

Abstract
Objective
Treatments for recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) have limited efficacy. One potential therapeutic target for HNSCC is the RAS/RAF/MEK/ERK cascade, which is one of the major signaling pathways for HNSCC cell survival. In HNSCC, RAS can be activated either by HRAS mutation or upstream signaling. The ABL inhibitor nilotinib acts as a weak RAF inhibitor that induces RAF dimerization and subsequent activation of MEK/ERK in other cancer cell lines with activated RAS, leading to an unexpected dependence on MEK/ERK for cell survival. We hypothesized that nilotinib and the MEK inhibitor MEK162 would be synergistic in HNSCC cell lines owing to the frequent activation of RAS.
Methods
We treated HNSCC cell lines with nilotinib and performed immunoblotting and cell viability experiments. We used an orthotopic mouse model to assess synergistic effects in vivo.
Results
Nilotinib induced significant BRAF-CRAF heterodimerization and ERK activation irrespective of RAS mutation status. In cell viability assays, nilotinib synergized with MEK162. MEK162 alone induced G1 arrest that was minimally enhanced by nilotinib. In the mouse model, treatment with MEK162 alone or combined with nilotinib led to tumor growth inhibition.
Conclusions
In HNSCC, nilotinib-induced RAF dimerization is independent of RAS mutation status, but this dimerization does not lead to MEK dependence for cell survival in all HNSCC cell lines. MEK inhibition alone leads to decreased proliferation both in vitro and in vivo. Although nilotinib has some synergistic effects with MEK162, other agents may be more effective against HNSCC when combined with MEK162.
Keywords: Head and neck cancer, RAF, MEK, kinase inhibition
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide, and few treatment options are available for recurrent or metastatic disease. HNSCC is a disabling, disfiguring cancer that is frequently lethal. Targeted therapy for cancer can be very effective if it inhibits the key oncogenic drivers. For example, in chronic myeloid leukemia (CML), inhibition of BCR-ABL with nilotinib is a markedly effective therapy. However, the only targeted therapy used for HNSCC is the anti-epidermal growth factor receptor antibody cetuximab, which is used in unselected patients with a response rate of less than 15%. Highly effective targeted therapy for HNSCC does not yet exist owing to a lack of easily druggable oncogenic drivers[1,2].
Increasingly we and others have recognized the power of combinations of targeted therapy that take advantage of our knowledge of signaling pathways. In this regard, Packer et al showed that treatment with nilotinib leads to the unexpected activation of MEK and ERK through induction of robust RAF dimerization in melanoma, pancreatic cancer, and colon cancer cell lines with mutant RAS [3]. We also showed that several drugs that are weak RAF inhibitors (nilotinib, dasatinib, and imatinib) induce robust RAF dimerization [4]. Nilotinib is a potent ABL inhibitor that inhibits BCR-ABL, leading to decreased RAS/RAF/MEK downstream signaling in CML cell lines. However, in CML cell lines with a gatekeeper mutation (BCR-ABLT315I), nilotinib does not inhibit BCR-ABLT315I and RAS remains active. Activated RAS signaling is critical for paradoxical activation of the MEK/ERK pathway in CML cells. When MEK and ERK are activated via drug-induced RAF dimerization, this activation of RAF leads the cancer cells to have an unexpected dependence on MEK/ERK for survival. Consequently, the combination of nilotinib and MEK inhibition is synergistic in BCR-ABLT315I-expressing CML cells [3].
The RAS/RAF/MEK/ERK pathway plays an important role in the progression of many human cancers. Once activated by surface receptors, RAS recruits RAF, a serine/threonine kinase, to the cell membrane and activates it. RAF then phosphorylates MEK, which in turn phosphorylates and activates ERK, leading to cancer progression or senescence depending on the degree of activation and presence of concurrent tumor suppressor pathways. The RAS/RAF/MEK/ERK cascade is frequently activated in HNSCC via upstream activation of epidermal growth factor receptor and other receptor tyrosine kinases. HRAS is mutated in 5–10% of cases of human HNSCC [1,2] and 3% of HNSCC cell lines [5], and this mutation predicts resistance to epidermal growth factor receptor inhibition [6]. Other RAS mutations are rarely described in HNSCC [7].
