

Abstract
Background
Acetaldehyde, the toxic ethanol metabolite, disrupts intestinal epithelial barrier function. Aldehyde dehydrogenase (ALDH) detoxifies acetaldehyde into acetate. Sub populations of Asians and Native Americans show polymorphism with loss of function mutations in ALDH2. We evaluated the effect of ALDH2 deficiency on ethanol-induced disruption of intestinal epithelial tight junctions and adherens junctions, gut barrier dysfunction and liver injury.
Methods
Wild type and ALDH2 deficient mice were fed (1–6%) in Lieber-DeCarli diet for 4 weeks. Gut permeability in vivo measured by plasma-to-luminal flux of FITC-inulin, tight junction and adherens junction integrity analyzed by confocal microscopy and liver injury was assessed by analysis of plasma transaminase activity, histopathology and liver triglyceride.
Results
Ethanol feeding elevated colonic mucosal acetaldehyde, which was significantly greater in ALDH2 deficient mice. ALDH2−/− mice showed a drastic reduction in the ethanol diet intake. Therefore, this study was continued only in wild type and ALDH2+/− mice. Ethanol feeding elevated mucosal inulin permeability in distal colon, but not in proximal colon, ileum or jejunum of wild type mice. In ALDH2+/− mice, ethanol-induced inulin permeability in distal colon was not only higher than that in wild type mice, but inulin permeability was also elevated in the proximal colon, ileum and jejunum. Greater inulin permeability in distal colon of ALDH2+/− mice was associated with a more severe redistribution of tight junction and adherens junction proteins from the intercellular junctions. In ALDH2+/− mice, but not in wild type mice, ethanol feeding caused a loss of junctional distribution of tight junction and adherens junction proteins in the ileum. Histopathology, plasma transaminases and liver triglyceride analyses showed that ethanol-induced liver damage was significantly greater in ALDH2+/− mice compared to wild type mice.
Conclusion
These data demonstrate that ALDH2 deficiency enhances ethanol-induced disruption of intestinal epithelial tight junctions, barrier dysfunction and liver damage.
Introduction
Alcoholic liver disease (ALD) is associated with the disruption of intestinal epithelial barrier function and increased permeability to bacterial toxins from the colonic lumen into the systemic circulation (Rao, 2009, Rao et al., 2004). Endotoxemia was shown not only in alcoholics with the symptoms of liver disease (Bode et al., 1984, Rao et al., 2004, Schafer et al., 2002), but also in experimental models of ALD (Jokelainen et al., 2001, Mathurin et al., 2000, Nanji et al., 2002, Rao et al., 2004, Rao, 2009). Endotoxin-mediated Kupffer cell activation and inflammatory reactions are considered as key steps in the mechanisms involved in the development of ALD (Duryee et al., 2004). Therefore, alcohol-induced gut barrier disruption is an important event in the initiation and progression of ALD.
Epithelial tight junctions confer barrier function in the intestinal mucosa by forming a diffusion barrier to toxins, allergens and pathogens from the gut lumen into the tissue and systemic circulation (Anderson et al., 1991). Tight junctions are multi protein complexes assembled at the apical end of epithelial cells. Occludin, claudins, tricellulin and junctional adhesion molecules are the transmembrane proteins of tight junctions (Anderson and Van Itallie, 2009), the intracellular domains of which bind to adapter proteins such as ZO-1, ZO-2 and ZO-3 (Furuse et al., 1994). Adapter proteins interact with numerous plaque proteins and anchor the tight junction protein complexes into the actin cytoskeleton (Rodgers and Fanning, 2011, Turner, 2006, Madara, 1987). Adherens junctions that lie beneath the tight junctions do not form physical barrier to diffusion of macromolecules, but they indirectly regulate the integrity of tight junctions (Gumbiner et al., 1988a). E-cadherin is the transmembrane protein of adherens junctions. The intracellular domain of E-cadherin interacts with catenins such as α-catenin, β-catenin and γ-catenin (Lilien et al., 2002). Ethanol consumption is known to disrupt both tight junctions (Atkinson and Rao, 2001, Basuroy et al., 2005, Sheth et al., 2004) and adherens junctions (Atkinson and Rao, 2001, Basuroy et al., 2005, Sheth et al., 2007).
Ethanol is metabolized by alcohol dehydrogenase (ADH) to toxic acetaldehyde, which is oxidized to non-toxic acetate by aldehyde dehydrogenase (ALDH) (Seitz and Stickel, 2010). A significant body of evidence indicates that ethanol metabolism into acetaldehyde plays a crucial role in ethanol-induced tissue injury (Boffetta and Hashibe, 2006, Seitz and Stickel, 2007). Acetaldehyde, but not ethanol, disrupts tight junctions in Caco-2 cell monolayers (Atkinson and Rao, 2001, Rao, 2009, Rao, 1998, Seth et al., 2004, Sheth et al., 2004), and contributes to barrier dysfunction in rat colon (Ferrier et al., 2006). Acetaldehyde modulates drug permeability in vitro and bioavailability in vivo (Fisher et al., 2010). Therefore, factors that affect acetaldehyde levels in tissues may have significant impact on ethanol toxicity. Polymorphism in ADH and ALDH in subpopulations of Asians and Native Americans (Morimoto and Takeshita, 1996, Yokoyama et al., 2003, Eriksson, 2001, Wall et al., 1997) raised the concern over the sensitivity to and toxicity of ethanol consumption in these populations. Indeed, loss of function mutations of ALDH2 in Asian subpopulations has shown an increased sensitivity to alcohol consumption. Deletion of ALDH2 gene in mice was found to exhibit alcohol avoidance and higher accumulation of acetaldehyde in brain and liver (Isse et al., 2002).
In the present study, we evaluated the effect of ethanol on gut permeability, tight junction and adherens junction integrity, and liver injury in ALDH2 deficient mice using chronic ethanol feeding model. Results presented do support our hypothesis that ALDH2 deficiency increases sensitivity to gut and liver injury by chronic alcohol feeding.
Materials and Methods
Chemicals
Maltose dextrin, feeding tubes and holders were purchased from Bioserv (Flemington, NJ). Lieber DeCarli liquid diet (Dyet #717780) was purchased from Dyets Inc. (Bethlehem, PA). EnzyChrom Alanine transaminase (EALT-100) and EnzyChrom Aspartate transaminase (EAST-100) assay kits were purchased from BioAssay systems (Hayward, CA). Triglyceride reagent kit set was purchased from Pointe Scientific Inc., (Canton, MI). Hoechst 33342 dye was from Life technologies (Grand Island, NY). All other chemicals were purchased from either Sigma Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Tustin, CA).
