

Abstract
Anaplasma phagocytophilum(Ap) is a tick-borne pathogen, which can cause granulocytic anaplasmosis in humans and animals. In vivo this obligate intracellular pathogen is primarily located in circulating mature granulocytes, but it also infects endothelial cells. In order to study the interaction between Ap-infected endothelial cells and human granulocytes under conditions similar to those found naturally in the infected host, an in vitro model that mimics physiological flow conditions in the microvasculature was established. Cell-to-cell interactions were then visualized by microscopy, which showed that granulocytes adhered strongly to Ap-infected endothelial cells at a shear stress of 0.5 dyne/cm2. In addition, Ap-transmission assays under flow conditions showed that the bacteria transferred from infected endothelial cells to circulating granulocytes and were able to establish infection in constantly moving granulocytes. Cell surface analysis showed that Ap induced up-regulation of the cell adhesion molecules ICAM-1 and VCAM-1 on infected endothelial cells in a dose-dependent manner. Furthermore, IL-8 secretion by endothelial cells indicated that the presence of Ap induced a pro-inflammatory response. In summary, the results of this study suggest that endothelial cells of the microvasculature (1) provide an excellent site for Ap dissemination to peripheral blood granulocytes under flow conditions and therefore may play a crucial role in the development of persistent infection, and (2) are stimulated by Ap to express surface molecules and cytokines that may lead to inflammatory responses at the site of the infection.
Keywords: Anaplasma phagocytophilum, Transmission, Endothelial cells, Granulocytes adhesion, Shear flow
Introduction
Anaplasma phagocytophilum (Ap) is a tick-borne pathogen that is able to infect many different animal species and humans worldwide. Ap can cause sometimes a severe clinical illness called granulocytic anaplasmosis in humans, domestic dogs, cats, horses or tick-borne fever in ruminants [3, 7, 8, 46]. The clinical signs are non-specific, including fever, leucopenia, thrombocytopenia and anorexia. During the acute phase of granulocytic anaplasmosis, the causative organism is visible in peripheral granulocytes and forms ‘bacteria-filled vacuoles’ known as morulae [4, 36].
Like other intracellular organisms, Ap is able to modulate host cell gene expression to favor its own survival. It uses differential gene expression to maintain the transmission cycle between tick vector and vertebrate host [29, 33, 40]. Feeding ticks carrying the organisms release bacteria into surrounding host tissue via salivary secretion. Interaction and invasion of mammalian cells are probably facilitated by salivary factors [20]. Polymorphonuclear leukocytes (PMNs) are recruited to the feeding lesion by pro-inflammatory cytokines, but the events leading to their invasion remain undefined. Adhesion to and infection of human neutrophil granulocytes by Ap during the acute stage of the disease are specifically mediated by tetrasaccharide sialyl Lewisx (sLex or CD15s) on P-selectin glycoprotein ligand 1 (PSGL-1) [19, 22]. However, PMNs do not return to the circulatory system after extravasation into tissue. Consequently, these cells cannot serve as a source for subsequent Ap dissemination in the host. It has been suggested that endothelial cells can serve as reservoirs for the bacterium and to pass them on to PMNs under in vivo conditions. Microvascular endothelial cells probably represent the essential link between infectious Ap organisms and circulating PMNs [31]. Likewise, the closely related agent of bovine heartwater disease, Ehrlichia ruminantium, colonizes microvascular endothelium of the brain and heart in naturally infected ruminants and experimentally infected mice, respectively, as well as neutrophil granulocytes [10, 48]. Furthermore, A. marginale (the agent of bovine anaplasmosis) can infect endothelial cells in vivo [11, 30]. Needless to say, the physiological barrier formed by vascular endothelial cells (ECs), and particularly its breach, is important for the pathogenesis of infections with different representatives of the Anaplasmataceae family. This cell layer regulates the passage of immune molecules and immune cells from blood vessel into surrounding tissue with a complex system of molecules [34]. ECs also serve as important antigen-presenting cells for the immune system [17, 37]. Importantly, due to their access to the lumen of the blood vessels, endothelial cells easily interact with circulating blood cells. We therefore hypothesized that endothelial cells might be a well-suited niche for initial replication or that they could serve as a reservoir for Ap during persistent infection.
Over decades, most in vitro adhesion assays were performed under static conditions to analyze the interaction between ECs and PMNs. Static assays provide valuable information regarding the mechanisms of cell adhesion, but they are clearly limited models to understand adhesive processes in circulating fluids [6, 47]. Transmission of Ap from endothelial cells to PMNs was previously observed under static conditions [21]. However, if this behavior constitutes a key element of disease pathogenesis, it must also function under flow conditions. In this study, an in vitro model was utilized to mimic the microvascular environment at physiological shear stress. The aims of this project were (1) to investigate the adhesion of PMNs to Ap-infected ECs under flow conditions; (2) to evaluate the transmission of Ap between ECs and PMNs under flow conditions; and (3) to analyze the production of cell adhesion molecules and human interleukin-8 secretion by Ap-infected endothelial cells during the infection process.
Materials and methods
Ap culture, propagation and purification
The HL-60 (Human promyelocytic leukemia cells) cell line (ATCC® CCL-240) was obtained from American Type Culture Collection (LGC Standards GmbH, Wesel, Germany) and used to propagate the mCherry-transformed Ap strain HGE1 (mCherry/HGE1) [18]. All experiments described in this manuscript were performed with this Ap organism. Uninfected and infected HL-60 cells were cultured in RPMI-1640 medium (GE Healthcare Europe GmbH, Freiburg, Germany) buffered with 25 mM HEPES, 0.1 % NaHCO3 and supplemented with 10 % heat-inactivated fetal bovine serum (Sigma-Aldrich Chemie GmbH, Munich, Germany), and 2 mM L-Glutamine in a humidified 5 % CO2 atmosphere at 37 °C. Trypan blue (0.5 %) was used to determine cell viability. Giemsa staining was routinely used to check the percentage of Ap-infected cells in the cultures by counting 100 cells per slide using a light microscope (Leica DM5000; Leica Microsystems GmbH, Wetzlar, Germany) [9]. Ap cultures were harvested when ~80 % cells were infected.
Ap were purified from mechanically disrupted host cells. Briefly, infected HL-60 cells (1.0 × 106 or 1.0 × 107 cells) were concentrated in 1.5-ml culture medium in a 2.0-ml sterile tube containing 0.2 ml of autoclaved rock tumbler grit (60/90 grit silicon carbide; Lortone, Inc., Mukilteo, WA, USA). Cell suspensions were vortexed vigorously for 30 s, the grit was allowed to settle, and the supernatants were transferred to a 10-ml Luer lock syringe and passed through a 2.0-μm pore size filter (Puradisc™ 25 GD; GE Healthcare Europe GmbH) into a sterile 2.0-ml tube. Host cell-free Ap were collected by centrifugation at 11,000×g for 5 min at 4 °C. The Ap pellet was washed twice with 1× PBS containing 0.5 % fetal bovine serum and suspended in 150 or 200 μl of cold basic MCDB 131 medium (Life Technologies, Darmstadt, Germany).