In the current study, we hypothesized that in HNSCC cell lines, nilotinib would activate MEK and ERK owing to RAF dimerization, as a result of frequent activation of RAS in HNSCC by mutation or upstream signaling. We tested the effect of the combination of nilotinib and a clinically relevant MEK inhibitor (MEK162) on cell proliferation in vitro using HNSCC cell lines and tumor growth in vivo using an orthotopic mouse model.
Materials and Methods
Reagents
Antibodies used in this study were anti-pERK, total ERK, pMEK, total MEK (Cell Signaling Technology, Danvers, MA), total CRAF (BD Biosciences, San Jose, CA), agarose-conjugated CRAF, total BRAF, and β-actin (Sigma-Aldrich, St Louis, MO). Drugs included nilotinib (Santa Cruz Biotechnology, Dallas, TX) and MEK162 (Selleck Chemicals, Houston, TX).
Cell culture
Human HNSCC cell lines OSC19, UMSCC47, HN31, and UMSCC17A were obtained from Dr. Jeffrey Myers [8] and cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum as described previously [9].
Western blot analysis and immunoprecipitation
Western blot analysis and immunoprecipitation were performed as previously described [10]. Briefly, for Western blot analysis, cells were lysed on ice and the lysates were centrifuged at 20,000g for 5 minutes at 4°C and then boiled with 1X sample buffer for 5 minutes. Equal amounts of protein aliquots were resolved with SDS–polyacrylamide gel electrophoresis. For immunoprecipitation, equal amounts of protein lysates were incubated with agarose-conjugated CRAF antibody overnight. Immunoprecipitates were then washed 3 times and boiled with sample buffer for 8 minutes and resolved using SDS PAGE. For both Western blot analysis and immunoprecipitation, proteins were transferred to nitrocellulose membranes, immunoblotted with a primary antibody, and detected with a horseradish peroxidase–conjugated secondary antibody (Bio-Rad Laboratories, Hercules, CA) and ECL reagent (Pierce Biotech, Rockford, IL).
Measurement of cell cycle and apoptosis
Cell cycle analysis and apoptosis assays were done as described previously [4]. Briefly, cells were treated with 1 μmol/L nilotinib, 150 nmol/L MEK162, or both for 72 hours. To measure cell cycle, we fixed and stained cells with BrdU FITC using the BrdU Flow Kit (BD Pharmingen, San Jose, CA) according to kit’s manual and then sorted the cells using fluorescence-assisted cell sorting (FSCScan, Becton Dickinson, San Jose, CA). To measure apoptosis, we subjected fixed cells to the TUNEL assay using the Apo BrdU kit (Phoenix flow systems, City, ST San Diego, CA) and then sorted them using fluorescence-assisted cell sorting.
Cell viability assay
Cell viability was measured using the 3-(4, 5 dimethylthiazol-2-Y1)-2, 5-diphenyltetrazoliumbromide (MTT) assay as described previously [11]. The half maximal inhibitory concentration (IC50) values for each drug were calculated using the Chou-Talley equation in the Calcusyn software program (Biosoft, Cambridge, UK).
Colony formation assay
Cells were seeded in 6-well plates (100–200 cells per well) and treated with 1 μmol/L nilotinib, 150 nmol/L MEK162, a combination of nilotinib and MEK, or vehicle alone for 2–3 weeks. Cells were then washed with phosphate-buffered saline and fixed with 4% paraformaldehyde, followed by staining with 0.05% crystal violet for 20 minutes. After staining, cells were washed thoroughly with water to wash out any unbound crystal violet. Colony area was calculated using Image J software as previously described [12]. The amount of crystal violet per well was measured using a spectrophotometer after the colonies were dissolved in methanol. A standard curve with known concentrations of crystal violet was used to calculate unknown concentrations.