Antibodies
Anti-ZO-1, anti-occludin, and anti-claudin-3 antibodies were purchased from Invitrogen (Carlsbad, CA). Anti E-Cadherin and anti β-catenin antibodies were purchased from BD Biosciences (Billerica, MA). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG, HRP-conjugated anti-rabbit IgG and anti-β-actin antibodies were obtained from Sigma Aldrich (St. Louis, MO). AlexaFlour-488-conjugated anti-mouse IgG and Cy3-conjugated anti-rabbit IgG were purchased from Molecular Probes (Eugene, OR).
Animals
All animal experiments were performed according to the protocol approved by the University of Tennessee Health Science Center (UTHSC) Institutional Animal Care and Use Committee (IACUC). ALDH2 knockout mice, (C57BL/6 background) generated as described before (Isse et al., 2002), were bred and genotyped to obtain wild type, ALDH2+/− and ALDH2−/− mice. Animals were housed in institutional animal care facility with 12 hours light and dark cycles, and were fed regular laboratory chow until the start of experiments. All mice had free access to standard rodent diet and water before the study.
Ethanol feeding
Adult female (12–14 weeks age) wild type, ALDH2+/− and ALDH2−/− mice were fed Lieber-DeCarli liquid diet that contained alcohol or isocaloric dextrin for 4 weeks. Animals were gradually adapted to an ethanol-containing diet (0% for 2 days, 1% for 2 days and 2% ethanol for 2 days, followed by 4% ethanol for one week, 5% for one week and 6% for one week). Control animals are pair-fed with diets with isocalorically substituted maltose dextrin for ethanol. For acetaldehyde measurement, three days after 4% ethanol diet animals were fasted for 16 hours with ad libitum access to water. Following the fasting period, animals were fed liquid diet with or without 5% ethanol for 2 hours. In all experiments animals were maintained in pairs to facilitate body temperature maintenance.
At the end of the experiment, the gut permeability was measured as described before. Blood samples were collected by cardiac puncture (into heparinized tubes) and were centrifuged 3000 x g for 10 min at 4°C to prepare plasma samples. Pieces (approximately 0.5 cm long) of intestinal segments were fixed in buffered formalin or cryo-fixed in OCT, and rest of the tissues was snap frozen in liquid nitrogen for preparation of detergent-insoluble and soluble fractions of mucosa.
Acetaldehyde assay
To evaluate acetaldehyde levels, plasma and luminal contents were extracted in cold perchloric acid (0.6 M). Colonic mucosa was snap frozen in liquid nitrogen and sonicated in 0.6 M perchloric acid to prepare the mucosal extract. All samples were snap frozen until further analysis. Acetaldehyde levels in different perchloric acid extracts were evaluated by head space Gas Chromatograph (GC) Clarus 500 equipped with Elite BAC-2 capillary column and Flame Ionization Detector (FID), (PerkinElmer Inc. Waltham, MA) (Jokelainen et al., 1996). Samples were melted at room temperature and transferred immediately into gas chromatography vials, which were further heated to 40°C in headspace sampler Turbomatrix 110 (PerkinElmer Inc.) before analysis. Measurements in each sample were done in duplicates. Using TotalChrom software, acetaldehyde calibration curve and the dilution factors acetaldehyde concentrations in samples were calculated.
Gut permeability in vivo
At the end of 4 weeks of ethanol feeding, mice were intravenously injected with FITC-inulin (50 mg/ml solution; 2 μl/g body weight) via tail vein using a restrainer. One hour after injection, blood samples collected by cardiac puncture under isoflurane anesthesia to prepare plasma. Mice were euthanized by cervical dislocation under isoflurane anesthesia. Luminal contents of intestinal segments were flushed with 0.9% saline. Fluorescence in plasma and luminal flushing was measured using a fluorescence plate reader. Fluorescence values in the luminal flushing were normalized to fluorescence values in corresponding plasma samples and calculated as percent of amount injected per gram body weight. This in vivo method to assess intestinal mucosal permeability measures serosal-to-lumen flux of inulin, which involves no physical manipulation of the intestinal segment. Since the permeability through epithelial tight junctions is a matter of simple diffusion, vascular-to-lumen and lumen-to-vascular permeability should be comparable. In general, several in vitro and in vivo methods have been used to assess intestinal permeability, each with its own advantages and disadvantages. Therefore, it is important to confirm permeability changes with microscopic analysis of tight junction integrity by immunofluorescence microscopy. We found that the microscopic results are in full agreement with the permeability results obtained from the method we applied.
Immunofluorescence microscopy
Cryo-sections (10 μm thickness) were fixed in acetone: methanol (1:1) at −20°C for 2 min and rehydrated in PBS (137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM disodium hydrogen phosphate and 1.8 mM potassium dihydrogen phosphate). Sections were permeabilized with 0.2% Triton X-100 in PBS for 10 min and blocked in 4% non-fat milk in Triton-Tris buffer (150 mM sodium chloride containing 10% Tween-20 and 20 mM Tris, pH 7.4). It was then incubated for one hour with the primary antibodies (mouse monoclonal anti-occludin and rabbit polyclonal anti-ZO-1 antibodies or mouse monoclonal E-cadherin and rabbit polyclonal anti-β-catenin antibodies), followed by incubation for one hour with secondary antibodies (AlexaFluor-488-conjugated anti-mouse IgG and cy3-conjugated anti-rabbit IgG antibodies). The fluorescence was examined by using a Zeiss 710 confocal microscope and images from x-y sections (1 μm) were collected using LSM 5 Pascal software. Images were be stacked by using the software, Image J (NIH) and processed by Adobe Photoshop (Adobe Systems Inc., San Jose, CA). All images for tissue samples from different group were collected and processed under identical conditions.
Liver histopathology
Liver tissue was fixed in 10% buffered formalin and 8 μm thick paraffin embedded sections were stained with hematoxylin and eosin. Stained sections were imaged in a Nikon 80Ti microscope using 10X objective lens and a color camera.
Plasma transaminase assay
Plasma aspartate transaminase (AST) and alanine transaminase (ALT) activities were measured by colorimetric assay using EnzyChrom ALT/AST assay kits according to vendor’s instructions.
Oil Red-O staining
To detect fat deposition in the liver, frozen liver sections from both control and ethanol treated groups were fixed for 10 min in 4% Paraformaldehyde, stained with Oil Red O (Sigma-Aldrich, St. Louis, MO, USA) and then rinsed with 60% isopropanol. Nuclei were lightly stained with hematoxylin stain. Images collected in a Nikon 80Ti microscope using 10X objective lens and a color camera.
