

Abstract
Patients with heart failure continue to suffer adverse health consequences despite advances in therapies over the last two decades. Identification of novel therapeutic targets that may attenuate disease progression is therefore needed. The inflammasome may play a central role in modulating chronic inflammation and in turn affecting heart failure progression. The inflammasome is a complex of intracellular interaction proteins that trigger maturation of pro-inflammatory cytokines interleukin-1beta and interleukin-18 to initiate the inflammatory response. This response is amplified through production of tumor necrosis factor α and activation of inducible nitric oxide synthase. The purpose of this review is to discuss recent evidence implicating this inflammatory pathway in the pathophysiology of heart failure.
Keywords: Heart failure, Inflammasome, Inflammation
The syndrome of heart failure (HF) results from various structural or functional impairments in cardiac function leading to an inability to maintain cardiac output at normal filling pressures.1,2 HF remains a major cause of morbidity and mortality in the United States and is the leading cause of hospitalization among individuals over age 65, leading to costs of care exceeding 31 billion dollars annually.3 Over the past two decades, advances in pharmacological and device therapies for HF have significantly improved prognosis for HF patients with low ejection fraction, however, the overall prognosis continues to be poor for these patients with mortality rates approaching 50% in 5 years.4 Therefore, attenuating HF disease progression remains an important goal. Identification of novel pathways and effectively intervening on potential therapeutic targets may slow HF disease progression. It is known that HF is associated with a low-grade chronic inflammation leading to adverse cardiac remodeling.5 In this review, we discuss advances and recent evidence regarding the inflammatory pathway in the pathophysiology of HF.
Importance of Inflammation in Heart Failure
Studies with ACE inhibitors, beta-blockers, and aldosterone antagonists all showed benefit in HF patients with low ejection fraction.6 However, the persistent high risk for mortality among these patients suggest that neurohormonal activation does not fully explain HF progression. Inflammatory cytokines, such as tumor necrosis factor alpha (TNFα), interleukin 1 (IL-1) and 6 (IL-6), and C-reactive protein (CRP) are all increased in HF and their levels are related to HF severity and prognosis.7 These cytokines are thought to modulate myocardial remodeling, myocyte hypertrophy and apoptosis, decreased contractility, increased fibrosis, and other adverse structural changes.8-10 These findings have led to the “cytokine hypothesis” of HF progression.7,10 Originally it was felt that inflammatory cytokines in HF represents an epiphenomenon, however, recent evidence is suggestive of its mechanistic role.11 Initial HF studies focused on individual cytokines, however, uncovering pathophysiological processes of myocardial remodeling requires further study of the inflammatory pathways and the underlying mechanisms of cytokine activation.
Danger-associated molecular patterns (DAMPs)
Sterile inflammation in HF is initiated by danger-associated molecular patterns (DAMPs), which are host-derived molecules indicative of cellular damage and has been shown to modulate irreversible myocardial changes, such as fibrosis, apoptosis and hypertrophy.12-14 Proposed mechanisms of DAMP formation in HF include mitochondrial dysfunction, cellular death, ischemia, cardiac load and oxidative stress.15-19 Mitochondrial dysfunction and necrotic or apoptotic cardiomyocyte death lead to the release of cellular components such as nuclear and mitochondrial nucleic acids, extracellular ATP, protein aggregates, and other debris.15,16 Transient ischemia and reperfusion injury, myocardial under perfusion, and other sources of oxidative stress lead to the production of reactive oxygen and nitrogen products, which are powerful DAMPs associated with ventricular remodeling.1,20 Increased ventricular filling pressures, cavity distension, congestion, shear stress, and other alterations in loading leads to myocardial injury. Byproducts of this injury are detected by myocytes and immune cells as DAMPs and lead to an accelerated sterile inflammation in HF.17,19 The inflammatory response amplifies the production of DAMPs, resulting in a positive-feedback loop accelerating HF pathophysiology.16 Increased cardiac pressure and poor pump function directly trigger activation of inflammatory cells, such as peripheral monocytes, which aggregate in the heart and are released into circulation.18,19 Activated inflammatory cells release pro-inflammatory cytokines, such as TNFα, which magnify the inflammatory process and contribute to fibrotic changes in the myocardium and progressive remodeling.5,14,19,21,22
NLRP3 Inflammasome
DAMP-activated inflammation occurs via the NLRP3 inflammasome, a complex of intracellular interaction proteins that recognize DAMPs and triggers maturation of pro-inflammatory cytokines to initiate and amplify the inflammatory response.23-25 The inflammasome is composed of a NOD (nucleotide binding oligomerization domain)-like receptor, ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), and pro-caspase-1 (Figure 1).26-28 The activated inflammasome cleaves pro-caspase-1 into the active enzyme caspase-1.29 Caspase-1 in turn activates IL-1 family proinflammatory cytokines IL-1β and IL-18, by cleavage of pro-IL-1β and pro-IL-18 into active forms.24,30-32 Thus, the inflammasome is a powerful mediator of the immune response via caspase-1 activation of IL-1β and IL-18. The NLRP3 inflammasome can also induce pyroptosis in a caspase-1-dependent manner.27 Loss of cardiomyocytes via pyroptosis reduces contractile reserve leading to HF progression.33 In addition, as cytosolic components are released with pyroptosis, extracellular ASC becomes a danger signal and functions to initiate further inflammasome formation. ASC in extracellular space continues to activate caspase-1, propagating the inflammatory cascade.33
Figure 1. The NLRP3 inflammasome.

