

Abstract
Background
Aspergillus nomius is an opportunistic pathogen and one of the three most important producers of aflatoxins in section Flavi. This fungus has been reported to contaminate agricultural commodities, but it has also been sampled in non-agricultural areas so the host range is not well known. Having a similar mycotoxin profile as A. parasiticus, isolates of A. nomius are capable of secreting B- and G- aflatoxins.
Results
In this study we discovered that the A. nomius type strain (NRRL 13137) has a genome size of approximately 36 Mb which is comparable to other Aspergilli whose genomes have been sequenced. Its genome encompasses 11,918 predicted genes, 72 % of which were assigned GO terms using BLAST2GO. More than 1,200 of those predicted genes were identified as unique to A. nomius, and the most significantly enriched GO category among the unique genes was oxidoreducatase activity. Phylogenomic inference shows NRRL 13137 as ancestral to the other aflatoxigenic species examined from section Flavi. This strain contains a single mating-type idiomorph designated as MAT1-1.
Conclusions
This study provides a preliminary analysis of the A. nomius genome. Given the recently discovered potential for A. nomius to undergo sexual recombination, and based on our findings, this genome sequence provides an additional evolutionary reference point for studying the genetics and biology of aflatoxin production.
Keywords: Aspergillus nomius, Genome sequence, Gene ontology, Phylogenomics, Mating-type locus
Background
Aspergillus nomius is an aflatoxigenic fungal species, grouped in the genus’ section Flavi, which is often overshadowed by the more readily-sampled aflatoxigenic species such as A. flavus and A. parasiticus. The relatively little research attention applied to A. nomius likely relates to a historical belief that its occurrences are rare [1, 2]; however, more recent reports suggest that this fungus has a wider ecological distribution and a greater economic potential than once believed [3, 4]. Most samplings of A. nomius have been reported from infected insects [5–8] and non-agricultural soils [4]. Reports also exist that show A. nomius’ potential as a human pathogen [9] as well as its indirect potential for animal and human impact through aflatoxin contamination of agricultural commodities such as maize [4], wheat [6], sugar cane [10], and tree nuts [6, 11].
In culture, A. nomius appears similar to the A. flavus L morphotype (L-type), so differentiation between these closely-related species can be fairly subjective and open to misidentification. The A. flavus L-type produces fewer large sclerotia and abundant conidia while the A. flavus S-type produces abundant small sclerotia and fewer conidia, but these are not considered different species [12]. Historically, aflatoxin profiles have been used to distinguish between A. flavus and A. nomius since their macro- and micro-morphologies were highly similar. If an isolate produced only B-aflatoxins it was classified as A. flavus, and if B- and G- aflatoxins were produced, it was considered to be A. nomius. Whereas A. parasiticus has several distinct morphological characteristics, this is not the case for distinguishing between A. flavus and A. nomius. For example, A. parasiticus typically produces asexual spore forming structures (conidiophores) with a single layer of spore-forming cells (phialides) and lacking an intermediate layer of cells (metulae), and has dark green conidia which are noticeably rough-walled [13]. These differences are not observed when comparing A. flavus and A. nomius whose colony diameters and coloration are very similar, tending towards yellow-green on Czapek’s Dox (CZ) agar. The size and texture (smooth to finely roughened) of their conidia are also very similar. The sclerotia of A. nomius are reported to be more elongated than those of A. flavus, and it has been observed that A. flavus grows well at 42 °C while A. nomius growth is slowed [6]. Due to the number of similarities between A. flavus and A. nomius, genomic comparisons may be more useful than traditional methods for species identifications.
There have been additional cryptic aflatoxin producing species recently characterized that are related to, and potentially indistinguishable from, A. flavus (L- or S-type), A. nomius and/or A. parasiticus such as A. arachidicola, A. minisclerotigenes and A. parvisclerotigenus [14–16]; therefore, a holistic approach to fungal identification and classification is required to make certain what is being characterized is not a naturally-occurring mutant or a hybrid offspring exhibiting a recombinant phenotype. This will involve comparisons of species morphology, extrolite production, host/niche range, and genomics [17]. A population study of A. nomius isolates sampled throughout Thailand revealed three potential clades or lineages for this species [3]. Within the last few years the sexual (teleomorphic) state was discovered and assigned to the genus Petromyces, and although the fungus is heterothallic, some strains contain both mating-type (MAT) idiomorphs and exist as functionally bisexual- meaning that either idiomorph is capable of outcrossing [4]. Its bisexuality may allow for more rampant sexual recombination in the environment regardless of the dominant mating type present. With the potential for hybridization between aflatoxigenic species (unpublished observations), A. nomius might serve as a mycotoxin reservoir in non-agricultural areas that are near agricultural fields [3], and assist in the generation of offspring (i.e., new ‘species’) with greater host ranges and novel chemotype profiles.
In this study we sequenced the genome of A. nomius to improve our understanding of its evolutionary place among the aflatoxigenic fungi, to ascertain those features that distinguish it from other closely-related species, and to promote research interest for this organism since it may offer more insights into the speciation and development of novel aflatoxigenic species.