Orthotopic xenograft experiments
All animal studies were approved by the animal care and use committee. We injected 5 × 106 UMSCC47 cells into the tongues of nude mice as previously described [13]. After 2 weeks, the tumor burden was measured and mice bearing tongue tumors were randomized into 4 treatment groups: vehicle alone (control), 75 mg/kg nilotinib, 100 mg/kg MEK162, or both drugs (combination). Drugs were administered by oral gavage, 4 to 7 days per week. The tumor volume was measured periodically until euthanasia.
Statistical analysis
Mean values were calculated from n=3 independent experiments and expressed as mean + sd. Student’s t-test was used to evaluate differences for significance and P values of <0.05 was considered as statically significant.
Results
Nilotinib leads to BRAF-CRAF dimerization and MEK/ERK activation in HNSCC cells independent of HRAS mutation
To investigate the effect of nilotinib on MEK/ERK pathway activation in HNSCC, we treated HNSCC cell lines that had HRAS mutations (HN31 and UMSCC17A) and HNSCC cell lines that did not (OSC19 and UMSCC47) with 1 μmol/L nilotinib [5]. The mean peak plasma concentration of nilotinib is 3.6 μmol/L [14], demonstrating that experiments were all done within physiologic ranges. All 4 cell lines showed robust BRAF-CRAF dimerization and activation of MEK and ERK at 4 to 24 hours after treatment was completed (Fig. 1A).
Figure 1.

Nilotinib leads to BRAF-CRAF heterodimerization and subsequent MEK-dependent ERK activation in head and neck squamous cell carcinoma (HNSCC) cells. (A) HNSCC cells were treated with 1 μmol/L nilotinib for the indicated times and then lysed. RAF dimerization was measured using immunoprecipitation (IP) with an anti-CRAF antibody followed by immunoblotting for total BRAF and CRAF (upper panels). To check MEK and ERK activation, we subjected lysates to immunoblotting with the indicated antibodies (lower panels). (B) Cells were treated with 1 μmol/L nilotinib, 150 nmol/L MEK162, or both for 3 hours and lysed. Cell lysates were resolved by SDS PAGE and Western blot analysis was performed to measure levels of pMEK, pERK, total MEK, ERK, and μ-actin.
To determine whether nilotinib-induced ERK activation was MEK-dependent, we treated the cells with 1 μmol/L nilotinib, the MEK inhibitor MEK162 (150 nmol/L), or both for 4 hours. MEK162 inhibited both basal and nilotinib-induced ERK activation (Fig. 1B). We observed an increase in phosphorylated MEK after treatment with MEK162, as reported earlier, owing to relief of upstream feedback. Despite the increase in pMEK, MEK is still potently inhibited by MEK162 [15]. Taken together, these results indicate that nilotinib leads to RAF dimerization and subsequent MEK-dependent ERK activation in HNSCC cells.
Nilotinib and MEK inhibition is synergistic in HNSCC cell lines independent of HRAS mutation status
We hypothesized that MEK activation by nilotinib would result in dependence of HNSCC cells on MEK signaling, as occurs in CML cells [3]. We investigated the effect of treatment with the combination of nilotinib and MEK162 on cell viability in 4 different HNSCC cell lines using MTT assays (Fig. 2A). The combination of nilotinib and MEK162 was synergistic in all 4 HNSCC cell lines (Table 1). In colony formation assays, nilotinib alone did not inhibit growth in any cell line except UMSCC17A. In OSC19 cells, nilotinib led to a significant increase in colonies, consistent with MEK/ERK activation. Single-agent MEK162 inhibited cell growth significantly (p < 0.05) in UMSCC17A, HN31, and OSC19 cells. In HN31 and OSC19 cells, the addition of nilotinib led to a further decrease in the number of cells (Fig. 2B and Supplementary Fig. S1).
Figure 2.

Nilotinib and MEK162 synergistically induce synthetic lethality in HNSCC cells independent of RAS mutation status. (A) Cells were treated with different doses of nilotinib only, MEK162 only, or both for 72 hours. Cell viability was measured using the MTT assay. Error bars indicate standard deviation. (B) Cell growth was estimated using the colony formation assay in cells treated with 1 μmol/L nilotinib only, 150 nmol/L MEK162 only, or both drugs. *p < 0.05 compared with untreated control; †p < 0.05 compared with MEK162 alone. Error bars indicate standard deviation.
