

Abstract
There is increasing evidence implicating HER3 in several types of cancer. But the development of targeted therapies to inactivate HER3 function has been a challenging endeavor. Its kinase domain functions in allostery not catalysis, and the classical ATP-analog class of tyrosine kinase inhibitors fail to inactivate it. Here we describe a novel approach that eliminates HER3 expression. The small-molecule cotransin CT8 binds the Sec61 translocon and prevents the signal peptide of the nascent HER3 protein from initiating its cotranslational translocation, resulting in the degradation of HER3 but not the other HER proteins. CT8 treatment suppresses the induction of HER3 that accompanies lapatinib treatment of HER2-amplified cancers and synergistically enhances the apoptotic effects of lapatinib. The target selectivities of cotransins are highly dependent on their structure and the signal sequence of targeted proteins and can be narrowed through structure-function studies. Targeting Sec61-dependent processing identifies a novel strategy to eliminate HER3 function.
Keywords: HER3, ErbB3, Sec61, cotranslational localization, cotransins
Introduction
The human epidermal growth factor receptor (HER) family is comprised of four members, EGFR, HER2, HER3, and HER4. These are highly homologous type I transmembrane tyrosine kinase receptors consisting of a ligand-binding extracellular domain, a transmembrane region, an intracellular tyrosine kinase domain and a C-terminal signaling tail. Ligand binding stabilizes an open conformation of the extracellular region exposing a dimerization interface that mediates the formation of receptor dimers and possibly oligomers. Dimerization or oligomerization of receptors in turn leads to the allosteric activation of one kinase domain by another, and subsequent phosphorylation of C-terminal tails. Phosphorylated C-terminal tails recruit numerous second messenger proteins leading to the generation of numerous intracellular signaling cascades including the Ras/MAPK and PI3K/Akt signaling pathways. HER receptors can generate signals through homo- or hetero-dimerization. While EGFR and HER4 are fully competent receptors capable of signaling through homo- or hetero-dimerization, HER2 and HER3 lack the full complement of functionalities and are committed partners for heterodimerization.
The HER family receptors are frequently implicated in the biology of many types of human cancers. This occurs through the amplification of EGFR or HER2 as seen in cancers of the breast, lung, stomach, endometrium, head & neck, or brain 28, 30, 38, 44, 50, or through mutational activation of the extracellular domain of EGFR in gliomas 12, or the kinase domain of EGFR in lung cancers 41, or the kinase domain of HER2 in cancers of the lung or breast 8, 43. In many of these cancers, EGFR or HER2 are disease-driving oncogenes and agents that target them show considerable efficacy in the treatment of these cancers 4, 18, 31, 45. These agents include small molecule inhibitors of their tyrosine kinase catalytic functions, or monoclonal antibodies that interfere with the ligand-activation or dimerization functions embodied within their extracellular domains, or that can mediate immunologic responses against cancers with amplification and massive overexpression of these receptors.
Although the catalytically inactive HER3 lacks the transforming potential inherent in the catalytically competent HER family members, there is increasing evidence that HER3 plays a key orthogonal role in many types of human cancers, either as an obligate partner for EGFR or HER2, or promiscuous partner for MET, or in other cancers where its catalytic partner remains to be defined. HER3 is essential for HER2-driven tumorigenesis as demonstrated in experimental models with HER2-amplified human cancer cells or mouse genetic models 21, 25, 49. Furthermore, HER3 is not just a requisite downstream substrate of HER2 in these cancers. It has critical functions both upstream and downstream of HER2. It functions upstream because its kinase domain, although catalytically inactive, is a key allosteric activator of the HER2 kinase domain 23. It functions downstream of HER2 because its signaling tail contains six consensus binding sites for the regulatory subunit of PI3K, and when phosphorylated, HER3 is one of the strongest known activators of PI3K/Akt signaling, providing a strong cellular survival signal, important in many cancers 36, 46. Attempts to inhibit HER2 signaling in HER2-amplified cancers results in a robust upregulation of HER3 that restores HER2-HER3 signaling and undermines the efficacy of all current HER2-targeting pharmaceutical agents 2, 14, 40. These findings have redefined the HER2-HER3 signaling complex as the functionally relevant driver of HER2-amplified cancers and the inactivation of this signaling activity as the new bar for the highly effective therapy of this disease. Even the most potent and selective inhibitors of the HER2 tyrosine kinase lack the requisite therapeutic index and have only modest clinical activities. The highly effective treatment of this disease requires agents that eliminate the functions of HER3.