Results and discussion
Genome details for A. nomius NRRL 13137
We sequenced the genome of A. nomius type strain NRRL 13137 to 42x coverage. This gave us 5 million reads with a median read length of 306 bp, 2,713 contigs of data (950 of which were ≥ 1000 bp), and a N50 length of 66,657 bp. The raw sequence reads have been deposited in the NCBI Sequence Read Archive as SRR1297207. The genome assembly of A. nomius NRRL 13137 is approximately 36 Mb in size and includes 11,918 protein-encoding genes (Table 1). Comparatively, A. nomius has a similar genome size to the other sequenced species from section Flavi. For example, assemblies for A. flavus NRRL 3357 and A. oryzae RIB40 are approximately 37 Mb [18, 19]. Table 2 shows various morphological, toxigenic, and genomic differences between A. flavus, A. nomius and A. parasiticus. Reports of the genome sizes of other sequenced Aspergillus species indicate that A. niger CBS 513.88, A. nidulans FGSC A4, and A. fumigatus Af293 have genome assemblies that include approximately 34 Mb, 30 Mb, and 29 Mb, respectively [20–22]. The reason for section Flavi species having larger genomes than other sequenced Aspergilli remains uncertain, but one reported possibility is gene acquisition [18]. The A. oryzae genome is one of the larger genomes of the section Flavi examined, but it also has a higher percentage of repetitive DNA than the others (Table 2). Of the nearly 12,000 predicted genes for A. nomius, 39 % (4,674) were assigned as hypothetical with no functional annotation. Although similar numbers of predicted genes were found in other sequenced species, due to limits in sequencing the A. nomius genome it should be noted that genes may not have been annotated if they happened to exist between contigs that have no overlap. The number of genes considered unique to A. nomius is 1,124 or only about 9 % of the total number of its predicted genes. SMURF* predicted A. nomius having an estimated 44 secondary metabolite clusters (SMCs) while antiSMASH predicted 53 SMCs (Table 3). The difference between these counts is likely related to stringency of predictions between the two programs; however, it is still recommended that both tools be used since they incorporate different algorithms in cluster prediction [23]. For example, SMURF* predictions include DMAT backbone genes whereas antiSMASH does not, but even without considering DMAT clusters antiSMASH predicted a greater number of SMCs than SMURF*. Beyond section Flavi, our 44 SMURF*-predicted SMCs in A. nomius was comparable to the 40 in A. nidulans, and noticeably higher than the 25 in P. chrysogenum. Andersen et al. reported a putative count of 51 SMCs in A. nidulans which was also based on SMURF* but may have also included “-like” genes (e.g., NRPS-like) [24]. According to antiSMASH predictions, A. nomius had a much greater number of SMCs than both A. nidulans (35) and P. chrysogenum (31). Aspergillus parasiticus had the most predicted SMCs across all Aspergilli examined, based on predictions from both programs, while A. nidulans had the least number of SMCs (Table 3).
Table 1.
Genome characteristics for the A. nomius type strain NRRL 13137
| Genome characteristic | Value |
|---|---|
| General | |
| Assembly size (bp) | 36,156,695 |
| G + C (%) | 49 |
| Protein coding genes | 11,918 |
| Protein coding genes >100 amino acids | 11,672 |
| Predicted protein coding sequences >100 amino acids | |
| Coding (%) | 49 |
| Gene density (1 gene every n bp) | 3,034 |
| Median gene length (bp) | 1,447 |
| Mean gene length (bp) | 1,732 |
| Average number of exons per gene | 3.4 |
Table 2.
Morphological, phenotypic and genomic comparison of A. flavus, A. nomius and A. parasiticus
| Species | Morphology | Phenotype | Genomics | ||||
|---|---|---|---|---|---|---|---|
| Macroa | Microb | Toxic SMsc | Size (Mb)d | Genese | GC (%)f | Rep. DNA (%)g | |
| A. flavus L | 55–65 mm velvety to floccose olive green sclerotia (l + v) | Radiate to columnar 400–800 μm, rf/fr 20–45 μm, gl/el u/b 3–6 μm, gl/el, sm/fr | B1, B2, CPA | 36.89 | 13,485 | 48.22 | 1.17 |
| A. nomius | 40–70 mm velvety to floccose olive green sclerotia (e) | Radiate 300–1100 μm, rf/fr 25–65 μm, gl/el u/b 4.5–6.5 μm, gl/el, sm/fr | B1, B2, G1, G2 | 36.14 | 11,914 | 48.86 | 1.09 |
| A. parasiticus | 45–65 mm velvety to floccose dark green sclerotia (o) | Radiate 250–500 μm, fr/rf 20–35 μm, gl/el u/b 3.5–6 μm, gl, rf | B1, B2, G1, G2, OMST | 39.82 | 13,543 | 47.72 | 1.40 |
aColony characters on Czpaek’s medium, incubated at 25 oC for 7 days: diameter; texture; color. Sclerotia large and variable in shape (l + v), elongate (e), or occasionally formed (o)
bConidiophore characters: conidial head; stipe (rough = rf, finely-roughened = fr); vesicle (globose = gl, elongate = el); seriation (uniseriate = u, biseriate = b, both/either = u/b); conidia (globose = gl, elongate = el, smooth = sm, finely-roughened = fr, rough = rf)
cMajor toxic secondary metabolites: B and G aflatoxins; cyclopiazonic acid (CPA); O-methylsterigmatocystin (OMST)
dApproximate sizes of sequenced genomes
eEstimated gene counts based on annotation
fGC content for each genome
gPercentage of repetitive DNA
Table 3.