Preparation of human PMNs and DMSO-differentiated HL-60 cells (dHL-60)
Heparin anticoagulated human peripheral blood was collected from volunteers who did not show any clinical symptoms of febrile disease. Informed consent was obtained from volunteers prior to acquisition of samples. Human PMNs were isolated by discontinuous density gradient centrifugation with Histopaque-1077 and Histopaque-1119 (Sigma-Aldrich Chemie GmbH), as described elsewhere [16]. After isolation, the PMN pellet was resuspended in pre-equilibrated RPMI-1640 medium (37 °C, 5 % CO2 overnight) and adjusted to 5.0 × 105 cells/ml. The viability and purity of PMNs were determined by trypan blue (0.5 %) stain and Diff-Quick stain, respectively.
HL-60 cells were induced to differentiate into mature granulocytes by incubating the cells in growth medium containing 1.25 % DMSO for 6–7 days as reported [14, 15]. The level of CD11b expression on DMSO-differentiated HL-60 cells was measured by flow cytometry. DMSO-differentiated HL-60 cells were washed three times with 1× PBS prior tested. PMNs and DMSO-differentiated HL-60 cells were labeled with 20 μM CellTracker Green CMFDA dye (Life Technologies) at 37 °C for 30 min and then washed three times before they were used in transmission assays with endothelial cells.
Endothelial cell cultures under static and flow conditions
Two types of human microvascular endothelial cells were used in this study. Firstly, HMEC-1 cells (a human micro-vascular endothelial cell line) [1] were used between passages 20 to 35 and cultured in 25 mM HEPES and 0.25 % NaHCO3-buffered MCDB 131 medium (pH 7.5) (Life Technologies) supplemented with 10 mM L-glutamine, 10 % heat-inactivated fetal bovine serum, 1.0 μg/ml hydro-cortisone (all from Sigma-Aldrich Chemie GmbH) and 10 ng/ml epidermal growth factor (BD Biosciences, San Jose, CA, USA) in a humidified atmosphere with 5 % CO2 in air at 37 °C. Secondly, primary HDMEC cells (human dermal microvascular endothelial cells) were purchased from PromoCell GmbH, Heidelberg, Germany, and cultured in Endothelial Cell Growth Medium 2 (ECGM2; PromoCell GmbH) according to instructions. The manufacturer had confirmed the presence of endothelial cell-specific markers (e.g., vWF, CD31). Endothelial cells were detached with trypsin/EDTA (0.5 mg/ml and 0.22 mg/ml) and subcultured into new flasks at a density of 5.0 × 104–1.0 × 105 viable cells/ml. Cell viability was assessed with 0.5 % trypan blue.
For cultivation of endothelial cells under flow conditions, a pump culture system to generate a controlled unidirectional shear flow was applied (ibidi GmbH, Martinsried, Germany). All devices were assembled according to the manufacturer’s instructions and controlled by the Pump-Control software (version 1.5.0) to generate a laminar flow at defined shear stress in the channel slide. Briefly, 2 × 105 of HMEC-1 or HDMEC cells were seeded into a channel slide (μ-Slide l 0.6 Luer; ibidi GmbH) and incubated at 37 °C in a humidified atmosphere of air with 5 % CO2 for 2.5 h to allow attachment prior to connection with the perfusion set. Endothelial cells were further grown overnight at a shear stress of 2.0 dyne/cm2 and reached confluence after 24 to 48 h, which was suitable for infection or flow experiments.
Exposure of endothelial cells to Ap
Optimal infection time for Ap with endothelial cells under static conditions
To exclude the influence of hydrocortisone in MCDB 131 growth medium on endothelial cells adhesion expression, HMEC-1 endothelial cell layers were washed and cultured in hydrocortisone-free MCDB 131 growth medium in all infection assays in this study. At the beginning of this study, the optimal conditions to obtain low-level or high-level Ap infections of endothelial cell monolayers were assessed within a fixed time period under static conditions. HMEC-1 cells or HDMEC cells were seeded into a 4-well cell culture chamber (Sarstedt, Nümbrecht, Germany) and grown to 70–80 % confluency. Cell-free bacteria purified from 1.0 × 106 of infected HL-60 cells were then incubated with 2.0 × 105 HMEC-1 cells or HDMEC cells at a multiplicity of infection of 5:1 (‘MOI’ refers to the ratio of the number of Ap-infected HL-60 cells to the number of uninfected endothelial cells) for 24, 48 and 96 h in a humidified atmosphere with 5 % CO2 in air at 37 °C. After removal of unbound bacteria with three 1× PBS washes, HMEC-1 or HDMEC cell layers were subjected to Giemsa staining and immunofluorescence in situ.
Preparation of Ap-infected HMEC-1 cells for adhesion assay under flow conditions
For the adhesion assay, 2 × 105 HMEC-1 cells in channel slides were incubated with cell-free Ap purified from 2 × 105 or 1 × 106 (MOI of 1:1 or 5:1) infected HL-60 cells for 24 h. As a positive control, human TNF-α (100 ng/ml rh TNF-α; R&D Systems, Inc., Minneapolis, MN, USA), a strong stimulator of cell adhesion molecules expression, was used to stimulate HMEC-1 cell monolayers for at least 8 h [27]. HMEC-1 cell monolayers inoculated with cell lysates prepared from uninfected HL-60 cells by using the same purification procedure as described under ‘Ap culture, propagation and purification’ served as negative controls. HMEC-1 cells in slides for flow experiments were incubated in a humidified atmosphere of air with 5 % CO2 at 37 °C. After 24 h, all slides were washed with pre-equilibrated hydrocortisone-free MCDB 131 medium (at 37 °C, 5 % CO2 in air, overnight) and were then ready to be used for the adhesion assay under flow conditions.
PMN adhesion to Ap-infected HMEC-1 cell monolayer under flow conditions
Ap-infected HMEC-1 cell monolayers were prepared as described above. Similar experiments have been conducted under static conditions by one of the coauthors (UGM) [21]. The experiments described here, however, focused on PMN adhesion under flow conditions. A total volume of 8.0 ml RPMI-1640 medium containing 5 × 105 human PMNs/ml was added to the perfusion set, and PMNs were perfused over Ap-infected HMEC-1 cell monolayers at shear stresses ranging from 0.5 to 2.0 dyne/cm2. Positive and negative controls (see exposure of endothelial cells to Ap, part b) were handled in the same way.
