

Abstract
Acute kidney injury (AKI) is a clinical syndrome with multiple entities. Although AKI implies renal damage, functional impairment or both, diagnosis is solely based on the functional parameters of serum creatinine and urine output. The latest definition was provided by the Kidney Disease Improving Global Outcomes (KDIGO) working group in 2012. Independent of the underlying disease, and even in the case of full recovery, AKI is associated with an increased morbidity and mortality. Awareness of the patient's individual risk profile and the diversity of causes and clinical features of AKI is pivotal for optimization of prophylaxes, diagnosis and therapy of each form of AKI. A differentiated and individualized approach is required to improve patient mortality, morbidity, long-term kidney function and eventually the quality of life. In this review, we provide an overview of the different clinical settings in which specific forms of AKI may occur and point out possible diagnostic as well as therapeutic approaches. Secifically AKI is discussed in the context of non-kidney organ failure, organ transplantation, sepsis, malignancy and autoimmune disease.
Keywords: acute kidney injury, autoimmune disease, organ failure, sepsis
Acute kidney injury (AKI) is a clinical syndrome interfering with all kinds of medical disciplines. Appearing with an increasing incidence, AKI is still the most common and most expensive kidney disease in hospitals. A uniform definition of AKI has only been established since 2004 with the introduction of the Risk, Injury, Failure, Loss, and End stage renal disease (RIFLE) criteria. The latest definition of AKI was provided by the KDIGO guidelines in 2012. Since then, AKI is defined as any of the following [1]:
Increase in serum creatinine by at least 0.3 mg/dL (26.5 μmol/L) within 48 h; or
Increase in serum creatinine of at least 1.5 times baseline, which is known or presumed to have occurred within the prior 7 days; or
Urine volume <0.5 mL/kg/h for 6 h.
In addition, severity is staged by serum creatinine and urine output (Table 1).
Table 1.
Staging of AKI. Adapted from [1]
| Stage | Serum creatinine | Urine output |
|---|---|---|
| 1 | 1.5–1.9 times baseline OR at least 0.3 mg/dL (26.5 µmol/L) increase |
<0.5 mL/kg/h for 6–12 h |
| 2 | 2.0–2.9 times baseline | <0.5 mL/kg/h for at least 12 h |
| 3 | 3.0 times baseline OR increase in serum creatinine to at least 4.0 mg/dL (353.6 µmol/L) OR initiation of renal replacement therapy OR in patients aged below 18 years, decrease in eGFR to <35 mL/min/1.73 m2 |
<0.3 mL/kg/h for at least 24 h OR anuria for at least 12 h |
Despite a uniform definition, AKI remains a syndrome with multiple causes. Independent of the underlying disease, but depending on the extent and duration of renal dysfunction and the general condition of the patient, AKI is associated with a relative increase in mortality not only at the intensive care unit (ICU), but far beyond the acute situation itself [2, 3]. Incidence of AKI has been rising continuously during the past 25 years [4], and AKI according to KDIGO definitions is estimated to occur in 18% of general hospitalizations and up to 50% of ICU cases worldwide [5, 6]. Reasons for the increasing incidence might be found in an aging patient population accompanied by growing multi-morbidity. Already at admission to hospital, each patient brings along his own individual risk profile for the development of AKI during the further course of events. Susceptibility to AKI might be formed by diabetes mellitus, chronic kidney disease (CKD) or cardiac insufficiency as well as by pre-existing medications. During hospitalization, patients are exposed to events that eventually disturb the instable renal balance triggering AKI (Table 2).
Table 2.
Risk profile formation for AKI
| Exposures | Susceptibilities |
|---|---|
| • Sepsis • Critical illness • Circulatory shock • Burns • Trauma • Cardiac surgery (especially with cardio-pulmonary bypass) • Major non-cardiac surgery • Nephrotoxic drugs • Radiocontrast agents • Poisonous plants and animals • … |
• Dehydration and volume depletion • Advanced age • Female gender • Black race • CKD • Chronic diseases (heart, lung, liver) • Diabetes mellitus • Cancer • Anaemia • … |
Susceptibilities and exposures lead to formation of an individual risk profile for the development of AKI in each patient.
AKI can imply renal damage, functional impairment or both. Diagnosis of AKI is solely based on parameters of renal function. Manifestations of AKI include clinical asymptomatic, but prognosticly relevant kidney injuries, cases with aggravation of diagnostic and therapeutic procedures as well as life-threatening complications such as changes in fluid, electrolyte and acid–base balance. Independent of the underlying disease initiating hospitalization and despite full laboratory and clinical recovery, AKI is responsible for an increase in morbidity and mortality and an independent risk factor for the development of CKD [7, 8].
The use of serum creatinine as the only laboratory parameter or classic biomarker involved in the definition of AKI is unfortunate, as it is unreliable due to its large renal reserve that delays the diagnosis of AKI and contributes to overestimation of renal function in the early phases of the syndrome. Additionally, it underlies multiple outside influences and can only depict the loss of function but not indicate potential mechanisms or sites of damage. Different new biomarkers have evolved over the past few years to overcome these obstacles.
As the manifestations of AKI are very heterogeneous so are the causes. Besides the classic classification of identities into prerenal, renal and postrenal, AKI can also be regarded as a syndrome occurring in the context of multiple diseases.
Identification of individual risk profiles, prevention and fast diagnosis of AKI for initiation of optimal supportive therapy are of vital importance due to the abundant lack of causal therapeutic interventions.
This review points out special features of AKI in the context of frequent diseases such as cardiac insufficiency, sepsis, malignancies, liver insufficiency and autoimmune diseases as well as liver and kidney transplantation with regard to prevention, therapy and prognosis.
AKI and cardiac disorders
Pathophysiologic disorders of the cardiovascular system and the kidney can reciprocally initiate dysfunctions by haemodynamic and neuro-hormonal interactions. This relationship is reflected by the concept of the cardiorenal syndrome (CRS) [9] (Table 3). Up to 50% of patients with acute cardiac decompensation suffer from AKI (cardiorenal syndrome type I). In patients with cardiac shock, this proportion is increased to >70% [10].
Table 3.