Putative secondary metabolite clusters within various species included in our phylogenomic examination (Fig. 2)
| Species | SMURF* analysisa | antiSMASHb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NRPS | PKS | NRPS-PKS | DMAT | Total | NRPS | PKS | NRPS-PKS | Total | |
| A. flavus L | 14 | 24 | 2 | 7 | 47 | 18 | 24 | 3 | 45 |
| A. flavus S | 11 | 22 | 3 | 8 | 44 | 17 | 30 | 4 | 51 |
| A. nidulans | 10 | 25 | 1 | 4 | 40 | 11 | 22 | 2 | 35 |
| A. nomius | 14 | 20 | 3 | 7 | 44 | 17 | 28 | 8 | 53 |
| A. oryzae | 12 | 22 | 1 | 7 | 42 | 18 | 28 | 2 | 48 |
| A. parasiticus | 17 | 22 | 1 | 8 | 48 | 21 | 32 | 2 | 55 |
| P. chrysogenum | 8 | 15 | 1 | 1 | 25 | 10 | 19 | 2 | 31 |
aSMURF* predictions do not include “-like” backbone genes (NRPS-like, PKS-like, NRPS-PKS-like, DMAT-like)
bantiSMASH software does not consider DMAT backbone genes and its predictions do not include “-like” backbone genes
Gene ontology and enrichment for A. nomius NRRL 13137
A total of 8,599 genes were assigned gene ontology (GO) terms. Level-3 GO distribution results for each domain revealed that most gene products within the cellular component domain are involved in cell, membrane and organelle structure; within the biological process domain, most genes are involved in metabolic, cellular and single-organism processes; and analysis of the molecular function domain revealed that the greatest numbers of A. nomius genes are involved in ion binding, organic cyclic compound binding and heterocyclic compound binding (data not shown). For the 1,264 unique A. nomius genes, 13 of the 19 most significantly-enriched GO terms as shown by the Fisher’s Exact test were associated with molecular function, and six were associated with biological process. Significance levels, however, indicated that sequences associated with oxidoreductase activity had the highest enrichment (p-value = 9.67E-11) of all the GO terms/categories (Table 4). There were 227 protein sequences that were assigned to the “oxidoreductase activity” category of the molecular function domain. Of these proteins, only 43 were assigned functional annotation with 102 identified as hypothetical. The greatest number of any particular protein within the category of oxidoreductase activity was related to cytochrome P450 (n = 14). The epoxide metabolic process, and both fumagillin processes listed in Table 4, shared the same quantities and types of proteins (n = 12) which would indicate overlapping functions for these biological processes. Of the 255 proteins affiliated with the molecular function of heme binding, 66 were identified as hypothetical and only 21 of these were assigned functional annotation. As with oxidoreductase activity, the greatest number of any particular protein within the category of molecular function was related to cytochrome P450 (n = 93). Cytochrome P450 enzymes in fungi contribute to a multitude of complex bioconversions which are necessary for their adaptation and survival on various substrates [25].
Table 4.
Fisher’s exact test examining GO term enrichment for genes unique to A. nomius
| GO term | P-Value |
|---|---|
| Oxidoreductase activity | 9.67E–11 |
| Epoxide metabolic process | 3.55E–09 |
| Fumagillin biosynthetic process | 3.55E–09 |
| Fumagillin metabolic process | 3.55E–09 |
| Heme binding | 3.92E–08 |
| Tetrapyrrole binding | 4.50E–08 |
| Ether biosynthetic process | 1.23E–07 |
| Iron ion binding | 5.35E–07 |
| Catalytic activity | 1.17E–06 |
| Transition metal ion binding | 1.95E–06 |
| Oxidation-reduction process | 5.03E–06 |
| Modified amino acid binding | 8.85E–06 |
| Oxidoreductase activitya | 1.11E–05 |
| Ether metabolic process | 1.21E–05 |
| Phosphopantetheine binding | 1.94E–05 |
| Ion binding | 2.68E–05 |
| Amide binding | 4.10E–05 |
| Vitamin binding | 9.00E–05 |
| Amino acid binding | 9.63E–05 |
aActing on paired donors, with incorporation or reduction of molecular oxygen
Comparison of annotated genes and phylogenomics among sequenced Aspergillus genomes
An orthology analysis (Fig. 1) revealed that the five species examined from section Flavi share 7,497 genes. When A. nidulans and the outgroup taxa, P. chrysogenum, were added to the analysis the number of shared genes decreases to 4,977 (which were all used as input for our phylogenomic analysis). The count of 1,124 unique genes in A. nomius is not the highest number for the five section Flavi species examined, however, since A. flavus L-type (NRRL 3357) and A. parasiticus (SU-1) appear to harbor more unique genes (1,629 and 1,370, respectively). Aspergillus oryzae (RIB40) has 1,036 unique genes while S-type A. flavus (AF70) has the fewest unique genes (n = 991). The highest quantity of shared unique genes between A. nomius and each of the other species examined is with A. parasiticus (n = 297), and the lowest quantity shared is with A. oryzae (n = 49). The number of shared unique genes among A. flavus L, A. parasiticus and A. nomius is less than 1 % (n = 73) compared to those shared by all section Flavi species examined; however, the inclusion of another aflatoxin producer (A. flavus S) increased the number of shared unique genes by 11 % (n = 924). The finding that A. flavus S contained many of the unique genes found in the other aflatoxigenic species examined warrants further investigation since it shares more unique genes with three species that, among themselves, do not share as many (Fig. 1). The fewest numbers of shared genes involved an overlap of A. nomius and A. oryzae with A. parasiticus (n = 53), A. flavus L (n = 36), and A. flavus S (n = 30). Although closer examination of the unique genes within these closely-related species will result in better delimitation, it should be considered that the unique genes that separate A. flavus L, A. flavus S, and A. oryzae from one another may be more strain-specific than species-specific. Additionally, a closer examination of the unique genes within these fungi could offer a more refined diagnostic PCR methodology to aid in species delimitation.