The slides were placed in a heating chamber at constant 37 °C, which was controlled by a Temperature Controller (version 1.0.2; ibidi GmbH), and interactions were visualized using an inverted phase-contrast microscope equipped with a digital video camera (Leica Microsystems GmbH). Adherent PMNs were determined after 10 min of perfusion by photographing 10 randomly selected fields at 7× magnification. The numbers of adherent PMNs were counted using the software Image J (National Institute of Health, Bethesda, MD, USA), and the means values were calculated.
HMEC-1 cells in infection assays under static conditions with varying doses of Ap
Infection assays with varying doses of Ap were performed to verify dose-dependent effects of Ap organisms on cell adhesion molecule expression. A confluent monolayer of HMEC-1 in a 6-well tissue culture plate (approximately 8.0 × 105 cells/well) was incubated with different doses of Ap under static conditions. Ap were purified from 1.6 × 107 infected HL-60 cells and suspended in 200 μl of basic endothelial medium. Twofold serial dilutions of purified Ap were prepared and added to each well in order to obtain an MOI at 0.07:1–10:1. After 24 h, culture supernatants were collected and stored at −20 °C for IL-8 detection. ICAM-1 and VCAM-1 expression on HMEC-1 cells and the infection ratio of HMEC-1 cells were analyzed by flow cytometry.
Ap-transmission from endothelial cells to granulocytes
Primary HDMEC cells (PromoCell GmbH) were used in the Ap-transmission assay in order to maximally reflect physiological functions of endothelial cells. Ap-infected HDMEC cells were prepared as described for HMEC-1 cells under ‘exposure of endothelial cells to Ap, part b’ at an MOI of 1:1 or 5:1 for 24 h prior to the transmission assay. As negative control, uninfected HDMEC cells were incubated with lysates of uninfected HL-60 cells. The infection rates of HDMEC cells were measured by flow cytometry. Subsequently, 1.6 × 106 of uninfected human PMNs or DMSO-differentiated HL-60 cells (dHL-60 cells) in 8 ml of RPMI-1640 medium were added and co-cultured with uninfected (negative control) or Ap-infected HDMEC cells at 0.5 dyne/cm2.
At the time points indicated (e.g., 1 h, 3.5 h, 4.5 h, 24 h, 3 days, 5 days, 7 days), 100 μl of suspended cells was harvested and subjected to fluorescence microscopy, Giemsa staining and immunofluorescence in order to detect Ap infection in PMNs or DMSO-differentiated HL-60 cells.
Immunofluorescence analysis of Ap infection
Immunofluorescence was used to visualize Ap infection in endothelial cells, PMNs and DMSO-differentiated HL-60 cells (dHL-60 cells). PMNs and dHL-60 cells were deposited on glass slides by centrifugation prior to fixation. Endothelial cell monolayers were gently washed twice with ice-cold Dulbecco’s phosphate-buffered saline. Fixation was carried out with 2 % paraformaldehyde (PFA, in 1× PBS) at room temperature for 10 min. PMNs, dHL-60 or endothelial cell monolayers were permeabilized with 0.3 % (v/v) Triton X-100 for 10 min at room temperature followed by three 10-min washes with 1× Dulbecco’s PBS [13]. Non-specific binding sites were blocked using 5 % (w/v) bovine serum albumin (BSA) overnight at 4 °C followed by incubation with dog anti-Ap serum (diluted 1:200, pooled serum from naturally Ap-infected dogs) in 1× PBS with 1 % BSA for 1 h at room temperature. Afterwards, cells were washed three times and incubated with FITC-conjugated anti-dog serum (1:500; KPL, Inc., Gaithersburg, MD, USA) for 1 h at room temperature. After three washes with 1× Dulbec-co’s PBS, the cell nuclei were counterstained with DAPI nucleic acid stain (Life Technologies). Visualization was performed with a fluorescence microscope equipped with band-pass filters specific for DAPI, FITC and mCherry fluorophores, respectively (Leica Microsystems GmbH).
Cell adhesion molecule expression on HMEC-1 induced by Ap infection under flow and static conditions
Flow cytometric analysis was performed to detect inter-cellular cell adhesion molecule-1 (ICAM-1, CD54) and vascular cell adhesion molecule-1 (VCAM-1, CD106) expression on HMEC-1 cells in adhesion assays (flow) and dose-dependent infection assays (static). After 24 h of incubation, uninfected and infected HMEC-1 cell monolayers were washed twice with 1× Dulbecco’s PBS before enzymatic dissociation using Accutase™ (GE Healthcare Europe GmbH) at 37 °C for 5–10 min. Twenty microliters of normal human serum was added to 80 μl of cell suspensions (5 × 105–1 × 106 cells) in staining buffer (1× PBS containing 0.5–1.0 % (w/v) BSA and 0.09 % NaN3 at pH 7.2) followed by incubation on ice for 20 min. Cell suspensions were then washed twice with staining buffer at 4 °C and 350×g for 5 min. Afterwards, 2 μl of undiluted FITC antihuman CD54 (clone: HCD54; BioLegend, Inc., London, UK) and 2 μl of undiluted phycoerythrin (PE)-conjugated anti-human CD106 (clone: STA; BioLegend, Inc.) were added to 100 μl cell suspensions in staining buffer and incubated on ice for 15–20 min in the dark, followed by washing twice with ice-cold 1× PBS. Corresponding fluorochrome-labeled mouse IgG1 and κ isotype-matched control antibodies (FITC or PE-labeled; BioLegend, Inc.) were used to assess the level of background staining in cell–antibody binding. Dead cells were excluded with a fixable viability dye eFluor® 450 (eBioscience, Frankfurt am Main, Germany) using a violet laser (405 nm). After washing with staining buffer once, cells were fixed with ice-cold 2 % PFA (pH 7.2) before data acquisition in a MACS-Quant® VYB (Miltenyi Biotec GmbH, Bergisch-Gladbach, Germany). The infection ratio of HMEC-1 cells was evaluated by detecting mCherry-positive cells by flow cytometry. Ten thousand events were acquired and analyzed using FlowJo software (FlowJo, LLC., Ashland, OR, USA).
Measurement of interleukin-8 (IL-8) secretion by HMEC-1 cells infected with Ap
Culture supernatants of HMEC-1 cells infected with different doses of Ap as described under ‘exposure of endothelial cells to Ap’ were collected and stored at −20 °C. The concentration of IL-8 in the supernatants was measured with a commercial sandwich ELISA kit (Pierce Biotechnology, Inc., Rockford, IL, USA) according to the instructions of the manufacturer. Each sample was measured in triplicate.
