

Abstract
This paper reviews the anti-depressant actions of the N-methyl-D-aspartame glutamate receptor (NMDAR) antagonist, ketamine, and offers a potential neural mechanism for intranasal ketamine’s ultra-rapid actions based on the key role of NMDAR in the nonhuman primate prefrontal cortex (PFC). Although intravenous ketamine infusions can lift mood within hours, the current review describes how intranasal ketamine administration can have ultra-rapid antidepressant effects, beginning within minutes (5–40 minutes) and lasting hours, but with repeated treatments needed for sustained antidepressant actions. Research in rodents suggests that increased synaptogenesis in PFC may contribute to the prolonged benefit of ketamine administration, beginning hours after administration. However, these data cannot explain the relief that occurs within minutes of intranasal ketamine delivery. We hypothesize that the ultra-rapid effects of intranasal administration in humans may be due to ketamine blocking the NMDAR circuits that generate the emotional representations of pain (e.g. Brodmann Areas 24 and 25, insular cortex), cortical areas that can be overactive in depression and which sit above the nasal epithelium. In contrast, NMDAR blockade in the dorsolateral PFC following systemic administration of ketamine may contribute to cognitive deficits. This novel view may help to explain how intravenous ketamine can treat the symptoms of depression yet worsen the symptoms of schizophrenia.
Introduction
The following paper provides a review of the emerging clinical literature on the anti-depressant actions of the N-methyl-D-aspartate receptor (NMDAR) antagonist, ketamine, and presents a novel hypothesis for the potential neural mechanisms underlying the ultra-rapid relief of depression with intra-nasal delivery of ketamine. The hypothesis is based on the discovery that NMDAR actions are critical for generating mental representations in the dorsolateral prefrontal cortex (PFC) of the nonhuman primate, and parallel actions in the medial PFC circuits representing pain and suffering may contribute to symptoms of depression, and be rapidly alleviated by ketamine blockade.
All presently available FDA-approved antidepressants affect the biogenic amines dopamine, norepinephrine, and serotonin. These antidepressants are effective for some but not all persons with major depressive disorder. Patients are usually informed that it can take 4–6 weeks for an antidepressant to work, but given our inability to predict a priori either which antidepressant or what dose is right for which patient, in reality it takes months, not weeks, even when good outcomes are obtained. Further, many patients are “treatment refractory” because we have not yet found agents to treat their particular subtype of depression.
Recent evidence that ketamine is both an effective and rapid acting antidepressant in patients who have failed multiple antidepressant trials is providing hope that new antidepressants may be on the way.
A cautionary note: ketamine is not FDA-approved for the treatment of depression. Synthesized in 1962, ketamine was approved in 1970 as an anesthetic agent for diagnostic and surgical procedures not requiring muscle relaxation; for induction of anesthesia prior to the administration of other general anesthetic agents; and to supplement low-potency agents (e.g., nitrous oxide)[1]. Additionally, it has been used off label as an analgesic[2] and as a sedative[3].
In the 1980s Anis et al.[4] demonstrated that ketamine selectively reduced excitation of central neurons by NMDAR in cats and rats, and Morretti et al.[5] reported on clinical studies in healthy volunteers showing that at subanaesthetic doses intravenous ketamine was safe but did cause rapid and short-lived changes in mental status.
In the 1990s Skolnick et al.[6] suggested that adaptation of NMDARs might be involved in the pharmacotherapy of depression, setting the stage for several groups to start planning clinical trials to see if glutamatergic agents in general and ketamine in particular had antidepressant efficacy.
Given that efficacy and safety of ketamine for depression has not been definitively established, all studies conducted thus far have tested ketamine not as a first line treatment but in adults with “treatment-refractory depression” (TRD) operationalized as a major depressive episode that has failed to respond adequately to two or more antidepressants prescribed at adequate dose and duration[7].
Evidence that intravenous ketamine has antidepressant within hours
Placebo-controlled studies of intravenous ketamine have generally found optimal improvements in ratings of depression 24–72 hours after the infusion, with a significant decrease in depression ratings beginning at 110–240 min. These studies have reported a rapid “high” immediately after the infusion, as measured by a visual analog scale. It is not clear if this “high” represents a rapid relief in depressive mood (which might be experienced as a “high” given the rapidity of the change) or a drug-induced euphoria that is separable from a relief of depression. This challenge is confounded by the high rates of dissociative side effects that accompany intravenous ketamine infusions, which likely obscure changes in mood and its evaluation.
Berman et al.[8] conducted the first placebo controlled clinical trial of intravenous ketamine in the treatment of depression. Seven subjects meeting DSM-criteria for major depressive episodes received either 0.5 mg of ketamine or saline infused over 40 minutes on two different test days separated by at least 1 week. Hamilton Depression Rating Scale (HDRS) scores were taken at baseline as well as 40 minutes, 240 minutes, 24 hours, 48 hours, and 72 hours after the start of the infusion. Ketamine significantly improved HDRS ratings starting at 240 min and maintained through 72 hours, regardless of whether patients received placebo or ketamine first or second. Dissociative side effects were prominent in this study as measured by the BPRS positive symptoms scale, including perceptual disturbances.
Zarate et al.[9] subsequently carried out a randomized, placebo-controlled, double-blind, crossover study with 17 subjects receiving either ketamine followed by placebo or placebo followed by ketamine after one week. Compared to placebo, ketamine infusion resulted in significant decreases in HDRS scores within 110 minutes, which was still statistically significant after 1 week. As with Berman et al., perceptual disturbances and confusion were common side effects.
DiazGranados et al.[10] looked at suicidality in thirty-three patients with major depressive disorder receiving a single open-label infusion of ketamine. Suicidal ideation scores decreased significantly within 40 minutes and remained low up to 4 hours after ketamine infusion on the Scale for Suicide Ideation (SSI) as well as suicide subscales of the Montgomery-Asberg Depression Rating Scale (MADRS), the Beck Depression Inventory (BDI), and the HDRS.
Another study examined whether repeated ketamine infusions could produce sustained improvement in patients who had previously shown benefit from a single infusion. Nine of ten patients who had previously responded to a single dose of intravenous ketamine were enrolled in a protocol testing the safety and efficacy of repeated dose intravenous ketamine. They received an infusion on day 1, and five additional infusions on days 3, 5, 8, 10, and 12.[11]. Thereafter follow-up visits were conducted twice weekly for at least 4 weeks or until relapse. Three patients experienced significant but transient dissociative symptoms, but for all other patients side effects during and after each ketamine infusion were mild. The mean reduction in MADRS scores after the sixth infusion was 85%. Eight of the nine patients relapsed, on average, 19 days after the final infusions, with a side range of 6– 45 days. The remaining one patient had minimal depressive symptoms and required no antidepressant medication over three months after the final infusion.
Evidence that intranasal ketamine has antidepressant effects within minutes
In contrast to intravenous administration of ketamine, intranasal administration appears to produce fewer dissociative side effects, and an ultra-rapid relief within minutes that is not simply a drug-induced “high.”
