

Abstract
Background
Neuronal plasticity deficits are thought to underlie abnormal neurodevelopment in Fetal Alcohol Spectrum Disorders (FASD) and in animal models of this condition. Previously, we found that alcohol exposure during a period that is similar to the last months of gestation in humans disrupts ocular dominance plasticity (ODP), as measured in superficial cortical layers. We hypothesize that exposure to alcohol can differentially affect the potentiation and depression of responses that are necessary for activity dependent sprouting and pruning of neuronal networks. ODP is an established paradigm that allows the assessment of activity-dependent depression and potentiation of responses in vivo.
Methods
Mouse pups were exposed to 3.6 – 5g/kg of ethanol in saline daily or every other day between postnatal days 4 and 9. Visual cortex plasticity was then assessed during the critical period for ODP using two techniques that separately record in layers 4 (visual evoked potentials, VEPs) and 2/3 (optical imaging of intrinsic signals, OI).
Results
We discovered a layer-specific effect of early alcohol exposure. Recording of VEPs, from layer 4, showed that while the potentiation component of ODP (Pc-ODP) was disrupted in animals treated with alcohol when compared to saline controls, the depression component of ODP (Dc-ODP) was unaltered. In contrast, OI, from layers 2/3, showed that Dc-ODP was markedly disrupted in alcohol treated animals when compared to controls.
Conclusions
Combined with our previous work, these findings strongly suggest that developmental alcohol exposure has a distinct and layer-specific effect on the potentiation and depression of cortical responses after monocular deprivation.
Keywords: Ocular dominance, visual development, monocular deprivation, visual evoked potentials, fetal alcohol syndrome
INTRODUCTION
Alcohol consumption during pregnancy can lead to a wide variety of neurological problems in offspring. These alterations are collectively referred to as Fetal Alcohol Spectrum Disorders (FASD, Hannigan and Armant, 2000). FASD can be associated to deficits in visual processing(Burden et al., 2009; Coffman et al., 2013; Uecker and Nadel, 1996), impaired visual-motor integration (Chioco et al., 2009; Vaurio et al., 2011) and altered saccadic eye movement (Paolozza et al., 2013). Some of these deficits can be caused by major peripheral alterations such as microphtalmia and hypoplasia of the optic nerve (Stromland, 1998). However problems in visual acuity and contrast sensitivity may also be casued by alterations in the visual cortex resulting in amblyopia (Vernescu, 2012). Deficits in visual acuity and contrast sensitivity can be seen also in animal models of FASD (Hug et al., 2000; Lantz et al., 2014). There is growing evidence that impaired neuronal plasticity underlies many of the sensory deficits seen in FASD (see; Medina, 2011). For instance, recent studies by Sowel group showed that, during infancy and adolescence, control subjects presented a strikingly plastic cortex, with a robust increase in volume followed by loss, while FASD subjects displayed a less plastic cortex, with mostly volume loss (Lebel et al., 2012). One possible explanation of these findings is that developmental alcohol exposure preferentially disrupts processes related to the increase of neuronal connections rather than their elimination.
Neuronal plasticity encompasses the strengthening (potentiation) and weakening (depression) of neuronal responses, which can ultimately lead to an increase or elimination of neuronal connections, respectively. The ocular dominance plasticity (ODP) model relies on cortical changes that occur after monocular deprivation (MD) during a critical period of development (Hubel and Wiesel, 1970; Hubel et al., 1977). In normal animals, a few days of MD leads to depression of responses in neurons driven by the deprived (closed) eye (referred here as depression component of ODP or Dc-ODP), and a potentiation of responses in neurons driven by the experienced (open) eye (referred to here as the potentiation component of ODP or Pc-ODP) (Fig 1). The time-course of Pc-ODP and Dc-ODP in the mouse has been well described (Crozier et al., 2007; Smith et al., 2009). After 2–3 days of MD, predominantly Dc-ODP of the closed eye responses takes place. In contrast, Pc-ODP of the open eye responses is only seen after 5 days of MD (Fig 1b).
Figure 1.

Ocular dominance plasticity in mice. A) The mouse visual cortex is divided into two sections, a monocular section, and a binocular zone, responding to input from both eyes. In the binocular zone, neurons present different degrees of binocularity; from cells that are equally responsive to stimulation of either eye, to cells that respond almost exclusively to one eye. When both eyes are receiving normal input, the binocular zone is dominated by responses to the contralateral eye (light green). If this eye is sutured closed during the critical period, then the binocular zone shifts, responding primarily to input from the ipsilateral, open eye (blueish green). B) The shift in eye dominance in the binocular zone of V1 occurs through two temporally different mechanisms. First, there is a depression of inputs from the deprived eye that occurs after approximately 3 days of MD (Dc-ODP). This is followed by a potentiation of the non-deprived eye inputs (Pc-ODP), after approximately 5 days of MD.
Previously, recordings of visually evoked potentials (VEPs) using a single implanted electrode have been used to assess ODP and study plasticity deficits in mice before and after MD (Dolen et al., 2007; Yashiro et al., 2009). The recorded field potential represents the summation of the responses from many neurons, but is primarily composed of responses from layer 4, as indicated by the depth of the electrode implantation (~430nm), as well as the VEP size and polarity (largest negative polarity VEP is a result of direct thalamic input into layer 4 of the primary visual cortex; Heynen and Bear, 2001). Optical imaging of intrinsic signals has historically also been used to investigate ODP in rodents (Kalatsky and Stryker, 2003; Lantz et al., 2012; (Lehmann and Lowel 2008). The origin of these intrinsic signals lies in the changes in oxy-hemoglobin and hemoglobin with tissue activation, these signals are now considered to arise predominately from superficial cortical layers such as layer 2/3 (Frostig and Chen-Bee, 2009; Lee et al., 2012; McCurry et al., 2010; Smith et al., 2009; Bonhoeffer 1995).