Targeting HER3 function is more challenging than targeting EGFR or HER2. This is because of key differences that distinguish HER3 function from the other HER family members, and our more primitive understanding of how HER3 signaling is engaged in cancers. The HER3 kinase domain likely functions solely as an allosteric activator in signaling complexes, since it lacks catalytic kinase activity. Consistent with this, small molecule ATP-analog tyrosine kinase inhibitors that bind with high affinity within its kinase domain fail to inhibit its signaling function 29. Although it remains plausible that the signaling functions of HER3 may be modulated by yet unknown classes of small molecules that bind its kinase domain, this endeavor awaits deeper insights into the functions and structural attributes of its kinase domain. Numerous monoclonal antibodies and other protein therapeutics targeting its extracellular region have been developed that can interfere with aspects of its extracellular regions 7, 11, 13, 24, 35, 39. These may be of particular benefit in disease states driven by ligand-stimulation but they show decreased efficacy in cancers with HER2 amplification where HER3 signaling is likely engaged through ligand-independent mechanisms. In addition, it is unlikely that HER3-targeting antibody therapies can elicit strong immunological anti-tumor responses, since the expression of HER3 in cancers is not high enough to afford a reasonable therapeutic index for such immunological responses. This is in contrast to EGFR or HER2, which undergo gene amplification and massive protein overexpression in many cancers, allowing for such immunologically mediated effects. Here we describe a novel approach to targeting HER3. This involves interfering with mechanisms critical for its cotranslational translocation into the endoplasmic reticulum, resulting in the elimination of HER3 protein expression and loss of HER3 function.
Results
In a search for compounds that could suppress HER3 expression or signaling, we identified the cyclic peptide CT8. CT8 belongs to a class of cyclic heptadepsipeptides called “cotransins”, related to the fungal natural product HUN-7293. Cotransins inhibit the cotranslational translocation of a subset of secreted and transmembrane proteins, in particular TNF-α and VCAM-1 15, 34. When testing the effects of CT8 in HER2-amplified breast cancer cells, we observed the suppression of HER3 signaling, directly attributable to the suppression of HER3 expression (figure 1A). Expanded analysis in other cancer cells showed a similar reduction of HER3 expression, albeit with slightly different concentration dependencies (figure 1B). Interestingly, among the members of the HER family of receptor tyrosine kinases, HER3 is uniquely sensitive to CT8 and no similar effects are seen with other members of the HER family (figure 1C). Since HER3 is highly regulated through transcription, we tested whether CT8 inhibits the transcription of HER3. RT-qPCR analysis of the messenger RNA levels for HER3 shows that transcription is not downregulated by CT8 (figure 2A,B). Therefore, inhibition of HER3 expression occurs post-transcriptionally. To analyze whether suppression of HER3 expression is due to protein degradation, we used bortezomib to block the ubiquitin proteasome pathway responsible for the degradation of many proteins. In BT474 cells, the CT8-induced suppression of HER3 expression can be blocked by concomitant treatment with the proteasome inhibitor bortezomib (figure 2C). Together, these data show that the suppression of HER3 expression seen with CT8 treatment is due to the degradation of HER3 protein.
Figure 1. Cotransin suppresses the expression of HER3.

A. BT474 cells were treated with the indicated concentrations of CT8 for 24 or 48 hours and cell lysates were assayed by western blotting as indicated. B. Several HER2-amplified cancer cells were treated with increasing concentrations of CT8 for 24 hours and cell lysates were assayed for HER3 expression by western blotting. C. HEK-293 cells were transiently transfected with a pcDNA-DEST40 vector containing the open reading frames of EGFR, HER2, HER3, or HER4, in-frame with a c-terminal V5 tag. Transfectants were treated with 500nM CT8 for 24 hours and subjected to western blot analysis using anti-V5 and anti-actin antibodies.
Figure 2. CT8 induces the proteasome-dependent degradation of HER3.
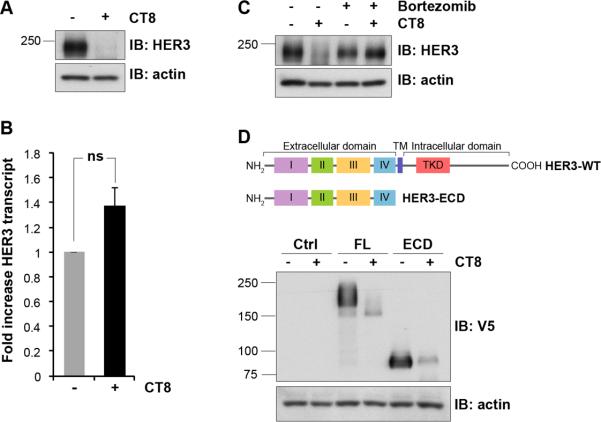
A. BT474 cells were treated with 300 nM CT8 or DMSO for 24 hours. The suppression of HER3 expression was confirmed by immunoblotting of cell lysates. B. Additional cells from the same experiment were used to purify RNA and the relative expression of HER3 mRNA was evaluated by real-time RTPCR analysis using HER3-specific primers and normalization with β-microglobulin. Results are expressed as fold change and are the average of triplicates. Error bars represent SEM. ns, non-significant induction compared with control cells. C. BT474 cells were either untreated or treated for 24h with 300 nM CT8 or 25nM bortezomib, or treated simultaneously with CT8 and bortezomib. The expression of HER3 was assayed by western blotting. D. HEK-293 cells were transiently transfected with the pcDNA-DEST40 vector containing the full length HER3 open reading frame or a HER3 mutant that is truncated after the extracellular domain, as shown in the schematic. Both constructs are in-frame with a C-terminal V5 tag. Following transfection, cells were treated with 500nM CT8 or DMSO for 24 hours and cell lysates were subjected to immunoblotting using V5 and actin antibodies. Molecular mass standards (kDa) are indicated on the left side of the figure.