Fig. 1.
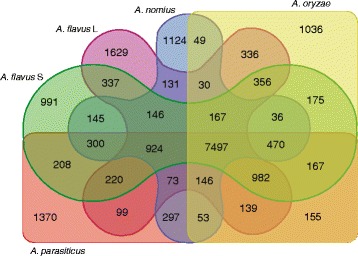
A Venn diagram quantifies unique and orthologous genes for multiple species in section Flavi. This diagram of overlapping shapes includes species names, gene counts, and color-shading: NRRL 13137 (A. nomius) is shaded blue, NRRL 3357 (A. flavus L) is shaded purple, AF70 (A. flavus S) is shaded green, SU-1 (A. parasiticus) is shaded red, and RIB40 (A. oryzae) is shaded yellow
Phylogenomic associations were used to infer a tree for multiple Aspergilli (Fig. 2). Using Penicillium chrysogenum as the outgroup taxa, we observed a divergence of the A. nidulans genome from those of section Flavi, which was expected. Although exhibiting a great deal of overall genomic similarity, A. nomius was observed to be the most basal genome of the species examined from section Flavi, followed by the A. parasiticus (SU-1) genome and then that of A. flavus S (AF70). The A. flavus L (NRRL 3357) and A. oryzae (RIB40) genomes appear to have diverged most recently. Based on our phylogenomic inference, A. nomius is confirmed as the ancestral organism compared to the other section Flavi species examined, and would suggest that it may be the progenitor to the other aflatoxigenic species in section Flavi. Previous conventional phylogenetic studies have given researchers an idea of the evolutionary relationships for the Aspergilli; therefore, a phylogenomic comparison including the genomes of all aflatoxigenic species would offer better resolution for the evolution of the aflatoxigenic phenotype. Similarly, phylogenomic comparison of the remaining species from section Flavi (e.g., A. alliaceus) would offer more insights into the relatedness of these fungi. For example, A. alliaceus is usually observed basal to the other species in many phylogenetic inferences involving section Flavi [15], but these are usually based on analyses of one to three individual loci. Ehrlich et al. [3] reported the possibility of multiple lineages of A. nomius that include genetically-distinct isolates, so a comparative phylogenomic study of strains from each lineage may ascertain whether or not these strains should all be considered A. nomius.
Fig. 2.
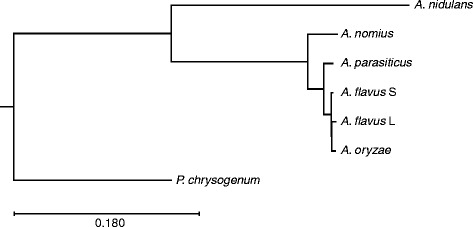
Phylogenomic comparison of sequenced Aspergillus species reveals patterns of ancestry. This tree was inferred from phylogenomic comparisons of multiple Aspergillus species (A. nidulans, A. nomius, A. parasiticus, A. flavus L, A. flavus S and A. oryzae) with Penicillium chrysogenum as the outgroup taxa
A. nomius NRRL 13137 MAT locus investigation revealed a single MAT1-1 idiomorph
According to Horn et al. [4], A. nomius is a heterothallic fungus that is incapable of self-fertilization despite some observed strains possessing both idiomorphs. The A. nomius type strain, NRRL 13137, is considered a MAT1-1 strain because only a single α-box idiomorph has been shown to be present using diagnostic PCR [26], and our sequencing results support this finding. BLAST query of nucleotide and protein sequences, for a MAT1-2 idiomorph, to the NRRL 13137 genome failed to uncover any homologs. The MAT1-1 gene of NRRL 13137 spans 1,165 bp, and is comprised of two exons adjoined by a single 52 bp intron. This MAT gene is flanked by two other genes, SLA2 and APN1, which have respective lengths of 4,138 bp and 2,189 bp, and are separated from the MAT gene by nucleotide distances of 1,492 bp and 1,778 bp, respectively (Fig. 3). These two flanking genes appear to have conserved locations upstream and downstream of the MAT genes in other species from section Flavi [26]. According to Metzenberg and Glass, MAT idiomorphs within heterothallic fungi will occupy a similar chromosomal location [27]; for A. oryzae, A. flavus and A. parasiticus, the idiomorphs are reported to reside on chromosome VI [26]. We cannot yet say which chromosome harbors the MAT gene in A. nomius, but the 65 kb contig that contains the SLA2, MAT1-1 and APN1 genes exhibits synteny to other section Flavi species and has sequence homology to chromosome VI in A. oryzae.