Lapidus et al.[12] recently completed the first published randomized, double-blind, controlled trial comparing intranasal ketamine vs. saline inhalation. Twenty subjects were recruited and eighteen completed two test days one week apart in which they received either ketamine on day 1 and saline placebo on day 7 or saline placebo on day 1 and ketamine on day 7, with change in MADRS scores 24 hours after inhalation being the primary outcome measure. The primary outcome measure was the 24 hour decrease in MADRS scores from baseline, which was a reduction greater than 40% (p<.001). Remarkably, the decrease in MADRS scores from baseline was already greater than 30% at only 40 min after intranasal administration (p<.05). Treatment was associated with minimal adverse effects.
Our own recent experience indicates that intranasal administration of ketamine can ameliorate symptoms of depression and anxiety in less than 20 minutes[13]. Some patients have had a very rapid positive response, e.g., reporting that their long-term serious depression had lifted as soon as 15 minutes after self-administering 20–40 mg of intranasal ketamine under supervision. Patients report that this is different from a drug “high”; rather, they feel calmer and less anxious. For example, one patient, who had failed to respond to either multiple medication trials or two ECT trials, said very calmly 10 minutes after inhaling 40 mg of ketamine, “I am not depressed.” A few minutes later he remarked, “I never noticed how beautiful the sunlight is shining through your drapes,” and asked if I would open the drapes. When I did he said, “That’s beautiful! I never noticed you had a river behind your office.” When the session ended about 45 minutes after he had inhaled ketamine, he said, “I was going to head home, but I think I will go meet some friends. I haven’t seen them in over a year.” Another patient, who had responded to a research study in which she received a single infusion of ketamine more than a year ago and was hoping that intranasal ketamine could replicate the beneficial response, said within 15 minutes of inhalation, “I am feeling better, like when I was in the intravenous ketamine study. I am less depressed. But for me what’s even better is I feel calm, and I was very anxious coming here this morning.” There is also the example of a patient whose depressive symptoms had necessitated her being on disability, unable to carry out a demanding job. Within 20 minutes of her first inhalation of ketamine, this patient said, “I feel lighter, less depressed. If this works maybe I can return to work after all.” Later that week she called her supervisor and asked him if she could try working again on a trial basis, part time. With continued intranasal ketamine treatment, she is now working full time. Assessments of these changes in mood and outlook may require the creation of new rating scales designed to capture these rapid transformations in mental state.
What is allowing this ultra-rapid improvement in mood? The work of Duman and Aghajanian[14] in rodents suggests that the antidepressant effects of ketamine treatment may involve increased synaptogenesis in the PFC. Although this may be important for longer-term amelioration of TRD, the rapid improvements in depression within minutes cannot be due to the formation of new synapses on spines, as new synapses were only evident after 2 hours, not within minutes[15]. We hypothesize that intranasal ketamine’s ultra-rapid antidepressant effects within minutes might be explained by the blockade of NMDAR circuits in the frontal circuits that represent the mental suffering aspects of pain (Brodmann areas 24 and 25 and the insular cortex), while more prolonged antidepressant actions may arise from new spine growth in the higher PFC regions that normally serve to regulate these pain pathways.
POTENTIAL NEURAL MECHANISMS
Glutamate actions at NMDA receptors
The neurotransmitter, glutamate, acts at a variety of receptors, including those that form ion channels to allow sodium entry into the cell (AMPA, kainate and NMDA receptors), and those that are coupled to G-proteins to have more indirect intracellular actions (the metabotropic glutamate receptors mGluR1–8). The NMDA receptor is of particular interest, as it passes calcium as well as sodium, thus engaging calcium-related signaling events. The NMDA receptor is also unique due to its voltage-dependent activation, as its ion channel is blocked by magnesium when the membrane is hyperpolarized. Orser at el.[16] proposed that ketamine has two distinct mechanisms: blocking the open calcium channel and decreasing the frequency of channel opening. Subsequent work has shown that the NMDA receptor is a heterotetramer consisting of two GluN1 and two GluN2 subunits; the latter can be either the rapid NR2A subunits or the slower NR2B subunits, which allow greater calcium entry[17]. Ketamine blocks both types of NMDA receptors[18], however, its effects in the prefrontal association cortex (PFC) likely involve NMDA-NR2B receptors, as described below.
The primate PFC regulates cognition and emotion
Increasing evidence indicates that the PFC plays a critical role in depression[19, 20]. The PFC provides top-down regulation of thought, action and emotion[21], and has extensive connections to either promote or inhibit these neural events[22–26]. The PFC expands greatly in primate evolution, with the ventral and medial PFC (vmPFC) specialized for the regulation of emotion (internal states), while the more dorsal and lateral regions of PFC (dlPFC) mediate cognition (external states)[22–24, 27]. Further forward in the frontal pole there are even PFC areas that subserve metacognition, e.g. insight about oneself and others[28]. These regions all interconnect to provide a holistic mental state.
The medial PFC includes the cingulate cortices that sit above the corpus callosum. The cingulate cortices in frontal lobe include the anterior cingulate cortex, also known as Brodmann Area (BA) 24, and the cingulate cortex under the genu of the corpus callosum, often called the subgenual cingulate. The subgenual cingulate is part of the vmPFC, and is often referred to as the subgenual vmPFC. This review will use these terms interchangeably, reflecting the literature cited. The most caudal part of the subgenual PFC is BA25, which plays a key role in depression (see Fig. 1). The differences in terminology between papers can be very confusing, especially as there are species differences that add to the ambiguity (e.g. in rodent medial PFC, these areas appear to be intermixed and undifferentiated compared to primates[23]). However, a cohesive picture is beginning to emerge, whereby circuits in the ventral and medial PFC can represent the emotional value of an event, while circuits in dorsolateral regions generate representations of external space and sensory features. For example, in the anterior cingulate cortex, BA24, there are neurons that fire to the expectation of punishment[29], while in the dlPFC there are neurons that can represent a position in visual space[30], all of these neurons capable of representing information in the absence of sensory stimulation.
Figure 1. Potential actions of intranasal ketamine on cortical areas BA24, BA25, and the insular cortex, and their relationship to the pathways mediating the emotional aspects of pain.