Some kinases, including HER3, interact directly with the Hsp90 molecular chaperone and are dependent on it for proper folding and stability and inhibitors of Hsp90 lead to the degradation of many such kinases including HER3 16. However a HER3 mutant lacking the entire intracellular domain remains fully sensitive to CT8 (figure 2D), and therefore CT8 does not degrade HER3 through mechanisms involving its kinase domain analogous to Hsp90 inhibitors.
Cotransins such as CT8 can interfere with the cotranslational movement of the transmembrane domains of nascent transmembrane proteins into the lipid bilayer of the endoplasmic reticulum, mediated through the Sec61 channel 33. However, since CT8 also degrades the secreted HER3 mutant lacking the transmembrane and intracellular domains, its effects on HER3 are unlikely to be mediated through this mechanism. More relevant, CT8 also interferes with cotranslational translocation initiated by N-terminal signal peptides, which target nascent secreted and type I membrane proteins, including all HER family members, to the endoplasmic reticulum. This is also mediated through inhibition of the Sec61translocon, but the extent to which a given secretory or type I membrane protein is sensitive to CT8 depends on the specific signal peptide sequences of the nascent protein emerging from the ribosome and captured by the signal recognition particle 34. To determine whether the sensitivity of HER3 to CT8 is contained within its signal peptide sequences, we generated a HER3 mutant with an altered signal peptide. The residues mutated were based on our best understanding of the mechanism by which cotransins block productive interactions between certain signal peptides and the Sec61 channel (figure 3A), as previous studies suggested that polar to hydrophobic amino acid substitutions within the central hydrophobic region of a signal peptide can confer cotransin resistance 15, 20. HEK-293 cells were transfected with wildtype HER3 or HER3 mutant and treated with CT8. The results show that the HER3 signal peptide mutant is resistant to CT8-induced degradation, confirming the critical role of the signal peptide in mediating the effects of CT8 (figure 3B). To further confirm the role of the HER3 signal peptide, we fused the N-terminal sequences of HER2 or HER3 to the N-terminus of a Gaussia luciferase expression construct, leading to the secretion of active luciferase into the media. As expected, secretion of the luciferase construct bearing the HER3 signal peptide is potently inhibited by CT8 , whereas the construct bearing the HER2 signal peptide is nearly 100-fold less sensitive (figure 3C). To confirm the role of the Sec61 translocon in mediating the CT8-driven degradation of HER3, we tested a CT8-resistant Sec61α R66I mutant. This mutant was identified through a mutation-prone biologic screen of cotransin resistance and found to properly assemble into functional translocons, but exhibits diminished binding to CT8 33. HEK-293 cells induced to express Sec61α WT (wildtype) or Sec61α R66I were transfected with HER3 and treated with CT8. As expected, CT8 effectively suppresses HER3 expression in Sec61a WT overexpressing cells. However, expression of the Sec61α R66I mutant rescues HER3 from the degradative effects of CT8 (figure 3D). These data confirm that CT8 blocks the translocation of the HER3 signal peptide helix into the Sec61 membrane channel as the nascent protein emerges from the ribosome, ultimately leading to its proteasome-dependent degradation (Figure 4).
Figure 3. HER3 suppression by CT8 is mediated through the signal peptide.

A. Schematic representation showing the site of the engineered mutations. B. HEK-293 cells were transiently transfected to express either WT HER3 or the indicated HER3 mutant shown in the schematic. Following transfection, cells were treated with 500nM CT8 for 24 hours and HER3 expression in cell lysates assayed by western blotting. The lanes were run on the same gel but were noncontiguous. C. HEK-293T cells transfected with plasmids encoding the mature domain of enhanced Gaussia luciferase (eGluc) fused to the signal sequence of HER2 or HER3 were incubated in doxycycline (1 μg/mL, to induce expression) and increasing concentrations of CT8 for 24 hours, after which luminescence was measured. D. HEK-293 cells with the tet-inducible induction of expression of wildtype Sec61α or the CT8-resistant R66I mutant Sec61α were incubated in doxycycline (1 μg/mL, to induce expression). The following day, cells were transfected with an empty vector or a HER3-expressing vector, and 4 hours later placed in media containing DMSO or 500nM CT8. After 24 hours of treatment, cell lysates were harvested and assayed for expression of HER3 by western blotting. Molecular mass standards (kDa) are indicated on the left side of the figure.
