

Abstract
Inflammatory diseases in mucosal organs as diverse as the lung, liver and intestine inevitably require the intimate interactions between neutrophils and epithelia. The physiologic consequences of such interactions often determine endpoint organ function, and for this reason, much recent interest has developed in identifying mechanisms and novel targets to promote the resolution of mucosal inflammation. Physiologically-relevant in vitro and in vivo model systems have aided in discovery of novel pathways to define basic inflammatory mechanisms and approaches to defining the concepts of inflammatory resolution. Here, we will review the recent literature regarding the contribution of neutrophils to inflammatory resolution, with an emphasis on the role of the tissue microenvironment, endogenous pathways for promoting resolution and the molecular determinants of neutrophil-epithelial cell interactions during ongoing inflammation. These recent studies highlight the dynamic nature of pro-resolving pathways and lend insight into the complexity of treating mucosal inflammation.
Keywords: metabolism, hypoxia-inducible factor, inflammation, nucleoside, nucleotidase, mucosa, colitis, epithelium, endothelium, murine model
Introduction
The presence of neutrophils (polymorphonuclear leukocyte, PMN) at sites of tissue injury and infection has long been recognized as a hallmark of acute inflammation. It has become increasingly appreciated that the presence of PMNs at sites of injury do not necessarily prove causation to tissue damage, and in fact, may provide clues into the initiation of inflammatory resolution. In fact, history has suggested this to be the case. As an example, the Roman gladiatorial surgeon Claudius Galen (130-200 AD) made the observation that pus formation in wounds of his patients was associated with more successful wound healing. He became widely known for “pus bonum et laudabile”, meaning “good and commendable pus”[1].
The contribution(s) of PMN to successful inflammatory resolution is an area of signifcant interest. Ongoing studies have revealed that infiltrating PMN communicate with the surrounding parenchymal tissues in ways which mold the microenvironment to promote tissue restitution, wound healing and homeostasis. In this review, we will summarize the current state of the art related to the role of PMN in the active resolution of inflammation, with a particular focus on the mucosa.
Inflammatory resolution: an active rather than passive process
The resolution of inflammation was historically conceived as a passive act of the healing process occuring independent of active biochemical pathways. This view has fundamentally shifted in the past decade [2-4]. It is now appreciated that uncontrolled inflammation is a unifying component in many diseases and new evidence indicates that inflammatory resolution is a biosynthetically proactive process [5]. These most recent findings implicate tissue decision processes wherein acute inflammation, chronic inflammation, or inflammatory resolution outcomes are dictated by endogenous processes employed to control the magnitude and duration of the acute response, particularly as they relate to the original cardinal signs of inflammation [6, 7]. It has now become evident that the resolution program of acute inflammation remains largely uncharted, particularly at mucosal surfaces, and that a complete understanding of these critical pathways will unquestionably direct new therapeutic options.
The mucosa serves as an excellent model for which to define many features of inflammatory resolution. Whether it be the gastrointestinal (GI) tract, the lung or the skin, a primary function of the mucosa is to provide a selective barrier to the outside. At these same surfaces exist the potential for infection by pathogenic organisms and the neccessity to control commensal microorganisms at homeostatic levels. In this regard, the tissue healing following injury occurs in conjunction with the constant flux of new antigenic material and require that the mucosal immune system appropriately dampen inflammatory and immunological reactions to harmless ingested antigens. The overlying epithelium plays an important role in coordinating both inflammation and resolution. The epithelium lies juxtaposed to the mucosal immune system and lines the entire gastrointestinal tract. Covering a surface area of approximately 300 m2, the human adult intestinal epithelium consists of a monolayer of cells with intercellular tight junctions, a complex three dimensional structure and a thick mucous gel layer that provides a dynamic and regulated barrier to the flux of the luminal contents to the lamina propria [8, 9]. It is widely understood that the gastrointestinal tract exists in a state of low-grade inflammation. Such a state results from the constant processing of luminal antigenic material during the development of oral tolerance and the priming of the mucosal immune system for rapid and effective responses to antigens or microbes that may penetrate the barrier[10].
There may also be significant differences between mucosal surfaces with regard to the contribution of PMN to the resolution process. For example, literature exists that the GI tract may differ from the lung in this regard. This particular aspect has been convincingly demonstrated in vivo. The depletion of circulating PMN using anti-Gr1 antibodies resulted in the exacerbation of symptoms in a number of different murine colitis models, strongly implicating PMN as a central protective factor in ongoing inflammation [11]. By contrast, the depletion of PMN in acute lung injury models appears to serve an anti-inflammatory role [12], although this idea has been revisited[13]. Nonetheless, these results suggest fundamental differences in mechanisms of inflammatory resolution between various mucosal organs. Below, we discuss some potential mechanisms that may contribute to the unique mucosal niche that drive the inflammatory resolution response.