Triglyceride assay
Quantitation of liver triglycerides was determined by using GPO method using an assay kit from Beckman Coulter (Brea, CA). Liver lipids were extracted by digesting the tissue with 3 M potassium hydroxide (in 65% ethanol) for 1 hour at 70°C at room temperature for 24 hours. Hepatic triglycerides were measured by enzymatic hydrolysis of triglycerides to glycerol and free fatty acids followed by colorimetric measurement (at 540 nm wavelength) of glycerol. Values for hepatic triglycerides were expressed as mg triglyceride/g liver tissue.
Statistical analyses
Values are expressed as mean ± SE of 4–8 animals. Statistical analysis was performed by Student’s t-test. Statistical significance was assessed at 95% confidence level.
Results
ALDH2 deficient mice accumulate acetaldehyde at a higher level in colonic mucosa
ALDH2 is an important enzyme involved in acetaldehyde metabolism into acetate in liver and gastrointestinal tissues (Seitz and Stickel, 2010). Polymorphism with loss function mutation in ALDH2 in subpopulations of Asians and Native Americans raised our interest in understanding the role of acetaldehyde in tissue injury. ALDH2 is the primary enzyme involved in acetaldehyde metabolism, and hence down regulation of ALDH2 is expected to raise the tissue level of acetaldehyde following alcohol consumption. We measured the levels of acetaldehyde in colonic tissue in wild type and ALDH2 deficient mice following ethanol diet feeding for 2 hours. Wild type, ALDH2+/− and ALDH2−/− mice were subjected to chronic ethanol feeding as described in methods section. Diet intake in ALDH2−/− mice was dramatically reduced when ethanol concentration in diet was increased to 4%. Therefore, this experiment was terminated at 3 days after 4% ethanol feeding.
After 3 days on 4% ethanol diet, animals were fasted overnight and allowed ad libitum access to liquid diet with or without 5% ethanol for 2 hours. Levels of ethanol in plasma and acetaldehyde in colonic tissue extracts and luminal contents were measured. Diet intake during two-hour period was slightly low in ethanol-fed ALDH2+/− and ALDH2−/− mice compared to ethanol-fed wild type mice (Fig. 1A). Plasma ethanol concentration was also significantly low in ethanol-fed ALDH2+/− and ALDH2−/− mice than that in ethanol-fed wild type mice (Fig. 1B). Plasma acetaldehyde concentration was also significantly higher in ALDH2+/− compared to that in wild type mice, and further higher in ALDH2−/− mice compared to that in ALDH2+/− mice. However, the background noise of assay in plasma was unusually high, likely due to plasma components interfering with the assay. Therefore, data is not included in this study. Ethanol feeding significantly increased acetaldehyde levels in colonic mucosa in wild type mice. Despite slight reduction in diet intake and plasma ethanol concentration, the levels of acetaldehyde in colonic tissue were several folds greater in ALDH2+/− and ALDH2−/− mice (Fig. 1C). Acetaldehyde level in colonic tissue was significantly greater even in non-ethanol ALDH2−/− mice. Acetaldehyde levels in colonic luminal contents in ethanol-fed wild type, ALDH2+/− and ALDH2−/− mice were not different from each other (Fig. 1D).
Fig. 1. ALDH2 deficient mice accumulate higher level of acetaldehyde in colonic mucosa.
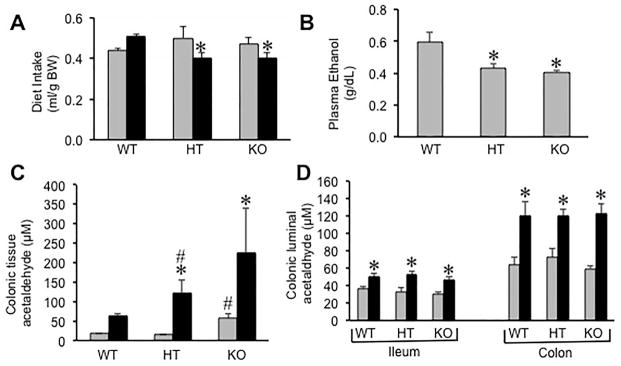
Wild type (WT), ALDH2+/− (HT) and ALDH2−/− (KO) mice were fed Lieber-DeCarli liquid diet with (black bars) or without (gray bars) 4% ethanol as described in the Methods section. After 3 days with 4% ethanol feeding, animals were fasted overnight followed by ad libitum feeding of 5% ethanol diet for two hours. Diet intake (A) and plasma ethanol concentration (B) were measured during the last 2 hours of feeding immediately after overnight fasting. Acetaldehyde levels in colonic mucosal extract (C) and colonic luminal contents (D) were estimated. Values are mean ± SE (n = 6). Asterisks indicate the values that are significantly (p<0.05) different from corresponding control values, and hash tags indicate values that are significantly (p<0.05) different from corresponding WT values.
ALDH2 deficiency enhances ethanol-induced gut permeability
Chronic ethanol feeding disrupts intestinal mucosal barrier function and increases mucosal permeability to macromolecules (Rao, 2009). Although the barrier function of Caco-2 cell monolayers aged for 3 weeks was compromised by ethanol (Elamin et al., 2014), other studies indicated that acetaldehyde, but not ethanol, is the primary mediator of increased paracellular epithelial permeability in Caco-2 cell monolayers (Atkinson and Rao, 2001, Rao, 1998, Rao et al., 2004, Suzuki et al., 2008) and rat colon (Ferrier et al., 2006). Therefore, we evaluated the effect of ALDH2 deficiency on gut permeability. This study compared wild type mice with the ALDH2 diet mice. ALDH2−/− mice were excluded as these mice were unable to consume ethanol. This is also true in human subjects who are homozygous for loss function mutation of ALDH2. Therefore, comparison of wild type with ALDH2+/− mice is physiologically relevant. Intestinal permeability evaluated by measuring plasma-to-luminal flux of FITC-inulin in vivo was significantly elevated by ethanol feeding in distal colon of wild type mice, and this effect of ethanol was about two-fold higher in distal colon of ALDH2+/− mice (Fig. 2A). Ethanol feeding showed no significant increase in mucosal inulin permeability in the proximal colon (Fig. 2B), ileum (Fig. 2C) or jejunum (Fig. 2D) in wild type mice. But in ALDH2+/− mice, ethanol induced a significant increase in inulin permeability in proximal colon, ileum and jejunum. Mucosal permeability to inulin was unaffected by ethanol in duodenum (Fig. 2E). Ethanol effect on intestinal permeability in wild type mice was >5-fold in the distal colon and the ethanol effect was confined to distal colon (Fig. 2F). In ALDH2+/− mice, the ethanol effect was nearly 14-fold in the distal colon, and 4–6-fold in jejunum, ileum and proximal colon.