Classification of cardiorenal syndrome
| Type I—acute cardiorenal syndrome | |
| Acute cardiac insufficiency induces AKI | Hypertensive lung oedema, acute decompensation of a pre-existing chronic cardiac insufficiency, cardiogenic shock, acute right heart failure |
| Type II—chronic cardiorenal syndrome | |
| Chronic cardiac insufficiency induces CKD | Chronic ischaemia induced by reduced peripheral perfusion, vasculopathy |
| Type III—acute renocardial syndrome | |
| AKI induces cardiac insufficiency | Hypervolaemia, lung oedema, cardiac arrhythmias due to electrolyte disturbance, uraemic pericarditis and myopathy |
| Type IV—chronic renocardial syndrome | |
| CKD induces cardiac insufficiency | Left ventricular hypertrophy and dysfunction, atherosclerosis due to disturbed calcium–phosphate homeostasis |
| Type V—secondary cardio-renal syndrome | |
| Systemic disease induces parallel, independent damage of the heart and the kidneys | Sepsis, systemic inflammatory response syndrome, septic shock, autoimmune diseases, diabetes mellitus |
Modified according to Ref. [100].
A reduced (left ventricular) cardiac pumping function marks the centre of pathogenesis of the cardiorenal syndrome. It leads to increased pressure in the venous system and/or activation of the renin–angiotensin–aldosterone system, changes in intrarenal blood flow and inflammatory processes [9]. Next to left ventricular dysfunction, right ventricular failure may consecutively lead to venous congestion in the liver and the kidney resulting in CRS [11]. A continuous control of volume status by daily measurement of body weight, regular monitoring of natriuretic peptide type B (BNP) or body composition measurement by bioimpedance is helpful in detection and prevention of the development of cardiorenal syndrome [9]. In patients with moderate elevations of BNP, bioelectrical impedance vector analysis (BIVA) might help to identify patients with acute heart failure and add valuable information with regard to hydration status. Thereby, BIVA might help to identify patients being at risk for development of AKI as early as during presentation in the emergency department [12].
Cardiological diagnosis and interventions such as catheter examinations regularly involve exposition to iodine-containing contrast media. Patients with pre-existing cardiac dysfunctions often show a risk profile including hypertension, diabetes mellitus and/or arteriosclerosis and medications that also influence the susceptibility for renal dysfunction and are especially vulnerable to contrast-induced AKI [13]. AKI following percutaneous coronary angiography occurs in about 10% of patients and has been associated with adverse short- and long-term effects on cardiovascular morbidity, mortality and progression to end-stage renal disease [14]. In a recent observational study including almost 6000 Canadian patients, Leung et al. were able to show that statins, beta-blockers and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers were less likely prescribed in patients who experienced mild or severe AKI especially if they were of older age despite the well-established beneficial effects on cardiovascular outcome. The use of these medications was associated with lower long-term mortality even in patients after AKI. Therefore, the decision to prescribe cardiovascular medications seems to be consistently modified by kidney function and age even in patients at high risk for life-threatening cardiovascular events [14–16].
Patients undergoing cardiac surgery are also predisposed to develop AKI [17]. Even mild forms of AKI indicated by minimal rises in serum creatinine were shown to increase mortality [18] and to be independent risk factors for the development of CKD [7].
AKI and sepsis
Sepsis is the most common reason for AKI in critically ill patients [19, 20]. There is increasing evidence that sepsis-induced AKI is not predominantly ischaemia driven, but involves maladaptive (tubular) responses, renal inflammation and dysfunctions in microperfusion [20].
Septic AKI is often characterized by oliguria and results in an increased mortality, morbidity and prolonged hospitalization mainly by impairment of other organ systems with special focus on the lung [21]. On the other hand, patients already suffering from AKI triggered by other factors than sepsis are endangered to systemic infections with oliguria, higher fluid accumulation and need for renal replacement therapy among others being predictors for development of sepsis [22].
The increasing proportion of multiresistant bacteria enhances the use of potentially nephrotoxic antibiotics such as glycopeptides (e.g. vancomycin) and aminoglycosides (e.g. gentamicin) for severe systemic infections. Monitoring of drug levels for dose adjustment is suggested [1].
In patients with reduced cardiac function as well as in sepsis, optimization of fluid management is a vital part of therapy. While in the early phase of sepsis fast, intravenous fluid replacement is essential to ensure the minimally required blood pressure for sufficient organ perfusion, subsequent fluid accumulation can maintain or aggravate organ oedema and dysfunction [23]. Excessive or too early fluid removal by means of diuretics or renal replacement therapy compromises the risk of provoking prerenal, hypovolaemia-induced AKI. Optimal fluid management, therefore, includes the necessity of individualized treatment and constant re-evaluation of the patient and the course of disease. As any kind of fluid administered has to be considered, a drug attention has not only to be spent on the amount of fluid, but also on the type. Chloride-liberal intravenous fluid administration strategies are criticized for induction or exacerbation of hyperchloraemia and metabolic acidosis and might even decrease renal perfusion [24]. Multiple studies in critically ill and general surgical patients as well as in liver and kidney transplant recipients indicate an increased risk for AKI and associated morbidity in patients receiving higher volumes of chloride-liberal fluids than in patients treated chloride restrictively [25–28].
If life-threatening changes in fluid, electrolyte and acid–base balance require induction of renal replacement therapy, continuous rather than intermittent renal replacement therapy is often used in most severely affected, haemodynamically unstable patients, although no significant superiority with regard to mortality has been shown. An increased haemodynamic stability as well as an elimination of pro-inflammatory mediators with middle molecular size leading to a potentially faster regeneration of kidney function and a decrease in chronic, enduring dialysis-dependent kidney failure is postulated for continuous haemodiafiltration [29, 30]. New filter membranes for continuous renal replacement therapy (e.g. septeX™, oXiris™) [31], intermittent haemoperfusion (polymyxin B-immobilized fibre cartridge) [32] or adsorption (CytoSorb™) [33] are promising developments to reduce the load of inflammatory triggers and add to causal sepsis therapy. By use of these filters, key molecules of sepsis such as endo- and exotoxins, lipopolysaccharides and cytokines are meant to be eliminated. Interestingly, damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) do not only trigger inflammatory pathways in septic AKI. In AKI due to trauma, CRS, autoimmunity and sepsis, DAMPs are released due to end-organ damage, and increased levels of PAMPs, e.g. lipopolysaccharides, have been demonstrated. Pro-inflammatory cytokines released by leukocytes and/or parenchymal cells in response to DAMPs and PAMPS have a direct impact on the kidney. Pro-inflammatory cytokines such as tumour necrosis factor alpha (TNFα) or interleukin (IL)-6 impact intra-renal blood flow, and renal parenchymal tissue responds by upregulating chemoattractants, which may lead to leukocyte recruitment into renal tissue. Furthermore, tubular regeneration and mesangial cell survival are disturbed by a pro-inflammatory cytokine environment [34, 35]. Thus, reducing the load of inflammatory triggers may be a future general approach in AKI. However, besides first positive reports in sepsis-associated AKI, this treatment is not validated as standard therapy as controlled, randomized trials are missing.