Statistical analysis
All statistical analyses were carried out using un-paired Student’s t test with GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA). Data were represented as the means ± SE. Mean differences between the groups were considered statistically significant and highly significant at a p value of <0.05 (*) and <0.01 (**), respectively.
Results
Optimal time period for Ap to infect endothelial cells under static conditions
Microscopic evaluation revealed that Ap was able to invade endothelial cells in vitro within 24 h of incubation (Fig. 1a, b). Between 60 and 80 % of HMEC-1 cells were infected after incubation with isolated Ap at an MOI of 5:1 when evaluated after 24 h. Classical inclusions, so-called ‘morulae’, were observed at 24, 48 and 96 h in the cytoplasm of Giemsa-stained HMEC-1 cells (Fig. 1a). The fraction of infected cells decreased to 20–30 % at 48 and 96 h p.i. The 24-h incubation period was used for further experiments.
Fig. 1.

Detection of Ap infection in endothelial cells by Giemsa staining and immunofluorescence. a Giemsa-stained Ap-infected HMEC-1 cell monolayers. HMEC-1 cells were incubated with isolated Ap at an MOI of 5:1 for 24, 48 and 96 h. Morulae in the cytoplasm of HMEC-1 cells are indicated with black arrows. b Immunofluorescence staining of Ap-infected HMEC-1 and HDMEC. The HMEC-1 or HDMEC cells were incubated with isolated Ap at an MOI of 5:1. After 24 h, cells were fixed with 2 % PFA and permeabilized with 0.3 % Triton X-100 in 1× PBS. Dog anti-Ap serum was added; this was followed by incubation with FITC-conjugated anti-dog serum. The cell nuclei were visualized by DAPI-staining (blue). Fluorescence microscopy was carried out using band-pass filters specific for FITC, mCherry and DAPI. Scale bar 20 μm. Ap: mCherry/HGE1 (color figure online)
In order to verify the observations obtained with Giemsa staining, immunofluorescence was used to assess Ap infection in HMEC-1 cells after 24 h of incubation (Fig. 1b). Morulae varying in size were visible around cell nuclei of HMEC-1 cells (Fig. 1b, merge-panel). Epifluorescence microscopy was used to visualize mCherry-labeled Ap (Fig. 1b, mCherry-panel), and results were compared to those found with immunofluorescence staining (Fig. 1b, FITC-panel). Comparable results were obtained with primary HDEMC cells (Fig. 1b).
PMN adhesion to Ap-infected HMEC-1 cell monolayers under flow conditions
In the flow culture system, HMEC-1 cells formed a confluent monolayer after overnight growth at a shear stress of 2.0 dyne/cm2 (Fig. 2a, ECs). Infected HMEC-1 cells modified their morphology presenting enlarged cells or vacuolated cells; however, infected cells remained attached under shear stress (Fig. 2a, infected ECs). Harvested HMEC-1 cells analyzed with flow cytometry showed that 5.1 and 87.0 % of HMEC-1 cells carried Ap when exposed to the organisms at an MOI of 1:1 and 5:1, respectively (Fig. 2c). No tight adhesion of PMNs to the HMEC-1 cell monolayers was observed at 2.0 dyne/cm2. When the shear stress was adjusted to 0.5 dyne/cm2, tight adhesion of PMNs was observed on both Ap-infected HMEC-1 cell monolayers and TNF-α (100 ng/ml) stimulated monolayers. A shear stress of 0.5 dyne/cm2 was used for further experiments.
Fig. 2.
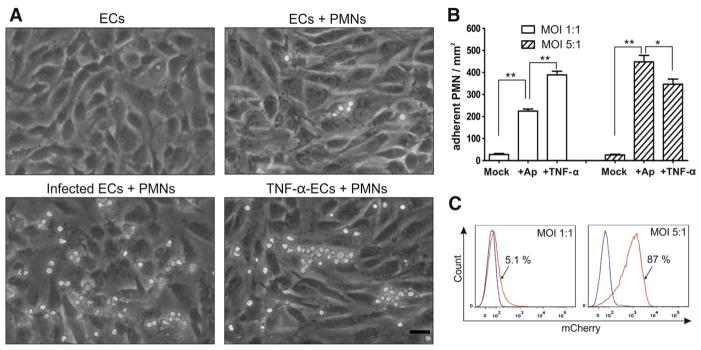
PMN adhesion to Ap-infected HMEC-1 cell monolayers. 4 × 106 PMNs isolated from volunteers in 8 ml medium was added to uninfected or Ap-infected HMEC-1 cells or to HMEC-1 cells treated with 100 ng/ml TNF-α and then perfused for 10 min at 0.5 dyne/cm2. Experiments were performed five times. a PMN adhesion after 10-min perfusion at 0.5 dyne/cm2. Scale bar 25 μm. b Number of adherent PMNs per mm2 on endothelial monolayer; means ± SE of three representative experiments are shown; * and ** indicate statistically significant differences: p < 0.05 and p < 0.01; Student’s t test. c Flow cytometry analysis of Ap infection ratio in endothelial cells. HMEC-1 cells exposed to Ap at an MOI of 1:1 (left panel) and 5:1 (right panel) and cultured for 24 h were harvested and subjected to Ap detection (fluorescence mCherry positive). A fraction of 5.1 and 87 % of HMEC-1 cells visible in the red spectrum in both panels were shown to be infected. Blue lines show the fraction of uninfected cells. Representative results of one out of three experiments are shown. ECs endothelial cells; Ap: mCherry/HGE1 (color figure online)
Within 10 min of interaction, Ap infection of HMEC-1 cells (MOI of 1:1) significantly enhanced PMNs adhesion (224.9 ± 9.4 PMNs/mm2, Fig. 2b) to the cells when compared to uninfected HMEC-1 cells (27.9 ± 4.5 PMNs/mm2, p < 0.01). Many more PMNs adhering to high-level infected HMEC-1 cells (MOI of 5:1) were observed (448.3 ± 29.5 PMNs/mm2, Fig. 2b, p < 0.01). PMN interaction with infected HMEC-1 (MOI of 5:1) was significantly more prominent than observed with cells stimulated with TNF-α (346.5 ± 23.8 PMNs/mm2, Fig. 2b, p < 0.01).
Ap-transmission from endothelial cells to granulocytes
In order to maximally reflect physiological functions of endothelial cells, primary HDMEC cells were used in the transmission assay. Two HDMEC cell populations infected with varying doses of Ap were investigated under flow conditions. Before the Ap-transmission assay was initiated, flow cytometric analysis showed that approximately 10 % (low-level infection) or 70 % (high-level infection) of HDMEC cells carried Ap when exposed to the bacterium at an MOI of 1:1 or 5:1, respectively (data not shown).