Fig. 3.
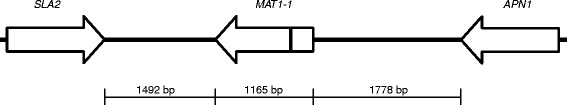
The mating-type locus of A. nomius NRRL 13137 contains a single MAT1-1 idiomorph. This schematic diagram for the mating-type locus of NRRL 13137 shows the orientation and physical distance shared among the MAT1-1 idiomorph and the adjacent upstream and downstream genes. The black vertical line in the MAT1-1 arrow represents the approximate location of a 52 bp intron
Within section Flavi, comparison of the A. nomius amino acid sequence with other MAT1-1 idiomorphs revealed 79 % identity to A. alliaceus and 91 % identity to both A. flavus and A. parasiticus (Fig. 4). Of the 374 amino acids aligned, 34 % were conserved, 32 % involved semi-conserved substitutions, and 34 % involved non-synonymous substitutions. The maximum likelihood (ML) tree, inferred from the MAT1-1 amino acid alignment of six Aspergillus species, shows homothallic E. nidulans to be more basal to the other species, with homothallic A. alliaceus being the basal organism to A. nomius. However, this inference was based on a single locus. Aspergillus alliaceus was not included in our phylogenomic analysis because its genome has yet to be sequenced, so it is unknown whether or not this species would maintain its basal position compared to the rest of section Flavi.
Fig. 4.

Phylogeny for MAT1-1 idiomorphs of various Aspergillus species reveals patterns of ancestry for this locus. A maximum likelihood phylogeny was inferred for the MAT1-1 gene of six Aspergillus species (top image). Also included is an amino acid alignment of the MAT1-1 gene for the same six species that was generated using CLUSTAL-W (bottom image). Beneath each column are characters that relate to the level of conservation in amino acid substitutions among the different species. An asterisk (*) indicates that an identical amino acid residue exists for all species examined; a colon (:) indicates that at least one highly conserved amino acid substitution exists; a period (.) indicates that at least one semi-conserved substitution exists; and no character indicates that at least one un-conserved amino acid substitution exists. The black inverted arrow indicates the location of a 52 bp intron separating the two coding regions that comprise the MAT1-1 gene in these organisms
Perhaps the evolutionary relationship of homothallic and heterothallic breeding systems can be inferred from this ML tree. For example, the MAT1-1 ancestor to heterothallic Aspergilli diverged from E. nidulans and its remnant locus organization can be found in A. alliaceus, the only known Aspergillus species for which both MAT idiomorphs are adjacent to one other at a single locus [26]. Interestingly, A. nomius strains that have the MAT1-1/MAT1-2 genotype are capable of bisexuality [4], but the chromosomal positioning of their MAT locus/loci has yet to be determined. Most strains of A. flavus and A. parasiticus are of a single mating type; however, some strains have been discovered to contain both genes (e.g., NRRL 3357) although only one of them is functional (Ignazio Carbone, personal communications). Aspergillus nomius strain NRRL 13137 contains only the MAT1-1 gene, yet other strains have been discovered that contain both idiomorphs although incapable of self-fertilization [4]. Mapping the mating-type locus/loci in a bisexual strain of A. nomius, for comparison to other MAT1-1/MAT1-2 strains, as well as inter-specific mating tests between A. nomius and closely-related species, should also be undertaken.
Conclusions
Many avenues remain to be explored regarding this aflatoxigenic fungus. Aspergillus nomius appears to be the ancestral aflatoxigenic species based on genomic comparisons to other section Flavi. Future genomic comparisons should include more species within section Flavi as well as expand beyond section Flavi to encompass other aflatoxin-producing Aspergilli. Further comparisons are warranted for A. nomius strains that thrive in distinct ecological niches and that exhibit genomic differences. By elucidating its ecological, evolutionary and genomic differences, we may finally understand the reasons for speciation of other aflatoxigenic species as well as the purpose for aflatoxin production.