The Medina lab pioneered the use of ODP to investigate neuronal plasticity deficits in FASD. We showed that alcohol exposure from postnatal day 10 (P10) to P30 in ferrets or between P5-P9 in mice, periods that are roughly equivalent to the last months of human gestation, leads to a permanent impairment in ODP (Medina et al., 2003; Lantz et al., 2012). However it is still unknown whether the neuronal plasticity impairments seen after alcohol exposure are due to a specific disruption of Pc-ODP, Dc-ODP, or both. Because potentiation and depression rely on different mechanisms, it is critical to tease apart alcohol’s effect on each of these processes to understand the underlying causes of neuronal plasticity deficits in FASD.
To address this question, mice were exposed to alcohol or saline, using a binge drinking like paradigm, during a period roughly equivalent to the third trimester of human gestation. After an ethanol-free period of ~2 weeks we recorded VEPs and optical imaging of intrinsic signal responses to assess changes in Pc-ODP and Dc-ODP.
METHODS
Experiment 1 (carried out in the Medina Lab)
Animals
All procedures described here were approved by the IACUC. Visibly pregnant C57/BL6 female mice were obtained from a commercial supplier (Harlan), and singularly housed in the University of Maryland animal housing. Pregnant dams were checked daily until pups were born. Day of birth was designated as P0. A total of 27 animals were used from 6 litters.
Ethanol Exposure
Pups received a single injection of 5g/kg of alcohol (25% ethanol in normal saline i.p.) or saline as a control on days P5, 7 and 9. According to our previously published studies, this protocol leads to blood alcohol levels of 411 mg/dl (± 43) at 1 hour post injection (Lantz et al., 2012).
Electrode Implantation
Electrode implantation was done as described previously (Lantz et al., 2014) Electrodes were implanted in P21 – 22 mice. Mice were anesthetized with i.p. ketamine 120 mg/kg (Bioniche Pharma, Lake Forest, IL) and xylazine 9mg/kg (Akorn, Inc, Decatur, IL). Once anesthetized, 2% lidocaine jelly (Akorn, Inc, Decatur, IL) was applied locally on the scalp at the incision site. Burr holes were drilled 1.0 mm caudal from bregma and 2.0 mm lateral from the midline and ground electrodes were implanted. Tungsten microelectrodes (FHC, impedance 0.3–0.5 M ohms) were implanted in bilateral burr holes drilled at 3.00 mm lateral of midline and 0.00 mm of lambda, at a depth of 0.43 mm. Electrodes were secured with cyanoacrylate glue (Elmers, Westerville, OH). A nail glued over the rostral portion of the skull was used to secure each animal’s head during recording. After surgery, each animal was monitored until recovery of righting reflexes and was then given 0.05mg/kg of buprenorphine (Stokes Pharmacy, Mt. Laurel, NJ) for post-surgical analgesia.
Assessment of Ocular dominance
Awake animals were habituated on the experimental setup for 45 minutes one day prior to the experiment. VEPs were recorded using XCell-3 amplifiers (FHC inc., Bowdoin ME; one for each recording electrode), a 1401 digitizer (CED, Cambridge, England) and Spike 2 software (Cambridge Electronics Design, Cambridge, UK). Xcell-3 amplifiers were set with a low cut-off of 0.1 Hz, and a high cut-off of 100Hz. Visual stimulations were presented to each eye individually using a monitor placed 18 cm from the nose of the animal (mean luminance 27cd/m2, area of 15x31cm) and controlled by a custom program using MATLAB (MathWorks, Natick, MA). Stimuli consisted of full field ordinal sine wave 2 Hz reversing gratings, at 0.05 cycles per degree with 100% contrast. To avoid stimulus response potentiation (Cooke and Bear, 2010), drifting gratings were presented at different angles in the experiments performed before (45o) and after (135o) MD. VEP responses were then averaged from 100 stimulation presentations, and amplitudes were recorded using peak to trough measurements. At the end of the first day of recording, animals were anesthetized using vaporized isoflurane (Baxter, Deerfield, IL) and small portions of the upper and lower right eyelids were trimmed. The eyelids were then sutured and covered with tissue-glue (CP Medical, Portland, OR). Animals were then returned to the animal colony and remained monocularly deprived for 3, or 5 -10 days, until post-MD VEPs were recorded. After deprivation, animals were briefly anesthetized using vaporized isoflurane (Baxter, Deerfield, IL), and the sutured eye was opened. Animals were then placed in the experimental setup and post-MD VEP recordings were done immediately after recovery from light anesthesia. Eyelids were checked daily for any sign of opening during the period of MD. Animals with partial eye lid opening during the period of deprivation were discarded. We also discarded animals where responses from both eyes decreased or increased more than 2 fold from the pre-MD day of recording.
Statistics
Data are reported as mean ± standard error. Statistical analysis was performed using SPSS (IBM), univariate ANOVAs were used to compare CBI measurements within experimental groups. For comparison of ipsilateral and contralateral VEP amplitude within each group paired t-tests were employed.