Figure 4. CT8 interferes with the movement of HER3 into the Sec61 channel.
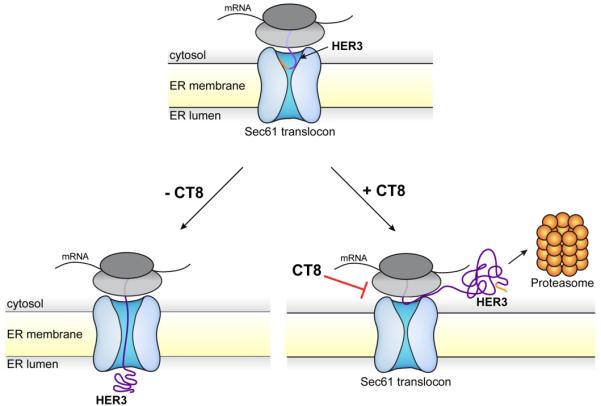
Schematic model showing the mode of action of CT8. CT8 blocks the movement of the HER3 signal peptide helix into the Sec61 membrane channel as the nascent protein emerges from the ribosome, ultimately leading to its proteasome-dependent degradation.
The functions of HER3 are particularly important in HER2-amplified cancer cells and our inability to inactivate HER3 function underlies the limited activities of all forms of HER2-targeted therapies to date. A drug that can inactivate HER3 would be of significant benefit in the treatment of these cancers in combination with a HER2 inhibitor. We tested this in BT474 HER2-amplified breast cancer cells. Treatment of these cancers with the HER2 tyrosine kinase inhibitor lapatinib fully inactivates HER2-HER3 signaling, but this is subsequently restored through the upregulation of HER3 expression (figure 5A, left panels). However, the addition of CT8 counteracts the upregulation of HER3 expression, preventing the restoration of p-HER3 signaling (figure 5A, right panels). The failure to rescue HER3 signaling leads to apoptotic cell death. This occurs at concentrations of lapatinib and CT8 that are only minimally pro-apoptotic by themselves, but that show substantial apoptotic effects in combination (figure 5B). Neither lapatinib or CT8 or their combination have any significant effects on the survival of cancer cell lines that are not dependent on HER2 or HER3 (supplementary figure 1).
Figure 5. CT8 treatment enhances the efficacy of HER2 inhibitor therapy.
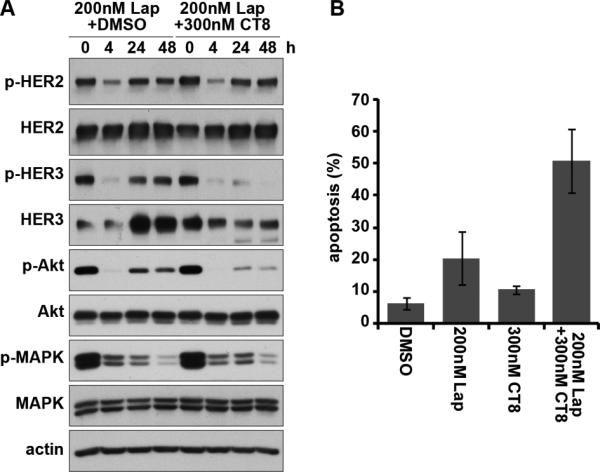
A. BT474 cells were treated with lapatinib with/without CT8 for the indicated durations of time up to 48 hours. The effects on HER2-HER3 signaling were assayed by western blotting as indicated. B. BT474 cells were treated with the indicated drugs for 72 hours, and the fraction of apoptotic cells quantified by FACS analysis of DNA degradation.
Discussion
There is increasing evidence implicating HER3 in the pathogenesis of several types of human cancers and in mediating resistance to a number of targeted cancer therapies. The best characterized are HER2-amplified breast cancers where HER3 plays an essential role as a co-driver of the disease. HER3 likely plays a similar role in other HER2-amplified cancers such as HER2-amplified gastric, esophageal, and endometrial cancers, although these have been less well studied. There is some evidence supporting a role for HER3 in triple negative breast cancers 48. HER3-driven autocrine ligand signaling loops are seen in some cancers including ovarian cancers 42. There is ample evidence supporting a role for HER3 in the pathogenesis of colon cancers 5, 17, 51 and potentially in mediating resistance to EGFR or MEK inhibitors in colon cancers 47, 52. HER3 signaling mediates at least some modes of resistance to RAF kinase inhibitors in BRAF-driven melanomas 1, 10, 37 and may have a more primary function in melanomas 37. These lines of evidence have made a compelling case for the development of inhibitors of HER3 function for the treatment of cancer.