PMN adhesion to the HDMEC monolayer was observed microscopically after leukocytes were added to the chamber slide. Less than 1 % of the PMNs isolated from volunteers were infected with Ap after 4.5 h co-culture with Ap-infected HDMEC cells under flow conditions (Fig. 3a, left panel). Interestingly, additional infected PMNs were not observed for up to 24 h, and PMNs isolated from volunteers lysed quickly within 24 h under flow conditions. This compares well to the reported half-life of 7 h for PMNs in peripheral blood [38].
Fig. 3.

Ap transmission from infected endothelial cells to PMNs and DMSO-differentiated HL-60 cells. dHL-60 cells were prepared by incubating the cells in the presence of 1.25 % DMSO in growth medium for 6–7 days. 1.6 × 106 CMFDA-labeled or CMFDA-unlabeled PMNs or dHL-60 cells in 8 ml of RPMI-1640 medium were added and co-cultured with uninfected or infected HDMEC cells (at 10 or 70 % infection level) at 0.5 dyne/cm2. At time points indicated, PMNs or dHL-60 cells were harvested and subjected to fluorescence microscopy, immunofluorescence assay and Giemsa staining. a Representative fluorescence microscopy images for PMNs (4.5 h) and dHL-60 cells (24 h) from the high-level infection group (HDMEC cells at 70 % infection level) after co-culture with Ap-infected HDMEC cells. b Flow cytometric analysis of CD11b expression on DMSO-differentiated HL-60 cells (dHL-60 cells). Undifferentiated and differentiated HL-60 cells were washed and stained with FITC-conjugated anti-human CD11b antibody (blue and orange line) or FITC-conjugated mouse IgG1 (black line). c Images of Giemsa-stained dHL-60 cells in the high-level infection group at Day 3 and Day 5. d Proportions of Ap-infected dHL-60 cells at Days 0, 3, 5 and 7. Results of one of two independent experiments are shown. e Representative micrographs of immunofluorescence stained dHL-60 cells from the high-level infection group at Day 3 and Day 5. Scale bar 20 μm (color figure online)
DMSO-differentiated HL-60 cells (dHL-60 cells) mimicking functional PMNs [14, 15] were therefore used for additional co-culture experiments involving Ap-infected HDMEC-1 cells. Figure 3b shows that 92 % of all HL-60 cells expressed CD11b on their surface for 7 days in the presence of 1.25 % DMSO. After co-culture of endothelial and dHL-60 cells for 1 or 3.5 h, no morulae were observed in dHL-60 cells by Giemsa staining and fluorescence microscopy. After prolonged co-culture for 24 h, however, less than 1 % of dHL-60 cells interacting with the high-level infected HDMEC cells carried Ap (Fig. 3a, right panel). Two days later (day 3 after exposure to Ap), approximately 10 % of dHL-60 cells and at day 5, 80 % of the cells were infected with the bacterium (Fig. 3c). Most of dHL-60 cells were lysed by day 7. Comparable results were obtained when a HDMEC population was used as an Ap source and the bacteria were available at a low level (10 % infection rate in HDMEC cells; Fig. 3d). Immunofluorescence analysis was performed in addition to verify results seen after Giemsa staining (Fig. 3e).
Cell adhesion molecule expression on HMEC-1 cells induced by Ap infection: analyses under flow and static conditions
ICAM-1 and VCAM-1 expressions on HMEC-1 cells were evaluated under flow conditions and under static conditions. After 10 min of interaction with PMNs under flow conditions, high-level infected HMEC-1 cells (87.0 %) demonstrated a significant up-regulation of ICAM-1 from 24.7 % (baseline level expression of HMEC-1) to 91.7 % (Fig. 4a, ECs + Ap). In comparison, a slight increase to 37.7 % was observed when only 5.1 % of HMEC-1 cells were infected with Ap (Fig. 4a, ECs + Ap). Almost 100 % of HMEC-1 cells were capable of expressing ICAM-1 in the presence of 100 ng/ml TNF-α (positive control) after 24-h incubation (Fig. 4a, ECs + TNF-α). In contrast to the baseline level of ICAM-1 expression, VCAM-1 was not constitutively expressed on the HMEC-1 cells. Moreover, VCAM-1 expression was only inducible on HMEC-1 cells with a high Ap infection rate (Fig. 4a, ECs + Ap). A fraction of HMEC-1 cells (30.5 or 56.6 %) was capable of expressing VCAM-1 after stimulation with 100 ng/ml TNF-α for 24 h (Fig. 4a, ECs + TNF-α).
Fig. 4.

ICAM-1 and VCAM-1 expression on HMEC-1 cells. A Flow cytometry analysis of ICAM-1 and VCAM-1 expression on HMEC-1 cells after interaction with PMNs under flow conditions. Uninfected, TNF-α (100 ng/ml) treated and Ap-infected HMEC-1 cells (5.1, 87.0 % infection rate) that were co-cultured with PMNs (5 × 105) at 0.5 dyne/cm2 for 10 min were harvested and subjected to flow cytometric analysis. Results of one of three representative flow experiments are shown. B Expression of ICAM-1 and VCAM-1 on HMEC-1 cells by Ap infection under static conditions as function of the infection dose. Surface expressions are calculated as the percentages of positive cells in the gated cell population. Means ± SE from one of three independent experiments are shown. Each measurement was performed in triplicate. The infection ratios of HMEC-1 cells were determined by flow cytometry using mCherry-labeled Ap. * and ** indicate statistically significant differences: p < 0.05 and p < 0.01; Student’s t test (a: ‘group 0.6:1’ vs. ‘group HMEC-1 + HL-60’; b: any of ‘group 1.3:1, 2.5:1, 5:1 and 10:1’ vs. ‘group HMEC-1 + HL-60’). Ap: mCherry/HGE1. ICAM-1: intercellular cell adhesion molecule 1; VCAM-1: vascular cell adhesion molecule 1
Results of experiments under flow conditions showed that the number of Ap in the culture influenced ICAM-1 and VCAM-1 expression in a dose-dependent manner. To verify this effect, both ICAM-1 and VCAM-1 expressions were repeatedly examined under static conditions in a less complex in vitro system. As shown in Fig. 4b, ICAM-1 expression was induced in a dose-dependent way on infected HMEC-1 cells when the infection ratio was increased (Fig. 4b, left panel). Similar results were obtained for VCAM-1 (Fig. 4b, right panel). The addition of HL-60 cell lysate had no effect on ICAM-1 and VCAM-1 expression when compared to untreated HMEC-1 cells (Fig. 4b, column HL-60 cell lysate).