Fig. 2. ALDH2 deficiency promotes ethanol-induced increase in intestinal permeability in mice.
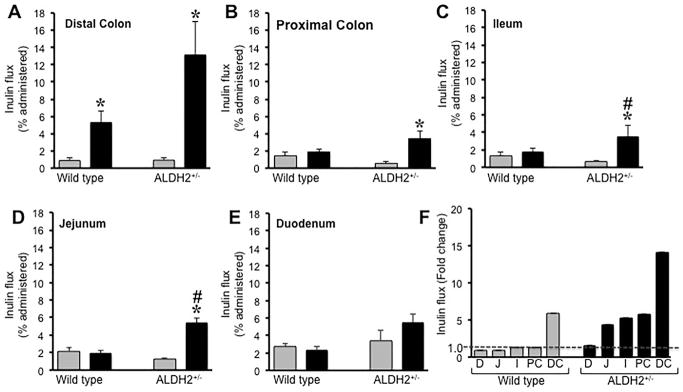
A–E: Wild type and ALDH2+/− mice were fed with Lieber-DeCarli liquid diet with (black bars) or without (gray bars) ethanol (up to 6% as described in the Methods section). Intestinal mucosal barrier function was evaluated by measuring plasma-to-lumen flux of FITC-inulin in distal colon (A), proximal colon (B), ileum (C), jejunum (D) and duodenum (E) as described in Methods section. Values are mean ± SE (n = 8). Asterisks indicate the values that are significantly (p<0.05) different from corresponding values for non-ethanol group, and hash tags indicate the values that are significantly (p<0.05) different from corresponding values for wild type group. F: Inulin permeability in duodenum (D), jejunum (J), ileum (I), proximal colon (PC) and distal colon (DC) was calculated as fold change by dividing values for ethanol group by corresponding values for non-ethanol groups.
ALDH2 deficiency enhances ethanol-induced disruption of tight junctions and adherens junctions
Tight junctions form the primary component of intestinal epithelial barrier function and adherens junctions are known to indirectly regulate the integrity of tight junctions and the epithelial barrier function. Increase in mucosal permeability to inulin suggested that ethanol feeding may disrupt the intestinal epithelial tight junctions and adherens junctions in the current model of ethanol feeding. Therefore, cryosections of distal colon were stained for tight junction (occludin and ZO-1) and adherens junction (E-cadherin and β-catenin) proteins. In the absence of ethanol feeding, occludin and ZO-1 were co-localized at the intercellular junctions of colonic epithelium. Ethanol feeding resulted in a reduction in the levels of junctional localization of tight junction proteins, occludin and ZO-1 in wild type mice (Fig. 3). The loss of junctional localization of occludin and ZO-1 by ethanol feeding was much greater in ALDH2+/− mice. Similarly, ethanol feeding reduced the levels of E-cadherin and β-catenin at the epithelial junctions of distal colon in wild type mice, and this effect was more severe in ALDH2+/− mice (Fig. 4).
Fig. 3. Ethanol-induced disruption of tight junctions in distal colon is more severe in ALDH2 deficient mice.
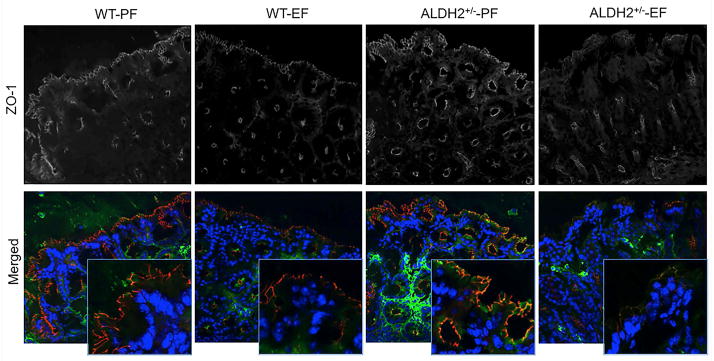
Wild type and ALDH2+/− mice were fed Lieber-DeCarli liquid diet with or without ethanol (up to 6% as described in the Methods section). Cryosections of distal colon were stained for occludin (green) and ZO-1 (red) by immunofluorescence method. Images collected by confocal microscopy. These are the representative images from at least 4 different mice.
Fig. 4. Ethanol-induced disruption of adherens junctions in distal colon is more severe in ALDH2 deficient mice.

Wild type and ALDH2+/− mice were fed Lieber-DeCarli liquid diet with or without ethanol (up to 6% as described in the Methods section). Cryosections of distal colon were stained for E-cadherin (green) and β-catenin (red) by immunofluorescence method. Images collected by confocal microscopy. These are the representative images from at least 4 different mice.
ALDH2 deficiency promotes ethanol-induced adherens junction disruption in mouse ileum
To determine the effect of ethanol feeding on the integrity of tight junctions and adherens junctions of the ileal mucosal epithelium in wild type and ALDH2+/− mice, we stained cryosections of ileum for occludin, ZO-1, E-cadherin and β-catenin. Ethanol feeding did not alter the junctional distribution of occludin and ZO-1 in the ileum of wild type mice, whereas ethanol feeding resulted in a dramatic loss of junctional occludin and ZO1 in the ileum of ALDH2+/− mice (Fig. 5). Similarly, ethanol feeding caused no effect on junctional distribution of E-cadherin and β-catenin in the ileum of wild type mice, but induced a loss of these proteins in the ileum of ALDH2+/− mice (Fig. 6).
Fig. 5. ALDH2 deficiency promotes ethanol-induced disruption of tight junctions in mouse ileum in vivo.
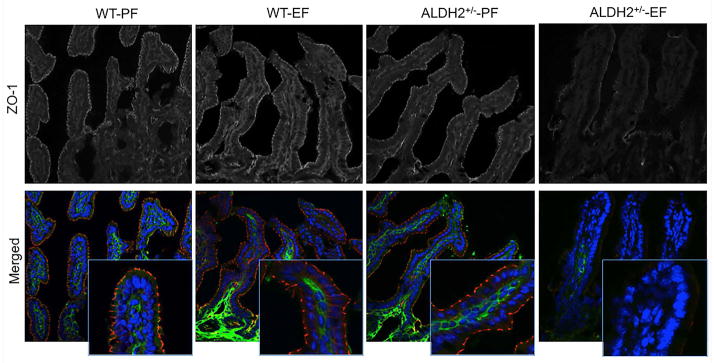
Wild type and ALDH2+/− mice were fed Lieber-DeCarli liquid diet with or without ethanol (up to 6% as described in the Methods section). Cryosections of ileum were stained for occludin (green) and ZO-1 (red) by immunofluorescence method. Images collected by confocal microscopy. These are the representative images from at least 4 different mice.