Methods
Sequencing the genome of A. nomius NRRL 13137
The genome of NRRL 13137 was sequenced using an Ion Torrent Personal Genome Machine (PGM) from Life Technologies (Grand Island, New York). To obtain genomic DNA, 75 ml of potato dextrose broth was inoculated with 500 μl of fungal spore suspension and placed in an orbital shaker set at 30 °C and 125 rpm overnight. Resulting mycelial pellets were then vacuum-filtered, with two sterile water washes, blot dried, and approximately 200 mg was used to extract DNA using a Masterpure Yeast DNA Purification kit (Epicentre Biotechnologies, Madison, Wisconsin). The template was first subjected to fragmentation using the Ion Xpress Plus Fragment Library kit. Template purification steps were performed using AgenCourt AmPure XP beads (Beckman Coulter, Inc., Indianapolis, Indiana) and a magnetic rack, followed by re-suspension with Low TE buffer. Size selection of DNA fragments was performed using the E-Gel system (Life Technologies) and library qualification was performed with an Agilent 2100 Bioanalyzer instrument and High Sensitivity DNA kit (Agilent Technologies, Santa Clara, California). A OneTouch 2 (OT2) instrument and OT2 400 kit (Life Technologies) were used to prepare template-positive ion sphere particles (ISPs). The ISPs were qualified using a Qubit fluorometer and then enriched using the Ion OneTouch ES system with its respective component of the OT2 400 kit (Life Technologies). Sequencing was performed using the Ion PGM Sequencing 400 kit, an Ion 318 Chip v2, and the PGM instrument (Life Technologies) following manufacturer’s instructions.
Genome annotation
Newbler software [28] was used to assemble the A. nomius genome, and the annotations were created using the MAKER program [29]. MAKER produced ab initio gene predictions from the repeat-masked genomic sequence using Genemark [30] and Augustus [31]. Hint-based predictions were also generated from Augustus. Genemark was trained for A. nomius through its internal self-training module. An initial run of MAKER was performed to develop a set of genes that were used to train Augustus with predicted proteins from A. oryzae RIB40, A. flavus NRRL 3357 (L-type), A. flavus AF70 (S-type), and A. parasiticus SU-1 with the “protein2genome” option set. Both A. oryzae and A. flavus NRRL 3357 genome sequences and annotations were acquired from NCBI. The genome sequences and annotations for A. flavus AF70 and A. parasiticus were acquired from JCVI [32]. A subsequent run of MAKER was performed using Genemark and Augustus with the Uniref50 protein database [33] and the predicted proteins from A. flavus AF70 and A. parasiticus as evidence of protein homology. The predicted genes, from the second MAKER run, were then used to retrain Augustus, and MAKER was run a third time with the updated gene prediction file. Repetitive elements were identified within MAKER using the RepBase program [34] repeat library for fungi along with MAKER’s built-in repeat database known as RepeatMasker [35]. Functional annotation was added to the MAKER gene predictions using BLASTP against the NCBI fungal RefSeq protein database [36]. Gene names were transferred from the best BLAST hits if they had an e-value ≤ 1e–10, identity ≥ 50 %, and coverage ≥ 70 %. The genome annotation was converted to NCBI submission format using Genome Annotation Generator [37] and deposited at NCBI under BioSample #02768702.
Genome comparison
Genomic comparisons between A. nomius and other Aspergilli; particularly those from section Flavi, were performed using various types of analyses. The Secondary Metabolite Unique Regions Finder (SMURF) and the Antibiotics & Secondary Metabolite Analysis Shell (antiSMASH) programs [38, 39] were used to predict secondary metabolite clusters in the five Aspergilli from section Flavi with sequenced genomes. SMURF and antiSMASH vary in the quantities of backbone genes they explore, so both were modeled to look for the same three backbone genes: PKS, NRPS and PKS-NRPS hybrid. We also performed a phylogenomic investigation, which first involved detecting orthologous proteins within other fungi using Proteinortho [40], aligning them using MUSCLE [41], and concatenating them into a 2.2 Mb amino acid alignment using GBLOCKS [42]. The phylogenetic tree was inferred using RAxML-HPC [43] with the rtREV amino acid substitution matrix and Penicillium chrysogenum as the outgroup organism. The protein sequences for A. nidulans and P. chrysogenum were retrieved from AspGD and JGI, respectively. To quantify the genes unique to A. nomius as well as those genes it shares with closely-related species from section Flavi, orthologs identified by Proteinortho were put into a Venn diagram using software found at [44].
Gene ontology and function
GO terms were assigned to protein sequences within the BLAST2GO pipeline [45] using BLAST+ against the NCBI fungal database and InterProScan. A Fisher’s Exact test was run within BLAST2GO to identify GO terms that were over-represented among gene products unique to A. nomius with the reference set being GO terms for all non-unique A. nomius gene products. The data generated from the Fisher’s Exact test included only those GO terms that are enriched below a threshold of p ≤ 0.05, showed the percentage of sequences associated with each listed term, and ranked those terms from the highest to lowest level of enrichment.
MAT locus investigation
A portion of the MAT1-1 idiomorph for A. nomius NRRL 13137 had been previously sequenced and accessioned in GenBank (HQ001936). We then BLAST-queried the nucleotide sequence against our A. nomius genome and determined the respective contig for which we had 100 % identity and coverage, as well as a large span of sequence both upstream and downstream of our known sequence. Ramirez-Prado et al. [26] reported that there are conserved regions that flank the MAT idiomorph in certain Aspergilli. These regions include a gene for cytoskeleton assembly control (SLA2) and a gene for DNA lyase (APN1). Mapping of these loci, in relation to the idiomorph, involved the use of both nucleotide and amino acid sequences through BLAST query, and manual alignment using the Sequencher 5.4 program (Gene Codes Corporation, Ann Arbor, Michigan). Another component to analyzing the MAT1-1 idiomorph for A. nomius involved using ClustalW [46] to create an amino acid alignment and to weigh substitutions for the same idiomorph across other Aspergillus species: A. alliaceus, A. flavus, A. fumigatus, A. parasiticus, and E. nidulans. Except for A. nomius, the protein sequences were obtained from the NCBI database. Additionally, a phylogenetic association was inferred for the Aspergillus MAT1-1 idiomorph, based on the amino acids of these same species, using a software program known as Randomized Axelerated Maximum Likelihood [47].