Measurement of human IL-8 secretion by HMEC-1 cells infected with Ap
To verify whether IL-8 was released by endothelial cells, the concentration of IL-8 in the culture supernatants was assessed. ELISA results showed that secretion of IL-8 by HMEC-1 cells was dose-dependent and increased with the numbers of Ap organisms in the culture (Fig. 5). Again, the addition of HL-60 cell lysate had no effect on IL-8 secretion (184.3 ± 12.6 ng/ml, p > 0.05) when compared to untreated HMEC-1 cells (244.8 ± 58.9 ng/ml). When the multiplicity of infection was raised to 10:1, an extremely high level of IL-8 was detected in the supernatants (1,264.1 ± 377.2 ng/ml, p < 0.01) compared to untreated HMEC-1 cells. However, the level of IL-8 secretion by Ap-infected HMEC-1 cells was significantly lower than that observed after TNF-α stimulation (3,473.1 ± 539.9 ng/ml, p < 0.05).
Fig. 5.

Induction of IL-8 secretion by endothelial cells in response to Ap infection IL-8 concentration measured after 24 h exposure of HMEC-1 cells to different doses of Ap under static conditions. Each sample was measured in triplicate, and data are shown as means ± SE. *p < 0.05 and **p < 0.01 indicate statistically significant differences; Student’s t test
Discussion
Besides PMNs, microvascular endothelial cells have been shown to be susceptible target cells for Ap infection in vitro [21, 31]. However, the role of microvascular endothelial cells in the pathogenesis, especially during the initial transmission period of Ap infection, is not fully understood. It is unknown how Ap migrates from tick attachment sites to peripheral blood granulocytes, which is the most commonly detected host cell type in vertebrates. The reason for the lack of information might be (1) due to Ap’s low-abundance in endothelial cells during the early stages of the infection; consequently, Ap is difficult to detect with conventional microscopic methods in clinical samples; thus, endothelial cells as a source for Ap dissemination have remained undetected, (2) and due to the lack of suitable in vitro models that allow a precise dissection of the interactions necessary to transfer Ap from endothelial cells to circulating PMNs.
In this study, a cell culture system mimicking physiological shear flow was established in order to explore the likely natural interaction between Ap-infected endothelial cells and PMNs as well as to analyze expression of human IL-8 and two cell adhesion molecules (ICAM-1 and VCAM-1) that are main contributors to the tight adhesion of PMNs in vivo [26, 39]. Using this flow culture system, different flow rates were applied to generate defined shear stress ranging from 0.5 to 2 dyne/cm2. Interactions with endothelial cells such as rolling and tight adhesion of PMNs were clearly visible at the lowest chosen shear stress of 0.5 dyne/cm2 (Fig. 2). Almost no tight adhesion was observed at 2.0 dyne/cm2. Shear forces in the blood vessels should be considered when the interaction between Ap, endothelial cells and PMNs is investigated, as the forces exerted by moving fluids are capable of modulating cytoskeletal rearrangement, cell morphology and gene expression in cells as well as influencing leukocyte-endothelial adhesion [12, 44]. Typical values of shear stress in human blood vessels are in the range of 0.1–13 dyne/cm2 depending on the type of blood vessel, e.g., large arteries, small arteries or, as in our case, capillaries [35].
Our transmission assays showed that Ap uptake by circulating PMNs occurred within 4.5 h after the addition of PMNs to the endothelial cell culture (Fig. 3b). Additional infected cells, however, were not observed in the following 19.5 h when PMNs isolated from volunteers were used. These results reflect the low number of infected PMNs seen in human patients. In order to obtain longer culture periods, dHL-60 cells, which have been shown to resemble mature neutrophils [14, 15], were used. The results of these transmission assays showed that Ap organisms transfer to circulating granulocytes from infected endothelial cells and finally establish infections in circulating, moving granulocytes under flow culture conditions. Infection in dHL-60 cells was detectable after 3 days, and subsequently, the infection level increased during the following 4 days of co-culture with infected endothelial cells (Fig. 3d). Given that the replication cycle of Ap is roughly 24 h [43], it is likely that the bacteria replicated in infected host cells (endothelial cells or dHL-60 cells) and that released Ap organisms were the source for further ongoing infection of host cells. Interestingly, studies in sheep have revealed that each cycle of bacteremia is followed by a period of a few days in which Ap cannot be detected in blood [41]. This might indicate that the bacterium resides in tissue-bound cells only to return to the peripheral blood under optimal conditions. In this context, microvascular endothelial cells could serve as a ‘safe-haven’ for Ap during the early or chronic phase of the persistent infection.
Adhesion of circulating leukocytes to endothelial cells is an essential process of inflammatory responses of the innate immune system [26], which is mediated by specific endothelial-leukocyte adhesion molecules including selectins (e.g., P- and E-selectin), integrins (e.g., LFA-1, Mac-1) and the immunoglobulin superfamily (e.g., ICAM-1, VCAM-1 and PECAM-1) resulting in capture, rolling, tight adhesion of leukocytes, passage across the endothelial wall and subsequent migration to the inflammatory site [23, 26, 28]. The up-regulation of cell adhesion molecules on endothelial cells allows functional PMNs to reach the site of infection and exert their critical functions to eliminate foreign agents by phagocytic and cytotoxic activities [32, 45]. The results of this study showed that infected microvascular endothelial cells that up-regulated ICAM-1 and VCAM-1 molecules enabled PMNs to adhere to the endothelium under flow conditions. Furthermore, ICAM-1 and VCAM-1 up-regulation on HMEC-1 cells was induced by Ap infection in a dose-dependent manner and not by cell debris of other cultured cells. This suggests that endothelial cells can be activated by Ap organisms and that this process probably is the first step of an inflammatory response, which results in severe damage of the endothelial lining seen after infections with several rickettsial organisms [43].
IL-8, also known as neutrophil chemotactic factor (NCF), is a member of the CXC chemokine family. It attracts and activates neutrophils in inflammatory response [25]. A previous study demonstrated that IL-8 secretion is inducible in human neutrophils by Ap infection [2]. Our results showed a similar effect: Ap induced a substantial IL-8 secretion in cultured endothelial cells (Fig. 5). This indicated a strong inflammatory stimulation of endothelial cells triggered by Ap infection and might also be associated with tissue damage seen after infections with other rickettsial organisms [5, 24].