Fig. 6. ALDH2 deficiency promotes ethanol-induced disruption of adherens junctions in mouse ileum in vivo.
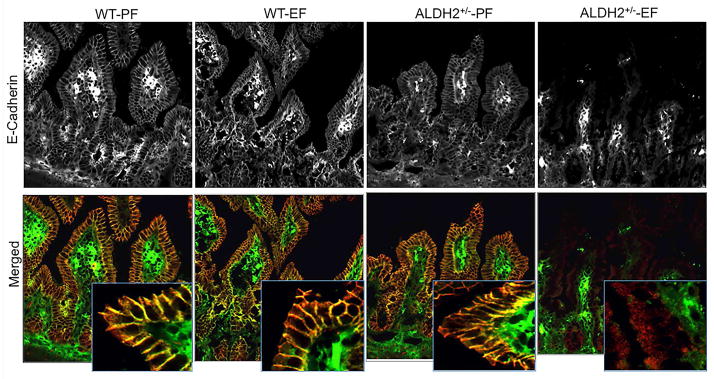
Wild type and ALDH2+/− mice were fed Lieber-DeCarli liquid diet with or without ethanol (up to 6% as described in the Methods section). Cryosections of ileum were stained for E-cadherin (green) and β-catenin (red) by immunofluorescence method. Images collected by confocal microscopy. These are the representative images from at least 4 different mice.
Ethanol-induced liver injury is enhanced by ALDH2 deficiency
Alcoholic liver disease is associated with increased intestinal permeability. Endotoxemia caused by increased intestinal mucosal barrier dysfunction is suggested to play a role in the mechanism of alcoholic liver injury. A greater increase in ethanol-induced intestinal mucosal permeability in ALDH2+/− mice raised the question whether ethanol-induced liver injury is greater in ALDH2+/− mice. Therefore, we evaluated the effect of ethanol feeding on liver injury in wild type and ALDH2+/− mice. Ethanol feeding significantly increased liver weight in ALDH2+/− mice (Fig. 7A), whereas there was no significant change in liver weight in wild type mice. Activities of plasma transaminases, ALT (Fig. 7B) and AST (Fig. 7C), were significantly elevated in ethanol fed wild type mice compared to plasma of pair fed control mice. Ethanol-induced elevation of plasma ALT and AST activities were significantly greater in ALDH2+/− mice compared to those in wild type mice. Histopathology by H & E staining of paraffin sections showed lesions in the liver of ethanol fed wild type mice, and this effect of ethanol was much more severe in ethanol fed ALDH2+/− mice (Fig. 7D).
Fig. 7. Chronic ethanol-induced liver injury is more severe in ALDH2 deficient mice.
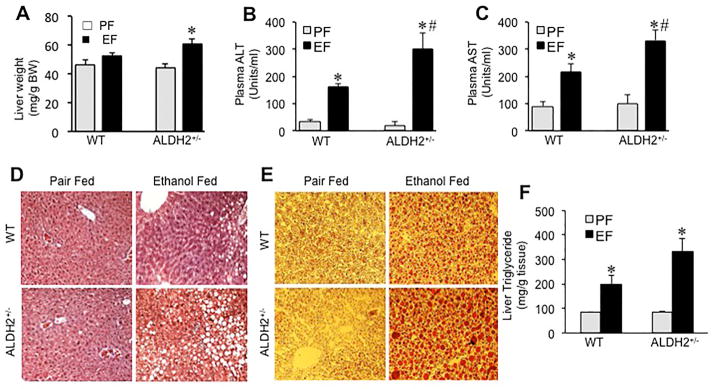
Wild type (WT) and ALDH2+/− mice were fed Lieber-DeCarli liquid diet with (black bars) or without (gray bars) ethanol (up to 6% as described in the Methods section). Liver weights (A) were recorded and H&E stained paraffin sections of liver were imaged by light microscopy (B). Plasma was analyzed for ALT (C) and AST (D) activities. Values are mean ± SE (n = 7). Asterisks indicate the values that are significantly (p<0.05) different from corresponding values for non-ethanol pair fed group, and hash tags indicate the values that are significantly (p<0.05) different from corresponding values for wild type group. E: Cryosections of liver were stained with Oil Red-O and imaged by light microscopy. These are the representative images from at least 3 different mice. F: Liver extracts were assayed for triglyceride content. Values are mean ± SE (n = 6). Asterisks indicate the values that are significantly (p<0.05) different from corresponding values for non-ethanol pair fed group, and the hash tag indicates that the value is significantly (p<0.05) different from corresponding value for wild type group.
An initial stage of alcoholic liver damage is fatty liver. Chronic ethanol feeding has been repeatedly demonstrated to cause fatty liver. We investigated the effect of ALDH2 deficiency on ethanol-induced fat accumulation in liver. Staining of liver sections with Oil Red-O confirmed ethanol-induced fatty liver in wild type mice (Fig. 7E). Stain for fat deposit was much greater in liver sections of ethanol-fed ALDH2+/− mice. This was confirmed by quantitative analysis of triglycerides in liver tissue. Ethanol feeding increased triglyceride levels by 1.3-fold in wild type mice, whereas in ALDH2+/− mice, it was elevated by 2.7-fold (Fig. 7F).
Discussion
ALDH2 polymorphism in subpopulations of Asians and Native Americans has raised the question whether these individuals are sensitive to acetaldehyde-mediated tissue injury in alcohol-related diseases (Morimoto and Takeshita, 1996, Wall et al., 1997, Yokoyama et al., 2003). In this study, we used ALDH2 deficient mice to evaluate the role of ALDH2 and acetaldehyde in alcohol-induced gut barrier dysfunction and liver injury in vivo. We provide evidence to show that ALDH2 deficiency indeed increases the sensitivity to alcohol-induced gut barrier dysfunction and liver damage.
Accumulation of acetaldehyde in colonic mucosa following ethanol feeding at a greater level in ALDH2 deficient mice suggests that polymorphism with loss of function ALDH2 mutation is likely to accumulate higher levels of acetaldehyde in tissues following alcohol consumption. This is consistent with the previous report that ALDH2 deficiency leads to higher acetaldehyde accumulation in brain and liver (Isse et al., 2002). Interestingly, ALDH2−/− mice showed a dramatic decline in intake of ethanol-containing diet 2 days after increasing the ethanol concentration to 4%. This is in agreement with the fact that human subjects homozygous for loss of function ALDH2 mutation are also unable to tolerate alcohol. Acetaldehyde levels in the luminal contents of ileum and colon were unaffected by ALDH2 deficiency. This indicates that ethanol metabolism in the intestinal lumen is predominantly mediated by intestinal micro flora. Intestinal micro flora expresses high levels of ADH and a very low level of ALDH. A significantly high level of acetaldehyde in the colonic mucosa of ALDH2−/− mice in the absence of ethanol feeding suggest that polymorphic humans with homozygous ALDH2 mutation may maintain a higher level of acetaldehyde in the colonic mucosa even without alcohol consumption. Ethanol produced by bacterial fermentation may be the source of acetaldehyde under such conditions. Humans with heterozygous ALDH2 polymorphism do tolerate alcohol, but are characterized by flushing after alcohol consumption. These subjects may be at a higher risk for alcohol-related diseases. Therefore, further studies were focused on understanding the differences between wild type and ALDH2+/− mice in alcohol-induced gut and liver injury.