But our understanding of precisely how HER3 mediates tumorigenic functions is not as well developed as EGFR or HER2 or other receptor tyrosine kinase oncogenes. In particular, the structural features underlying its tumor-promoting activities are not well described. Its kinase domain is clearly important in its signaling function, although targeting it awaits much deeper insights into its mode of function, as the classical approach to target the active site using ATP-analog small molecule inhibitors does not affect the functions of this catalytically inactive kinase 29. A number of antibodies targeting the extracellular domain of HER3 are in development, with different functional properties. This approach may be effective in cancers that depend on the classical ligand-induced dimerization function of HER3 such as cancers that are driven by autocrine ligand loops. But in other cancers such as those driven by massive overexpression of HER2, interfering with the ligand-induced activation of HER3 has modest effects at best 35, 39. Eliminating the expression of HER3 is certainly an effective means of suppressing its functions in tumorigenesis. Here we present a novel small-molecule approach to targeting HER3 expression. This involves inhibiting its cotranslational localization at the Sec61 membrane channel leading to its degradation. This was shown here using a cyclic heptadepsipeptide of the cotransin family which suppresses HER3 expression in all cell types examined to date, and with profound effects on the HER2-HER3 tumor driver in HER2-amplified cancer cells when combined with the HER2-inhibitor lapatinib. The results are consistent with the notion that dual inhibition of HER2 and HER3 is required for effective treatment of HER2-amplified cancers, and provides an effective mechanism to target HER3. More structure-function work is needed to explore the chemical space in this arena, understand and optimize the selectivities involved in Sec61-targeting molecules, and to develop candidates more suitable for clinical development.
The effect of CT8 on phosphorylation of HER3 occurs later and at higher concentrations than its effect on total HER3 expression (figure 1A). This is consistent with the fact that CT8 directly affects HER3 expression, not its signaling activity. The signaling activity of HER3 is not only related to its expression level, but also related to a number of mechanisms linked with downstream feedback loops that function to preserve HER3 signaling when jeopardized by drugs 3. The more resilient nature of p-HER3 decline compared with total HER3 decline reflects the multitude of variables involved in preserving HER3 signaling beyond its expression levels.
Understanding the target selectivities of cotransins is a matter of ongoing work. The experience to date confirms that the inhibition of cotranslational translocation is not an all-or-none phenomenon. As such, cotransins do not interfere with the Sec61-mediated translocation and expression of all secreted or transmembrane proteins. Moreover, even small changes in the side chains of the cotransin cyclopeptide can profoundly affect substrate selectivity. A comparison of the cotransins CT8 and CT9, which differ in only two side chains, showed vastly different selectivity profiles and much higher selectivity for CT8 compared to CT9 34. In a panel of 25 secreted and type I and II transmembrane proteins, CT8 showed potent inhibition of only TNFα and VCAM-1. It remains distinctly possible that further structure-activity studies can identify additional Sec61 binding molecules with even higher selectivities, towards HER3 or other secreted or transmembrane proteins of interest. The basis for the observed selectivities of cotransins lies in the specific amino acid sequences of the N-terminal signal peptide, as shown by signal sequence swapping studies 6, 15. The best evidence to date regarding the mode of action of cotransins suggests that they prevent certain nascent proteins from accessing the lumen of the endoplasmic reticulum in a manner that is dependent on Sec61 and the specific residues on the hydrophobic N-terminal signal peptide or the transmembrane domain 33, 34.
The four members of the HER family of type I transmembrane receptor tyrosine kinases are highly homologous proteins with many similarities in structure and function. All have cleavable N-terminal signal peptide sequences and are localized to the plasma membrane. As such, it is surprising that HER3 is uniquely sensitive to the cotransin CT8. This suggests that somehow the movement of the HER3 N-terminal sequences through the Sec61 channel differs from the other HER family members. Why HER3 should be processed differently is not self-evident. However it is interesting to note that among the HER family, the HER3 gene is unique in that it expresses several truncated alternative transcripts that lack the intracellular and transmembrane domains and encode for secreted isoforms of HER3 26. These soluble HER3 extracellular proteins appear to have decoy functions in the negative regulation of neuregulin signaling 27. Perhaps the observed differences in HER3 signal processing through the Sec61 channel compared with other HER family proteins is related to its wider localization and distribution requirements, which encompass secreted soluble isoforms as well as transmembrane receptor isoforms, thus imparting upon it certain similarities with other secreted proteins including sensitivity to Sec61 modulators.