Experiment 2 (carried out in the Majewska lab)
Animals
All procedures were conducted in strict accordance with the University of Rochester’s Committee on Animal Resources. C57/BL6 breeding pairs were maintained in-house under a 12 hours light/dark cycle, and supplied chow and water ad libitum. Breeders were checked daily until the birth of pups (P0), when the male was separated from the dam and litter. A total of 19 mice from 4 litters were used for experiments. Litter size was between 6 and 9 pups.
Ethanol Exposure
Pups (P4) were weighed, toe-clipped, and randomly assigned to a saline vehicle control or alcohol (ethanol; EtOH) treatment group. From P4 through P9, pups received two daily subcutaneous (s.c.) injections, spaced 2 hours apart. Pups were returned to the dam immediately following each injection. EtOH treated animals received a total of 3.6g/kg EtOH in two doses of 1.8g/kg EtOH, delivered using 20% v/v 200 proof EtOH in 0.9% NaCl. The 20% EtOH was prepared fresh within 30 minutes of each dose. Control animals received an equivalent volume of 0.9% NaCl. Blood alcohol levels at 1.5 hours after the second dose on P4 were (356 ± 10 mg/dL, mean ± SEM). Animals were weighed on P4-P9, P14, P21 (weaning), and P27. Weights between saline vehicle control and alcohol treated animals were not significantly different at any time point sampled (data not shown).
Optical Imaging of Intrinsic Signals
On P28 ± 1 day, animals were separated into non-deprived (ND) or monocularly deprived (MD) cohorts. MD animals were anesthetized using vaporized isoflurane (5% induction, 3% maintenance) and right upper and lower eyelids were resected and sutured together using 2 mattress sutures (5.0 Vicryl, Ethicon, Inc., Portland, OR). After 4 days (P31± 1 day), ND or MD animals were anesthetized with isoflurane (5% induction, 3% maintenance) and intraperitoneal injection of chloroproxithene (2mg/kg). MD animal eyelids were reopened. The skull over the contralateral visual cortex was exposed, cleared of membranes, covered with 0.5% agarose, and sealed with a coverslip to create an imaging window. Intrinsic signal optical imaging was performed using a DALSA 2M30 CCD camera and custom acquisition software (Kalatsky and Stryker, 2003). Anesthesia was maintained with isoflurane (0.75%) throughout imaging. Vasculature was illuminated using a green LED (550nm) and a region of interest containing binocular visual cortex was selected based on characteristic vascular patterns. Intrinsic signal images were then collected using illumination from a red LED (700nm). Visual stimuli were presented, consisting of white horizontal square-wave bar gratings on a black background moving upwards (90°) or downwards (270°) at a frequency of 8°/s for 6 minutes. Visually evoked responses to stimuli moving in both directions, presented to the contralateral and ipsilateral eyes individually, were collected. The amplitude of the fast fourier transform component of the intrinsic signal was averaged for each eye from responses to both stimulus directions and compared between eyes offline using MatLab to determine ocular dominance (OD) (Kalatsky and Stryker, 2003;Tropea et al., 2010). A contralateral bias index (CBI) was calculated by the following equation: CBI = average contralateral response / average ipsilateral response. Monocular deprivation, intrinsic signal optical imaging, and OD analysis were all done by an investigator blind to animal treatment.
Statistics
Data are reported as mean ± standard error. Comparisons were performed using Prism VI statistical analysis software (Graphpad). CBI values were analyzed using a two-way ANOVA with Bonferroni post-hoc comparisons.
RESULTS
Mouse pups were exposed to 5g/kg of alcohol or saline on P5, P7 and P9, mimicking binge alcohol drinking during the third trimester equivalent of human gestation. At P25 ocular dominance was assessed by calculating peak to trough measures of VEPs resulting from stimulation of each eye individually (Fig. 2).
Figure 2.
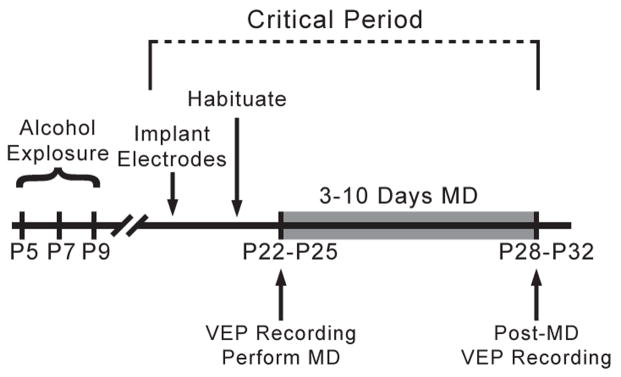
Experimental design. Animals were exposed to 5g/kg of ethanol or control saline on P5, P7, and P9. Animals were then implanted with recording electrodes on P21–P24, and habituated to the recording apparatus 24hrs following surgical recovery. After MD for a short period (3 days) or a longer period (5–10 days), the deprived eye was opened and post-MD VEPs were recorded.
After the initial evaluation of ocular dominance, each saline and alcohol treated animal was monocularly deprived for 5–10 days. It is well established that in mice 5 days of MD is sufficient to produce a decrease and an increase in the responses of the deprived and experienced eye respectively (Frenkel and Bear, 2004). At the end of this period, VEPs were recorded to assess the effect of MD on ocular dominance.