Gut barrier function was evaluated by measuring plasma-to-luminal flux of fluorescently labeled inulin, so that the location of gut leakiness could be spotted. A greater inulin permeability in the distal colon of ethanol-fed wild type mice indicated that chronic ethanol feeding disrupts colonic mucosal barrier function. In wild type mice, this effect of ethanol was confined to distal colon. Barrier function of proximal colon, ileum, jejunum and duodenum was unaffected. The reason for confinement of leakiness in the distal colon is unclear. One likely explanation is that micro flora resides in the distal colon at several folds greater density compared to the proximal colon and ileum. Alternatively, mucosal defense mechanism against injurious factors is stronger in other segments of intestine compared to that in the distal colon. In ALDH2+/− mice, ethanol-induced increase in inulin permeability in the distal colon was several folds greater than that in wild type mice, indicating that ALDH2 deficient mice are more sensitive to ethanol effect on gut permeability. Additionally, our data indicate that ethanol feeding disrupts mucosal barrier function, also in the proximal colon, ileum and jejunum. An elevation of acetaldehyde accumulation in the intestinal mucosa of ALDH2 deficient mice may be responsible for this wide spread damage to intestinal barrier function.
The present study shows that the ethanol-induced increase in mucosal permeability is confined to distal colon. But, a recent study showed an ethanol-induced increase in permeability in mouse ileum (Kirpich et al., 2012). The study by Kirpich et al showed no significant increase in ileal permeability by ethanol in regular diet, which is in agreement with the results of our present study. Significant increase in permeability in ileum was observed when ethanol was administered in high unsaturated-fat diet. Additionally, there were several other differences in the experimental conditions between our study and the study by Kirpich et al. Ethanol feeding was performed for 8 weeks in their study, while only for 4 weeks in our study. They have studied the permeability by introducing fluid into the lumen of intestinal sacs in vitro. Therefore, differences in the intestinal segments involved in ethanol-induced permeability in different laboratories may be caused by multiple factors.
Our study showed no significant change in plasma endotoxins due to high variability, and therefore, data not presented in the manuscript. Similarly, a recent study also showed no significant change in plasma endotoxin by ethanol feeding in regular Lieber DeCarli diet (Kirpich et al., 2012). Endotoxemia is more obvious than in severe ALD models such as intra-gastric alcohol administration. Normally, endotoxin diffuses into mesenteric circulation due to increased tight junction permeability and is delivered to liver, where endotoxins detoxified by Kupffer cells. Only, when liver gets saturated with endotoxins, it spills into plasma. Therefore, low or lack of endotoxin in plasma does not rule out endotoxin delivery into the liver. Elevated intestinal permeability to macromolecule is considered as an indication of changes in endotoxin permeability.
The gross morphology of intestinal mucosa was unaffected, nor indicated a sign of mucosal inflammation (data not shown). But, staining the cryosections of distal colon showed a significant loss of tight junction proteins at the epithelial junctions of distal colon. This effect of ethanol was more severe in the distal colon of ALDH2 deficient mice. Confocal microscopy also showed a loss of junctional distribution of tight junction proteins in the ileum of ethanol fed ALDH2+/− mice, but it was unaffected in those of wild type mice. These results are consistent with the changes in inulin permeability data. Epithelial tight junctions are the primary component of mucosal barrier function. Tight junctions form a physical barrier to the diffusion of macromolecules including bacterial lipopolysaccharides. A loss of tight junction barrier and its exacerbation by ALDH2 deficiency indicates a higher level of endotoxin absorption and endotoxemia in alcoholics with ALDH2 loss of function polymorphism.
Previous studies showed that acetaldehyde rather than ethanol is responsible for disruption of tight junctions and barrier dysfunction in Caco-2 cell monolayers (Atkinson and Rao, 2001, Rao, 2009, Rao, 1998). Acetaldehyde was also found to disrupt tight junctions in rat colon (Ferrier et al., 2006) and human colonic mucosa (Basuroy et al., 2005). An exacerbation of ethanol-induced tight junction disruption in mouse colon by ALDH2 deficiency establishes the role of acetaldehyde in intestinal epithelial tight junction disruption in vivo. Occludin and claudin-3 are the transmembrane proteins of tight junctions. Ethanol-induced depletion of these proteins from the detergent-insoluble fractions of distal colon is more severe in ALDH2 deficient mice, indicating that acetaldehyde accumulation may have a direct impact on the functions of these tight junction proteins.
Previous studies demonstrated that acetaldehyde disrupts adherens junctions in Caco-2 cell monolayers (Atkinson and Rao, 2001, Sheth et al., 2007) and human colonic mucosa by increasing tyrosine phosphorylation of E-cadherin and β-catenin. Loss of junctional distribution of E-cadherin and β-catenin and decline in their levels in detergent-insoluble fractions of distal colon in ethanol fed wild type mice indicated that ethanol feeding disrupts adherens junctions. Ethanol-induced disruption of adherens junction was more severe in ALDH2+/− mice, demonstrating the potential role of acetaldehyde accumulation in adherens junction disruption in vivo. Previous studies in Caco-2 cell monolayers demonstrated that acetaldehyde disrupts adherens junctions prior to disruption of tight junctions, indicating that adherens junction disruption may indirectly contribute to destabilization of tight junctions in acetaldehyde-treated intestinal epithelium (Gumbiner et al., 1988b, Madara et al., 1986). The adherens junction disruption by ethanol in mouse colon may support the role of adherens junction disruption in ethanol-induced destabilization of tight junctions in vivo.