AKI and malignancy
AKI is also present in patients suffering from hemato-oncological diseases where it might appear as a complication of the primary disease or associated to therapy.
AKI caused by light chain-associated tubulointerstitial nephropathy is a common manifestation in patients with multiple myeloma [36]. This so-called cast nephropathy is associated with reduced 1-year survival and limitation of therapeutical options [37]. There is hope that high cut-off haemodialysis in combination with new chemotherapy is an effective option to avoid dialysis-dependent CKD.
A great number of chemotherapies are characterized by potential nephrotoxicity. Cisplatin, carboplantin and imatinib show acute tubulotoxicity, interferones can induce glomerulonephritis and mitomycine treatment might go along with thrombotic microangiopathy (TMA). In addition, many chemotherapeutics have to be dose adjusted to glomerular filtration rates (GFRs).
Early identification of patients being at risk for AKI is relevant for optimization of volume status, existing medication and exposition to further nephrotoxic agents such as iodinated contrast media.
In the context of chemotherapy, especially during induction, clinicians have to be aware of the tumour lysis syndrome common in haematological malignancies (e.g. acute myeloic or lymphatic leukaemia) as well as in fast melting tumours (e.g. non-small cell lung carcinoma, Burkitt's lymphoma). Tumour lysis syndrome goes along with hyperuricaemia, hyperkalaemia, hyperphosphatemia and secondary hypocalcaemia and can lead to severe complications such as AKI, cardiac arrhythmias and pulmonary oedema [38]. Between 40 and 71% of patients suffering from tumour lysis syndrome develop a need for renal replacement therapy [38]. Furthermore, patients with no need for renal replacement therapy benefit from a continuous monitoring of volume and acid–base status, electrolytes and medication by an experienced nephrologist. Sophisticated nephrological advice might prevent complication-associated delays in cytoreductive therapy or dose reductions, which have an impact on long-term prognosis.
AKI and liver insufficiency
AKI can occur as hepatorenal syndrome in advanced liver insufficiency. Distinct vasodilatation in the splanchnic area resulting in portal hypertension and a decrease in effective arterial volume and blood pressure leads to vasoconstriction of the renal artery with a consecutive disturbance of the renin–angiotensin–aldosterone–prostaglandin system [39]. The acute form of hepato-renal syndrome (Type I) is often triggered by a bacterial infection such as spontaneous bacterial peritonitis, but can also arise spontaneously. The need for renal replacement therapy in patients with liver insufficiency is associated with an increased mortality [40, 41]. Liver transplantation should be considered as causal treatment in the case of hepatorenal syndrome. Diuretics must be used carefully and with adequate monitoring of plasma electrolytes [42]. Diuretics can help to achieve fluid withdrawal, but cannot be considered to be a treatment for the hepatorenal syndrome itself. Paracentesis can reduce pressure on the renal veins and, therefore, ameliorate renal perfusion as long as post-interventional haemodynamic instability is prevented by substitution of fluid and/or human albumin [42]. Application of the vasopressin analogue terlipressin, which is often combined with human albumin, can improve kidney function and at least in the short term reduce mortality [43]. Fluid withdrawal by intermittent haemodialysis is often limited by hypotension.
AKI and autoimmune diseases
Different autoimmune diseases such as vasculitis and systemic lupus erythematosus are associated with glomerulonephritis (GN) causing AKI (Table 4). Pathomechanisms are diverse, and renal damage is mediated by autoantibodies and/or cellular immunity [44, 45]. Patients may initially present with a variety of extrarenal symptoms [46]. The presence of a nephritic sediment and—depending on the disease entity—a moderate to severe proteinuria indicates renal involvement. Microhaematuria might be the only hint at renal involvement. However, progression to severe AKI may occur rapidly [47–49]. If GN is suspected, a renal biopsy should be taken to guide diagnosis and therapy. The autoantibody profile is relevant for diagnosis and provides useful additional information [50]. Immunosuppressive treatment should be initiated as soon as possible after the diagnosis has been made. Depending on the disease entity, steroids are administered in combination with other immunosuppressants such as cyclophosphamide, mycophenolate mofetil or rituximab [49, 51, 52]. In cases of severe AKI, patients may benefit from extracorporeal procedures such as plasmapheresis or immunoadsorption [53, 54]. If renal function is already severely reduced at diagnosis, there is usually no full recovery from AKI and renal function remains impaired [47, 48, 55, 56]. Thus, flares following disease onset have to be avoided, and immunosuppressive treatment to maintain remission is necessary [1, 51, 57]. The patients have to be monitored closely as relapses may also occur under maintenance therapy [55, 56, 58]. Regular urinary examination for new onset or worsening haematuria is useful to detect active renal disease in vasculitis; in selected diseases, serial measurements of autoantibody titres may help to identify patients at risk [59, 60]. Rises of titres are reported to precede renal flare in anti-neutrophile cytoplasmatic antibodies (ANCA)-associated vasculitis.
Table 4.