Oxygen consumption by PMN as a driver of resolution during acute inflammation
Recent studies have indicated that a contributing factor to differences in the inflammatory resolution response between mucosal tissues is oxygen metabolism. The intestinal mucosa, for example, exhibits a particularly unique oxygenation profile, experiencing profound fluctuations in blood flow and metabolism. For instance, less than 5% of total blood volume is present in the gut during fasting, but following ingestion of a meal, approximately 30% of total blood volume is shunted to the gastrointestinal tract [14]. These fluctuations in blood flow result in a relatively low baseline pO2 (<40 mmHg [15, 16]) under physiologic conditions. By comparison to the lung where baseline pO2 can be as high as 110 mmHg [17], it is perhaps not surprising that mucosal tissues have evolved a number of adaptive features to cope with these austere metabolic changes. Studies comparing functional responses between epithelial cells from different tissues have revealed that intestinal epithelia appear to be uniquely resistant to hypoxia and that even very low levels of O2 within the normal mucosa (so-called “physiologic hypoxia”) may be a regulatory adaptation mechanism to the steep oxygen gradient across the intestinal mucosa[18]. A key discovery was the observation that epithelial cells of the distal gut basally regulate hypoxia-inducible factor (HIF) [19], the master regulator of oxygen homeostasis [20]. Kelly et al, recently demonstrated that the low-O2 conditions of the distal GI tract that enable microbial short chain fatty acid (SCFA) production (e.g. butyrate), promotes epithelial O2 consumption to the extent that HIF is stabilized and functionally maintains mucosal barrier function [21] and the expression of certain antimicrobial peptides [22].
This aspect of oxygen metabolism is exacerbated during inflammation. It was recently demonstrated that during acute inflammatory disease, infiltrating neutrophils mold the tissue microenvironment in ways that significantly promote the stabilization of HIF[23] (Figure 1). Microarray analysis of epithelial cells following PMN transmigration identified the induction of a prominent cohort of HIF target genes. Utilizing HIF reporter mice, Gp91phox-/- mice (lack a respiratory burst) and PMN depletion strategies in acute colitis models, these studies revealed that transmigrating neutrophils rapidly deplete the microenvironment of molecular oxygen in an NADPH-oxidase-dependent manner and “transcriptionally imprint” a molecular fingerprint that significantly reflects PMN induction of HIF target genes onto the surrounding tissue (Figure 1). Importantly, this molecular signature promotes effective HIF-dependent inflammatory resolution. Indeed, Gp91phox-/- mice developed highly accentuated colitis relative to controls with exaggerated PMN infiltration, diminished inflammatory hypoxia and increased microbial invasion. In this regard, a clinical corollary to these findings have indicated that patients which lack a functional NADPH oxidase (i.e. chronic granulomatous disease) often present with an inflammatory bowel disease (IBD)-like syndrome[24]. Interestingly, chronic granulomatous disease (CGD) patients exhibit congenital defects in genes coding the subunits comprising the neutrophil NADPH oxidase complex (i.e. mutations in: CYBA, CYBB, NCF1, NCF2, NCF4, RAC1 and RAC2). This NADPH oxidase complex is responsible for the generation of reactive oxygen species (ROS) and used by innate immune cells (esp. PMN) to kill invading pathogens. Approximately 40% of CGD patients develop IBD-like symptoms [25]. Such clinical observations suggest that CGD-associated IBD could represent a failure to resolve acute intestinal insults.
Figure 1. PMN activation-dependent O2 consumption stabilizes mucosal HIF and promotes antimicrobial activity.
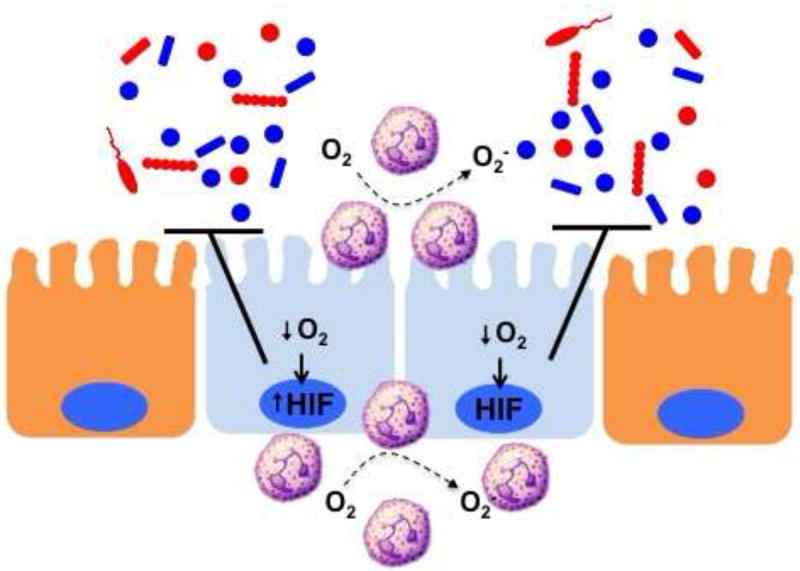
PMN accumulation at sites of mucosal damage or infection become activated to consume large amounts of O2 via the NADPH oxidase (NOX-2 complex) As a result, the localized microenvironment becomes deplete of molecular O2 and culminates in the stabilization of HIF within the epithelium and surrounding parenchymal cells. The activation of multiple HIF target genes (see text) promotes the active resolution of inflammation within the mucosa, particularly related to barrier and antimicrobial function.