Acknowledgements
The authors would like to acknowledge the referees who took the time to review and help to improve this body of work.
Abbreviations
- NRRL
Northern Regional Research Laboratory
- GO
Gene ontology
- CZ
Czapek’s
- CPA
Cyclopiazonic acid
- OMST
O-methylsterigmatocystin
- NCBI
National Center for Biotechnology Information
- SMURF
Secondary metabolite unique regions finder
- antiSMASH
Antibiotics & Secondary Metabolite Analysis Shell
- BLAST
Basic local alignment search tool
- MAT
Mating-type
- bp
Base pairs
- SLA2
Cytoskeleton assembly control gene
- APN1
DNA lyase gene
- ML
Maximum likelihood
- PGM
Personal genome machine
- JCVI
J. Craig Venter Institute
- PSK
Polyketide synthase
- NRPS
Non-ribosomal peptide synthase
- RAxML
Randomized axelerated maximum likelihood
- AspGD
Aspergillus genome database
- JGI
Joint Genome Institute
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GGM conceived of the study, created its design, performed investigation of MAT locus, drafted most the manuscript, and created the tables and figures. BMM assisted in the genome sequencing, performed annotation of the genomic sequence as well as various genomic analyses, and helped to draft the Methods section of the manuscript. SBB helped with the genome sequencing preparations and helped to draft the Background section of the manuscript. All authors read, edited and approved the final manuscript.
Contributor Information
Geromy G. Moore, Email: geromy.moore@ars.usda.gov
Brian M. Mack, Email: brian.mack@ars.usda.gov
Shannon B. Beltz, Email: shannon.beltz@ars.usda.gov
References
- 1.Horn BW, Dorner JW. Soil populations of Aspergillus species from section Flavi along a transect through peanut growing regions of the United States. Mycologia. 1998;90:767–776. doi: 10.2307/3761317. [DOI] [Google Scholar]
- 2.Doster MA, Cotty PJ, Michailides TJ. Description of a distinctive aflatoxin-producing strain of Aspergillus nomius that produces submerged sclerotia. Mycopathologia. 2009;168:193–201. doi: 10.1007/s11046-009-9214-8. [DOI] [PubMed] [Google Scholar]
- 3.Ehrlich KC, Kobbeman K, Montalbano BG, Cotty PJ. Aflatoxin-producing Aspergillus species from Thailand. Int J Food Microbiol. 2007;114:153–159. doi: 10.1016/j.ijfoodmicro.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Horn BW, Moore GG, Carbone I. Sexual reproduction in aflatoxin-producing Aspergillus nomius. Mycologia. 2011;103:174–183. doi: 10.3852/10-115. [DOI] [PubMed] [Google Scholar]
- 5.Hesseltine CW, Shotwell OL, Smith M, Ellis JJ, Vandegraft E, Shannon G. Production of various aflatoxins by strains of the Aspergillus flavus series. In: Herzberg M, editor. Proceedings of the first U.S.-Japan conference on toxic micro-organisms: 7–10 October 1968; Honolulu. Washington, DC: UJNR Joint Panels on Toxic Micro-organisms and US Department of Interior; 1970. pp. 202–210. [Google Scholar]
- 6.Kurtzman CP, Horn BW, Hesseltine CW. Aspergillus nomius, a new aflatoxin-producing species related to Aspergillus flavus and Aspergillus tamarii. A Van Leeuw J Microb. 1987;53:147–158. doi: 10.1007/BF00393843. [DOI] [PubMed] [Google Scholar]
- 7.Ito Y, Peterson SW, Goto T. Isolation and characterization of Aspergillus nomius from Japanese soil and silkworm excrement. Mycotoxins. 1998;46:9–15. doi: 10.2520/myco1975.1998.9. [DOI] [Google Scholar]
- 8.Rojas MG, Morales-Ramos JA, Klich MA, Wright M. Three fungal species isolated from Coptotermes formosanus (Isoptera: Rhinotermitidae) bodies, carton material, and infested wood. Fla Entomol. 2001;84:156–158. doi: 10.2307/3496679. [DOI] [Google Scholar]
- 9.Manikandan P, Varga J, Kocsubé S, Samson RA, Anita R, Revathi R, et al. Mycotic keratitis due to Aspergillus nomius. J Clin Microbiol. 2009;47:3382–3385. doi: 10.1128/JCM.01051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumeda Y, Asao T, Takahashi H, Ichinoe M. High prevalence of B and G aflatoxin-producing fungi in sugarcane field soil in Japan: Heteroduplex panel analysis identifies a new genotype within Aspergillus section Flavi and Aspergillus nomius. FEMS Microbiol Ecol. 2003;45:229–238. doi: 10.1016/S0168-6496(03)00154-5. [DOI] [PubMed] [Google Scholar]
- 11.Olsen M, Johnsson P, Möller T, Paladino R, Lindblad M. Aspergillus nomius, an important aflatoxin producer in Brazil nuts? World Mycotoxin J. 2008;1:123–126. doi: 10.3920/WMJ2008.1032. [DOI] [Google Scholar]
- 12.Barros GG, Torres AM, Rodriguez MI, Chulze SN. Genetic diversity within Aspergillus flavus strains isolated from peanut-cropped soils in Argentina. Soil Biol Biochem. 2006;38:145–152. doi: 10.1016/j.soilbio.2005.04.028. [DOI] [Google Scholar]