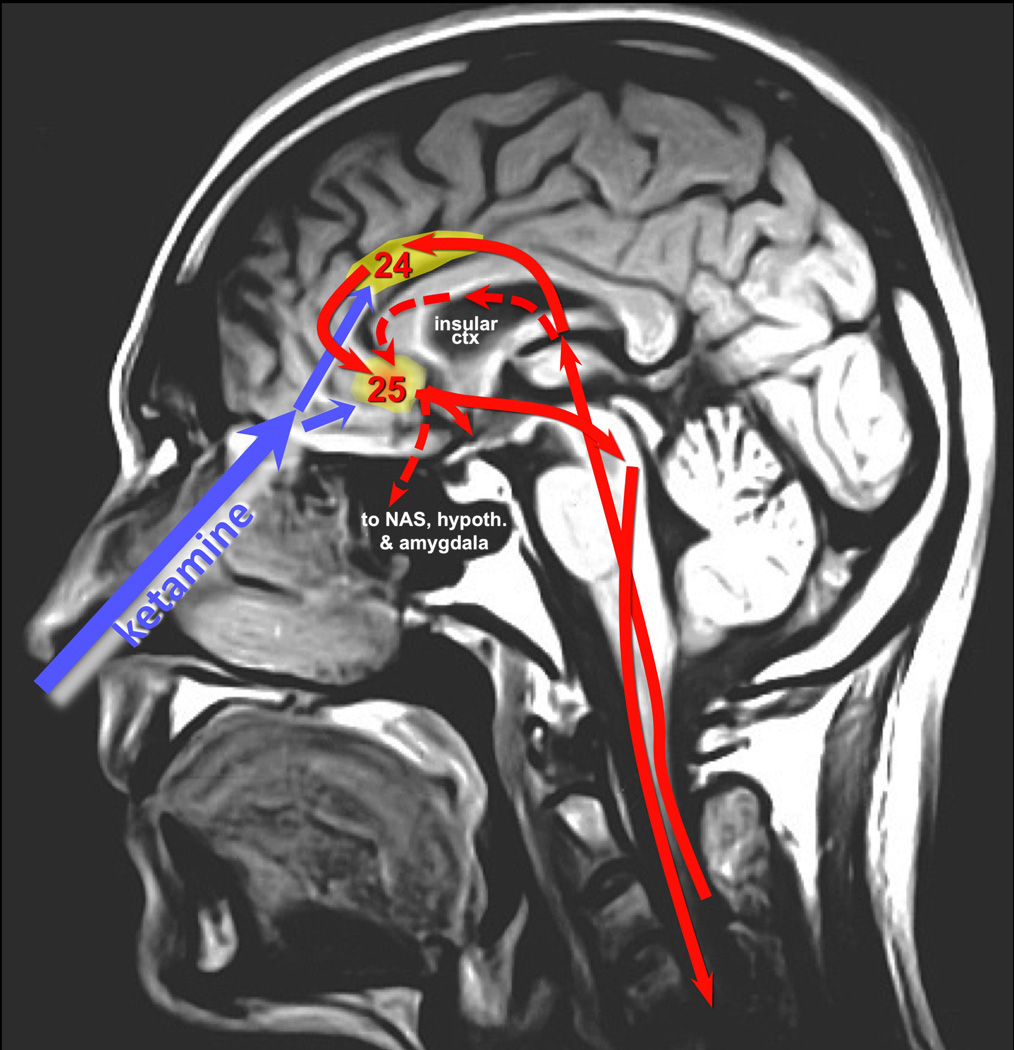
A midsagittal MRI of the human head showing the relationship between intranasal delivery of drug and its proximity to structures of the vmPFC. Some of the pathways mediating the emotional aspects of pain are represented in red, with projections from medial thalamus to insular cortex and anterior cingulate cortex (BA24), both of which project to subgenual BA25. BA25 in turn projects back down to brainstem, as well as to amygdala, hypothalamus and the nucleus accumbens. Brain structures not located near the midline are indicated with dashed lines.
The key role of vmPFC Brodmann Area 25 in Major Depressive Disorder
Imaging studies and ensuing deep brain stimulation treatments have revealed that the subgenual vmPFC is particularly relevant to Major Depressive Disorder (MDD)[19, 20, 31]. Within the vmPFC, BA25 is of particular interest, as this region is the “head ganglion” of the visceromotor system[23], positioned to control much of the limbic and autonomic nervous system (see below). The work of Mayberg and colleagues has shown that BA25 is activated in healthy individuals when they think of sad events, and this same region is overactive in patients with MDD[32]. Conversely, the activity of BA25 is reduced in MDD under conditions of effective treatment, including psychotherapy[20, 32]. Interestingly, BA25 has an exceptional density of serotonin transporters, suggesting it may be a site for selective serotonin reuptake inhibitor antidepressant actions[32]. Based on these data, deep brain stimulation (DBS) has been used to quiet the activity of BA25 in patients with intractable depression, and many have reported immediate relief. For example, one patient described the experience as follows: "All of a sudden they hit the spot, and I feel so calm and so peaceful. It was overwhelming to be able to process emotion on somebody's face. I'd been numb to that for so long."[33] These comments are remarkably similar to those of patients experiencing intranasal ketamine administration described above, suggesting that the two treatments may influence common brain circuitry.
Overlap with pain circuits
BA24, BA25 and the insular cortex are all key parts of a circuit processing the emotional aspects of pain (Fig. 1;[34, 35]). The sensory vs. emotional aspects of pain are processed by parallel pathways[36, 37] where the arousing and suffering aspects of pain are processed by diffuse projections arising through the brainstem and medial thalamus, which then projects to the insular cortex and the anterior cingulate BA24[35, 38], which both project to BA25 (Fig. 1;[23]). Lesion studies in rats have shown that the anterior cingulate cortex is a key area for processing the emotional aspects of pain[39]. Similarly, physiological recordings in monkeys have shown that neurons in BA24 respond to the expectation of punishment[29], while neurons in the dorsal region of BA25 respond to aversive events[40]. BA25 in turn projects to a number of brain structures that mediate emotional responding[23, 41], including the amygdala (unconscious primitive emotional associations[42, 43]), hypothalamus (vegetative functions[44]), and nucleus accumbens (changes in emotional habits, loss of feeling of reward[45]), and to the brainstem for control of reflexes and the autonomic nervous system[46], thus completing the body-brain-body loop that can maintain vicious cycling. For example, BA25 projections to brainstem can excite columns of neurons in the peri-aqueductal gray that generate coordinated responses to painful events, e.g. the freezing response (“mental paralysis”), or other body-wide responses to pain and stress[46]., The overactivation of this medial pain system in depression may lead to the generation of mental suffering normally caused by a painful stimulus, yet in the absence of an actual sensory event. This view of depression emphasizes why it is indeed a mental disorder.
Researchers have hypothesized that the effects of intravenous ketamine may involve alterations in glutamate signaling in these circuits[47, 48]. For example, the depressed patients who respond best to ketamine treatment are those with overactivity of BA24 before treatment[49], suggesting that ketamine may be acting by reducing the firing of an overactive system generating mental anguish. Similar results have been seen in patients with bipolar depression, where response to ketamine correlated best with activity in the BA25[50]. Additional support can be found in imaging studies of the ketamine response in both healthy subjects and patients with depression. For example, i.v. infusions of ketamine reduced the activity of the vmPFC in healthy volunteers[51], and reduced the activity of the insular cortex in patients with intractable depression[52]. Carlson et al. also found reduced activity in the lateral habenula, a brain region that responds to aversive stimulation and inhibits the dopamine reward system[53]. Thus, the human imaging studies suggest that ketamine alters the activity of the circuits mediating the emotional response to pain.
Research in animals also links NMDA receptors in these brain regions with response to pain. For example, studies in rats have shown that increased sensitivity to visceral pain is associated with increased NMDA-NR2B receptor expression within the anterior cingulate cortex, and is reduced by NMDA-NR2B blockade[54, 55]. These results may have parallels to ketamine’s actions in depression. Although NMDA-NR2B receptors have often been thought to be extra-synaptic or more potent in mediating GABAergic actions, more recent data show that they are essential for the generation of mental representations by pyramidal cell circuits in the primate PFC.