Diverse causes and clinical findings of AKI
| Clinical findings | Important laboratory values | Urinary findings | |
|---|---|---|---|
| Acute cardiorenal/renocardial syndrome | Renal and cardiac failure, lung oedema/hypervolemia | ↑BNP | – |
| Sepsis | Systemic inflammatory response syndrome | ↑C-reactive protein, ↑procalcitonin, leukocytosis | (Leukocyturia) |
| Tumour lysis syndrome | ↑Uric acid ↑LDH, ↑CK, ↑K+, Phosphate ↓Ca2+ |
(Urate crystals) | |
| Hepatorenal syndrome | Ascites, cholestasis, oedema, portale hypertension | Hyponatraemia, ↑ GOT/GPT |
Proteinuria <0.5 g/day urinary Na+ <10 mmol/l urinary osmolarity > serum osmolarity |
| Systemic autoimmune diseases | |||
| ANCA vasculitis | Constitutional symptoms, arthralgia, purpura, sinusitis, epistaxis, haemoptysis | Inflammation, ↑ANCA, ↑anti-PR3/-MPO | Glomerular erythrocyturia, proteinuria |
| Systemic lupus erythematosus | Constitutional symptoms, arthralgia, (butterfly) rash, alopecia, oral ulcerations | Inflammation, leukopenia, thrombopenia, ↓C3, C4, ↑ANA, ↑anti-dsDNA | Glomerular erythrocyturia, proteinuria |
| IgA vasculitis | Constitutional symptoms, arthralgia, abdominal pain, purpura, bloody stool | Inflammation | Glomerular erythrocyturia, proteinuria |
| Thrombotic microangiopathy | |||
| TTP | Constitutional symptoms, neurologic symptoms, bleeding signs | Thrombopenia ↓Hb, ↓haptoglobin, ↑LDH, ↓ADAMTS13 activity |
|
| STEC-HUS | Constitutional symptoms, diarrhoea, bleeding signs, renal failure | Thrombopenia, ↓Hb, ↓haptoglobin, ↑LDH, Shiga toxin in stool | – |
| aHUS | Constitutional symptoms, bleeding signs, renal failure | Thrombopenia, ↓Hb, ↓haptoglobin, ↑LDH, complement abnormalities | – |
| AIN | |||
| Drug induced | Fever, nausea, rash, flank pain | Inflammation, ↑eosinophils | Haematuria, leukocyturia, eosinophiluria, mild proteinuria |
| Infectious agents | Depending on the type of infection | Inflammation | Haematuria, leukocyturia, proteinuria |
| ‘TINU’ | Constitutional symptoms, uveitis | Inflammation | Haematuria, leukocyturia, proteinuria |
| IgG4-related kidney disease | Extrarenal manifestations: lymph node swelling, salivary gland swelling, etc. | ↑Total IgG, IgG4, ↑IgE, ↑eosinophils, ↓C3, C4 | Haematuria, leukocyturia, proteinuria |
| Renal allograft failure | |||
| Rejection | ↑Serum creatinine, antibody-mediated rejection: donor-specific antibodies | – | |
| Polyoma virus-nephropathy | Viral load in serum | – | |
| cytomegaly disease | Viral load in serum | – | |
| Urinary tract infection/pyelonephritis | Dysuria, fever | Inflammation | Leukocyturia, nitrite positive, bacteria |
| Recurrence of primary disease | Depending on primary disease | Depending on primary disease | Depending on primary disease |
| CNI toxicity (acute) | (Neurotoxic side effects) | ↑CNI trough levels | – |
AKI and thrombotic microangiopathy
Three different entities of primary TMA can be distinguished: (i) thrombotic thrombocytopenic purpura (TTP), (ii) Shiga toxin-mediated haemolytic uraemic syndrome (STEC-HUS) and (iii) atypical haemolytic uraemic syndrome (aHUS) [61]. Microangiopathic, Coombs-negative, haemolytic anaemia accompanied by thrombocytopenia is the common clinical feature of all three forms of TMA [61]. The pathophysiology of each form is different. TTP is caused by a hereditary (genetic) or acquired (autoantibodies) ADAMTS13 deficiency, whereas aHUS is caused by an abnormal activation of the alternative complement pathway due to defective regulation of specific complement factors [50, 61–63]. Shiga toxin produced by Escherichia coli is the pathogenic factor in STEC-HUS [64–66]. It is toxic to endothelial and renal parenchymal cells. Shiga toxin has a prothrombotic effect that may be mediated by increased endothelial release of von Willebrand factor [65, 67–69]. Severe AKI is usually found in STEC-HUS and aHUS, whereas patients with TTP lack AKI or develop only milder forms [61]. In addition, patients with TTP may present with neurologic symptoms. Further diagnostic is necessary to distinguish the different forms of TMA (Table 4). If AKI is suspected to be caused by TMA, plasmapheresis should be performed in the first place [70, 71]. It is very important to obtain diagnostic material before the extracorporeal therapy is initiated. The therapy should be modified once the exact diagnosis is made. Blockade of the terminal complement pathway with eculizumab has been proved to be efficacious in aHUS [72–74]. In cases of TTP or aHUS where autoantibodies to ADAMTS13 or regulatory complement factors are found, B-cell depletion may be an option [75, 76]. The optimal therapy for STEC-HUS is not clear yet; the value and timing of antibiotic therapy are still controversial.
AKI and acute interstitial nephritis
Acute interstitial nephritis (AIN) is an important cause of AKI with increasing incidence especially in elderly patients where biopsy studies reveal a prevalence of more than 10% [77]. There are many different forms and causes triggering AIN. The most common form seen in more than two-thirds of patients presenting with AIN is due to a hypersensitivity reaction against specific drugs [77]. Drug-induced AIN may be associated with proton pump inhibitors, antibiotics, diuretics and non-steroidal anti-inflammatory drugs [77–80]. It usually develops within weeks after first exposure or within days after re-exposure. Patients may present with unspecific clinical symptoms like rash, nausea, emesis, fever and flank pain. The urine analysis reveals leukocyturia including eosinophilia, haematuria and mild proteinuria. The classic triad consisting of rash, fever and eosinophiluria is found in about 10% of all cases [81]. The suspected drug(s) should be stopped, and glucocorticoids can be administered if renal function does not recover within days. In about 15% of patients with AIN, the underlying cause is an infectious agent. AIN may occur in leptospirosis, legionellosis or in the context of streptococcal infections [82]. Patients with AIN due to Hantavirus infection can present with high fever, abdominal pain, myalgia and nausea. Signs and symptoms of haemorrhagic fever may be present [82]. AIN may also be caused by systemic, non-infectious diseases [78, 83–85], IgG4-related disease and sarcoidosis [77, 85, 86]. TINU syndrome (tubulointerstitial nephritis with uveitis) is a relatively rare cause of AIN in adolescents; ophthalmologic symptoms usually manifest within weeks or months after AIN [87]. The prognosis is usually good, and the disease is steroid sensitive.
AKI and liver transplantation
AKI after liver transplantation is a frequent complication occurring in up to two-thirds of patients [88, 89], leads to at least temporary need of renal replacement therapy in up to 30% of these cases [90], is a risk factor for CKD [89] and associated with increased mortality. The most common reasons for AKI after liver transplantation are ischaemia-induced acute tubular necrosis, nephrotoxic drug reactions especially due to immunosuppressive agents, a pre-existing hepatorenal syndrome and a poor preoperative condition of the patient [88].
Patients after liver transplantation often suffer from coagulopathies and thrombocythaemia. Especially in the early postsurgical phase, patients are at risk of major bleedings due to bleeding disorders and the large intra-abdominal surgical wound. Due to haemodynamic instability, continuous renal replacement therapy is often applied to those patients when needed. This requires anticoagulation for the avoidance of clotting in the extracorporeal circuit. Heparin is commonly used, but associated with increased bleeding risks. Regional citrate anticoagulation has proved to be a safe alternative even in patients after liver transplantation and can be performed without larger metabolic complications if adequate monitoring is guaranteed [90].