Significant evidence indicates that the large amounts of localized oxygen consumption associated with acute inflammation signals epithelial restitution and inflammatory resolution through the stabilization of HIF[26] (see Figure 1). Numerous studies have demonstrated that such “inflammatory hypoxia” stabilizes the transcription factor HIF[27]. Once stabilized, HIF triggers the transcription of a cohort of genes that enable intestinal epithelial cells to resolve defective barrier function[18, 28-30]. Originally studies by microarray of intestinal epithelial cells subjected to low O2 revealed profound influences on barrier-related genes[31] that have now been validated in a number animal models of intestinal inflammation [19, 32-36] and in human intestinal tissues [23, 37-39]. The functional proteins encoded by HIF targets genes include those that localize primarily to the most luminal aspect of polarized epithelia that contribute fundamentally to effective barrier function. These target genes include mucins,[40], molecules that modify mucins (e.g. intestinal trefoil factor[18]), antimicrobials [22], xenobiotic clearance,[28] and nucleotide signaling [30] / metabolism (e.g. ecto-5’-nucleotidase)[30, 31].
It is noteworthy that one of the more prominent epithelial genes induced by PMN transmigration was cyclooxygenase-2 (COX-2) [23]. COX-2 contributes fundamentally to both inflammation and resolution [6, 41]. During epithelial cell-PMN interactions, pro-resolving lipid mediators (e.g. lipoxins, resolvins and protectins) are amplified by transcellular biosynthesis through the interactions of two or more cell types, each contributing an enzymatic product[42]; in this case epithelial cell COX-2 generates 18-HEPE from dietary omega-3 fatty acids and PMN-expressed 5-LO to generate resolvins [3]. Such locally generated resolvins is then made available to activate surface expressed ChemR23 receptor, which in turn has been shown to activate a number of antimicrobial pathways within the mucosa[43].
The identification of HIF as a central component to the resolution of mucosal inflammation has guided the development pharmacologic compounds that function to stabilize HIF and drive the expression of HIF target genes[44]. In most instances, the pharmacologic approach to achieve HIF stabilization in normoxia has involved the inhibition of HIF prolyl-hydroxylases (PHDs), originally described as products of genes related to C. elegans egl-9[45] and subsequently cloned in mammals as three isoforms of PHDs (PHD1, PHD2 and PHD3) that were shown to hydroxylate HIF-α in vitro[45, 46]. Targeting the catalytic domain of PHDs was initially achieved by a screen of molecules that interfere with critical cofactors such as the 2-oxoglutarate mimicry (e.g. dimethyloxalylglycine, DMOG)[47]. The original studies using PHD inhibition in mucosal inflammation demonstrated a protective role for HIF stabilizers in different models of intestinal inflammation. The use of DMOG for the treatment of intestinal inflammation in chemically-induced colitis proved highly effective in promoting the resolution of inflammation at least in part through anti-apoptotic mechanisms[32]. A parallel study published at the same time utilized a HIF stabilizer (FG-4497) that was based on a screen to identify erythropoietin inducers. Similar to DMOG, FG-4497 blocks the active site of PHDs[47]. In both studies, HIF stabilizer treatment was associated with profound pro-resolving functions, particularly related to mucosal barrier function [32, 35]. The recent use of more HIF-1 selective stabilizers, such as AKB-4924[23, 48, 49], holds promise in such models and indicates that IBD may be one of the more promising applications for PHD-inhibitor therapy.
Taken together, these studies have provided new molecular insight into the role of PMN in inflammatory resolution. “Transcriptional imprinting” of select molecular pathways, exemplified by HIF, provides mucosal memory of trafficked PMN that elicits functional resolution response central to tissue homeostasis. While PMN accumulation in the form of crypt abscesses has served as a longstanding hallmark of mucosal inflammation, these findings reveal that PMN infiltration is a necessity for limited disease severity. These studies also show that the PMN respiratory burst is a fundamental component for initiating a hypoxic niche, impeding further PMN infiltration and inducing HIF target genes sufficient to overcome the resolution deficit.