Although there is ample evidence that HER3 plays a role in many types of cancer, it mediates its functions without catalytical activity, and at least in some circumstances without ligand stimulation, making it a more challenging drug target compared with other receptor tyrosine kinases. This work reveals an entirely new mechanism by which HER3 function can be disrupted, paving a pathway for a new class of HER3-targeting drugs.
Materials and Methods
Cell culture and drugs
Cell lines (BT474, SkBr3, HCC1954, HCC1569, TE-4, N87, H2170, HEK-293) were obtained from the American Type Culture Collection (ATCC) and cultured at 37°C, 5% CO2 in DMEM/HamF12 media supplemented with 10% fetal bovine serum, penicillin, streptomycin, and L-glutamine. Lapatinib was purified from commercial Tykerb tablets by organic extraction as previously described 2. CT8 was synthesized using modifications of published protocols 9, 32. Bortezomib was obtained from Selleckchem.com.
Western blotting
Cell lysates were prepared using modified RIPA lysis buffer (150 mM NaCl, 0.1% SDS, 1% Nonidet P40, 1% sodium deoxycholate and 10 mM sodium phosphate, pH 7.2) supplemented with protease and phosphatase inhibitors. Western blots were performed using antibodies purchased from SantaCruz Biotechnologies (HER3, actin), Cell Signaling (p-HER2, pAKTthr308, pAKTser473, p-MAPK, MAPK), and Invitrogen (V5).
qPCR
Total cellular RNA was isolated using the Qiagen RNeasy kit following the manufacturer's instructions. RT and PCR amplification of ERBB3 was performed using primers and methods that have been described previously 2. Quantitative PCR assays were performed in triplicate for HER3 and the housekeeping gene β-microglobulin from cDNA prepared from mock- or CT8-treated cells for 24h. We used the ΔΔCT methodology to obtain the ratios after normalizing to β-microglobulin. The ratios were obtained against the average for the mock readout. The mean ratio and S.E.M.was determined on the triplicate values obtained. Student's t test was performed on the triplicate values using two-tailed unequal variance parameters.
Apoptosis
For the analysis and quantitation of apoptotic cells, ethidium bromide stained nuclei were prepared according to the method of Nusse 19 and their DNA content was assayed by fluorescence activated cell sorting (FACS). Apoptotic cells were identified by their sub-G1 DNA content.
DNA constructs and transfections
The EGFR, HER2, HER3, and HER4 cDNAs were transiently expressed in HEK-293 cells in pcDNA-DEST40 vectors (Life Technologies) using Lipofectamine 2000 according to standard transfection protocols.
The HER3 truncated and signal peptide mutants were generated by fragment gene synthesis and restriction enzyme cloning at Genewiz and all mutated vectors were confirmed by sequencing the entire cDNA insert. The generation of HEK-293 cells expressing inducible wildtype or CT8-resistant Sec61α was previously described 33.
The luciferase assays were performed in HEK-293T T-rex cells. The mature domain of secreted luciferase from Gaussia princeps (eGLuc2, courtesy of Jeffery W. Kelly, The Scripps Research Institute) was cloned into pcDNA5/FRT/TO. The signal sequence plus four amino acids of the mature domain from HER2 and HER3 were cloned upstream of eGLuc2 mature domain 22 and used in transient transfection experiments (referred as HER2ss-eGLuc and HER3ss-eGLuc, respectively). HEK-293T T-rex cells (constitutively expressing the Tet repressor, Life Technologies) were cultured in DMEM with 10% FBS at 5% CO2. Cells were plated 1×106 cells/well in a 6-well plate and incubated for 6 hours. Cells were transiently transfected with HER2ss-eGluc and HER3ss-eGLuc using lipofectamine 2000 (Invitrogen). After overnight incubation, transfected cells were re-plated in a clear-bottomed, black 96-well plate at a density of 20,000 cells/well. Six hours after platting, the cells were treated with doxycycline (1 μg/mL) and the indicated concentrations of CT8 for 24 hrs. Secreted luciferase in the conditioned media was quantified by luminescence using the BioLux Gaussia luciferase assay kit (New England Biolabs).
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health CA122216 & CA112970 (MMM), Howard Hughes Medical Institute (JT), and a Fundación Ramón Areces Fellowship (ARS).
Footnotes
Conflicts of Interest: The authors have no conflicts to disclose.