AKI and kidney transplantation
A specific definition for AKI in kidney transplant recipients has not been created. Especially in the early post-transplant phase, there is no stable creatinine value that can be used for diagnosis. Besides well-known prerenal, renal and postrenal causes for AKI, further specific causes such as calcineurin-inhibitor (CNI) toxicity, delayed graft function, acute rejections and recurrence of the primary disease have to be considered when talking about AKI in renal transplant recipients [91].
In the context of the transplantation itself, every organ is exposed to ischaemia and reperfusion injury. If this inevitable AKI results in delayed graft function, mostly defined by the necessity of renal replacement therapy during the first week after transplantation, it is associated with a decreased transplant and patient survival [91].
Not much data is available on the incidence of AKI in transplant recipients during later phases. About 20% of Japanese patients developed AKI within a follow-up period of 4 years [92]. AKI in transplant recipients seems to be mainly associated to rejections, infections and cardiovascular events [92, 93]. It is associated with more transplant losses and an increased mortality [93]. A continuous nephrological care of kidney transplant recipients, especially during ongoing viral or bacterial infections or during interventions imposed by other medical disciplines (exposure to contrast media, surgery) in a specialist transplant centre, should be suggested.
Biomarkers and AKI
As AKI is a clinical syndrome of multiple entities, searching for the one true biomarker implying early diagnosis, localization of damage, prediction of acute and long-term outcomes seems more like an unaccomplishable desire. It is clear that urine output and serum creatinine are only surrogate parameters for the declining renal function, while most of the new biomarkers are indicators of renal damage (Figure 1).
Fig. 1.
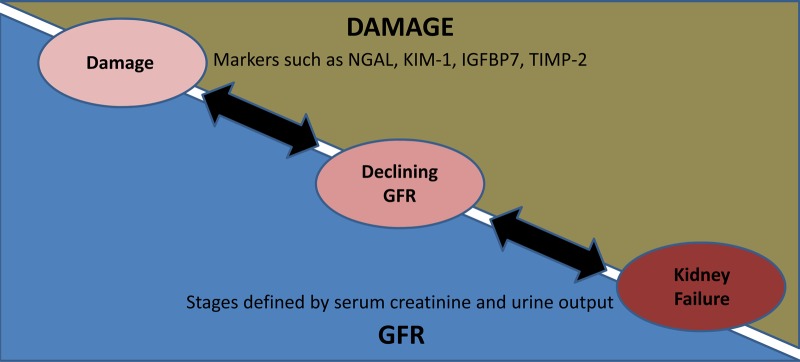
Conceptual model of damage and function in AKI. Modified [1].
Different biomarkers in urine and serum are in clinical testing. Cystatin C, IL-18, kidney injury molecule 1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) are considered to be earlier and more sensitive markers for AKI when compared with serum creatinine [94]. Some of those new makers also reflect renal function (e.g. cystatin C), while others depict structural renal damage (e.g. NGAL). Unlike serum creatinine, cystatin C is independent of muscular mass. It reveals a loss of GFR 1–2 days before a rise in creatinine becomes measurable [95]. Increased NGAL levels seem to have prognostic value with regard to need for renal replacement therapy [96]. However, NGAL is a protein that is not specific to the kidney, but can also be found in many cells of the human body as in the lung and the intestine. Serum NGAL is significantly increased in patients with AKI and sepsis when compared with patients with non-septic AKI [97]. The rise of NGAL due to systemic inflammation indicates a declining clinical condition of the patient, but loses specificity for early detection and prognosis of AKI.
Two big, multicentre studies led to the discovery and validation of two new, promising urinary biomarkers for AKI in 2013: insulin-like growth factor-binding protein 7 (IGFBP7) and tissue inhibitor of metalloproteinases-2 (TIMP-2) [98]. These two biomarkers out of 340 evaluated proteins in blood and urine showed the best predictive value for development of AKI in three different patient cohorts. IGFBP7 and TIMP-2 are inductors of cell cycle arrest during the post-mitotic G1 phase occurring during the very early phase of cellular damage. The combination of IGFBP7 and TIMP-2 was superior to all other described biomarkers with regard to prediction of AKI KDIGO Stage 2 or 3 within 12 h after sampling. In addition, values for the risk evaluation with regard to need for renal replacement therapy, persistence of declined renal function and mortality within 30 days after AKI KDIGO Stage 2 or 3 could be determined. A long-term evaluation of these damage-associated markers with a combined end point of end-stage renal disease and death showed that as long as there was no AKI with decline in renal function as to KDIGO criteria, there was no worsening of prognosis [99]. This points out that the loss of function as implied in the KDIGO criteria is a validated predictor for long-term outcome.
As AKI is very heterogeneous, maybe researchers and clinicians have to get away from the search for the ‘troponin equivalent’ and accept the challenge of applying the right set of biomarkers to their patients as part of individualized care. This might also involve ‘non-renal’ biomarkers of the underlying diseases. AKI is a very heterogeneous clinical syndrome, which cannot be fully depicted by this article. Prophylaxes, diagnosis and therapy of each form of AKI call for a differentiated and individualized approach in order to improve patient's mortality, morbidity, long-term kidney function and eventually the quality of life.
Conflict of interest statement
None declared.