Purine nucleoside signaling in inflammatory resolution
One molecule of significant interest to the pro-resolving function of PMN at inflammatory sites is the purine nucleoside adenosine [50] (Figure 2) A number of studies have indicated that PMN serve as a prominent reservoir for adenosine precursors and that adenosine and its analogs can ameliorate the course of a variety of inflammatory diseases [51]. During inflammation, PMN actively release adenine nucleotides, particularly in the form of ATP or ADP[52]. PMN release ATP in an activation-dependent manner through a mechanism involving connexin 43 (C×43) hemichannels expressed on the surface of PMN [52]. Subsequent studies demonstrated that human PMNs release ATP predominantly from the leading edge of their cell surface as a mechanism to amplify chemotactic signals and direct cell orientation by feedback through P2Y2 nucleotide receptors [53].
Figure 2. Nucleotide metabolism and signaling promotes mucosal resolution.
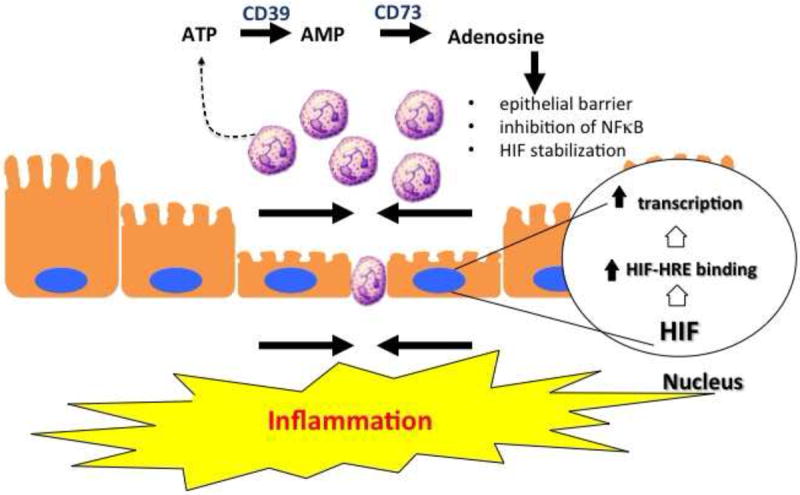
Activated PMN represent an important source of extracellular nucleotides during inflammation. Local metabolism of ATP/ADP by CD39 and CD73 generates high concentrations of adenosine. Activation of surface adenosine receptors promotes mucosal inflammatory resolution through a number of mechanisms, including epithelial restitution, inhibition of NF-κB (via Cullin-1 deneddylation) and stabilization of HIF (via Cullin-2 deneddylation).
It is now well documented that that inhibition of adenosine kinase and the dephosphorylation of ATP and AMP by surface apyrases (e.g. CD39) and ecto-5’-nucleotidase (CD73), respectively, represent the major pathways for accumulation of extracellular adenosine [30, 31, 54]. Once liberated in the extracellular space, adenosine is either recycled (e.g. through dipyridamole-sensitive carriers) or interacts with cell surface Ado receptors [55]. Presently, four subtypes of G protein-coupled Ado receptors exist, designated AA1R, AA2AR, AA2BR or AA3R and are classified according to utilization of pertussis toxin-sensitive (A1 and A3) or insensitive (A2A and A2B) pathways [55]. Recent work has specifically implicated the AA2BR in pro-resolving responses, wherein activation of this receptor elicits potent inhibition of inflammatory signaling cascades mediated by NF-κB [56]. Work in various cell types has shown that adenosine inhibits NF-κB activation through a number of distinct mechanisms, including elevation of intracellular cyclic adenosine monophosphate (cAMP) and activation of protein kinase A (PKA) which blocks IκB phosphorylation thus inhibiting NF-κB activation[57], inhibition of tumor necrosis factor (TNF)-α-induced NF-κB activity and subsequent gene expression by inhibition of nuclear translocation of active NF-κB without influencing IκB phosphorylation or degradation[58], and increased SUMO-1 modifications of IκBα by adenosine inhibition of phosphorylation and degradation of IκBα, which attenuates NF-κB activation[59].
Platelets provide a rich source of extracellular ATP as a pro-resolving signature during mucosal inflammation. Platelets release nucleotides at high concentration upon activation by ADP or collagen via dense granule release [60]. It was shown that interactions between platelets and PMN provide important signals for the resolution of intestinal inflammation and fluid transport via in an adenosine-dependent mannewr [61]. Indeed, these studies showed that platelets migrate across intestinal epithelia in a PMN-dependent manner. Furthermore, platelet-PMN comigration was observed in intestinal tissue derived from human patients with IBD. The translocated platelet-PMN clusters were found to release large quantities of ATP, which was sequentially metabolized to adenosine via a 2-step enzymatic reaction involving CD73 and CD39-like molecules expressed on IEC (Figure 2)[62]. These studies identified a mechanism involving adenosine-mediated activation of electrogenic chloride secretion, with concomitant water movement into the intestinal lumen, originally discovered by Madara et al [63]. This physiologic response has been demonstrated to serve as a protective flushing mechanism for mucosal-associated bacteria[64].