After MD, saline treated animals exhibited a decrease in CBI values from 1.80 ±0.36 to 0.76 ±0.32 (n = 7). This change indicates a shift from contralateral (deprived) to ipsilateral (experienced) eye dominance. Surprisingly, a reduction in CBI values was also seen in the ethanol exposed animals, from an average CBI value of 1.46 ±0.35 to 1.11 ±0.33 (n = 11, F=32.309, df=1, p < 0.001, ANOVA). Despite this difference, the average CBI value for ethanol treated animals remained above 1, indicating a continued dominance of the contralateral eye (interaction between ethanol and MD, F = 1.137, df =1, p = 0.004; ANOVA, Fig. 3a). We calculated the percentage of change in CBIs before and after MD. Fig. 3b shows that the reduction in CBI after MD was higher in saline (57% ± 7) than in alcohol treated animals (26% ± 6; t=3.24, df = 15, p = 0.005), indicating that MD of ethanol exposed animals resulted in a less plastic ocular dominance response.
Figure 3.

Effect of 5–10 days of MD on contralateral bias (CBI). A) Both alcohol (1.46 ±0.35; 1.11 ±0.33; n = 11) and saline (1.80 ±0.36; 0.76 ±0.32; n = 7) treated animals exhibited a significant decrease in CBI values after MD. B) However, the magnitude of this change was larger in saline (57% ± 7) treated animals than ethanol (26% ± 6). * p = 0.029 ** p < 0.001.
After examining the changes in CBI, we compared the individual changes in eye responses (Fig 4). In saline exposed animals both potentiation and depression components of ODP were observed. This was illustrated by the capacity of MD to reduce contralateral (deprived) eye responses from 156.9μV ±13.8 to 94.21μV ±7.89 and to increase ipsilateral eye responses from 87.12μV ±4.90 to 131.52μV ±11.37. Paired t-tests indicated that both changes reached statistical significance (contra, t=5.21, df=6, p=0.001; ipsi, t=−3.67, df=6, p=0.01). In the alcohol-treated group, we also observed a reduction in contralateral (deprived) eye responses (t=2.96, df=9, p=0.01), with VEPs decreasing from 125.74μV ±13.49 to 96.6μV ± 6.05. However, this change was not accompanied by a significant increase in ipsilateral eye responses (t = −1.16, df=9, p = 0.27), as amplitudes changed only from 89.69μV ± 12.4 to 97.53μV ± 10.26. These findings suggest that early alcohol exposure leads to a remarkable impairment in Pc-ODP, but not Dc-ODP. This difference appears to be the cause of the smaller CBI changes seen in alcohol exposed animals.
Figure 4.

Changes in individual eye responses after 5–10 days of MD. After 5–10 days of MD saline treated animals (n = 9) exhibited a significant increase in ipsilateral (87.12μV ±4.90 to 131.52μV ±11.37) eye responses, as well as a significant decrease in contralateral eye responses (156.9μV ±13.8 to 94.21μV ±7.89) (A). These bidirectional changes can be seen in traces from a representative animal. In contrast, ethanol exposed animals (n = 14) demonstrated no change in ipsilateral eye responses (89.69μV ± 12.4 to 97.53μV ± 10.26), yet showed a significant decrease in contralateral eye response (125.74μV ±13.49 to 96.6μV ± 6.05) (B). * p = 0.01 ** p < 0.001
It is well established that the two components of ODP do not have the same time course (Frenkel and Bear, 2004). The potentiation component is observed after 5 days of MD while the depression component can start 24 hours after MD with decreased VEPs reliably recorded after 3 days (Smith, et al. 2009). Therefore, we considered the possibility that alcohol could delay the early stages of the depression component. To test this we recorded from mice after only 3 days of MD (Fig. 5). When we examined CBI values for alcohol exposed animals, we observed a significant shift in CBI values from 1.67 ± 0.2 to 1.07 ± 0.23 (n = 4). The magnitude of these CBI shifts closely mirrored those observed in saline treated animals with 3 days of MD. The CBIs of these animals shifted from 1.62 ±0.49 to 0.94 ±0.18 (n = 5, F= 16.995, df=1, p=0.001, ANOVA, Fig. 5b). Indeed, when we compared the percent change between pre- and post MD, we saw no significant difference between control and alcohol exposed animals (t= −0.46, df= 8, p= 0.65). These results indicate that after 3 days of MD, alcohol and saline treated animals demonstrate a similar magnitude of response changes (Fig. 5).
Figure 5.

Effect of 3 days of MD on CBI. Both alcohol (1.67± 0.2; 1.07 ± 0.23; n = 4) and saline (1.62 ± 0.49 to 0.94 ±0.18; n = 5) treated animals exhibited a significant decrease in CBI values (A). The magnitude of this change was similar in both groups (B). * p = 0.001
To see if these changes in responses were indeed similar, we looked at the effect of 3 days of MD on the individual eye responses (Fig. 6). This period of MD caused a decrease in contralateral eye responses in alcohol animals, with contralateral eye amplitudes shifting from 126.5μV ±7.56 to 90.7μV ±4.44 (t=4.15, df=3, p = 0.02). In contrast, the increase in ipsilateral (experienced) eye VEP amplitudes after 3 day MD was not significant, 77.43μV ±6.09 to 85.01μV ±10.09 (t=−0.04, df=3, p= 0.96, Fig. 6b). Curiously, in saline treated animals, 3 day MD did not significantly alter ipsilateral eye VEP amplitudes; 133.73μV ±44.35 to 163.75μV ±33.71 (t= −0.78, df= 4, p =0.47), or contralateral eye VEP amplitudes (169.51μV ±44.3 to 146μV ±22.6; t= 1.42, df = 4, p = 0.22). There is some variation in the VEP amplitude, and MD induced changes in VEP amplitude within these groups. This can be attributed to our use of animals from multiple litters (litter number = 6), as well as minor differences in electrode implant tolerance, which is normal for this type of chronic implant recording. To account for this variability, most studies using 3 days of MD normalize contralateral responses to the ipsilateral ones (Frenkel and Bear 2004). This normalization is based on the assumption that the ipsilateral eye responses do not change after short periods of MD. This type of normalization cannot be done in the 5 day MD paradigm, since contralateral and ipsilateral eye responses change. Fig. 7 shows the responses of saline treated animals after normalization. Note that all animals presented a decrease in contralateral responses. Our results indicate that early ethanol exposure results in a disruption of the potentiation, but not the depression component of ODP, as assessed by VEP recordings.