Table 1.
Median effects of nilotinib and MEK162 as single agents and in combination on head and neck squamous cell carcinoma cells in vitro.
| Cell line | Single agent, IC50* | Combination, IC50* | Combination index simulations, Fa = 0.5 | Interpretation | ||
|---|---|---|---|---|---|---|
|
| ||||||
| Nilotinib, nmol/L | MEK162, nmol/L | Nilotinib, nmol/L | MEK162, nmol/L | |||
| UMSCC47 | 33.3 | >200 | 1115 | 28.0 | 0.334 ± 96 | Not conclusive |
| OSC19 | 7027 | >200 | 472 | 11.8 | 0.089 ± 0.04 | Synergistic |
| HN31 | >8000 | >200 | 710 | 17.7 | 0.085 ± 0.08 | Synergistic |
| UMSCC17A | >8000 | >200 | 802 | 20.0 | 0.011 ± 0.07 | Synergistic |
IC50, half maximal inhibitory concentration.
MEK inhibition leads to G1 cell cycle arrest and reduction in cell proliferation that is not enhanced by nilotinib
To characterize the biological effects of nilotinib and MEK162 as single agents and in combination, we performed cell cycle analysis in 3 HNSCC cell lines. Consistent with the results of the MTT and colony formation assays, single-agent nilotinib did not show any effect after 72 hours of treatment, but MEK162 alone significantly increased the number of cells in the G1 phase and reduced the number of cells in the S phase of the cell cycle (p < 0.05), consistent with G1 arrest. The combination of the two drugs led to enhanced arrest (p < 0.05) only in UMSCC17A cells (Fig. 3). We did not observe any substantial apoptosis or senescence with any of the treatments (all values less than 5%).
Figure 3.
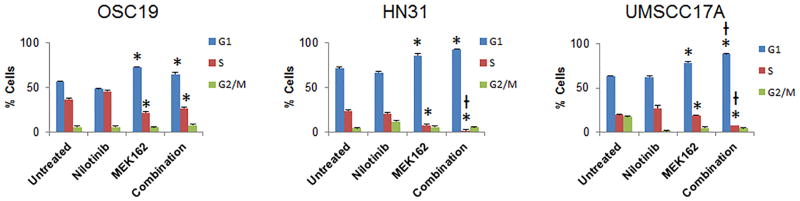
MEK inhibition leads to G1 cell cycle arrest in head and neck squamous cell carcinoma. Cells were treated with 1 μmol/L nilotinib, 150 nmol/L MEK162, or both for 72 hours. Cell cycle analysis was performed using Brdu fluorescence-assisted cell sorting. *p < 0.05 compared with untreated control, †p < 0.05 compared with MEK162 alone. Error bars indicate standard deviation.
MEK162 inhibits tumor growth in vivo in an orthotopic mouse model of HNSCC
To determine the efficacy of nilotinib and MEK162 in vivo, we used an orthotopic mouse model of human HNSCC with UMSCC47 cells [13]. Daily dosing with both drugs for 7 days per week (Fig. 4A) was not well tolerated, so we later reduced the dosing schedule to 4 days per week. We examined the growth of tumors for up to 21 days (Fig. 4A) or 40 days (Fig. 4B). Growth of tongue tumors was strongly suppressed by MEK162 alone and also in combination with nilotinib. However, owing to variability in tumor growth in this model, we found a significant effect (p < 0.05) only with the dosing schedule of 7 days per week (Fig. 4C). Nilotinib did not enhance the efficacy of MEK162. A lack of synergy may be due to the significant effect of MEK162 as a single agent particularly in the more intense dosing schedule.
Figure 4.