It is worth noting that the Ap load in HMEC-1 cells varied considerably in our experiments even when the same infection dose was used to infect the endothelial cells (Figs. 2c vs. 4b). Only dense-cored cell forms of Ap (DCs) are infectious for HL-60 cells as shown previously [42]. For our experiments, we determined the number of infected HL-60 cells prior to Ap purification. Because of the varying numbers of Ap organisms present in a single HL-60 cell, we assumed that the variability seen in our experiments was likely due to the varying numbers of infectious DCs found in different batches of infected HL-60 cell cultures.
In conclusion, data obtained with this in vitro model demonstrated that granulocytes strongly adhered to Ap-infected endothelial cells under flow conditions at shear stress of 0.5 dyne/cm2. The close proximity of endothelial cells and PMNs resulting from the adhesion may further facilitate the transfer of Ap from endothelial cells to circulatory granulocytes. Up-regulations of the intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) on endothelial cells and the secretion of IL-8 by endothelial cells were induced by Ap infection in a dose-dependent manner. Here, we showed for the first time that transmission of Ap from microvascular endothelial cells to granulocytes occurred under flow conditions.
Acknowledgments
The authors thank Anke Schiller and Stephanie Hiereth for their assistance in cell and bacterial culture; Dr. Silke Kadlez-Gebhardt, Dr. Winfried Kapfhammer and Dr. Vollert Lüthje for collecting blood samples from our donors; and Dr. Sonja Härtle for her useful discussion about flow cytometric data analysis. The authors thank Dr. Evelyn Overzier and Dr. Anna Rettinger for their careful reading of our manuscript. J.W. is a research fellow of the China Scholarship Council (CSC).
Contributor Information
Jinyong Wang, Institute for Infectious Diseases and Zoonoses, Department of Veterinary Sciences, Faculty of Veterinary Medicine, LMU Munich, Veterinärstr. 13, 80539 Munich, Germany.
Viktor Dyachenko, Institute for Infectious Diseases and Zoonoses, Department of Veterinary Sciences, Faculty of Veterinary Medicine, LMU Munich, Veterinärstr. 13, 80539 Munich, Germany.
Ulrike G. Munderloh, Department of Entomology, University of Minnesota, St. Paul, MN 55108, USA
Reinhard K. Straubinger, Email: R.Straubinger@lmu.de, Institute for Infectious Diseases and Zoonoses, Department of Veterinary Sciences, Faculty of Veterinary Medicine, LMU Munich, Veterinärstr. 13, 80539 Munich, Germany
References
- 1.Ades EW, Candal FJ, Swerlick RA, et al. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 2.Akkoyunlu M, Malawista SE, Anguita J, et al. Exploitation of interleukin-8-induced neutrophil chemotaxis by the agent of human granulocytic ehrlichiosis. Infect Immun. 2001;69:5577–5588. doi: 10.1128/IAI.69.9.5577-5588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnez M, Petrovec M, Lotric-Furlan S, et al. First European pediatric case of human granulocytic ehrlichiosis. J Clin Microbiol. 2001;39:4591–4592. doi: 10.1128/JCM.39.12.4591-4592.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakken JS, Dumler S. Human granulocytic anaplasmosis. Infect Dis Clin N Am. 2008;22:433–448. doi: 10.1016/j.idc.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Bechah Y, Capo C, Raoult D, et al. Infection of endothelial cells with virulent Rickettsia prowazekii increases the transmigration of leukocytes. J Infect Dis. 2008;197:142–147. doi: 10.1086/523649. [DOI] [PubMed] [Google Scholar]
- 6.Bevilacqua MP, Pober JS, Wheeler ME, et al. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest. 1985;76:2003–2011. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjoersdorff A, Bagert B, Massung RF, et al. Isolation and characterization of two European strains of Ehrlichia phagocytophila of equine origin. Clin Diagn Lab Immunol. 2002;9:341–343. doi: 10.1128/CDLI.9.2.341-343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjoersdorff A, Svendenius L, Owens JH, et al. Feline granulocytic ehrlichiosis–a report of a new clinical entity and characterisation of the infectious agent. J Small Anim Pract. 1999;40:20–24. doi: 10.1111/j.1748-5827.1999.tb03249.x. [DOI] [PubMed] [Google Scholar]
- 9.Borjesson DL. Culture, isolation, and labeling of Anaplasma phagocytophilum for subsequent infection of human neutrophils. Methods Mol Biol. 2008;431:159–171. doi: 10.1007/978-1-60327-032-8_13. [DOI] [PubMed] [Google Scholar]
- 10.Brown CC, Skowronek AJ. Histologic and immunochemical study of the pathogenesis of heartwater (Cowdria ruminantium infection) in goats and mice. Am J Vet Res. 1990;51:1476–1480. [PubMed] [Google Scholar]
- 11.Carreno AD, Alleman AR, Barbet AF, et al. In vivo endothelial cell infection by Anaplasma marginale. Vet Pathol. 2007;44:116–118. doi: 10.1354/vp.44-1-116. [DOI] [PubMed] [Google Scholar]
- 12.Chiu JJ, Lee PL, Chang SF, et al. Shear stress regulates gene expression in vascular endothelial cells in response to tumor necrosis factor-alpha: a study of the transcription profile with complementary DNA microarray. J Biomed Sci. 2005;12:481–502. doi: 10.1007/s11373-005-4338-4. [DOI] [PubMed] [Google Scholar]
- 13.Coling D, Kachar B. Principles and application of fluorescence microscopy. Curr Protoc Mol Biol Unit. 2001;14:10. doi: 10.1002/0471142727.mb1410s44. [DOI] [PubMed] [Google Scholar]
- 14.Collins SJ, Ruscetti FW, Gallagher RE, et al. Normal functional characteristics of cultured human promyelocytic leukemia cells (HL-60) after induction of differentiation by dimethylsulfoxide. J Exp Med. 1979;149:969–974. doi: 10.1084/jem.149.4.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins SJ, Ruscetti FW, Gallagher RE, et al. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc Natl Acad Sci USA. 1978;75:2458–2462. doi: 10.1073/pnas.75.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.English D, Andersen BR. Single-step separation of red blood cells. Granulocytes and mononuclear leukocytes on discontinuous density gradients of Ficoll-Hypaque. J Immunol Methods. 1974;5:249–252. doi: 10.1016/0022-1759(74)90109-4. [DOI] [PubMed] [Google Scholar]
- 17.Epperson DE, Pober JS. Antigen-presenting function of human endothelial cells. Direct activation of resting CD8 T cells. J Immunol. 1994;153:5402–5412. [PubMed] [Google Scholar]
