue (BALT): structure and function. In: Sidonia F, Andrea C, editors. Advances in Immunology. Vol. 107. Academic Press; San Diego, CA: 2010. pp. 187–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiley JA, Richert LE, Swain SD, Harmsen A, Barnard DL, Randall TD, Jutila M, Douglas T, Broomell C, Young M. Inducible bronchus-associated lymphoid tissue elicited by a protein cage nanoparticle enhances protection in mice against diverse respiratory viruses. PLoS One. 2009;4:e7142. doi: 10.1371/journal.pone.0007142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richert LE, Harmsen AL, Rynda-Apple A, Wiley JA, Servid AE, Douglas T, Harmsen AG. Inducible bronchus-associated lymphoid tissue (iBALT) synergizes with local lymph nodes during antiviral CD4+ T cell responses. Lymphat Res Biol. 2013;11:196–202. doi: 10.1089/lrb.2013.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2011;134:2139–2147. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 46.Zabel F, Kündig TM, Bachmann MF. Virus-induced humoral immunity: on how B cell responses are initiated. Curr Opin Virol. 2013;3:357–362. doi: 10.1016/j.coviro.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 47.Bright RA, Carter DM, Daniluk S, Toapanta FR, Ahmad A, Gavrilov V, Massare M, Pushko P, Mytle N, Rowe T. Influenza virus-like particles elicit broader immune responses than whole virion inactivated influenza virus or recombinant hemagglutinin. Vaccine. 2007;25:3871–3878. doi: 10.1016/j.vaccine.2007.01.106. [DOI] [PubMed] [Google Scholar]
- 48.Lua LHL, Connors NK, Sainsbury F, Chuan YP, Wibowo N, Middelberg APJ. Bioengineering virus-like particles as vaccines. Biotechnol Bioeng. 2014;111:425–440. doi: 10.1002/bit.25159. [DOI] [PubMed] [Google Scholar]
- 49.Richert LE, Servid AE, Harmsen AL, Rynda-Apple A, Han S, Wiley JA, Douglas T, Harmsen AG. A virus-like particle vaccine platform elicits heightened and hastened local lung mucosal antibody production after a single dose. Vaccine. 2012;30:3653–3665. doi: 10.1016/j.vaccine.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambert LC, Fauci AS. Influenza vaccines for the future. N EnglJ Med. 2010;363:2036–2044. doi: 10.1056/NEJMra1002842. [DOI] [PubMed] [Google Scholar]
- 51.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 52.Ekiert DC, Bhabha G, Elsliger M-A, Friesen RHE, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanekiyo M, Wei C-J, Yassine HM, McTamney PM, Boyington JC, Whittle JRR, Rao SS, Kong W-P, Wang L, Nabel GJ. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature. 2013;499:102–106. doi: 10.1038/nature12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cubas R, Zhang S, Kwon S, Sevick-Muraca EM, Li M, Chen C, Yao Q. Virus-like particle (VLP) lymphatic trafficking and immune response generation after immunization by different routes. J Immunother (Hagerstown, Md) 2009;32:118–128. doi: 10.1097/CJI.0b013e31818f13c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ichinohe T, Tamura S-I, Kawaguchi A, Ninomiya A, Imai M, Itamura S, Odagiri T, Tashiro M, Takahashi H, Sawa H. Cross-protection against H5N1 influenza virus infection is afforded by intranasal inoculation with seasonal trivalent inactivated influenza vaccine. J Infect Dis. 2007;196:1313–1320. doi: 10.1086/521304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bachmann MF, Kündig TM, Freer G, Li Y, Kang CY, Bishop DH, Hengartner H, Zinkernagel RM. Induction of protective cytotoxic T cells with viral proteins. Eur J Immunol. 1994;24:2228–2236. doi: 10.1002/eji.1830240944. [DOI] [PubMed] [Google Scholar]
- 57.Storni T, Lechner F, Erdmann I, Bächi T, Jegerlehner A, Dumrese T, Kündig TM, Ruedl C, Bachmann MF. Critical role for activation of antigen-presenting cells in priming of cytotoxic T cell responses after vaccination with virus-like particles. J Immunol. 2002;168:2880–2886. doi: 10.4049/jimmunol.168.6.2880. [DOI] [PubMed] [Google Scholar]
- 58.Parent MA, Berggren KN, Kummer LW, Wilhelm LB, Szaba FM, Mullarky IK, Smiley ST. Cell-mediated protection against pulmonary Yersinia pestis infection. Infect Immun. 2005;73:7304–7310. doi: 10.1128/IAI.73.11.7304-7310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.An P, Digard P. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol. 2002;83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- 60.Patterson DP, Rynda-Apple A, Harmsen AL, Harmsen AG, Douglas T. Biomimetic antigenic nanoparticles elicit controlled protective immune response to influenza. ACS Nano. 2013;7:3036–3044. doi: 10.1021/nn4006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braun M, Jandus C, Maurer P, Hammann-Haenni A, Schwarz K, Bachmann MF, Speiser DE, Romero P. Virus-like particles induce robust human T-helper cell responses. Eur J Immunol. 2012;42:330–340. doi: 10.1002/eji.201142064. [DOI] [PubMed] [Google Scholar]
- 62.Speiser DE, Schwarz K, Baumgaertner P, Manolova V, Devevre E, Sterry W, Walden P, Zippelius A, Conzett KB, Senti G. Memory and effector CD8 T-cell responses after nanoparticle vaccination of melanoma patients. J Immunother. 2010;33:848–858. doi: 10.1097/CJI.0b013e3181f1d614. [DOI] [PubMed] [Google Scholar]
- 63.Goldinger SM, Dummer R, Baumgaertner P, Mihic-Probst D, Schwarz K, Hammann-Haenni A, Willers J, Geldhof C, Prior JO, Kündig TM, et al. Nano-particle vaccination combined with TLR-7 and -9 ligands triggers memory and effector CD8 + T-cell responses in melanoma patients. Eur J Immunol. 2012;42:3049–3061. doi: 10.1002/eji.201142361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zolnik BS, González-Fernández Á , Sadrieh N, Dobrovolskaia MA. Minireview: nanoparticles and the immune system. Endocrinology. 2010;151:458–465. doi: 10.1210/en.2009-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]