References
- 1. Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012; 2: 1–138. [Google Scholar]
- 2.Rodrigues F, Bruetto R, Torres U, et al. Incidence and mortality of acute kidney injury after myocardial infarction: a comparison between KDIGO and RIFLE criteria. PLoS One 2013; 8: e69998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singbartl K, Kellum J. AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int 2012; 81: 819–825 [DOI] [PubMed] [Google Scholar]
- 4.Lameire N, Bagga A, Cruz D, et al. Acute kidney injury: an increasing global concern. Lancet 2013; 382: 170–179 [DOI] [PubMed] [Google Scholar]
- 5.Zeng X, McMahon G, Brunelli S, et al. Incidence, outcomes, and comparisons across definitions of AKI in hospitalized individuals. Clin J Am Soc Nephrol 2014; 9: 12–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoste E, Schurgers M. Epidemiology of acute kidney injury: how big is the problem? Crit Care Med 2008; 36: S146–S151 [DOI] [PubMed] [Google Scholar]
- 7.Coca S, Singanamala S, Parikh C. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 2012; 81: 442–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai T, Patel U, Chang T, et al. Contemporary incidence, predictors, and outcomes of acute kidney injury in patients undergoing percutaneous coronary interventions: insights from the NCDR Cath-PCI registry. JACC Cardiovasc Interv 2014; 7: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronco C, House A, Haapio M. Cardiorenal and renocardiac syndromes: the need for a comprehensive classification and consensus. Nat Clin Pract Nephrol 2008; 4: 310–311 [DOI] [PubMed] [Google Scholar]
- 10.Zannad F, Mebazaa A, Juillière Y, et al. EFICA Investigators. Clinical profile, contemporary management and one-year mortality in patients with severe acute heart failure syndromes: the EFICA study. Eur J Heart Fail 2006; 8: 697–705 [DOI] [PubMed] [Google Scholar]
- 11.Damman K, Testani JM. The kidney in heart failure: an update. Eur Heart J 2015; 36: 1437–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Somma S, Lalle I, Magrini L, et al. Additive diagnostic and prognostic value of BIVA to brain natriuretic peptide ‘grey-zone’ in patients with acute heart failure in the emergency department. Eur Heart J Acute Cardiovasc Care 2014; 3(2): 167–175 [DOI] [PubMed] [Google Scholar]
- 13.Legrand M, Mebazaa A, Ronco C, et al. When cardiac failure, kidney dysfunction, and kidney injury intersect in acute conditions: the case of cardiorenal syndrome. Crit Care Med 2014; 42: 2109–2117 [DOI] [PubMed] [Google Scholar]
- 14.Leung K, Pannu N, Tan Z, et al. Contrast-associated AKI and use of cardiovascular medications after coronary syndrome. Clin J Am Soc Nephrol 2014; 9: 1840–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winkelmayer W, Levin R, Setoguchi S. Associations of kidney function with cardiovascular medication use after myocardial infarction. Clin J Am Soc Nephrol 2008; 3: 1415–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang T, Desai M, Solomon D, et al. Kidney function and long-term medication adherence after myocardial infarction in the elderly. Clin J Am Soc Nephrol 2011; 6: 864–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aspromonte N, Cruz D, ROnco C, et al. Role of bioimpedance vectorial analysis in cardio-renal syndromes. Semin Nephrol 2012; 32: 93–99 [DOI] [PubMed] [Google Scholar]
- 18.Feldkamp T, Kribben A. Contrast media induced nephropathy: definition, incidence, outcome, pathophysiology, risk factors and prevention. Minerva Med 2008; 99: 177–196 [PubMed] [Google Scholar]
- 19.Bagshaw S, Uchino S, Bellomo R, et al. Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol 2007; 2: 431–439 [DOI] [PubMed] [Google Scholar]
- 20.Zarbock A, Gomez H, Kellum J. Sepsis-induced acute kidney injury revisited: pathophysiology, prevention and future therapies. Curr Opin Crit Care 2014; 20: 588–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vieira JJ, Castro I, Curvello-Neto A, et al. Effect of acute kidney injury on weaning from mechanical ventilation in critically ill patients. Crit Care Med 2007; 35: 184–191 [DOI] [PubMed] [Google Scholar]
- 22.Metha R, Bouchard J, Soroko S, et al. Sepsis as a cause and consequence of acute kidney injury: Program to Improve Care in Acute Renal Disease. Intensive Care Med 2011; 37: 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prowle J, Kirwan C, Bellomo R. Fluid management for the prevention and attenuation of acute kidney injury. Nat Rev Nephrol 2014; 10: 37–47 [DOI] [PubMed] [Google Scholar]
- 24.Chowdhury A, Cox E, Francis S, et al. A randomized, controlled, double-blind crossover study on the effects of 2-L infusions of 0.9% saline and plasmalyte®148 on renal blood flow velocity and renal cortical tissue perfusion in healthy volunteers. Ann Surg 2012; 256: 18–24 [DOI] [PubMed] [Google Scholar]
- 25.Yunos N, Bellomo R, Hegarty C, et al. Association between a chloride-liberal vs chloride-restrictive intravenous fluid administration strategy and kidney injury in critically ill adults. JAMA 2012; 308: 1566–1572 [DOI] [PubMed] [Google Scholar]
- 26.Nadeem A, Salahuddin N, ElHazmi A, et al. Chloride-liberal fluids are associated with acute kidney injury after liver transplantation. Crit Care 2014; 18: 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw A, Bagshaw S, Goldstein S, et al. Major complications, mortality, and resource utilization after open abdominal surgery: 0.9% saline compared to Plasma-Lyte. Ann Surg 2012; 255: 821–829 [DOI] [PubMed] [Google Scholar]
- 28.O'Malley C, Frumento R, Hardy M, et al. A randomized, double-blind comparison of lactated Ringer's solution and 0.9% NaCl during renal transplantation. Anesth Analg 2005; 100: 1518–1524 [DOI] [PubMed] [Google Scholar]
- 29.Sun Z, Ye H, Shen X, et al. Continuous venovenous hemofiltration versus extended daily hemofiltration in patients with septic acute kidney injury: a retrospective cohort study. Crit Care 2014; 18: R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wald R, Shariff S, Adhikari N, et al. The association between renal replacement therapy modality and long-term outcomes among critically ill adults with acute kidney injury: a retrospective cohort study. Crit Care Med 2014; 42: 868–877 [DOI] [PubMed] [Google Scholar]