Figure 6.

Changes in individual eye responses after 3 days of MD. After 3 days of MD, saline treated animals exhibited no change in response amplitude of the ipsilateral eye (133.73μV ±44.35; 163.75μV ±33.71; n = 5), yet the contralateral eye (169.51μV ±44.3; 146μV ±22.6; n = 5) exhibited a trend toward decreased response amplitude (A). This change can be seen in VEP responses from a representative animal. Ethanol exposed animals also showed a significant decrease in their contralateral eye responses (126.5μV ±7.56 to 90.7μV ±4.44, n=4) after MD, but no changes were seen in ipisilateral responses (77.43μV ±6.09; 5.01μV ±10.09; n= 5) (B). This change can be seen in VEP traces from a representative animal. * p = 0.01.
Figure 7.

Normalized contralateral eye responses after 3 days of MD. When contralateral eye VEPs are normalized to ipsilateral eye responses, the decrease in contralateral eye responses for all groups becomes apparent (A). With this data transformation, saline and alcohol treated animals exhibit a similar percent change in VEP amplitude from before to after MD (B). * p = 0.04, ** p = 0.007
The results obtained from VEP recording demonstrated that early alcohol exposure affects Dc-ODP but not Pc-ODP. This finding was intriguing because in our previous study using optical imaging of intrinsic signals (OI), alcohol treated animals did not show an OD shift after 10 days of MD (Lantz, et a; 2012). While these prior findings strongly suggested an effect of alcohol on both Pc-ODP and Dc-ODP, it is difficult to tease apart the contributions of these individual components using OI. To test whether alcohol affects Dc-ODP using optical imaging of intrinsic signals, shorter periods of MD (less than 5 days) would be required. This is particularly relevant since VEPs and intrinsic signals record from different layers (2/3 and 4 respectively) which could indicate a layer-specific effect of alcohol exposure.
In an independent experiment, our collaborators in the Majewska laboratory tested the effect of early alcohol exposure on Dc-ODP using optical imaging of intrinsic signals (See methods, experiment 2). Saline injected control animals and alcohol injected animals were either monocularly deprived for 4 days or left non-deprived. A 2-way ANOVA showed a significant main effect of deprivation (F=13.85, df=1, p=0.002) but not of treatment (F=0.1429, df=1, p=0.71) or an interaction (F=2.316, df=1, p=0.15). Post-hoc analysis demonstrated that control animals monocularly deprived for 4 days exhibited a significantly lower CBI value (1.134 ± 0.05 n=5) compared to non-deprived (ND) control animals (1.967 ± 0.32, n=3, Bonferroni test, p=0.02), indicative of Dc-OPD. In contrast, a reduction in CBI values was not observed in animals previously exposed to alcohol. The CBI values were 1.785 ± 0.07 n= 4 for non-deprived EtOH mice and 1.436 ± 0.15 n=7 for 4 day MD EtOH mice (Bonferroni test, p=0.67) (Fig. 8).
Figure 8.
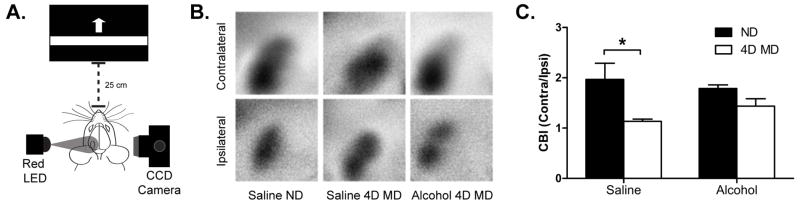
Changes in ocular dominance after 4 days of MD, assessed by optical imaging of intrinsic signals. Mice were treated with 1.8g/kg x 2 of ethanol or saline vehicle control daily from P4–P9. Note that in all previous figures animals were exposed to 5g/kg of ethanol or control saline on P5, P7, and P9. A) Diagram of intrinsic signal optical imaging set-up. B) Representative average contralateral response and average ipsilateral response amplitude maps used for CBI calculation. C) Compared to non-deprived saline animals (1.97 ± 0.32, n=3), 4 day MD saline animals exhibited a significantly lower CBI (1.134 ± 0.05 n=5). In contrast, after alcohol treatment, 4 days of MD did not result in a significant decrease in CBI (1.785 ± 0.07, n= 4; 1.44 ± 0.15, n=7). * p < 0.05. Graphed mean ± SEM.