NLRP3 inflammasome is comprised of three proteins: NLRP3, ASC and pro-caspase-1. NRLP3 has three domains: leucine-rich repeats (LRR), NACHT domain, and a PYD domain. The adaptor protein ASC pairs with NLRP3 via PYD domains and with pro-caspase-1 via CARD domains. DAMP (danger-associated molecular patterns) activation via LRR triggers transcription of NLRP3 and pro-IL-1β. The fully assembled NRLP3 inflammasome activates caspase-1, leading to the activation and release of IL-1β and IL-18.
Inflammasome Activation
Both exogenous (pathogens, toxins) and endogenous (DAMPs) molecules activate NLRP3 inflammasome.28,34,35 Inflammasome formation and activation requires two signals: a priming and an activating signal.36 First, danger signals (DAMPs) activate the transcription factor NF-κB, leading to the production of NLRP3 and pro-IL-1β (Figure 1).34 Other components of the NLRP3 inflammasome pathway (ASC, pro-caspase-1, and pro-IL-18) are readily available in steady state.29 While the definitive mechanism of NLRP3 inflammasome activation has yet to be uncovered, proposed mechanisms include cation movement (such as K+ efflux or Ca2+ influx), mitochondrial membrane dysfunction, frustrated phagocytosis, production of reactive oxygen species, and direct binding of oxidized mitochondrial DNA to NLRP3 itself.28,29,36,37 This two-step process likely represents a regulatory checkpoint in inflammasome activation. Assembly of the inflammasome occurs when the N-terminal pyrin domain (PYD) of NLRP3 interacts with the PYD of ASC in a homotypic fashion, leading to recruitment and activation of caspase-1 (Figure 1).14,22,23,36 Further work is needed to elucidate mechanisms of inflammasome activation in HF.
NLRP3
NLR family, pyrin domain-containing 3 (NLRP3, aka NALP3 or cryopyrin) is a pattern recognition receptor (PRR) implicated in the pathogenesis of inflammation in chronic disease.35 NLRP3 transcription is regulated by nuclear factor κB (NF-κB) in the presence of a danger signal, e.g. DAMPs. NLRP3 is made up of 3 functional domains: C-terminal leucine-rich repeats (LRR) with regulatory function, a NACHT domain that has ATPase activity, and PYD that serves as a death-fold domain.22,30,35 NLRs are believed to play a role in the cytosol analogous to that of the toll-like receptors (TLRs) in the plasma membrane. Like other NLR family receptors, NLRP3 functions to guard the intracellular environment to maintain homeostasis.27 NLRP3 differs from other nod-like receptors in that physiological expression levels are not sufficient for inflammasome activation. NLRP3 expression is up regulated via NF-κB upon the sensing of a danger signal. NLRP3 functions as both an intracellular PRR, surveying the cytosol for danger signals, and as a platform protein in inflammasome formation, initiating an inflammatory response.27 NLRP3 is expressed in a number of cells, e.g. leukocytes, myocytes, and cardiac fibroblasts.38 NLRP3 knockout mice have smaller areas of infarct in an experimental model of acute myocardial infarction;13 constitutively active NLRP3 leads to uncontrolled activation of the inflammasome.39 There is no evidence of transcriptional control of NLRP3 to date.