References
- 1.Abel EV, Basile KJ, Kugel CH, 3rd, Witkiewicz AK, Le K, Amaravadi RK, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. The Journal of clinical investigation. 2013;123:2155–2168. doi: 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amin DN, Sergina N, Ahuja D, McMahon M, Blair JA, Wang D, et al. Resiliency and vulnerability in the HER2-HER3 tumorigenic driver. Sci Transl Med. 2010;2:16ra17. doi: 10.1126/scitranslmed.3000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amin DN, Sergina N, Lim L, Goga A, Moasser MM. HER3 signalling is regulated through a multitude of redundant mechanisms in HER2-driven tumour cells. Biochem J. 2012;447:417–425. doi: 10.1042/BJ20120724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beji A, Horst D, Engel J, Kirchner T, Ullrich A. Toward the prognostic significance and therapeutic potential of HER3 receptor tyrosine kinase in human colon cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:956–968. doi: 10.1158/1078-0432.CCR-11-1186. [DOI] [PubMed] [Google Scholar]
- 6.Besemer J, Harant H, Wang S, Oberhauser B, Marquardt K, Foster CA, et al. Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature. 2005;436:290–293. doi: 10.1038/nature03670. [DOI] [PubMed] [Google Scholar]
- 7.Blackburn E, Zona S, Murphy ML, Brown IR, Chan SK, Gullick WJ. A monoclonal antibody to the human HER3 receptor inhibits Neuregulin 1-beta binding and co-operates with Herceptin in inhibiting the growth of breast cancer derived cell lines. Breast Cancer Res Treat. 2012;134:53–59. doi: 10.1007/s10549-011-1908-1. [DOI] [PubMed] [Google Scholar]
- 8.Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–237. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Bilban M, Foster CA, Boger DL. Solution-phase parallel synthesis of a pharmacophore library of HUN-7293 analogues: a general chemical mutagenesis approach to defining structure-function properties of naturally occurring cyclic (depsi)peptides. J Am Chem Soc. 2002;124:5431–5440. doi: 10.1021/ja020166v. [DOI] [PubMed] [Google Scholar]
- 10.Fattore L, Marra E, Pisanu ME, Noto A, de Vitis C, Belleudi F, et al. Activation of an early feedback survival loop involving phospho-ErbB3 is a general response of melanoma cells to RAF/MEK inhibition and is abrogated by anti-ErbB3 antibodies. Journal of translational medicine. 2013;11:180. doi: 10.1186/1479-5876-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foreman PK, Gore M, Kobel PA, Xu L, Yee H, Hannum C, et al. ErbB3 Inhibitory Surrobodies Inhibit Tumor Cell Proliferation In Vitro and In Vivo. Molecular cancer therapeutics. 2012;11:1411–1420. doi: 10.1158/1535-7163.MCT-12-0068. [DOI] [PubMed] [Google Scholar]
- 12.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer research. 2000;60:1383–1387. [PubMed] [Google Scholar]
- 13.Garner AP, Bialucha CU, Sprague ER, Garrett JT, Sheng Q, Li S, et al. An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or neuregulin. Cancer research. 2013;73:6024–6035. doi: 10.1158/0008-5472.CAN-13-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sanchez V, Chakrabarty A, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci U S A. 2011;108:5021–5026. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrison JL, Kunkel EJ, Hegde RS, Taunton J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature. 2005;436:285–289. doi: 10.1038/nature03821. [DOI] [PubMed] [Google Scholar]
- 16.Gerbin CS, Landgraf R. Geldanamycin selectively targets the nascent form of ERBB3 for degradation. Cell stress & chaperones. 2010;15:529–544. doi: 10.1007/s12192-009-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gespach C. Increasing potential of HER3 signaling in colon cancer progression and therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:917–919. doi: 10.1158/1078-0432.CCR-11-3143. [DOI] [PubMed] [Google Scholar]
- 18.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 19.Giaretti W, Nusse M. Light scatter of isolated cell nuclei as a parameter discriminating the cell-cycle subcompartments. Methods in cell biology. 1994;41:389–400. doi: 10.1016/s0091-679x(08)61730-6. [DOI] [PubMed] [Google Scholar]
- 20.Harant H, Lettner N, Hofer L, Oberhauser B, de Vries JE, Lindley IJ. The translocation inhibitor CAM741 interferes with vascular cell adhesion molecule 1 signal peptide insertion at the translocon. The Journal of biological chemistry. 2006;281:30492–30502. doi: 10.1074/jbc.M607243200. [DOI] [PubMed] [Google Scholar]
- 21.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hulleman JD, Brown SJ, Rosen H, Kelly JW. A high-throughput cell-based Gaussia luciferase reporter assay for identifying modulators of fibulin-3 secretion. Journal of biomolecular screening. 2013;18:647–658. doi: 10.1177/1087057112469405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human HER3 receptor. Proc Nat Acad Sci USA. 2009;106:21608–21613. doi: 10.1073/pnas.0912101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kugel CH, 3rd, Hartsough EJ, Davies MA, Setiady YY, Aplin AE. Function-Blocking ERBB3 Antibody Inhibits the Adaptive Response to RAF Inhibitor. Cancer research. 2014 doi: 10.1158/0008-5472.CAN-14-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer research. 2008;68:5878–5887. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 26.Lee H, Maihle NJ. Isolation and characterization of four alternate c-erbB3 transcripts expressed in ovarian carcinoma-derived cell lines and normal human tissues. Oncogene. 1998;16:3243–3252. doi: 10.1038/sj.onc.1201866. [DOI] [PubMed] [Google Scholar]