Disruption of intestinal mucosal barrier function and increased permeability to endotoxins appear to play a crucial role in the pathogenesis of ALD (Rao, 2009). Therefore, exacerbation of ethanol-induced tight junction and adherens junction disruption and elevated gut permeability in ALDH2 deficient mice raised the question whether ALDH2 deficiency also increased the severity of ethanol-induced liver damage. The results of this study show that ethanol-induced liver injury is indeed greater in ALDH2+/− mice. Histopathology indicates that ALDH2 deficiency promotes liver injury, which was associated with greater increase in plasma ALT/AST activity in ethanol fed ALDH2+/− mice. ALDH2 deficiency also increased ethanol-induced fat accumulation in the liver. These results demonstrate that ALDH2 deficiency exacerbates ethanol-induced liver damage. A recent study showed that ethanol-induced inflammation in the liver is more severe in ALDH2 knockout mice (Kwon et al., 2014). However, this study showed a slight decline in ethanol-induced fatty liver. This is in contrast to our study, showing an increased ethanol-induced fatty liver. This discrepancy may be due to one or more of the following differences in the experimental models. First, a combination of chronic and binge ethanol feeding was used in the previous study, but only chronic ethanol feeding was applied in the present study. Second, male mice were used in the previous, whereas female mice were used in the present study. Finally, previous study used ALDH2−/− mice, but ALDH2+/− mice were used in the present study. Combination of these differences may have caused this discrepancy.
From the results of this study we can only conclude that acetaldehyde promotes alcoholic liver disease, and cannot rule out the potential role of ethanol itself. In fact, it is quite likely that both ethanol and acetaldehyde contribute to the pathogenesis of alcoholic liver disease. Previous studies have shown that ADH inhibitor, 4-methyl pyrazole, ameliorate alcoholic liver damage in rats and mice (Ronis et al., 2010), supporting the hypothesis that ethanol metabolism and acetaldehyde generation play at least partial role in alcoholic liver disease. ALDH2 deficiency in subsets of Asian population is likely to enhance the severity of alcoholic liver disease. Genetic deletion of ALDH2 is the most specific and physiologic approach to determine the role of ethanol metabolism in the pathogenesis of ALD. ALDH2+/− model is a true representation of human cases of heterozygous ALDH2 polymorphism. Homozygous ALDH2 polymorphics are unable to drink alcohol, and therefore ALDH2−/− or total inhibition of ALDH activities may not represent physiologic conditions.
In summary, the present study shows that a partial decrease in ALDH2 elevates ethanol-induced intestinal epithelial tight junction and adherens junction disruption, exacerbates gut barrier dysfunction and promotes liver injury. These data suggest that human subjects with heterozygous ALDH2 polymorphism may be at a higher risk for developing alcoholic gut and liver injury.
Acknowledgments
This study was supported by NIH grants, AA12307 (RR), DK55532 (RR), P20 AA017837 (LEN), R01 AA019673 (LEN) and 1U01AA021890 (LEN)
Authors acknowledge Ms. Ernestine Hayes at the department of Comparative Medicine, UTHSC for her help with tail vein injections and cardiac puncture for collection of blood.
Abbreviations
- ADH
alcohol dehydrogenase
- ALD
alcoholic liver disease
- ALDH
aldehyde dehydrogenase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ECL
enhanced chemiluminescent
- FITC
fluorescein isothiocyanate
- HRP
horse radish peroxidase
- ZO-1
zona occludens-1
- ZO-2
zona occludens-2
- ZO-3
zona occludens-3
References
- ANDERSON C, ANDERSSON T, MOLANDER M. Ethanol absorption across human skin measured by in vivo microdialysis technique. Acta Derm Venereol. 1991;71:389–93. [PubMed] [Google Scholar]
- ANDERSON JM, VAN ITALLIE CM. Physiology and function of the tight junction. Cold Spring Harbor perspectives in biology. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATKINSON KJ, RAO RK. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1280–8. doi: 10.1152/ajpgi.2001.280.6.G1280. [DOI] [PubMed] [Google Scholar]
- BASUROY S, SHETH P, MANSBACH CM, RAO RK. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and L-glutamine. American journal of physiology Gastrointestinal and liver physiology. 2005;289:G367–75. doi: 10.1152/ajpgi.00464.2004. [DOI] [PubMed] [Google Scholar]
- BODE JC, BODE C, HEIDELBACH R, DURR HK, MARTINI GA. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology. 1984;31:30–4. [PubMed] [Google Scholar]
- BOFFETTA P, HASHIBE M. Alcohol and cancer. Lancet Oncol. 2006;7:149–56. doi: 10.1016/S1470-2045(06)70577-0. [DOI] [PubMed] [Google Scholar]
- DURYEE MJ, KLASSEN LW, FREEMAN TL, WILLIS MS, TUMA DJ, THIELE GM. Lipopolysaccharide is a cofactor for malondialdehyde-acetaldehyde adduct-mediated cytokine/chemokine release by rat sinusoidal liver endothelial and Kupffer cells. Alcohol Clin Exp Res. 2004;28:1931–8. doi: 10.1097/01.alc.0000148115.90045.c5. [DOI] [PubMed] [Google Scholar]
- ELAMIN E, MASCLEE A, DEKKER J, JONKERS D. Ethanol disrupts intestinal epithelial tight junction integrity through intracellular calcium-mediated Rho/ROCK activation. American journal of physiology Gastrointestinal and liver physiology. 2014;306:G677–85. doi: 10.1152/ajpgi.00236.2013. [DOI] [PubMed] [Google Scholar]
- ERIKSSON CJ. The role of acetaldehyde in the actions of alcohol (update 2000) Alcoholism, clinical and experimental research. 2001;25:15S–32S. doi: 10.1097/00000374-200105051-00005. [DOI] [PubMed] [Google Scholar]
- FERRIER L, BERARD F, DEBRAUWER L, CHABO C, LANGELLA P, BUENO L, FIORAMONTI J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol. 2006;168:1148–54. doi: 10.2353/ajpath.2006.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FISHER SJ, SWAAN PW, EDDINGTON ND. The ethanol metabolite acetaldehyde increases paracellular drug permeability in vitro and oral bioavailability in vivo. J Pharmacol Exp Ther. 2010;332:326–33. doi: 10.1124/jpet.109.158642. [DOI] [PubMed] [Google Scholar]