Given its long history, it is somewhat surprising how little is known about the molecular mechanisms of adenosine responses. While signaling mechanisms through the various adenosine receptors is well characterized, less is known about post-receptor events. One potentially important mechanism has revealed that adenosine stabilizes HIF-α [65] and inhibits NF-κB [56, 66] through actions on Cullin neddylation pathways. These findings were based on original studies work implicating commensal bacterial inhibition of NF-κB through Cullin-1 (Cul-1) deneddylation [67]. Bacterial fermentation products (e.g. butyrate) can increase intracellular ROS and lead to impaired neddylation of Cullins thus influencing NFκB signaling, implicating an important role for the commensal intestinal flora for the regulation of inflammatory processes[68]. Studies with adenosine analogs revealed that adenosine signaling potently deneddylates Cul-1 and impacts the proteosomal degradation of IκB proteins that hold NF-κB in check [56]. The E3 SCF ubiquitin ligase specific to IκB-family members, comprised of SKP1, CUL1, and the F-box domain of β-TrCP, is responsible for the polyubiquitination of IκB[69]. The active E3 SCF requires the COP9 signalosome (CSN) to bind Nedd8 to Cul-1, and deneddylated Cul-1 is incapable of ubiquitination of IκB[70]. Deneddylation reactions on Cullin targets via CSN-associated proteolysis is increasingly implicated as a central point for Cullin-mediated E3 ubiquitylation [71].
Curtis, et al, recently revealed a mechanism in addition to PHD inhibition for HIF-α isoform stabilization, namely targeting Cul-2 neddylation[65]. It is notable that an analogous E3 ligase complex exists upstream of HIF-α proteins, which is comprised of Cul-2, Elongin B/C and the von Hippel-Lindau protein (pVHL). Similar to the Cul-1-IκB-NF-κB axis, neddylation of Cul-2 is required for the ubiquitination of hydroxylated HIF-1α in normoxia[72]. In vitro revealed robust HIF-1α stabilization and Cul-2 deneddylation following acute treatments with adenosine analogs and low concentrations of MLN4924, a structural analog of adenosine monophosphate (AMP) that functions by inhibiting the Nedd8-activating enzyme [73]. The protective role of HIF and the contribution of the neddylation pathway were further confirmed in a murine DSS-colitis model. Colitic mice treated with MLN4924 displayed decreased percent body weight loss, decreased colon shortening and decreased histologic injury scores[74]. A parallel pathway for deneddylation of Cullin proteins has been reported. The identification of the Nedd8-specific protease DEN1/SENP8 has provided new insight into this emerging field. DEN1 contains isopeptidase activity capable of directly deneddylating Cullin targets [75, 76]. Knockdown of DEN-1 in epithelial and endothelial cells using lentivirus shRNA revealed decreased Cul-1 and Cul-2 neddylation at baseline and significantly abrogated barrier dysfunction in vitro[65, 66].
The most prominent signal provided to the mucosa by PMN-derived adenosine is the elevation of intracellular cAMP. It is notable that there is significant interest in harnessing cAMP signaling in pro-resolution pharmacology[4]. Studies in the mucosa have shown that increased cAMP promotes tissue barrier function through mechanisms involving increased expression of tight junction proteins ZO-1 and occludin [77] and an increase in the number of tight-junction strands [78]. Increased cAMP is also accompanied by increased actin polymerization actin increased phosphorylation of intermediate filaments, implicating cAMP-mediated changes in permeability at the level of the cytoskeleton[8]. Because of the actin-binding and cross linking functions of vasodilator-stimulated phosphoprotein (VASP), it has been demonstrated that protein kinase A (PKA)-mediated VASP phosphorylation may be crucial in this pathway (Figure 3). In fact, VASP localizes with ZO-1 at the tight junction and appears as phospho-VASP at the junction following adenosine-mediated PKA activation[79]. Loss and gain of function studies have shown prominent influences on endothelial and epithelial permeability in response to PKA agonists[29, 79]. Given the transient increase in epithelial permeability associated with PMN transmigration, these studies indicate PMN-derived adenosine signaling to the epithelial tight junction provides as signal to “close the door after leaving” and as such, serves an important role in mucosal resolution (Figure 2).
Figure 3. PMN-mediated epithelial restitution.