Nilotinib does not enhance MEK162 inhibition of tumor growth in an orthotopic mouse model. Mice were injected with UMSCC47 cells in the tongue. After 2 weeks, tumor-bearing mice were grouped randomly and treated with nilotinib only, MEK162 only, a combination of nilotinib and MEK162, or vehicle only for 7 days per week (A) or 4 days per week (B). Tumor volume was measured periodically and plotted against time for the mice individually with each color representing an individual mouse (left 4 graphs). The average fold change in tumor volume over time (upper right) and at day 21 or at the time of euthanasia (lower right) was compared with day 0 (treatment start date). Error bars indicate standard deviation. *p < 0.05 compared with untreated control.
Discussion
In the current study we demonstrated that clinically relevant concentrations of nilotinib led to robust and sustained BRAF-CRAF heterodimerization and activation of MEK and ERK in HNSCC cell lines with both mutant and wild-type HRAS. MEK162 completely abrogated both basal and nilotinib-induced ERK activation. Nilotinib alone had little effect on cell viability, but was synergistic with MEK162 as measured by MTT assays. However, MEK inhibition alone inhibited colony formation and produced cell cycle arrest in vitro and these effects were enhanced by nilotinib in only 2 cell lines. Likewise, nilotinib alone had little effect on tumor growth in vivo but MEK162 alone reduced tumor growth. Taken together, these results indicate that in HNSCC, nilotinib induces RAF dimerization independent of RAS mutation, but this dimerization does not lead to MEK dependence for cell survival. Consequently, although nilotinib has some synergistic effects with MEK inhibition, other agents may be more effective than nilotinib when combined with a MEK inhibitor.
As in melanoma, colon cancer, and pancreatic cancer cell lines, nilotinib produced robust RAF dimerization in HNSCC cell lines [3]. Although nilotinib-induced RAF dimerization in HNSCC was independent of RAS mutation status, our findings are still consistent with the model proposed by Packer et al [3]. They proposed that RAS activation (caused by mutation or upstream signaling) in the presence of drug-induced RAF dimerization leads to downstream MEK and ERK activation, which our results also showed. However, unlike in the BCR-ABLT315I CML cells in that study, in the HNSCC cells in our study, treatment with nilotinib did not result in cell dependence on MEK for survival. MEK inhibition alone or in combination with nilotinib did not result in apoptosis. Likewise, robust MEK inhibition alone does not lead to substantial apoptosis in most cancer cell lines with mutant RAS [16]. This finding suggests that other pathways downstream of RAF dimerization are MEK-independent and contribute to cell death. Although MEK is the only well-established RAF substrate, RAF proteins have kinase-independent roles in cell signaling [17–19].
Although nilotinib-induced MEK activation did not lead to MEK dependence in HNSCC cells, PI3K inhibitors may do so. We recently demonstrated that following incubation with a PI3K inhibitor, pERK levels increased in HNSCC cell lines. The combination of MEK and PI3K inhibitors led to additive or synergistic effects in most HNSCC cell lines. This combination also led to enhanced cell cycle arrest. In HNSCC cell lines with HRAS mutations, the drug combination was profoundly synergistic, with combination indices of <0.1 [5].
Potent ATP-competitive BRAF inhibitors (e.g., dabrafenib, vemurafenib) also cause RAF dimerization in melanoma with wild-type BRAF and mutant NRAS [20]. Pouilikakos et al demonstrated that for drug-induced ERK activation to occur, the drug needed to bind to the ATP-binding site of one RAF molecule in the dimer, leading to activation of the non–drug-bound protomer [21]. This effect may underlie the induction of squamous tumors in patients treated with BRAF inhibitors, as well as drug resistance in BRAF mutant tumors.
In conclusion, we found that in HNSCC, nilotinib induced RAF dimerization independent of RAS mutation, but this dimerization did not lead to MEK dependence for survival. Instead, MEK inhibition alone led to cell cycle arrest and decreased tumor growth in vivo. Future research in HNSCC should focus on MEK inhibitors combined with other agents, such as inhibitors of the PI3K/AKT pathway.
Supplementary Material
Nilotinib and MEK162 affect colony formation in head and neck squamous cell carcinoma cells independent of RAS mutation status. Cell growth was estimated using the colony formation assay for cells treated with 1 μmol/L nilotinib only, 150 nmol/L MEK162 only, or both.