- 27.Lee H, Akita RW, Sliwkowski MX, Maihle NJ. A naturally occurring secreted human ErbB3 receptor isoform inhibits heregulin-stimulated activation of ErbB2, ErbB3, and ErbB4. Cancer research. 2001;61:4467–4473. [PubMed] [Google Scholar]
- 28.Licitra L, Mesia R, Rivera F, Remenar E, Hitt R, Erfan J, et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2011;22:1078–1087. doi: 10.1093/annonc/mdq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Littlefield P, Moasser MM, Jura N. An ATP-competitive inhibitor modulates the allosteric function of the HER3 pseudokinase. Chemistry & biology. 2014;21:453–458. doi: 10.1016/j.chembiol.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Lopez-Gines C, Cerda-Nicolas M, Gil-Benso R, Pellin A, Lopez-Guerrero JA, Callaghan R, et al. Association of chromosome 7, chromosome 10 and EGFR gene amplification in glioblastoma multiforme. Clin Neuropathol. 2005;24:209–218. [PubMed] [Google Scholar]
- 31.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 32.MacKinnon AL, Garrison JL, Hegde RS, Taunton J. Photo-leucine incorporation reveals the target of a cyclodepsipeptide inhibitor of cotranslational translocation. J Am Chem Soc. 2007;129:14560–14561. doi: 10.1021/ja076250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackinnon AL, Paavilainen VO, Sharma A, Hegde RS, Taunton J. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. eLife. 2014;3:e01483. doi: 10.7554/eLife.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maifeld SV, MacKinnon AL, Garrison JL, Sharma A, Kunkel EJ, Hegde RS, et al. Secretory protein profiling reveals TNF-alpha inactivation by selective and promiscuous Sec61 modulators. Chemistry & biology. 2011;18:1082–1088. doi: 10.1016/j.chembiol.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Molecular cancer therapeutics. 2012;11:582–593. doi: 10.1158/1535-7163.MCT-11-0820. [DOI] [PubMed] [Google Scholar]
- 36.Prigent SA, Gullick WJ. Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 1994;13:2831–2841. doi: 10.1002/j.1460-2075.1994.tb06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reschke M, Mihic-Probst D, van der Horst EH, Knyazev P, Wild PJ, Hutterer M, et al. HER3 is a determinant for poor prognosis in melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:5188–5197. doi: 10.1158/1078-0432.CCR-08-0186. [DOI] [PubMed] [Google Scholar]
- 38.Santin AD, Bellone S, Van Stedum S, Bushen W, Palmieri M, Siegel ER, et al. Amplification of c-erbB2 oncogene: a major prognostic indicator in uterine serous papillary carcinoma. Cancer. 2005;104:1391–1397. doi: 10.1002/cncr.21308. [DOI] [PubMed] [Google Scholar]
- 39.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer research. 2010;70:2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 42.Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer cell. 2010;17:298–310. doi: 10.1016/j.ccr.2009.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer research. 2005;65:1642–1646. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 44.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 45.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 46.Soltoff SP, Carraway KL, 3rd, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell reports. 2014;7:86–93. doi: 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- 48.Tao JJ, Castel P, Radosevic-Robin N, Elkabets M, Auricchio N, Aceto N, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Science signaling. 2014;7:ra29. doi: 10.1126/scisignal.2005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaught DB, Stanford JC, Young C, Hicks DJ, Wheeler F, Rinehart C, et al. HER3 is required for HER2-induced preneoplastic changes to the breast epithelium and tumor formation. Cancer research. 2012;72:2672–2682. doi: 10.1158/0008-5472.CAN-11-3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yano T, Doi T, Ohtsu A, Boku N, Hashizume K, Nakanishi M, et al. Comparison of HER2 gene amplification assessed by fluorescence in situ hybridization and HER2 protein expression assessed by immunohistochemistry in gastric cancer. Oncology reports. 2006;15:65–71. [PubMed] [Google Scholar]
- 51.Yao YL, Shao J, Zhang C, Wu JH, Zhang QH, Wang JJ, et al. Proliferation of colorectal cancer is promoted by two signaling transduction expression patterns: ErbB2/ErbB3/AKT and MET/ErbB3/MAPK. PloS one. 2013;8:e78086. doi: 10.1371/journal.pone.0078086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang L, Castanaro C, Luan B, Yang K, Fan L, Fairhurst JL, et al. ERBB3/HER2 signaling promotes resistance to EGFR blockade in head and neck and colorectal cancer models. Molecular cancer therapeutics. 2014;13:1345–1355. doi: 10.1158/1535-7163.MCT-13-1033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.