The key role of NMDA receptors in dlPFC circuits generating mental representations
How might ketamine blockade of NMDAR change the vmPFC circuits involved in depression? Although the role of glutamate NMDAR has not yet been studied in the primate vmPFC, anatomical and physiological studies of the primate dlPFC have shown that NMDAR play a critical role in the circuits that generate mental representations of visual space[56]. NMDAR-NR2B are found exclusively within the post-synaptic density in layer III circuits in the dlPFC, in contrast to the extrasynaptic location seen in many other circuits, e.g. hippocampus. Thus, NMDAR-NR2B are positioned to mediate synaptic communication in these highly evolved circuits. Electrophysiological data confirmed this hypothesis. Recordings were made from the dlPFC of monkeys performing a visuospatial working memory task in which the monkey had to remember an ever-changing spatial position over a brief delay period (Fig. 2A). Goldman-Rakic and colleagues first discovered that there are neurons in the dlPFC that are able to represent spatial position over the Delay period by maintaining persistent firing in the absence of sensory stimulation, but only following a cue at their “preferred direction” ([30]; Fig. 2B). Goldman-Rakic[57] uncovered the layer III dlPFC microcircuits that generate these neural representations, including glutamatergic pyramidal cells that excite each other to maintain persistent firing (Fig. 2C). This persistent firing arises from glutamate stimulation of NMDAR-NR2B on pyramidal cells spines (Figs. 2D–E). In contrast to sensory cortex, NMDAR-NR2B are found exclusively within the post-synaptic density in these layer III synapses on spines, and are not at extra-synaptic sites (Fig. 2E;[56]. Blockade of these NMDAR-NR2B, e.g. with local NMDA-NR2B blockade or by systemic administration of ketamine, decreases the firing of Delay cell circuits and erodes the neural representation of visual space ([56]; Figs. 2D).
Figure 2. Glutamate NMDA receptor circuits in dorsolateral prefrontal cortex play a key role in mental representations of visual space.
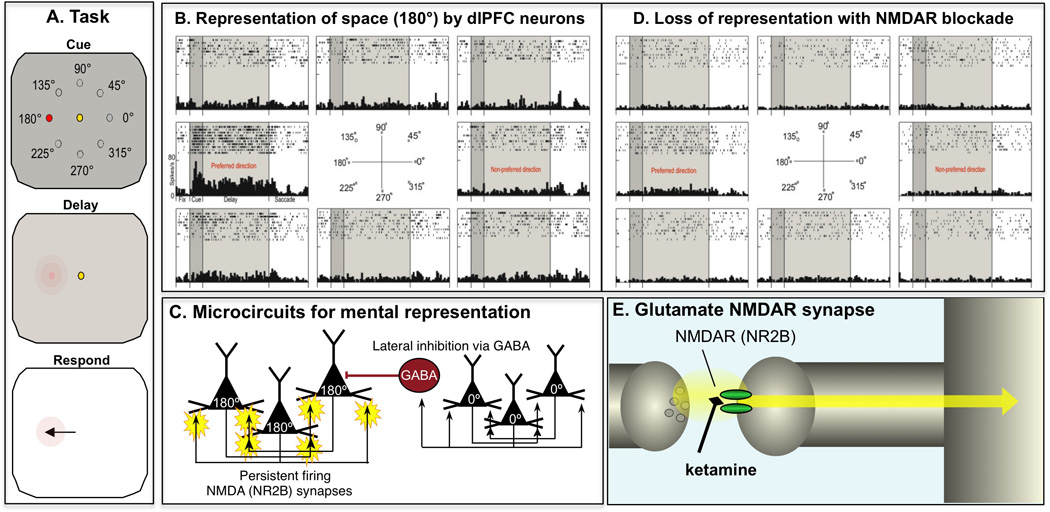
A. Monkeys perform a visuospatial working memory task in which they have to remember an ever-changing spatial position over a brief delay period. The monkey fixates on a central spot as a cue is briefly flashed at one of eight locations (e.g. 180°). The monkey then remembers that position for several seconds during the Delay period. When the fixation spot extinguishes, the monkey responds by moving its eyes to the remembered location, and if correct receives a juice reward. The position of the cue changes randomly over hundreds of trials, thus requiring the constant updating of the contents of working memory. B. An example of a neuron in the dlPFC that represents spatial position over the Delay period. These neurons often fire to the Cue, and then continue to generate persistent firing across the Delay period, but only for their preferred spatial position, not for other locations. C. Goldman-Rakic[57] uncovered the layer III dlPFC microcircuits that generate these neural representations. Pyramidal cells receive highly processed visuospatial information from the parietal association cortex, with clusters of cells receiving similar locations. Pyramidal cells with a similar “preferred direction” excite each other to generate the persistent firing needed to maintain information in the absence of sensory stimulation. The spatial specificity is enhanced through lateral inhibition from GABAergic interneurons; e.g. when the Cue is at 0°, the 0° pyramidal cells inhibit the 180° pyramidal cells by engaging GABAergic projections. D. Blockade of NMDAR-NR2B in the dlPFC markedly weakens the ability of dlPFC circuits to generate neural representations of visual space. Similar effects were found with systemic administration of ketamine. E. NMDAR-NR2B in the dlPFC are found exclusively within the post-synaptic density on pyramidal cell spines, and not at extra-synaptic sites. Based on data from[56].
Although there have not yet been studies of NMDAR actions in primate vmPFC, studies of the rodent vmPFC suggest that NMDAR actions are especially important for these circuits as well[58], including NMDAR-NR2B mediation of pain in rat medial PFC[54]. If similar actions occur in humans, we hypothesize that ketamine may block the representation of pain and suffering by vmPFC circuits, temporarily relieving mental anguish. A rapid decline in the firing of BA24, BA25 and/or insular cortex might shift mental state and be experienced as a “lifting” or a “high”. This respite in firing may provide a “foot in door” to allow higher PFC circuits to provide more sustained regulation for longer-term relief, i.e. true antidepressant actions.
Intranasal ketamine is ideally positioned to target higher pain circuits
Intranasal administration has been established as an effective method for drug delivery to brain[59], reaching brain via the olfactory system[60]. These studies suggest that intranasally-applied ketamine enters the brain through the olfactory system, where the olfactory nerves traverse holes in the cribiform plate into the olfactory bulb, which sits immediately below the vmPFC (Fig. 1). Thus, intranasal administration may direct drug to those brain regions where ketamine may have its therapeutic actions (schematically illustrated in Fig. 1). Indeed, intranasal administration of ketamine is given in the emergency room for the rapid relief of pain, providing effective analgesia within 5–20 minutes[2]. These data suggest that ketamine may have similar effects in depression, relieving the mental aspects of pain. The findings from the emergency room analgesia studies further suggest that intranasal ketamine may be particularly helpful in the subset of depressed patients who experience physical as well as mental pain.