DISCUSSION
Here we showed that early alcohol exposure leads to long lasting effects on Pc-ODP but not Dc-ODP in layer 4. Surprisingly, the effect of early alcohol exposure on ODP as assessed by VEPs seems to be much less robust than what our laboratory has previously observed using optical imaging of intrinsic signals (Lantz et al., 2012). In our previous study, 10 days of MD did not result in any distinguishable change in CBIs in alcohol exposed mice, suggesting that both Pc- ODP and Dc-ODP were disrupted. In the present study we confirmed that early alcohol exposure disrupts Dc-ODP assessed by optical imaging of intrinsic signals after 4 days of MD (Fig. 8). Importantly, this result was obtained through an independent experiment carried out by the Majewska lab and with a slightly different method of alcohol exposure. In both alcohol administration paradigms alcohol was given between P4 and P9; in the Majewska lab 3.6g/kg of alcohol was administered every day while in the Medina lab 5g/kg was administered every-other-day during this time frame. Although the paradigms differ slightly, they both result in abnormal ODP. Both the Medina and Majewska ethanol paradigms were chosen to specifically induce aberrant apoptosis via BAC levels over 200 mg/dL for an extended period of time (Ikonomidou, et al. 2000; Lantz, et al. 2012) during the brain growth spurt. It’s possible that there was more ethanol induced apoptosis in the Majewska paradigm via the daily treatment with ethanol which may make their paradigm more severe, we believe this difference in daily versus every-other-day treatment is compensated for by the increased ethanol dose given by the Medina lab. Interestingly, the fact that optical imaging of intrinsic signals and VEPs register responses from layers 2/3 and 4 respectively (Cooke and Bear, 2010;Frenkel and Bear, 2004;Frostig and Chen-Bee, 2009; Lee et al., 2012; McCurry et al., 2010; Smith et al., 2009), allows us to hypothesize that alcohol may affect extragranular layers more than the granular layer. This hypothesis is also supported by prior studies demonstrating that Dc-ODP relies on different mechanisms in layer 2/3 in layer 4 (Crozier, et al. 2007). This work can be further extended and confirmed using laminar recordings of VEP and single units in the primary visual cortex to further confirm layer specific deficits induced by early alcohol exposure.
Dc-ODP begins during the first 24hrs after MD, and is clear after 3 days (Frenkel and Bear 2004). This effect is mechanistically different across cortical layers. For instance, in layer 2/3, Dc-ODP relies on cannabinoid receptors and is independent of AMPA receptor internalization. In contrast, in layer 4 the depression component is independent of cannabinoid receptors, and appears to rely on traditional LTD mechanisms, involving clathrin dependent AMPA receptor internalization (Crozier, et al. 2007, Smith, et al. 2009).
Conversely, Pc-ODP is consistently detected after 5 days of MD (Frenkel and Bear 2004). In contrast to the depression component, potentiation of non-deprived eye responses appears to share similar mechanisms in layer 2/3 and layer 4, as both rely on AMPA receptor insertion at the synapse (Heynen and Bear 2001).
We observed that early alcohol exposure affects Pc-ODP but not Dc-ODP in layer 4. The fact that VEPs recordings are derived mostly from layer 4 (Cooke and Bear 2010), where potentiation and depression rely on insertion and internalization of AMPARs respectively suggests that alcohol may affect glutamatergic transmission (Bellinger, et al. 2002, Rema and Ebner 1999, Savage, et al. 1991), possibly disrupting the NR2A/B ratio which is crucial in maintaining normal ODP (Cho, et al, 2010). Ethanol’s effects on Dc-ODP in layer 2/3, but not in layer 4, suggest a possible action on endocannabinoid transmission. In fact a series of studies by Basavarajappa’s group suggest a strong effect of developmental alcohol on anandamide-CB1r signaling (Basavarajappa et al., 2008; Subbanna et al., 2013).
Pc-ODP and Dc-ODP share similar mechanisms with LTP and LTD respectively (Crozier et al., 2007; Heynen and Bear, 2001; Yoon et al., 2009). Most studies on the effects of early alcohol exposure in LTP and LTD have been done in rats using hippocampal slice preparations. Studies using a variety of alcohol doses, regimens and time periods showed unequivocally that alcohol can disrupt LTP in hippocampus (Izumi et al., 2005; Puglia and Valenzuela, 2010b; Puglia and Valenzuela, 2010a; Richardson et al., 2002; Savage et al., 2010; Sutherland et al., 1997). Interestingly, in this region, pharmacological potentiation of AMPA receptor responses reverses the deficits of alcohol exposed animals in the Morris water maze, a behavior that is dependent on LTP in the hippocampus (Vaglenova et al., 2008). Surprisingly, few studies have investigated the effects of alcohol on LTD. Using rat hippocampal slice preparations, Izumi and collaborators demonstrated that exposure to alcohol at P0 or P7 (2 injections of 2.5g/kg s.c.; BEC = ~500 mg/dl) can disrupt LTD in rats at P30 (Izumi, et al. 2005). In contrast, according to a different study, exposure to moderate levels of alcohol (BEC= ~200mg/Kg) encompassing rat gestation did not affect LTD in vivo (Titterness and Christie 2008). Much less is known about the effects of early alcohol exposure on LTP and LTD in the neocortex. Our findings suggest that ethanol may severely affect LTP but that the effects on LTD are likely layer specific.
In conclusion, we have shown that early alcohol exposure impairs ODP in layer 4 by disrupting the potentiation of the experienced eye responses, while sparing the depression of the deprived eye responses. However, in layer 2/3, depression of responses was disrupted suggesting a layer specific effect of developmental alcohol exposure. Together with our previous study, these results indicate that animals exposed to early alcohol have impaired plasticity in layer 2/3 (as show by ODP and MD), while they maintain LTD-like plasticity in layer 4 (Lantz, 2012). This is the first study to report a disconnect between layer specific plasticity in the primary visual cortex. These findings contribute to our understanding of neuronal plasticity deficits in FASD.