- 31.Honore P, Jacobs R, Joannes-Boyau O, et al. Newly designed CRRT membranes for sepsis and SIRS—a pragmatic approach for bedside intensivists summarizing the more recent advances: a systematic structured review. ASAIO J 2013; 59: 99–106 [DOI] [PubMed] [Google Scholar]
- 32.Ronco C, Klein D. Polymyxin B hemoperfusion: a mechanistic perspective. Crit Care 2014; 18: 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hetz H, Berger R, Recknagel P, et al. Septic shock secondary to ß-hemolytic streptococcus-induced necrotizing fasciitis treated with a novel cytokine adsorption therapy. Int J Artif Organs 2014; 37: 422–426 [DOI] [PubMed] [Google Scholar]
- 34.Leemans JC, Kors L, Anders HJ, et al. Pattern recognition receptors and the inflammasome in kidney disease. Nat Rev Nephrol 2014; 10: 398–414 [DOI] [PubMed] [Google Scholar]
- 35.Colombo PC, Ganda A, Lin J, et al. Inflammatory activation: cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail Rev 2012; 17: 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutchison C, Batuman V, Behrens J, et al. The pathogenesis and diagnosis of acute kidney injury in multiple myeloma. Nat Rev Nephrol 2011; 8: 43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walther C, Podoll A, Finkel K. Treatment of acute kidney injury with cast nephropathy. Clin Nephrol 2014; 82: 1–6 [DOI] [PubMed] [Google Scholar]
- 38.Abu-Alfa A, Younes A. Tumor lysis syndrome and acute kidney injury: evaluation, prevention, and management. Am J Kidney Dis 2010; 55: S1–S13 [DOI] [PubMed] [Google Scholar]
- 39.Fagundes C, Gines P. Hepatorenal syndrome: a severe, but treatable, cause of kidney failure in cirrhosis. Am J Kidney Dis 2012; 59: 874–885 [DOI] [PubMed] [Google Scholar]
- 40.Weigand K, Bauer E, Encke J, et al. Prognostic value of standard parameters as predictors for long-term renal replacement therapy after liver transplantation. Nephron Clin Pract 2011; 119: c342–c347 [DOI] [PubMed] [Google Scholar]
- 41.Nadim M, Kellum J, Davenport A, et al. Hepatorenal syndrome: the 8th international consensus conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2012; 16: R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moller S, Krag A, Bendtsen F. Kidney injury in cirrhosis: pathophysiological and therapeutic aspects of hepatorenal syndromes. Liver Int 2014; 34: 1153–1163 [DOI] [PubMed] [Google Scholar]
- 43.Gluud L, Christensen K, Christensen E, et al. Systematic review of randomized trials on vasoconstrictor drugs for hepatorenal syndrome. Hepatology 2010; 51: 576–584 [DOI] [PubMed] [Google Scholar]
- 44.Couser WG. Basic and translational concepts of immune-mediated glomerular diseases. J Am Soc Nephrol 2012; 23: 381–399 [DOI] [PubMed] [Google Scholar]
- 45.Wilde B, Van Paassen P, Witzke O, et al. New pathophysiological insights and treatment of ANCA-associated vasculitis. Kidney Int 2011; 79: 599–612 [DOI] [PubMed] [Google Scholar]
- 46.Berden A, Goceroglu A, Jayne D, et al. Diagnosis and management of ANCA associated vasculitis. BJM 2012; 344: e344. [DOI] [PubMed] [Google Scholar]
- 47.Slot M, Tervaert J, Franssen C, et al. Renal survival and prognostic factors in patients with PR3-ANCA associated vasculitis with renal involvement. Kidney Int 2003; 63: 670–677 [DOI] [PubMed] [Google Scholar]
- 48.Sinico R, Di Toma L, Radice A. Renal involvement in anti-neutrophil cytoplasmic autoantibody associated vasculitis. Autoimmun Rev 2013; 12: 477–482 [DOI] [PubMed] [Google Scholar]
- 49.Segelmark M, Hellmark T. Autoimmune kidney diseases. Autoimmun Rev 2010; 9: A366–A371 [DOI] [PubMed] [Google Scholar]
- 50.Levy G, Nichols W, Lian E, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001; 413: 488–494 [DOI] [PubMed] [Google Scholar]
- 51.Mukhtyar C, Guillevin L, Cid M, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 2009; 68: 310–317 [DOI] [PubMed] [Google Scholar]
- 52.Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract 2012; 140: c179–c184 [DOI] [PubMed] [Google Scholar]
- 53.Walsh M, Casian A, Flossmann O, et al. Long-term follow-up of patients with severe ANCA-associated vasculitis comparing plasma exchange to intravenous methylprednisolone treatment is unclear. Kidney Int 2013; 84: 397–402 [DOI] [PubMed] [Google Scholar]
- 54.Jayne D, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 2007; 18: 2180–2188 [DOI] [PubMed] [Google Scholar]
- 55.Hilhorst M, Wilde B, van Breda Vriesman P, et al. Estimating renal survival using the ANCA-associated GN classification. J Am Soc Nephrol 2013; 24: 1371–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hilhorst M, Wilde B, van Paassen P, et al. Improved outcome in anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis: a 30-year follow-up study. Nephrol Dial Transplant 2013; 28: 373–379 [DOI] [PubMed] [Google Scholar]
- 57.Hellmich B, Flossmann O, Gross W, et al. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: focus on anti-neutrophil cytoplasm antibody-associated vasculitis. Ann Rheum Dis 2007; 66: 605–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flossmann O, Berden A, de Groot K, et al. Long-term patient survival in ANCA-associated vasculitis. Ann Rheum Dis 2011; 70: 488–494 [DOI] [PubMed] [Google Scholar]
- 59.Kemna M, Damoiseaux J, Austen J, et al. ANCA as a predictor of relapse: useful in patients with renal involvement but not in patients with nonrenal disease. J Am Soc Nephrol 2015; 26: 537–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Slot M, Tervaert J, Boomsma M, et al. Positive classic antineutrophil cytoplasmic antibody (C-ANCA) titer at switch to azathioprine therapy associated with relapse in proteinase 3-related vasculitis. Arthritis Rheum 2004; 51: 269–273 [DOI] [PubMed] [Google Scholar]
- 61.George J, Nester C. Syndromes of thrombotic microangiopathy. N Engl J Med 2014; 371: 654–666 [DOI] [PubMed] [Google Scholar]
- 62.Ståhl A, Vaziri-Sani F, Heinen S, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 2008; 111: 5307–5315 [DOI] [PubMed] [Google Scholar]
- 63.Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010; 5: 1844–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karmali M, Petric M, Steele B, et al. Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet 1983; 321: 619–620 [DOI] [PubMed] [Google Scholar]
- 65.Motto D, Chauhan A, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest 2005; 115: 2752–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buchholz U, Bernard H, Werber D, et al. German outbreak of Escherichia coli O104:H4 associated with sprouts. N Engl J Med 2011; 365: 1763–1770 [DOI] [PubMed] [Google Scholar]