Working hypothesis and the relationship to rTMS treatment of depression
We hypothesize that intranasal ketamine may help to ameliorate the symptoms of depression by very rapidly reducing the firing of neurons in BA24, BA25, and insular cortex, thus providing an opportunity for the left dlPFC to begin to regulate these pain circuits for longer-term anti-depressant actions. Studies in animals[61, 62] and then in humans[63] have shown that dlPFC circuits are taken “off-line” during stress exposure. With prolonged stress exposure there are additional architectural changes, with loss of spines and dendrites in those circuits that provide top-down regulation, and increased dendritic arborization in those circuits that drive the stress response[62, 64–68]. Thus, it may take time for spines to regrow in the higher PFC regions that provide top-down control and normalization of circuit strengths. Also, as ketamine impairs the functioning of the dlPFC[56], the drug would need to be cleared before these regulatory functions could be evident. Studies in rodents have observed new spines forming in PFC neurons in response to ketamine treatment, likely involving changes in mTor signaling pathways, the engagement of BDNF growth factors, and the inhibition of GSK-3 signaling, e.g. by lithium[15, 69, 70]. Spine growth is not evident for the first 2 hours after ketamine treatment, suggesting that these effects do not underlie the ultra-rapid changes in mood (within minutes), but likely contribute to the more prolonged mood stabilization that follows. The rodent PFC is undifferentiated, and thus one cannot know without explicit tract-tracing studies whether spine changes in rodent PFC are in neurons that excite or inhibit the stress response. However, accumulating data from human and nonhuman primates suggest that the prolonged anti-depressant effects may involve spine changes in higher PFC regions such as the dlPFC that are positioned to regulate the circuits that generate mental representations of pain.
Tract tracing studies in monkeys have shown that the dlPFC (e.g. areas BA9 and BA46) can influence BA25 via projections through BA10m and BA32 to BA25 (schematically illustrated in Fig. 3;[23, 71]), and can influence the insular cortex via projections via BA10m through a variety of orbital connections[23]. There are also extensive, direct connections between the dlPFC and BA24 (Fig. 3; e.g.[72]). Recent analyses of successful DBS treatments for intractable depression suggest that this anterior pathway involving BA10m must be activated in order to have persistent anti-depressant effects[73]. Thus, these circuits may be involved in the healthy, endogenous regulation of emotional state by higher PFC areas. Interestingly, BA10m appears to be involved in emotional regulation[74] and self-knowledge[28, 75], which may be needed for a person to have the self-awareness of emotional recovery.
Figure 3. Potential circuit interactions between dorsolateral prefrontal cortex and BA24 and BA25.
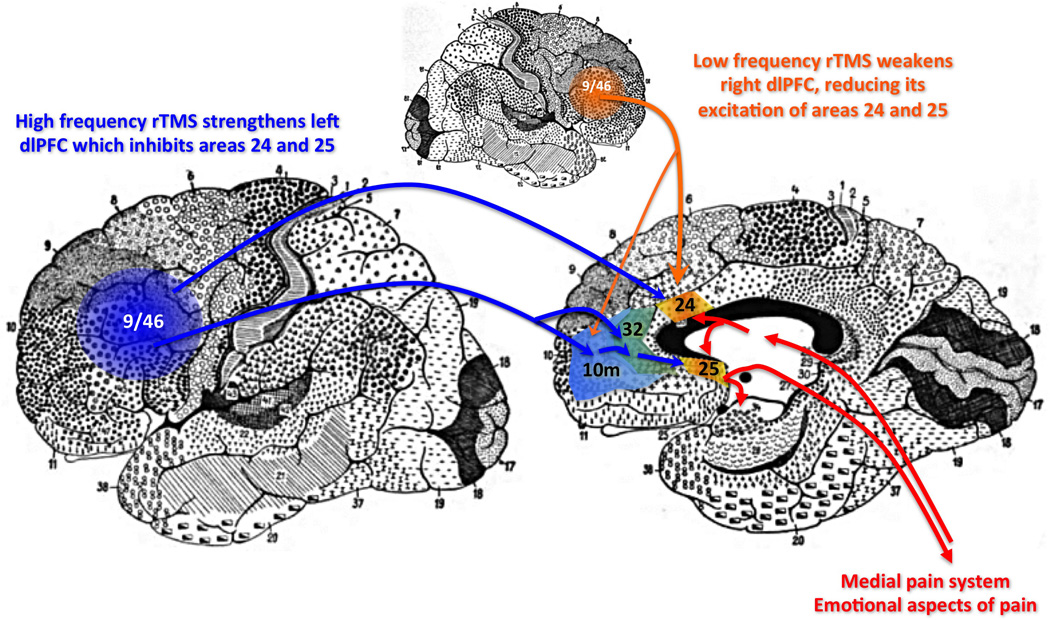
Tract-tracing studies in monkeys indicate that the dorsolateral PFC areas 9 and 46 can influence medial PFC circuits through direct projections to BA24, and indirect projections to BA25 via areas 10m and 32. They can also influence the insular cortex via 10m projections through orbital PFC (note: insular cortex is not evident on these Brodmann human brain maps, and thus is not shown for the sake of clarity). Studies of rTMS in patients with depression suggest that activation of the left dlPFC serves to inhibit BA25, while deactivation of the right dlPFC is helpful.
The human brain has hemispheric specialization that may also contribute to depression and its treatment. In an oversimplified manner one can say that the left PFC is the “Yes” hemisphere, while the right hemisphere is the “No” hemisphere. For example, there has been longstanding evidence that lesions to the left hemisphere are associated with depression[76] In contrast, mania is associated with reduced activity of the right PFC[77], which is specialized for inhibiting inappropriate actions and emotions[78]. Repetitive transcranial magnetic stimulation (rTMS) is increasingly used for the treatment of depression, using either high frequency rTMS applied to the left dlPFC to strengthen its function, or low frequency rTMS to the right dlPFC to weaken its influence[79]. rTMS may work in part by altering dlPFC influence on BA25, as there is evidence from functional connectivity studies that TMS sites in left dlPFC with better clinical efficacy were more negatively correlated with the subgenual cingulate, i.e. the region including BA25[80]. Conversely, the patients that respond best to low-frequency rTMS to the right dlPFC have the highest activity in subgenual vmPFC before treatment[81]. Thus, optimal treatment for intractable depression may be helped by a combination of intranasal ketamine and rTMS in order to help normalize the PFC circuits regulating emotion.
Discussion
The intranasal administration of ketamine may be especially effective in treating depressive symptoms, as it may target the circuits most relevant to depression, with fewer side effects, e.g. dissociation and confusion, than have been found with intravenous administration. Intranasal administration also has the great benefit that it can be self-administered, thus facilitating the multiple treatments often needed to produce sustained relief. However, it is important to note that ketamine treatment is not a panacea for depression: there are still patients for whom the treatment is ineffective or only effective for a brief time, or for whom the side effects are intolerable. Thus, it is important that ketamine is not viewed as a “miracle cure”, as it is often conveyed in the media.