Acknowledgments
This work was supported by NIH (NIAAA) grants R01AA022455 and R01AA13023 and to A.E.M, and R21 AA020855 to A.K.M. This work was also supported by NEI R01 EY019277 to A.K.M. and T32 ES007026 (E.L.W.). The Majewska laboratory thanks Judith D. Sharp and the laboratory of Linda P. Spear for assistance with blood alcohol analysis.
Reference List
- Basavarajappa BS, Nixon RA, Arancio O. Acute ethanol supress glutamatergic neurotransmission through endocannabinoids in hippocampal neurons. J Neurochem. 2008;107:1001–1013. doi: 10.1111/j.1471-4159.2008.05685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi N, Pizzorusso T, Ratto GM, Maffei L. Molecular basis of plasticity in the visual cortex. Trends Neurosci. 2003;26:369–378. doi: 10.1016/S0166-2236(03)00168-1. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T. Optical imaging of intrinsic signals as a tool to visualize the functional architecture of adult and developing visual cortex. Arzneimittelforschung. 1995;45:351–356. [PubMed] [Google Scholar]
- Burden MJ, Andrew C, Saint-Amour D, Meintjes EM, Molteno CD, Hoyme HE, Robinson LK, Khaole N, Nelson CA, Jacobson JL, Jacobson SW. The effects of fetal alcohol syndrome on response execution and inhibition: an event-related potential study. Alcohol Clin Exp Res. 2009 Nov;33(11):1994–2004. doi: 10.1111/j.1530-0277.2009.01038.x. [DOI] [PubMed] [Google Scholar]
- Chiodo LM, Janisse J, Delaney-Black V, Sokol RJ, Hannigan JH. A metric of maternal prenatal risk drinking predicts neurobehavioral outcomes in preschool children. Alcohol Clin Exp Res. 2009 Apr;33(4):634–44. doi: 10.1111/j.1530-0277.2008.00878.x. [DOI] [PubMed] [Google Scholar]
- Coffman BA, Kodituwakku P, Kodituwakku EL, Romero L, Sharadamma NM, Stone D, Stephen JM. Primary visual response (M100) delays in adolescents with FASD as measured with MEG. Hum Brain Mapp. 2013 Nov;34(11):2852–62. doi: 10.1002/hbm.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SF, Bear MF. Visual experience induces long-term potentiation in the primary visual cortex. J Neurosci. 2010;30:16304–16313. doi: 10.1523/JNEUROSCI.4333-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier RA, Wang Y, Liu CH, Bear MF. Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc Natl Acad Sci USA. 2007;104:1383–1388. doi: 10.1073/pnas.0609596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BSS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of Fragile X Syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel M, Bear MF. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron. 2004;44:917–933. doi: 10.1016/j.neuron.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Frostig RD, Chen-Bee C. Visualizing adult cortical plastcity using intrinsic signal optical imaging. In: Frostig RD, editor. In vivo optical imaging of brain function. Boca raton, FL: CRC Press; 2009. [PubMed] [Google Scholar]
- Hannigan JH, Armant DR. Alcohol in pregnancy and neonatal outcome. Semin Neonatol. 2000;5:243–254. doi: 10.1053/siny.2000.0027. [DOI] [PubMed] [Google Scholar]
- Heynen AJ, Bear MF. Long-term potentiation of thalamocortical transmission in the adult visual cortex in vivo. J Neurosci. 2001;21:9801–9813. doi: 10.1523/JNEUROSCI.21-24-09801.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol. 1970;206:419–436. doi: 10.1113/jphysiol.1970.sp009022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN, LeVay S. Plasticity of ocular dominance columns in monkey striate cortex. Philos Trans R Soc Lond B Biol Sci. 1977;278:377–409. doi: 10.1098/rstb.1977.0050. [DOI] [PubMed] [Google Scholar]
- Hug TE, Fitzgerald KM, Cibis GW. Clinical and electroretinographic findings in fetal alcohol syndrome. J AAPOS. 2000;4(4):200–410. doi: 10.1067/mpa.2000.105278. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Hörster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–60. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Kitabayashi R, Funatsu M, Izumi M, Yuede C, Hartman RE, Wozniak DF, Zorumski CF. A single day of ethanol exposure during development has persistent effects on bidirectional plasticity, N-methyl-D-aspartate receptor function and ethanol sensitivity. Neuroscience. 2005;136:269–279. doi: 10.1016/j.neuroscience.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Lantz CL, Pulimood NS, Rodrigues-Junior WS, Chen CK, Manhaes AC, Kalatsky VA, Medina AE. Visual defects in a mouse model of fetal alcohol spectrum disorder. Front Pediatr. 2014;2:1–8. doi: 10.3389/fped.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantz CL, Wang W, Medina AE. Early alcohol exposure disrupts visual cortex plasticity in mice. Int J Dev Neurosci. 2012;30:351–357. doi: 10.1016/j.ijdevneu.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, Mattson SN, Riley EP, Jones KL, Adnams CM, May PA, Bookheimer SY, O'Connor M, Narr KL, Kan E, Abaryan Z, Sowel EW. A longitudinal study of the long-term consequences of drinking during pregnancy: heavy in utero alcohol exposure disrupts the normal processes of brain development. J Neurosci. 2012;32:15243–15251. doi: 10.1523/JNEUROSCI.1161-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Koh D, Jo A, Lim HY, Jung YJ, Kim CK, Seo Y, Im CH, Kim BP, Suh M. Depth-dependent cerebral hemodynamic responses following direct cortical electrical stimulation (DCES) revealed by in vivo dual-optical imaging techniques. Optics Express. 2012;20:6932–6943. doi: 10.1364/OE.20.006932. [DOI] [PubMed] [Google Scholar]