- 18.Felsheim RF, Herron MJ, Nelson CM, et al. Transformation of Anaplasma phagocytophilum. BMC Biotechnol. 2006;6:42. doi: 10.1186/1472-6750-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodman JL, Nelson CM, Klein MB, et al. Leukocyte infection by the granulocytic ehrlichiosis agent is linked to expression of a selectin ligand. J Clin Invest. 1999;103:407–412. doi: 10.1172/JCI4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Granquist EG, Aleksandersen M, Bergstrom K, et al. A morphological and molecular study of Anaplasma phagocytophilum transmission events at the time of Ixodes ricinus tick bite. Acta Vet Scand. 2010;52:43. doi: 10.1186/1751-0147-52-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herron MJ, Ericson ME, Kurtti TJ, et al. The interactions of Anaplasma phagocytophilum, endothelial cells, and human neutrophils. Ann N Y Acad Sci. 2005;1063:374–382. doi: 10.1196/annals.1355.090. [DOI] [PubMed] [Google Scholar]
- 22.Herron MJ, Nelson CM, Larson J, et al. Intracellular parasitism by the human granulocytic ehrlichiosis bacterium through the P-selectin ligand, PSGL-1. Science. 2000;288:1653–1656. doi: 10.1126/science.288.5471.1653. [DOI] [PubMed] [Google Scholar]
- 23.Herter J, Zarbock A. Integrin regulation during leukocyte recruitment. J Immunol. 2013;190:4451–4457. doi: 10.4049/jimmunol.1203179. [DOI] [PubMed] [Google Scholar]
- 24.Kaplanski G, Teysseire N, Farnarier C, et al. IL-6 and IL-8 production from cultured human endothelial cells stimulated by infection with Rickettsia conorii via a cell-associated IL-1 alpha-dependent pathway. J Clin Invest. 1995;96:2839–2844. doi: 10.1172/JCI118354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008;13:2400–2407. doi: 10.2741/2853. [DOI] [PubMed] [Google Scholar]
- 26.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 27.Kucik DF. Measurement of adhesion under flow conditions. Curr Protoc Cell Biol Unit. 2009;9:6. doi: 10.1002/0471143030.cb0906s43. [DOI] [PubMed] [Google Scholar]
- 28.Langer HF, Chavakis T. Leukocyte-endothelial interactions in inflammation. J Cell Mol Med. 2009;13:1211–1220. doi: 10.1111/j.1582-4934.2009.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HC, Kioi M, Han J, et al. Anaplasma phagocytophilum-induced gene expression in both human neutrophils and HL-60 cells. Genomics. 2008;92:144–151. doi: 10.1016/j.ygeno.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Lepidi H, Bunnell JE, Martin ME, et al. Comparative pathology, and immunohistology associated with clinical illness after Ehrlichia phagocytophila-group infections. Am J Trop Med Hyg. 2000;62:29–37. doi: 10.4269/ajtmh.2000.62.29. [DOI] [PubMed] [Google Scholar]
- 31.Munderloh UG, Lynch MJ, Herron MJ, et al. Infection of endothelial cells with Anaplasma marginale and A. phagocytophilum. Vet Microbiol. 2004;101:53–64. doi: 10.1016/j.vetmic.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 32.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 33.Nelson CM, Herron MJ, Felsheim RF, et al. Whole genome transcription profiling of Anaplasma phagocytophilum in human and tick host cells by tiling array analysis. BMC Genomics. 2008;9:364. doi: 10.1186/1471-2164-9-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Razakandrainibe R, Combes V, Grau GE, et al. Crossing the wall: the opening of endothelial cell junctions during infectious diseases. Int J Biochem Cell Biol. 2013;45:1165–1173. doi: 10.1016/j.biocel.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 35.Reneman RS, Hoeks AP. Wall shear stress as measured in vivo: consequences for the design of the arterial system. Med Biol Eng Comput. 2008;46:499–507. doi: 10.1007/s11517-008-0330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rikihisa Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nat Rev Microbiol. 2010;8:328–339. doi: 10.1038/nrmicro2318. [DOI] [PubMed] [Google Scholar]
- 37.Rothermel AL, Wang Y, Schechner J, et al. Endothelial cells present antigens in vivo. BMC Immunol. 2004;5:5. doi: 10.1186/1471-2172-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saverymuttu SH, Peters AM, Keshavarzian A, et al. The kinetics of 111indium distribution following injection of 111indium labelled autologous granulocytes in man. Br J Haematol. 1985;61:675–685. doi: 10.1111/j.1365-2141.1985.tb02882.x. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt S, Moser M, Sperandio M. The molecular basis of leukocyte recruitment and its deficiencies. Mol Immunol. 2013;55:49–58. doi: 10.1016/j.molimm.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 40.Sultana H, Neelakanta G, Kantor FS, et al. Anaplasma phagocytophilum induces actin phosphorylation to selectively regulate gene transcription in Ixodes scapularis ticks. J Exp Med. 2010;207:1727–1743. doi: 10.1084/jem.20100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas RJ, Birtles RJ, Radford AD, et al. Recurrent bacteraemia in sheep infected persistently with Anaplasma phagocytophilum. J Comp Pathol. 2012;147:360–367. doi: 10.1016/j.jcpa.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Troese MJ, Carlyon JA. Anaplasma phagocytophilum dense-cored organisms mediate cellular adherence through recognition of human P-selectin glycoprotein ligand 1. Infect Immun. 2009;77:4018–4027. doi: 10.1128/IAI.00527-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker DH, Valbuena GA, Olano JP. Pathogenic mechanisms of diseases caused by Rickettsia. Ann N Y Acad Sci. 2003;990:1–11. doi: 10.1111/j.1749-6632.2003.tb07331.x. [DOI] [PubMed] [Google Scholar]
- 44.Walpola PL, Gotlieb AI, Langille BL. Monocyte adhesion and changes in endothelial cell number, morphology, and F-actin distribution elicited by low shear stress in vivo. Am J Pathol. 1993;142:1392–1400. [PMC free article] [PubMed] [Google Scholar]
- 45.Witko-Sarsat V, Rieu P, Descamps-Latscha B, et al. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest. 2000;80:617–653. doi: 10.1038/labinvest.3780067. [DOI] [PubMed] [Google Scholar]
- 46.Woldehiwet Z. The natural history of Anaplasma phagocytophilum. Vet Parasitol. 2010;167:108–122. doi: 10.1016/j.vetpar.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 47.Zen K, Liu Y, Mccall IC, et al. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol Biol Cell. 2005;16:2694–2703. doi: 10.1091/mbc.E05-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zweygarth E, Josemans AI, Steyn HC. In vitro isolation of Ehrlichia ruminantium from ovine blood into Ixodes scapularis (IDE8) cell cultures. Onderstepoort J Vet Res. 2008;75:121–126. [PubMed] [Google Scholar]