Understanding how ketamine has its beneficial actions will be a daunting task, as the circuits regulating and generating emotion are extraordinarily complex. However, the successful use of intranasal ketamine for treating physical pain suggests that the circuits that process the emotional aspects of the pain experience are likely involved. The ultra-rapid effects of intranasal ketamine suggest that NMDA receptor blockade may have immediate effects on pain circuits. Although previous research focused on the extra-synaptic effects of NMDAR-NR2B, it is now known that NMDAR-NR2B are essential synaptic elements in the PFC circuits generating mental representations in primates[56]. It is not known if these mechanisms extend to the primate vmPFC circuits that generate representations of suffering. If so, we propose a multi-step process, whereby 1) ketamine produces an initial, ultra-rapid reduction in the firing of these representational pain affect circuits, breaking their vicious cycle, followed by 2) strengthening of the higher dlPFC circuits that regulate these pain pathways, likely involving synaptogenesis, as suggested by Duman and colleagues[14, 82]. The addition of rTMS treatments may be helpful for some patients, as rTMS applied to the left dlPFC may strengthen the circuits that regulate emotion.
There are also measurement challenges to future clinical trials on intranasal ketamine and related interventions. Many studies conducted on ketamine have utilized standard measures of antidepressant efficacy developed decades ago. While existing scales have a vast literature supporting their validity and sensitivity to antidepressants (e.g. the MADRS and HDRS), there are some limitations to this approach. It is worth noting that these instruments have principally been conceptualized and tested under specific psychopharmacologic paradigms (e.g. monoamine oxidase inhibitors, tricyclic antidepressants, selective serotonin reuptake inhibitors), all of which have entirely different mechanisms of action and onset of effects than ketamine. Most critically, it is likely that these measures are inadequately describing and capturing patient experience, and thus are not capturing the underlying phenomena that are changing in those who respond to ketamine treatment. Further development of measures that meaningfully evaluate the effects of this novel class of treatment are essential for future research.
Finally, this approach may elucidate why ketamine helps symptoms of depression (reducing the pathological overactivity of BA25), but worsens or mimics the symptoms of schizophrenia (impairing the functioning of the dorsolateral PFC) e.g.[56, 83, 84]. Future research should explore whether there are any mechanisms that are differentially expressed in primate BA25 vs. dlPFC, in order to create anti-depressants that do not aggravate psychosis, and anti-psychotics that do not induce depressive symptoms.
Conclusion
Large numbers of patients with intractable depression require new approaches to treatment, and ketamine has shown promise. Intranasal administration of ketamine may be especially useful due to its ease of use, superior side effect profile, and ultra-rapid rate of onset. The intranasal route may have fewer side effects by preferentially targeting the vmPFC systems most relevant to depressive symptoms. We hypothesize that ketamine may have ultra-rapid beneficial effects within minutes by blocking NMDAR-mediated neural representations of suffering, thus easing a vicious cycle of mental anguish and paralysis. More sustained anti-depressant actions would require normalization of the higher PFC circuits that regulate mental pain circuits, e.g. through increased synaptogenesis in these higher regions. This approach can be coupled with rTMS applied to the dlPFC for those patients who require more comprehensive treatment for unremitting depressive symptoms.
Acknowledgements
This work is dedicated to the memory of Daniel Friedland, with the hope that we may discover more effective treatments to alleviate suffering. This work was funded in part by NIH Pioneer Award DP1AG047744 to AFTA. The authors would like to thank Drs. G. Aghajanian, R. Duman, J. Krystal, H. Mayberg, J. Murrough, G. Sanacora and J. Singh for informative and inspiring discussions.
List of Abbreviations
- BDI
Beck Depression Inventory
- BA
Brodmann Area
- DBS
Deep Brain Stimulation (which often is used to shut off a brain area)
- DSM
Diagnostic and Statistical Manual
- FDA
Food and Drug Administration
- PFC
prefrontal cortex
- dl
dorsolateral
- vm
ventromedial
- HDRS
Hamilton Depression Rating Scale
- MADRS
Montomery-Asberg Depression Rating Scale
- MDD
Major Depressive Disorder
- NMDAR-NR2B
N-methyl-D-aspartame glutamate receptor with NR2B subunits
- rTMS
Repetitive Transcranial Magnetic Stimulation
- TRD
treatment-resistant depression
Footnotes
Disclosures
Lewis Opler has the following disclosures: MultiHealth Systems, Inc., coauthor of the Positive and Negative Syndrome Scale (PANSS), royalties.
Amy Arnsten has the following disclosures: Arnsten and Yale University receive royalties from Shire Pharmaceuticals from the sales of Intuniv™ (extended release guanfacine) for the treatment of pediatric Attention Deficit Hyperactivity Disorder.
REFERENCES
- 1.Reich DL, Silvay G. Ketamine: an update on the first twenty-five years of clinical experience. Can J Anaesth. 1989;36(2):186–197. doi: 10.1007/BF03011442. [DOI] [PubMed] [Google Scholar]
- 2.Andolfatto G, Willman E, Joo D, et al. Intranasal ketamine for analgesia in the emergency department: a prospective observational series. Acad Emerg Med. 2013;20(10):1050–1054. doi: 10.1111/acem.12229. [DOI] [PubMed] [Google Scholar]
- 3.McCarty EC, Mencio GA, Walker LA, Green NE. Ketamine sedation for the reduction of children’s fractures in the emergency department. J Bone Joint Surg Am. 2000;82-A(7):912–918. doi: 10.2106/00004623-200007000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Anis NA, Berry SC, Burton NR, Lodge D. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurons by n-methyl-aspartate. Br J Pharmac. 1983;79(2):565–575. doi: 10.1111/j.1476-5381.1983.tb11031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moretti RJ, Hassan SZ, Goodman LI, Meltzer HY. Comparison of ketamine and thiopental in healthy volunteers: effects on mental status, mood, and personality. Anesth Analg. 1984;63(12):1087–1096. [PubMed] [Google Scholar]
- 6.Skolnick P, Layer RT, Popik P, et al. Adaptation of N-methyl-D-aspartate (NMDA) receptors following antidepressant treatment: implications for the pharmacotherapy of depression. Pharmacopsychiatry. 1996;29(1):23–26. doi: 10.1055/s-2007-979537. [DOI] [PubMed] [Google Scholar]
- 7.Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry. 2006;163(7):1161–1172. doi: 10.1176/ajp.2006.163.7.1161. [DOI] [PubMed] [Google Scholar]
- 8.Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 9.Zarate CAJ, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 10.DiazGranados N, Ibrahim LA, Brutsche NE, et al. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J Clin Psychiatry. 2010;71(12):1605–1611. doi: 10.4088/JCP.09m05327blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67(2):139–145. doi: 10.1016/j.biopsych.2009.08.038. [DOI] [PubMed] [Google Scholar]
- 12.Lapidus KA, Levitch CF, Perez AM, et al. A randomized controlled trial of intranasal ketamine in Major Depressive Disorder. Biol Psychiatry. 2014 Apr 3; doi: 10.1016/j.biopsych.2014.03.026. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Opler LA, Opler MG, Arnsten AFT. Ultrarapid response to intranasal ketamine in outpatients with long-term treatment-refractory depression. 2014 in preparation. [Google Scholar]