- FURUSE M, ITOH M, HIRASE T, NAGAFUCHI A, YONEMURA S, TSUKITA S. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol. 1994;127:1617–26. doi: 10.1083/jcb.127.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUMBINER B, STEVENSON B, GRIMALDI A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. The Journal of cell biology. 1988a;107:1575–87. doi: 10.1083/jcb.107.4.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUMBINER B, STEVENSON B, GRIMALDI A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J Cell Biol. 1988b;107:1575–87. doi: 10.1083/jcb.107.4.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISSE T, OYAMA T, KITAGAWA K, MATSUNO K, MATSUMOTO A, YOSHIDA A, NAKAYAMA K, KAWAMOTO T. Diminished alcohol preference in transgenic mice lacking aldehyde dehydrogenase activity. Pharmacogenetics. 2002;12:621–6. doi: 10.1097/00008571-200211000-00006. [DOI] [PubMed] [Google Scholar]
- JOKELAINEN K, MATYSIAK-BUDNIK T, MAKISALO H, HOCKERSTEDT K, SALASPURO M. High intracolonic acetaldehyde values produced by a bacteriocolonic pathway for ethanol oxidation in piglets. Gut. 1996;39:100–4. doi: 10.1136/gut.39.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOKELAINEN K, REINKE LA, NANJI AA. Nf-kappab activation is associated with free radical generation and endotoxemia and precedes pathological liver injury in experimental alcoholic liver disease. Cytokine. 2001;16:36–9. doi: 10.1006/cyto.2001.0930. [DOI] [PubMed] [Google Scholar]
- KIRPICH IA, FENG W, WANG Y, LIU Y, BARKER DF, BARVE SS, MCCLAIN CJ. The type of dietary fat modulates intestinal tight junction integrity, gut permeability, and hepatic toll-like receptor expression in a mouse model of alcoholic liver disease. Alcohol Clin Exp Res. 2012;36:835–46. doi: 10.1111/j.1530-0277.2011.01673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWON HJ, WON YS, PARK O, CHANG B, DURYEE MJ, THIELE GE, MATSUMOTO A, SINGH S, ABDELMEGEED MA, SONG BJ, KAWAMOTO T, VASILIOU V, THIELE GM, GAO B. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology. 2014;60:146–57. doi: 10.1002/hep.27036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LILIEN J, BALSAMO J, ARREGUI C, XU G. Turn-off, drop-out: functional state switching of cadherins. Dev Dyn. 2002;224:18–29. doi: 10.1002/dvdy.10087. [DOI] [PubMed] [Google Scholar]
- MADARA JL. Intestinal absorptive cell tight junctions are linked to cytoskeleton. Am J Physiol. 1987;253:C171–5. doi: 10.1152/ajpcell.1987.253.1.C171. [DOI] [PubMed] [Google Scholar]
- MADARA JL, BARENBERG D, CARLSON S. Effects of cytochalasin D on occluding junctions of intestinal absorptive cells: further evidence that the cytoskeleton may influence paracellular permeability and junctional charge selectivity. J Cell Biol. 1986;102:2125–36. doi: 10.1083/jcb.102.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHURIN P, DENG QG, KESHAVARZIAN A, CHOUDHARY S, HOLMES EW, TSUKAMOTO H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–17. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- MORIMOTO K, TAKESHITA T. Low Km aldehyde dehydrogenase (ALDH2) polymorphism, alcohol-drinking behavior, and chromosome alterations in peripheral lymphocytes. Environmental health perspectives. 1996;104(Suppl 3):563–7. doi: 10.1289/ehp.96104s3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NANJI AA, SU GL, LAPOSATA M, FRENCH SW. Pathogenesis of alcoholic liver disease--recent advances. Alcohol Clin Exp Res. 2002;26:731–6. [PubMed] [Google Scholar]
- RAO R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–44. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO RK. Acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Alcohol Clin Exp Res. 1998;22:1724–30. [PubMed] [Google Scholar]
- RAO RK, SETH A, SHETH P. Recent Advances in Alcoholic Liver Disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;286:G881–4. doi: 10.1152/ajpgi.00006.2004. [DOI] [PubMed] [Google Scholar]
- RODGERS LS, FANNING AS. Regulation of epithelial permeability by the actin cytoskeleton. Cytoskeleton. 2011;68:653–60. doi: 10.1002/cm.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RONIS MJ, KOROURIAN S, BLACKBURN ML, BADEAUX J, BADGER TM. The role of ethanol metabolism in development of alcoholic steatohepatitis in the rat. Alcohol. 2010;44:157–69. doi: 10.1016/j.alcohol.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAFER C, PARLESAK A, SCHUTT C, BODE JC, BODE C. Concentrations of lipopolysaccharide-binding protein, bactericidal/permeability-increasing protein, soluble CD14 and plasma lipids in relation to endotoxaemia in patients with alcoholic liver disease. Alcohol Alcohol. 2002;37:81–6. doi: 10.1093/alcalc/37.1.81. [DOI] [PubMed] [Google Scholar]
- SEITZ HK, STICKEL F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- SEITZ HK, STICKEL F. Acetaldehyde as an underestimated risk factor for cancer development: role of genetics in ethanol metabolism. Genes & nutrition. 2010;5:121–8. doi: 10.1007/s12263-009-0154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SETH A, BASUROY S, SHETH P, RAO RK. L-Glutamine ameliorates acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Am J Physiol Gastrointest Liver Physiol. 2004;287:G510–7. doi: 10.1152/ajpgi.00058.2004. [DOI] [PubMed] [Google Scholar]
- SHETH P, SETH A, ATKINSON KJ, GHEYI T, KALE G, GIORGIANNI F, DESIDERIO DM, LI C, NAREN A, RAO R. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem J. 2007;402:291–300. doi: 10.1042/BJ20060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHETH P, SETH A, THANGAVEL M, BASUROY S, RAO RK. Epidermal growth factor prevents acetaldehyde-induced paracellular permeability in Caco-2 cell monolayer. Alcohol Clin Exp Res. 2004;28:797–804. doi: 10.1097/01.alc.0000125358.92335.90. [DOI] [PubMed] [Google Scholar]
- SUZUKI T, SETH A, RAO R. Role of phospholipase Cgamma-induced activation of protein kinase Cepsilon (PKCepsilon) and PKCbetaI in epidermal growth factor-mediated protection of tight junctions from acetaldehyde in Caco-2 cell monolayers. J Biol Chem. 2008;283:3574–83. doi: 10.1074/jbc.M709141200. [DOI] [PubMed] [Google Scholar]
- TURNER JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169:1901–9. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALL TL, PETERSON CM, PETERSON KP, JOHNSON ML, THOMASSON HR, COLE M, EHLERS CL. Alcohol metabolism in Asian-American men with genetic polymorphisms of aldehyde dehydrogenase. Annals of internal medicine. 1997;127:376–9. doi: 10.7326/0003-4819-127-5-199709010-00007. [DOI] [PubMed] [Google Scholar]
- YOKOYAMA T, YOKOYAMA A, KATO H, TSUJINAKA T, MUTO M, OMORI T, HANEDA T, KUMAGAI Y, IGAKI H, YOKOYAMA M, WATANABE H, YOSHIMIZU H. Alcohol flushing, alcohol and aldehyde dehydrogenase genotypes, and risk for esophageal squamous cell carcinoma in Japanese men. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2003;12:1227–33. [PubMed] [Google Scholar]