- 67.Robinson L, Hurley R, Lingwood C, et al. Escherichia coli verotoxin binding to human paediatric glomerular mesangial cells. Pediatric Nephrol 1995; 9: 700–704 [DOI] [PubMed] [Google Scholar]
- 68.Hughes A, Ergonul Z, Stricklett P, et al. Molecular basis for high renal cell sensitivity to the cytotoxic effects of shigatoxin-1: upregulation of globotriaosylceramide expression. J Am Soc Nephrol 2002; 13: 2239–2245 [DOI] [PubMed] [Google Scholar]
- 69.Huang J, Motto D, Bundle D, et al. Shiga toxin B subunits induce VWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood 2010; 116: 3653–3659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Von Baeyer H. Plasmapheresis in thrombotic microangiopathy-associated syndromes: review of outcome data derived from clinical trials and open studies. Ther Apher 2002; 6: 320–328 [DOI] [PubMed] [Google Scholar]
- 71.Clark WF. Thrombotic microangiopathy: current knowledge and outcomes with plasma exchange. Semin Dial 2012; 25: 214–219 [DOI] [PubMed] [Google Scholar]
- 72.Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 2015; 87: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nürnberger J, Philipp T, Witzke O, et al. Eculizumab for atypical hemolytic–uremic syndrome. N Engl J Med 2009; 360: 542–544 [DOI] [PubMed] [Google Scholar]
- 74.Legendre C, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic–uremic syndrome. N Engl J Med 2013; 368: 2169–2181 [DOI] [PubMed] [Google Scholar]
- 75.Sankararaman S, Jeroudi M, Ibrahim H. Successful treatment of relapsing autoimmune thrombotic thrombocytopenic purpura with rituximab. Pediatr Int 2014; 56: 914–918 [DOI] [PubMed] [Google Scholar]
- 76.Froissart A, Buffet M, Veyradier A, et al. Efficacy and safety of first-line rituximab in severe, acquired thrombotic thrombocytopenic purpura with a suboptimal response to plasma exchange. Experience of the French Thrombotic Microangiopathies Reference Center . Crit Care Med 2012; 40: 104–111 [DOI] [PubMed] [Google Scholar]
- 77.Praga M, Sevillano A, Auñón P, et al. Changes in the aetiology, clinical presentation and management of acute interstitial nephritis, an increasingly common cause of acute kidney injury. Nephrol Dial Transplant 2014; pii: gfu326. [DOI] [PubMed] [Google Scholar]
- 78.Perazella M, Markowitz G. Drug-induced acute interstitial nephritis. Nature Rev Nephrol 2010; 6: 461–470 [DOI] [PubMed] [Google Scholar]
- 79.Klepser D, Collier D, Cochran G. Proton pump inhibitors and acute kidney injury: a nested case-control study. BMC Nephrol 2013; 14: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blank M-L, Parkin L, Paul C, et al. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int 2014; 86: 837–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baker R, Pusey C. The changing profile of acute tubulointerstitial nephritis. Nephrol Dial Transplant 2004; 19: 8–11 [DOI] [PubMed] [Google Scholar]
- 82.Muranyi W, Bahr U, Zeier M, et al. Hantavirus Infection. J Am Soc Nephrol 2005; 16: 3669–3679 [DOI] [PubMed] [Google Scholar]
- 83.Phillips E, Chung W-H, Mockenhaupt M, et al. Drug hypersensitivity: pharmacogenetics and clinical syndromes. J Allergy Clin Immunol 2011; 127: S60–S66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kardaun S, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Br J Dermatol 2013; 169: 1071–1080 [DOI] [PubMed] [Google Scholar]
- 85.Stone J, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012; 366: 539–551 [DOI] [PubMed] [Google Scholar]
- 86.Saeki T, Kawano M. IgG4-related kidney disease. Kidney Int 2014; 85: 251–257 [DOI] [PubMed] [Google Scholar]
- 87.Mackensen F, Smith J, Rosenbaum J. Enhanced recognition, treatment, and prognosis of tubulointerstitial nephritis and uveitis syndrome. Ophthalmology 2007; 114: 995–999e991 [DOI] [PubMed] [Google Scholar]
- 88.Karapanagiotou A, Kydona C, Dimitriadis C, et al. Acute kidney injury after orthotopic liver transplantation. Transplant Proc 2012; 44: 2727–2729 [DOI] [PubMed] [Google Scholar]
- 89.Umbro I, Tinti F, Piselli P, et al. Occurrence of chronic renal failure in liver transplantation: monitoring of pre- and posttransplantation renal function. Transplant Proc 2012; 44: 1956–1959 [DOI] [PubMed] [Google Scholar]
- 90.Saner F, Treckmann J, Geis A, et al. Efficacy and safety of regional citrate anticoagulation in liver transplant patients requiring post-operative renal replacement therapy. Nephrol Dial Transplant 2012; 27: 1651–1657 [DOI] [PubMed] [Google Scholar]
- 91.Cooper J, Wiseman A. Acute kidney injury in kidney transplantation. Curr Opin Nephrol Hypertens 2013; 22: 698–703 [DOI] [PubMed] [Google Scholar]
- 92.Nakamura M, Seki G, Iwadoh K, et al. Acute kidney injury as defined by the RIFLE criteria is a risk factor for kidney transplant graft failure. Clin Transplant 2011; 26: 520–528 [DOI] [PubMed] [Google Scholar]
- 93.Mehrotra A, Rose C, Pannu N, et al. Incidence and consequences of acute kidney injury in kidney transplant recipients. Am J Kidney Dis 2012; 59: 558–565 [DOI] [PubMed] [Google Scholar]
- 94.Coca SG, Yalavarthy R, Concato J, et al. Biomarkers for the diagnosis and risk stratification of acute kidney injury: a systematic review. Kidney Int 2008; 73: 1008–1016 [DOI] [PubMed] [Google Scholar]
- 95.Herget-Rosenthal S, Marggraf G, Hüsing J, et al. Early detection of acute renal failure by serum cystatin C. Kidney Int 2004; 66: 1115–1122 [DOI] [PubMed] [Google Scholar]
- 96.Haase M, Bellomo R, Devarajan P, et al. NGAL Meta-analysis Investigator Group. Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 2009; 54: 1012–1024 [DOI] [PubMed] [Google Scholar]
- 97.Bagshaw SM, Bennett M, Haase M, et al. Plasma and urine neutrophil gelatinase-associated lipocalin in septic versus non-septic acute kidney injury in critical illness. Intensive Care Med 2010; 36: 452–461 [DOI] [PubMed] [Google Scholar]
- 98.Kashani K, Al-Khafaji A, Ardiles T, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care 2013; 17: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Koyner JL, Shaw AD, Chawla LS, et al. on behalf of the Sapphire Investigators. Tissue Inhibitor Metalloproteinase-2 (TIMP-2) ⋅ IGF-Binding Protein-7 (IGFBP7) levels are associated with adverse long-term outcomes in patients with AKI. J Am Soc Nephrol 2015; 26: 1747–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ketteler M, Biggar P. Kardiorenales Syndrom. Der Nephrologe 2010; 5: 49–57 [Google Scholar]