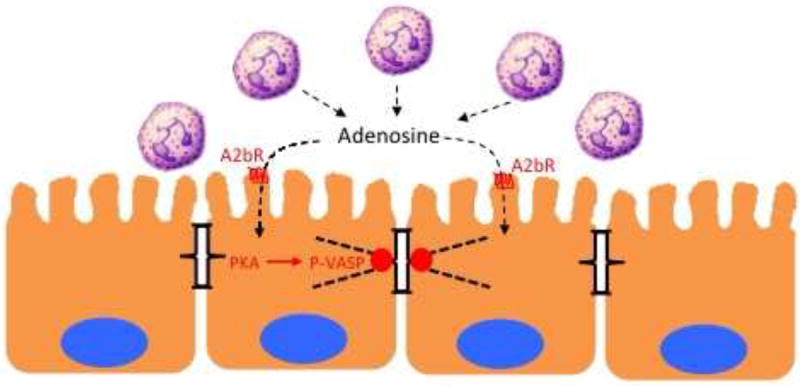
In acute mucosal inflammation, successful PMN transmigration results in transient increases in epithelial permeability. Nucleosides generated at the surface of the mucosa, such as adenosine, signal to the epithelium through adenosine receptors (e.g A2bR) to promote to the reassembly of apical junctional proteins through protein kinase A (PKA)-dependent phosphorylation of the actin-binding protein VASP. As such, this pathway represents a mechanism whereby PMN “close the door after leaving”.
Conclusion
The trafficking of PMN to sites of injury and infection represents an important initiation signal for resolution. In the mucosa, restitution of the epithelial barrier defines a critical determinant of inflammatory resolution. Recent studies investigating changes within the microenvironment of acute inflammation have revealed new important signaling pathways initiated by activated PMN, Of particular relevance is the shift in tissue oxygenation toward hypoxia, and specifically HIF-target pathways that are strongly associated with barrier function, altered nucleotide signaling and cellular bioenergetics which contribute fundamentally to inflammatory resolution. Studies in animal models have demonstrated an overall protective and anti-inflammatory influence of hydroxylase inhibition (i.e. HIF stabilization), identifying the mucosa as a strong candidate for targeted HIF-based therapy. In sum, the endogenous adaptive metabolic pathways activated in response to PMN infiltration represent potentially important therapeutic opportunities in mucosal inflammation.
Highlights.
Neutrophil accumulation is a hallmark of acute inflammation
In the mucosa, neutrophils signal the initiation of resolution
Activated neutrophils deplete local oxygen to the extent that HIF is stabilized
Mechanisms of inflammatory resolution include the local generation of multiple endogenous signals that culminate in restoration of epithelial barrier
Acknowledgments
This work was supported by National Institutes of Health Grants DK50189, DK104713 and DK95491 and by funding from the Crohn’s and Colitis Foundation of America.
Footnotes
The authors declare no financial interests in any of the work submitted here.
Disclosure statement: The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as influencing the objectivity of this review.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cohen IK. A Brief History of Wound Healing. 1. Yardley, PA: Oxford Clinical Communications; 1998. [Google Scholar]
- 2.Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med. 2013;5:661–674. doi: 10.1002/emmm.201202382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sousa LP, Alessandri AL, Pinho V, Teixeira MM. Pharmacological strategies to resolve acute inflammation. Curr Opin Pharmacol. 2013;13:625–631. doi: 10.1016/j.coph.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res. 2011;107:1170–1184. doi: 10.1161/CIRCRESAHA.110.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–524. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 10.Koch S, Nusrat A. The life and death of epithelia during inflammation: lessons learned from the gut. Annu Rev Pathol. 2012;7:35–60. doi: 10.1146/annurev-pathol-011811-120905. [DOI] [PubMed] [Google Scholar]
- 11.Kuhl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–1892. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 12.Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol. 2009;40:519–535. doi: 10.1165/rcmb.2008-0348TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Persson CG, Uller L. Resolution of cell-mediated airways diseases. Respir Res. 2010;11:75. doi: 10.1186/1465-9921-1111-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med. 2007;85:1295–1300. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 15.Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, et al. Correlation Between Intraluminal Oxygen Gradient and Radial Partitioning of Intestinal Microbiota. Gastroenterology. 2014;18:1055–1063. doi: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheridan WG, Lowndes RH, Young HL. Intraoperative tissue oximetry in the human gastrointestinal tract. Am J Surg. 1990;159:314–319. doi: 10.1016/s0002-9610(05)81226-7. [DOI] [PubMed] [Google Scholar]
- 17.Miller GW, Mugler JP, 3rd, Altes TA, Cai J, Mata JF, de Lange EE, et al. A short-breath-hold technique for lung pO2 mapping with 3He MRI. Magn Reson Med. 2010;63:127–136. doi: 10.1002/mrm.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford KM, Narravula S, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Ex Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semenza GL. Oxygen sensing, homeostasis, and disease. NEJM. 2011;365:537–547. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 21.Kelly CJ, Jheng L, C EL, Saeedi BJ, Scholz CC, Bayless AJ, et al. Host-microbe crosstalk between short-chain fatty acids and intestinal epithelial HIF provides a new mechanism to augment tissue barrier function. Cell Host Microbe. 2015 doi: 10.1016/j.chom.2015.03.005. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly CJ, Glover LE, Campbell EL, Kominsky DJ, Ehrentraut SF, Bowers BE, et al. Fundamental role for HIF-1alpha in constitutive expression of human beta defensin-1. Mucosal Immunol. 2013;6:1110–1118. doi: 10.1038/mi.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014;40:66–77. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang JS, Noack D, Rae J, Ellis BA, Newbury R, Pong AL, et al. Chronic granulomatous disease caused by a deficiency in p47(phox) mimicking Crohn’s disease. Clin Gastroenterol Hepatol. 2004;2:690–695. doi: 10.1016/s1542-3565(04)00292-7. [DOI] [PubMed] [Google Scholar]