- 14.Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338(6103):68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li N, Lee BT, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orser BA, Pennefather PS, MacDonald JF. Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology. 1997;86(4):903–917. doi: 10.1097/00000542-199704000-00021. [DOI] [PubMed] [Google Scholar]
- 17.Erreger K, Dravid SM, Banke TG, et al. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563(Pt. 2):345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu HT, Hollmann MW, Liu WH, et al. Modulation of NMDA receptor function by ketamine and magnesium: Part I. Anesth Analg. 2001;92(5):1173–1181. doi: 10.1097/00000539-200105000-00019. [DOI] [PubMed] [Google Scholar]
- 19.Murray EA, Wise SP, Drevets WC. Localization of dysfunction in major depressive disorder: Prefrontal cortex and amygdala. Biological Psychiatry. 2011;69(12):e43–e54. doi: 10.1016/j.biopsych.2010.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 21.Goldman-Rakic PS. The prefrontal landscape: implications of functional architecture for understanding human mentation and the central executive. Phil Trans R Soc London. 1996;351(1346):1445–1453. doi: 10.1098/rstb.1996.0129. [DOI] [PubMed] [Google Scholar]
- 22.Goldman-Rakic PS. Circuitry of the primate prefrontal cortex and the regulation of behavior by representational memory. In: Plum F, editor. Handbook of Physiology, The Nervous System, Higher Functions of the Brain. Bethesda: American Physiological Society; 1987. pp. 373–417. [Google Scholar]
- 23.Ongür D, Price JL. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex. 2000;10(3):206–219. doi: 10.1093/cercor/10.3.206. [DOI] [PubMed] [Google Scholar]
- 24.Ghashghaei HT, Barbas H. Pathways for emotion: interactions of prefrontal and anterior temporal pathways in the amygdala of the rhesus monkey. Neuroscience. 2002;115(4):1261–1279. doi: 10.1016/s0306-4522(02)00446-3. [DOI] [PubMed] [Google Scholar]
- 25.Barbas H, Medalla M, Alade O, et al. Relationship of prefrontal connections to inhibitory systems in superior temporal areas in the rhesus monkey. Cereb Cortex. 2005;15(9):1356–1370. doi: 10.1093/cercor/bhi018. [DOI] [PubMed] [Google Scholar]
- 26.Arnsten AFT, Wang M, Paspalas CD. Neuromodulation of thought: Flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron. 2012;76(1):223–239. doi: 10.1016/j.neuron.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbas H, Saha S, Rempel-Clower N, Ghashghaei T. Serial pathways from primate prefrontal cortex to autonomic areas may influence emotional expression. BMC Neurosci. 2003;4:25. doi: 10.1186/1471-2202-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amodio DM, Frith CD. Meeting of minds: the medial frontal cortex and social cognition. Nat Rev Neurosci. 2006;7(4):268–277. doi: 10.1038/nrn1884. [DOI] [PubMed] [Google Scholar]
- 29.Seo H, Lee D. Behavioral and neural changes after gains and losses of conditioned reinforcers. J Neurosci. 2009;29(11):3627–3641. doi: 10.1523/JNEUROSCI.4726-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Funahashi S, Bruce CJ, Goldman-Rakic PS. Mnemonic coding of visual space in the monkey's dorsolateral prefrontal cortex. J Neurophysiology. 1989;61(2):331–349. doi: 10.1152/jn.1989.61.2.331. [DOI] [PubMed] [Google Scholar]
- 31.Drevets WC, Price JL, Simpson JRJ, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 32.Mayberg HS. Targeted electrode-based modulation of neural circuits for depression. J Clin Invest. 2009;119(4):717–725. doi: 10.1172/JCI38454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobbs D. A depression switch? New York Times Magazine. 2006 [Google Scholar]
- 34.Vogt BA, Sikes RW. The medial pain system, cingulate cortex, and parallel processing of nociceptive information. Prog Brain Res. 2000;22:223–235. doi: 10.1016/s0079-6123(08)62141-x. [DOI] [PubMed] [Google Scholar]
- 35.Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14(7):502–511. doi: 10.1038/nrn3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willis WD, Al-Chaer ED, Quast MJ, Westlund KN. A visceral pain pathway in the dorsal column of the spinal cord. Proc Natl Acad Sci U S A. 1999;96(14):7675–7679. doi: 10.1073/pnas.96.14.7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willis WD, Westlund KN. Neuroanatomy of the pain system and of the pathways that modulate pain. J Clin Neurophysiol. 1997;14(1):2–31. doi: 10.1097/00004691-199701000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science. 2000;288(5472):1769–1772. doi: 10.1126/science.288.5472.1769. [DOI] [PubMed] [Google Scholar]
- 39.Yan N, Cao B, Xu J, et al. Glutamatergic activation of anterior cingulate cortex mediates the affective component of visceral pain memory in rats. Neurobiol Learn Mem. 2012;97(1):156–164. doi: 10.1016/j.nlm.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Monosov IE, Hikosaka O. Regionally distinct processing of rewards and punishments by the primate ventromedial prefrontal cortex. J Neurosci. 2012;32(30):10318–10330. doi: 10.1523/JNEUROSCI.1801-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghashghaei HT, Hilgetag CC, Barbas H. Sequence of information processing for emotions based on the anatomic dialogue between prefrontal cortex and amygdala. Neuroimage. 2007;34(3):905–923. doi: 10.1016/j.neuroimage.2006.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis M. The role of the amygdala in fear and anxiety. Annual Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 43.LeDoux J. Fear and the brain: Where have we been and where are we going? Biological Psychiatry. 1998;44(12):1229–1238. doi: 10.1016/s0006-3223(98)00282-0. [DOI] [PubMed] [Google Scholar]
- 44.Carmel PW. Vegetative dysfunctions of the hypothalamus. Acta Neurochir. 1985;75(1–4):113–121. doi: 10.1007/BF01406331. [DOI] [PubMed] [Google Scholar]
- 45.Everitt BJ, Belin D, Economidou D, et al. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philos Trans R Soc Lond B Biol Sci. 2008;363(1507):3125–3135. doi: 10.1098/rstb.2008.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neafsey EJ. Prefrontal control of the autonomic nervous system: Anatomical and physiological observations. Progress in Brain Res. 1990;85:147–165. doi: 10.1016/s0079-6123(08)62679-5. [DOI] [PubMed] [Google Scholar]
- 47.Maeng S, Zarate CAJ. The role of glutamate in mood disorders: Results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Current Psychiatry Reports. 2007;9(6):467–474. doi: 10.1007/s11920-007-0063-1. [DOI] [PubMed] [Google Scholar]
- 48.McCarthy DJ, Alexander R, Smith MA, et al. Glutamate-based depression GBD. Med Hypotheses. 2012;78(5) doi: 10.1016/j.mehy.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 49.Salvadore G, Cornwell BR, Colon-Rosario V, et al. Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry. 2009;65(4):289–295. doi: 10.1016/j.biopsych.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nugent AC, Diazgranados N, Carlson PJ, et al. Neural correlates of rapid antidepressant response to ketamine in bipolar disorder. Bipolar Disord. 2014;16(2):119–128. doi: 10.1111/bdi.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deakin JF, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65(2):154–164. doi: 10.1001/archgenpsychiatry.2007.37. [DOI] [PubMed] [Google Scholar]