- 13.Klich MA. Identification of common Aspergillus species. Utrecht: Centraalbureau voor Schimmelcultures; 2002. [Google Scholar]
- 14.Pildain MB, Frisvad JC, Vaamonde G, Cabral D, Varga J, Samson RA. Two novel aflatoxin-producing Aspergillus species from Argentinean peanuts. Int J Syst Evol Microbiol. 2008;58:725–735. doi: 10.1099/ijs.0.65123-0. [DOI] [PubMed] [Google Scholar]
- 15.Varga J, Frisvad JC, Samson RA. Two new aflatoxin producing species, and an overview of Aspergillus section Flavi. Stud Mycol. 2011;69:57–80. doi: 10.3114/sim.2011.69.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soares C, Rodrigues P, Peterson SW, Lima N, Venâncio A. Three new species of Aspergillus section Flavi isolated from almonds and maize in Portugal. Mycologia. 2012;104:682–697. doi: 10.3852/11-088. [DOI] [PubMed] [Google Scholar]
- 17.Samson RA, Varga J, Witiak SM, Geiser DM. The species concept in Aspergillus: recommendations of an international panel. Stud Mycol. 2007;59:71–73. doi: 10.3114/sim.2007.59.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, et al. Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005;438:1157–1161. doi: 10.1038/nature04300. [DOI] [PubMed] [Google Scholar]
- 19.Payne GA, Nierman WC, Wortman JR, Pritchard BL, Brown D, Dean RA, et al. Whole genome comparison of Aspergillus flavus and A. oryzae. Med Mycol. 2006;44:9–11. doi: 10.1080/13693780600835716. [DOI] [PubMed] [Google Scholar]
- 20.Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005;438:1105–1115. doi: 10.1038/nature04341. [DOI] [PubMed] [Google Scholar]
- 21.Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol. 2007;25:221–231. doi: 10.1038/nbt1282. [DOI] [PubMed] [Google Scholar]
- 22.Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature. 2005;438:1151–1156. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- 23.Federova ND, Moktali V, Medema MH. Bioinformatics approaches and software for detection of secondary metabolite gene clusters. In: Keller NP, Turner G, editors. Fungal secondary metabolism: methods and protocols. New York: Humana Press; 2012. pp. 23–45. [DOI] [PubMed] [Google Scholar]
- 24.Andersen MR, Nielsen JB, Klitgaard A, Petersen LM, Zachariasen M, Hansen TJ, et al. Accurate prediction of secondary metabolite gene clusters in filamentous fungi. P Natl Acad Sci USA. 2012;110:E99–107. doi: 10.1073/pnas.1205532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Brink HJM, van Gorcum RFM, van den Hondel CAMJJ, Punt PJ. Cytochrome P450 enzyme systems in fungi. Fungal Gent Biol. 1998;23:1–17. doi: 10.1006/fgbi.1997.1021. [DOI] [PubMed] [Google Scholar]
- 26.Ramirez-Prado JH, Moore GG, Horn BW, Carbone I. Characterization and population analysis of the mating-type genes in Aspergillus flavus and Aspergillus parasiticus. Fungal Genet Biol. 2008;45:1292–1299. doi: 10.1016/j.fgb.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Metzenberg RL, Glass NL. Mating type and mating strategies in Neurospora. Bioessays. 1990;12:53–59. doi: 10.1002/bies.950120202. [DOI] [PubMed] [Google Scholar]
- 28.454 sequencing analysis software. [http://www.454.com/products/analysis-software/].
- 29.Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33:6494–6506. doi: 10.1093/nar/gki937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl 2):ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 32.J. Craig Venter Institute A. flavus database. [ftp://ftp.jcvi.org/pub/data/a_flavus/].
- 33.Uniref50 protein database. [ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref50/uniref50.fasta.gz].
- 34.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 35.Tarailo‐Graovac M, Nansheng C. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;25(4.10):4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 36.RefSeq protein database. [ftp://ftp.ncbi.nlm.nih.gov/refseq/release/fungi/].
- 37.Genome annotation generator. [https://github.com/genomeannotation/GAG].
- 38.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, et al. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47:736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39(Suppl 2):W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (Co-) orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 43.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 44.Bioinformatics and evolutionary genomics: venn diagram software. [http://bioinformatics.psb.ugent.be/webtools/Venn/].
- 45.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 46.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]