- 25.Werlin SL, Chusid MJ, Caya J, Oechler HW. Colitis in chronic granulomatous disease. Gastroenterology. 1982;82:328–331. [PubMed] [Google Scholar]
- 26.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karhausen J, Haase VH, Colgan SP. Inflammatory Hypoxia: Role of Hypoxia-Inducible Factor. Cell Cycle. 2005;4:256–258. [PubMed] [Google Scholar]
- 28.Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer research. 2002;62:3387–3394. [PubMed] [Google Scholar]
- 29.Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. Faseb J. 2002;16:583–585. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- 30.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Ex Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–165. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Han IO, Kim HS, Kim HC, Joe EH, Kim WK. Synergistic expression of inducible nitric oxide synthase by phorbol ester and interferon-gamma is mediated through NF-kappaB and ERK in microglial cells. J Neurosci Res. 2003;73:659–669. doi: 10.1002/jnr.10706. [DOI] [PubMed] [Google Scholar]
- 34.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 35.Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–155. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134:2036–2048. 2048 e2031–2033. doi: 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giatromanolaki A, Sivridis E, Maltezos E, Papazoglou D, Simopoulos C, Gatter KC, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. Journal of clinical pathology. 2003;56:209–213. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mariani F, Sena P, Marzona L, Riccio M, Fano R, Manni P, et al. Cyclooxygenase-2 and Hypoxia-Inducible Factor-1alpha protein expression is related to inflammation, and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett. 2009;279:221–229. doi: 10.1016/j.canlet.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Matthijsen RA, Derikx JP, Kuipers D, van Dam RM, Dejong CH, Buurman WA. Enterocyte shedding and epithelial lining repair following ischemia of the human small intestine attenuate inflammation. PLoS One. 2009;4:e7045. doi: 10.1371/journal.pone.0007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Louis NA, Hamilton KE, Canny G, Shekels LL, Ho SB, Colgan SP. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem. 2006;99:1616–1627. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 41.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cycloxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 42.Serhan CN, Clish CB, Brannon J, Colgan SP, Gronert K, Chiang N. Anti-microinflammatory lipid signals generated from dietary N-3 fatty acids via cyclooxygenase-2 and transcellular processing: a novel mechanism for NSAID and N-3 PUFA therapeutic actions. J Physiol Pharmacol. 2000;51:643–654. [PubMed] [Google Scholar]
- 43.Campbell EL, Serhan CN, Colgan SP. Antimicrobial aspects of inflammatory resolution in the mucosa: a role for proresolving mediators. J Immunol. 2011;187:3475–3481. doi: 10.4049/jimmunol.1100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov. 2014;13:852–869. doi: 10.1038/nrd4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 46.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 47.Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8:139–152. doi: 10.1038/nrd2761. [DOI] [PubMed] [Google Scholar]
- 48.Keely S, Campbell EL, Baird AW, Hansbro PM, Shalwitz RA, Kotsakis A, et al. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol. 2014;7:114–123. doi: 10.1038/mi.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okumura CY, Hollands A, Tran DN, Olson J, Dahesh S, von Kockritz-Blickwede M, et al. A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J Mol Med. 2012;28:1079–1089. doi: 10.1007/s00109-012-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol. 2012;74:153–175. doi: 10.1146/annurev-physiol-020911-153230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eltzschig HK, Eckle T, Mager A, Kuper N, Karcher C, Weissmuller T, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. 2006;99:1100–1108. doi: 10.1161/01.RES.0000250174.31269.70. [DOI] [PubMed] [Google Scholar]
- 53.Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 54.Decking UK, Schlieper G, Kroll K, Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res. 1997;81:154–164. doi: 10.1161/01.res.81.2.154. [DOI] [PubMed] [Google Scholar]