- Lehmann K, Lowel S. Age-dependent ocular dominance plasticity in adult mice. PLoS One. 2008;3 doi: 10.1371/journal.pone.0003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurry CL, Shepherd JD, Tropea D, Wang KH, Bear MF, Sur M. Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat Neurosci. 2010;13:450–457. doi: 10.1038/nn.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina AE. Fetal Alcohol Spectrum Disorders and Abnormal Neuronal Plasticity. Neuroscientist. 2011;17 doi: 10.1177/1073858410383336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina AE, Krahe TE, Coppola DM, Ramoa AS. Neonatal alcohol exposure induces long-lasting impairment of visual cortical plasticity in ferrets. J Neurosci. 2003;23:10002–10012. doi: 10.1523/JNEUROSCI.23-31-10002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolozza A, Titman R, Brien D, Munoz DP, Reynolds JN. Altered accuracy of saccadic eye movements in children with fetal alcohol spectrum disorder. Alcohol Clin Exp Res. 2013 Sep;37(9):1491–8. doi: 10.1111/acer.12119. [DOI] [PubMed] [Google Scholar]
- Paul AP, Pohl-Guimaraes F, Krahe TE, Filgueiras CC, Lantz CL, Colello RJ, Wang W, Medina AE. Overexpression of serum response factor restores ocular dominance plasticity in a model of fetal alcohol spectrum disorders. J Neurosci. 2010;30:2513–2520. doi: 10.1523/JNEUROSCI.5840-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglia MP, Valenzuela CF. Ethanol acutely inhibits ionotropic glutamate receptor-mediated responses and long-term potentiation in the developing CA1 hippocampus. Alcohol Clin Exp Res. 2010a;34:594–606. doi: 10.1111/j.1530-0277.2009.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglia MP, Valenzuela CF. Repeated third trimester-equivalent ethanol exposure inhibits long-term potentiation in the hippocampal CA1 region of neonatal rats. Alcohol. 2010b;44:283–290. doi: 10.1016/j.alcohol.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DP, Byrnes ML, Brien JF, Reynolds JN, Dringenberg HC. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur J Neurosci. 2002;16:1593–1598. doi: 10.1046/j.1460-9568.2002.02214.x. [DOI] [PubMed] [Google Scholar]
- Savage DD, Rosenberg M, Wolff C, Akers K, El-Emawy A, Staples M, Varaschin R, Wright C, Seidel J, Caldwell KK, Hamilton DA. Effects of a novel cognition-enhancing agent on fetal ethanol induced learning deficits. Alcohol Clin Exp Res. 2010;34:1793–1802. doi: 10.1111/j.1530-0277.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GB, Heynen AJ, Bear MF. Bidirectional synaptic mechanisms of ocular dominance plasticity in visual cortex. Philos Trans R Soc Lond B Biol Sci. 2009;364:357–367. doi: 10.1098/rstb.2008.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strömland K. Ocular abnormalities in the fetal alcohol syndrome. Acta Ophthalmol Suppl. 1985;171:1–50. [PubMed] [Google Scholar]
- Stromland K, Pinazo-Duran M. Ophthalmic involvement in the fetal alcohol syndrome: clinical and animal model studies. Alcohol. 2002;37(1):2–810. doi: 10.1093/alcalc/37.1.2. [DOI] [PubMed] [Google Scholar]
- Subbanna S, Shivakumar M, Psychoyos D, Xie S, Basavarajappa BS. Anadamide-CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic and memory deficits. J Neurosci. 2013;33:6350–6366. doi: 10.1523/JNEUROSCI.3786-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland RJ, McDonald RJ, Savage DD. Prenatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal synaptic plasticity in adult offspring. Hippocampus. 1997;7:232–238. doi: 10.1002/(SICI)1098-1063(1997)7:2<232::AID-HIPO9>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Vaglenova J, Pandiella N, Wijayawardhane N, Vaithianathan T, Birru S, Breese C, Suppiramaniam V. Aniracetam reversed learning and memory deficits following prenatal ethanol exposure by modulating functions of synaptic AMPA receptors. Neuropsychopharmacology. 2008;33:1071–1083. doi: 10.1038/sj.npp.1301496. [DOI] [PubMed] [Google Scholar]
- Vernescu RM, Adams RJ, Courage ML. Children with fetal alcohol spectrum disorder show an amblyopia-like pattern of vision deficit. Dev Med Child Neurol. 2012 Jun;54(6):557–62. doi: 10.1111/j.1469-8749.2012.04254.x. [DOI] [PubMed] [Google Scholar]
- Uecker A, Nadel L. Spatial locations gone awry: object and spatial memory deficits in children with fetal alcohol syndrome. Neuropsychologia. 1996 Mar;34(3):209–23. doi: 10.1016/0028-3932(95)00096-8. [DOI] [PubMed] [Google Scholar]
- Vaurio L, Riley EP, Mattson SN. Neuropsychological comparison of children with heavy prenatal alcohol exposure and an IQ-matched comparison group. J Int Neuropsychol Soc. 2011 May;17(3):463–73. doi: 10.1017/S1355617711000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Riday TT, Condon KH, Roberts AC, Bernardo DR, Prakash R, Weinberg RJ, Ehlers MD, Philpot BD. Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci. 2009;12:777–783. doi: 10.1038/nn.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]