Acknowledgments
This work was supported by the National Cancer Institute Head & Neck SPORE P50CA097007 for The University of Texas MD Anderson Cancer Center (for J.N. Myers, B.S. Glisson, and F.M. Johnson). Flow cytometry was supported by the National Cancer Institute Cancer Center Support Grant P30CA016672 (to MD Anderson). We gratefully acknowledge Erica Goodoff and the Department of Scientific Publications for editorial assistance with the manuscript.
Footnotes
Conflicts of interest: FMJ has research funding from PIQUR Pharmaceuticals and has been on the scientific advisory boards for Novartis and Bristol-Myers Squibb. All others have no conflicts of interest.
References
- 1.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Packer LM, Rana S, Hayward R, O’Hare T, Eide CA, Rebocho A, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2012;20:715–727. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sen B, Peng S, Tang X, Erickson HS, Galindo H, Mazumdar T, et al. Kinase-impaired BRAF mutations in lung cancer confer sensitivity to dasatinib. Sci Transl Med. 2012;4:136ra170. doi: 10.1126/scitranslmed.3003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazumdar T, Byers LA, Ng PK, Mills GB, Peng S, Diao L, et al. A Comprehensive Evaluation of Biomarkers Predictive of Response to PI3K Inhibitors and of Resistance Mechanisms in Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther. 2014;13:2738–2750. doi: 10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hah JH, Zhao M, Pickering CR, Frederick MJ, Andrews GA, Jasser SA, et al. HRAS mutations and resistance to the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in head and neck squamous cell carcinoma cells. Head Neck. 2014;36:1547–1554. doi: 10.1002/hed.23499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker TP, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-13-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin Cancer Res. 2011;17:7248–7264. doi: 10.1158/1078-0432.CCR-11-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma WL, Jeng LB, Lai HC, Liao PY, Chang C. Androgen receptor enhances cell adhesion and decreases cell migration via modulating beta1-integrin-AKT signaling in hepatocellular carcinoma cells. Cancer Lett. 2014;351:64–71. doi: 10.1016/j.canlet.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 10.Sen B, Peng S, Woods DM, Wistuba I, Bell D, El-Naggar AK, et al. STAT5A-mediated SOCS2 expression regulates Jak2 and STAT3 activity following c-Src inhibition in head and neck squamous carcinoma. Clin Cancer Res. 2012;18:127–139. doi: 10.1158/1078-0432.CCR-11-1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–1965. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brannan JM, Dong W, Prudkin L, Behrens C, Lotan R, Bekele BN, et al. Expression of the receptor tyrosine kinase EphA2 is increased in smokers and predicts poor survival in non-small cell lung cancer. Clin Cancer Res. 2009;15:4423–4430. doi: 10.1158/1078-0432.CCR-09-0473. [DOI] [PubMed] [Google Scholar]
- 13.Myers JN, Holsinger FC, Jasser SA, Bekele BN, Fidler IJ. An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clin Cancer Res. 2002;8:293–298. [PubMed] [Google Scholar]
- 14.Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 15.Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74:2340–2350. doi: 10.1158/0008-5472.CAN-13-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgess MR, Hwang E, Firestone AJ, Huang T, Xu J, Zuber J, et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood. 2014;124:3947–3955. doi: 10.1182/blood-2014-05-574582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamilton G, Yee KS, Scrace S, O’Neill E. ATM regulates a RASSF1A-dependent DNA damage response. Curr Biol. 2009;19:2020–2025. doi: 10.1016/j.cub.2009.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2:232–260. doi: 10.1177/1947601911407323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matallanas D, Romano D, Al-Mulla F, O’Neill E, Al-Ali W, Crespo P, et al. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol Cell. 2011;44:893–906. doi: 10.1016/j.molcel.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 20.Kwong LN, Chin L. The brothers RAF. Cell. 2010;140:180–182. doi: 10.1016/j.cell.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 21.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nilotinib and MEK162 affect colony formation in head and neck squamous cell carcinoma cells independent of RAS mutation status. Cell growth was estimated using the colony formation assay for cells treated with 1 μmol/L nilotinib only, 150 nmol/L MEK162 only, or both.