- 52.Carlson PJ, Diazgranados N, Nugent AC, et al. Neural correlates of rapid antidepressant response to ketamine in treatment-resistant unipolar depression: a preliminary positron emission tomography study. Biol Psychiatry. 2013;73(12):1213–1221. doi: 10.1016/j.biopsych.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hikosaka O. The habenula: from stress evasion to value-based decision-making. Nat Rev Neurosci. 2010;11:503–513. doi: 10.1038/nrn2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fan J, Wu X, Cao Z, et al. Up-regulation of anterior cingulate cortex NR2B receptors contributes to visceral pain responses in rats. Gastroenterology. 2009;136(5):1732–1740. doi: 10.1053/j.gastro.2009.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Zhou L, Huang J, Gao J, et al. NMDA and AMPA receptors in the anterior cingulate cortex mediates visceral pain in visceral hypersensitivity rats. Cell Immunol. 2014;287(2):86–90. doi: 10.1016/j.cellimm.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 56.Wang M, Yang Y, Wang CJ, et al. NMDA receptors subserve working memory persistent neuronal firing In dorsolateral prefrontal cortex. Neuron. 2013;77(4):736–749. doi: 10.1016/j.neuron.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14(3):477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 58.Wang H, Stradtman GGr, Wang XJ, Gao WJ. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc Natl Acad Sci U S A. 2008;105(43):16791–16796. doi: 10.1073/pnas.0804318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel RB, Patel MR, Bhatt KK, et al. Evaluation of brain targeting efficiency of intranasal microemulsion containing olanzapine: pharmacodynamic and pharmacokinetic consideration. Drug Deliv. 2014 May 20;:1–9. doi: 10.3109/10717544.2014.912694. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 60.Wei Y, Ying M, Xu S, et al. Microdialysis pharmacokinetic study of scopolamine in plasma, olfactory bulb and vestibule after intranasal administration. Drug Deliv. 2014 May 28;:1–6. doi: 10.3109/10717544.2014.910565. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 61.Arnsten AFT. The biology of feeling frazzled. Science. 1998;280(5370):1711–1712. doi: 10.1126/science.280.5370.1711. [DOI] [PubMed] [Google Scholar]
- 62.Arnsten AFT. Stress signaling pathways that impair prefrontal cortex structure and function. Nature Reviews Neuroscience. 2009;10(6):410–422. doi: 10.1038/nrn2648. PMCID: PMC2907136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin S, Hermans EJ, van Marle HJF, et al. Acute psychological stress reduces working memory-related activity in the dorsolateral prefrontal cortex. Biological Psychiatry. 2009;66(1):25–32. doi: 10.1016/j.biopsych.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 64.Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22(15):6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Radley JJ, Rocher AB, Miller M, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006;16(3):313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- 66.Izquierdo A, Wellman CL, Holmes A. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci. 2006;26(21):5733–5738. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hains AB, Vu MA, Maciejewski PK, et al. Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc Natl Acad Sci U S A. 2009;106(42):17957–17962. doi: 10.1073/pnas.0908563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shansky RM, Hamo C, Hof PR, et al. Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb Cortex. 2009;106(42):17957–17962. doi: 10.1093/cercor/bhp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu RJ, Lee FS, Li XY, et al. Brain-Derived Neurotrophic Factor Val66Met Allele Impairs Basal and Ketamine-Stimulated Synaptogenesis in Prefrontal Cortex. Biol Psychiatry. 2012;71(11):996–1005. doi: 10.1016/j.biopsych.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu RJ, Fuchikami M, Dwyer JM, et al. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38(11):2268–2277. doi: 10.1038/npp.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barbas H, Pandya DN. Architecture and intrinsic connections of the prefrontal cortex in the rhesus monkey. J Comp Neurol. 1989;286(3):353–375. doi: 10.1002/cne.902860306. [DOI] [PubMed] [Google Scholar]
- 72.Selemon LD, Goldman-Rakic PS. Common cortical and subcortical targets of the dorsolateral prefrontal and posterior parietal cortices in the rhesus monkey: evidence for a distributed neural network subserving spatially guided behavior. J Neurosci. 1988;8(11):4049–4068. doi: 10.1523/JNEUROSCI.08-11-04049.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Riva-Posse P, Choi KS, Holtzheimer PE, et al. Defining Critical White Matter Pathways Mediating Successful Subcallosal Cingulate Deep Brain Stimulation for Treatment-Resistant Depression. Biol Psychiatry. 2014 Apr 13; doi: 10.1016/j.biopsych.2014.03.029. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ochsner KN, Silvers JA, Buhle JT. Functional imaging studies of emotion regulation: a synthetic review and evolving model of the cognitive control of emotion. Ann N Y Acad Sci. 2012;1251:E1–E24. doi: 10.1111/j.1749-6632.2012.06751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heatherton TF, Wyland CL, Macrae CN, et al. Medial prefrontal activity differentiates self from close others. Soc Cogn Affect Neurosci. 2006;1(1):18–25. doi: 10.1093/scan/nsl001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robinson RG, Lipsey JR, Bolla-Wilson K, et al. Mood disorders in left-handed stroke patients. Am J Psychiatry. 1985;142(12):1424–1429. doi: 10.1176/ajp.142.12.1424. [DOI] [PubMed] [Google Scholar]
- 77.Blumberg HP, Stern E, Ricketts S, et al. Rostral and orbital prefrontal cortex dysfunction in the manic state of bipolar disorder. American Journal of Psychiatry. 1999;156(12):1986–1988. doi: 10.1176/ajp.156.12.1986. [DOI] [PubMed] [Google Scholar]
- 78.Aron AR. From reactive to proactive and selective control: Developing a richer model for stopping inappropriate responses. Biological Psychiatry. 2011;69(12):e55–e68. doi: 10.1016/j.biopsych.2010.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen J, Zhou C, Wu B, et al. Left versus right repetitive transcranial magnetic stimulation in treating major depression: a meta-analysis of randomised controlled trials. Psychiatry Res. 2013;210(3):1260–1264. doi: 10.1016/j.psychres.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 80.Fox MD, Buckner RL, White MP, et al. Efficacy of transcranial magnetic stimulation targets for depression is related to intrinsic functional connectivity with the subgenual cingulate. Biol Psychiatry. 2012;72(7):595–603. doi: 10.1016/j.biopsych.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kito S, Hasegawa T, Koga Y. Cerebral blood flow in the ventromedial prefrontal cortex correlates with treatment response to low-frequency right prefrontal repetitive transcranial magnetic stimulation in the treatment of depression. Psychiatry Clin Neurosci. 2012;66(2):138–145. doi: 10.1111/j.1440-1819.2011.02312.x. [DOI] [PubMed] [Google Scholar]
- 82.Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73:1133–1141. doi: 10.1016/j.biopsych.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Javitt DC. Glutamatergic theories of schizophrenia. Isr J Psychiatry Relat Sci. 2010;47:4–16. [PubMed] [Google Scholar]
- 84.Driesen NR, McCarthy G, Bhagwagar Z, et al. The impact of NMDA receptor blockade on human working memory-related prefrontal function and connectivity. Neuropsychopharmacology. 2013;38:2613–2622. doi: 10.1038/npp.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]