- 55.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annual review of pharmacology and toxicology. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 56.Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest. 2007;117:703–711. doi: 10.1172/JCI30049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hong G, Zhang B, Harbrecht BG. Cyclic AMP inhibits IL-1beta plus IFNgamma-induced NF-kappaB translocation in hepatocytes by a PKA independent mechanism. J Surg Res. 2010;159:565–571. doi: 10.1016/j.jss.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Majumdar S, Aggarwal BB. Adenosine suppresses activation of nuclear factor-kappaB selectively induced by tumor necrosis factor in different cell types. Oncogene. 2003;22:1206–1218. doi: 10.1038/sj.onc.1206184. [DOI] [PubMed] [Google Scholar]
- 59.Liu Q, Li J, Khoury J, Colgan SP, Ibla JC. Adenosine signaling mediates SUMO-1 modification of IkappaBalpha during hypoxia and reoxygenation. J Biol Chem. 2009;284:13686–13695. doi: 10.1074/jbc.M809275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weissmuller T, Campbell EL, Rosenberger P, Scully M, Beck PL, Furuta GT, et al. PMNs facilitate translocation of platelets across human and mouse epithelium and together alter fluid homeostasis via epithelial cell-expressed ecto-NTPDases. J Clin Invest. 2008;118:3682–3692. doi: 10.1172/JCI35874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5’-nucleotidase (CD73) Purinergic Signal. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Madara JL, Patapoff TW, Gillece-Castro B, Colgan SP, Parkos CA, Delp C, et al. 5’-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J Clin Invest. 1993;91:2320–2325. doi: 10.1172/JCI116462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keely S, Kelly C, Weissmueller T, Burgess A, Wagner B, Robertson CE, et al. Activated fluid transport regulates bacterial-epithelial interactions and significantly shifts the murine colonic microbiome. Gut Microbes. 2012;3:250–260. doi: 10.4161/gmic.20529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Curtis VF, Ehrentraut SF, Campbell EL, Glover LE, Bayless AJ, Kelly CJ, et al. Stabilization of HIF through inhibition of Cullin-2 neddylation is protective in mucosal inflammatory responses. FASEB J. 2014 doi: 10.1096/fj.14-259663. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ehrentraut SF, Kominsky DJ, Glover LE, Campbell EL, Kelly CJ, Bowers BE, et al. Central role for endothelial human deneddylase-1/SENP8 in fine-tuning the vascular inflammatory response. J Immunol. 2013;190:392–400. doi: 10.4049/jimmunol.1202041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, et al. Prokaryotic regulation of epithelial responses by inhibition of IkappaB- alpha ubiquitination. Science. 2000;289:1560–1563. doi: 10.1126/science.289.5484.1560. [DOI] [PubMed] [Google Scholar]
- 68.Kumar A, Wu H, Collier-Hyams LS, Hansen JM, Li T, Yamoah K, et al. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. Embo J. 2007;26:4457–4466. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114:663–671. doi: 10.1016/s0092-8674(03)00722-0. [DOI] [PubMed] [Google Scholar]
- 70.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nature reviews. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 71.Parry G, Estelle M. Regulation of cullin-based ubiquitin ligases by the Nedd8/RUB ubiquitin-like proteins. Semin Cell Dev Biol. 2004;15:221–229. doi: 10.1016/j.semcdb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 72.Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004;18:3055–3065. doi: 10.1101/gad.1252404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 74.Cowley BP, Smith ME. Modulatin of E-cadherin expression and morphological phenotype in the intravascular component of adenocarcinomas. Int J Cancer. 1995;60:325–329. doi: 10.1002/ijc.2910600308. [DOI] [PubMed] [Google Scholar]
- 75.Mendoza HM, Shen LN, Botting C, Lewis A, Chen J, Ink B, et al. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–25643. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- 76.Wu K, Yamoah K, Dolios G, Gan-Erdene T, Tan P, Chen A, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–28891. doi: 10.1074/jbc.M302888200. [DOI] [PubMed] [Google Scholar]
- 77.Dye JF, Leach L, Clark P, Firth JA. Cyclic AMP and acidic fibroblast growth factor have opposing efffects on tight and adherens junctions in microvascular endothelial cells in vitro. Microvasc Res. 2001;62:94–113. doi: 10.1006/mvre.2001.2333. [DOI] [PubMed] [Google Scholar]
- 78.Adamson RH, Liu B, Fry GN, Rubin LL, Curry FE. Microvascular permeability and number of tight junctions are modulated by cAMP. The American journal of physiology. 1998;274:H1885–1894. doi: 10.1152/ajpheart.1998.274.6.H1885. [DOI] [PubMed] [Google Scholar]
- 79.Lawrence DW, Comerford KM, Colgan SP. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am J Physiol Cell Physiol. 2002;282:C1235–1245. doi: 10.1152/ajpcell.00288.2001. [DOI] [PubMed] [Google Scholar]