ASC
ASC is a vital component of the inflammasome and functions to recruit caspase-1 to the inflammasome complex.5,12,31,40 ASC is necessary for activation of pro-caspase-1 into caspase-1, which in turn is necessary for activation of IL-1 family cytokines, such as IL-1β and IL-18.5,12 ASC deficiency lessens inflammatory response to ischemia-reperfusion injury in the myocardium.14 ASC plays a vital role in the activation of the inflammasome and in the inflammatory response to danger signals.
Caspase-1
Caspase-1 is a cysteine protease that function as an effector protein in NLRP3 inflammasome. Caspase-1 is produced by the cell as a zymogen, pro-caspase-1, which undergoes autocatalysis for activation upon a homotypic interaction with the CARD domain on ASC.37,40 Activated caspase-1 is involved in the recruitment of innate immune cells to sites of inflammation, induces pyroptosis, and is the primary activator of IL-1β and IL-18.
Interleukin-1β and Interleukin-18
IL-1β is a pro-inflammatory cytokine and plays a role in cellular activities such as cell proliferation, differentiation and apoptosis 41,42. IL-1β induces calcium leakage from the sarcoplasmic reticulum in myocytes, impairing cardiac contractility 43,44. IL-1β stimulates nitric oxide production, as evidenced by a corresponding increase in circulating inducible nitric oxide synthase (iNOS), leading to cardiomyocyte apoptosis and tissue remodeling 44,45. IL-1β is increased in HF and is associated with poor exercise tolerance and with remodeling after ischemia-reperfusion injury 14,44. Modulation of IL-1β attenuates myocardial enlargement and ventricular dysfunction 46.
IL-1β is produced as a precursor protein in response to an inflammatory stimulus, while IL-18 is constitutively expressed as a biologically inactive precursor molecule lacking a signal peptide. 41,42,47 Both proform IL-1β and proform IL-18 require caspase-1 dependent proteolytic cleavage for activation.47,48 Increased IL-18 levels are correlated with functional class and mortality in HF.48 IL-18 is increased during acute HF and remains elevated after discharge. 1,49 In the non-failing myocardium, IL-18 is found in precursor form; in contrast, in the failing heart it is almost completely processed to active form. 48 IL-18 induces the production of TNFα. 49,50 While the inflammatory squelae of IL-18 in have been investigated; modifiable pathways that increase IL-18 production in HF are not well studied.
Inducible Nitric Oxide Synthase
Reactive nitrogen species (RNS) are molecules derived from a small uncharged molecule NO-, primarily produced by nitric oxide (NO) synthases, such as iNOS.17,51 While physiological levels of RNS are vital in the maintenance of vascular and cardiac cell function, excessive levels lead to toxicity and become highly reactive with other radicals.51 Excessive RNS levels lead to oxidative stress, an imbalance of free radical generation and detoxification.51 Oxidative stress is involved in the onset and progression of HF. Oxidative stress decreases cardiac contractility, myocardial Ca2+ regulation and mitochondrial function; the reactive species peroxynitrite has been shown to decrease myocardial contractility and disrupt the mitochondrial inner membrane in HF.51 Nitric oxide synthases are enzymes that catalyze the production of NO.51 iNOS produces NO, which is a key mediator in modulating microcirculatory changes and leukocyte-endothelial interactions 20,51 Over expression of iNOS leads to increased NO production and maladaptive ventricular remodeling and is implicated in the pathogenesis of HF.17,18 While NO may be cardioprotective in some forms, NO specifically produced by iNOS leads to myocardial injury.45 IL-1β is a powerful inducer of iNOS production.45 IL-18 has been shown to induce iNOS overexpression and the subsequent release of NO in the inflamed pancreas,50 however the relationship of IL-18 and iNOS production has not been studied in HF. The proposed NLRP3 inflammasome pathway links ASC methylation with decreased circulating IL-1β and IL-18 and subsequent decreased iNOS expression in persons with HF.
Tumor necrosis factor-alpha
TNFα is a pro-inflammatory cytokine that is not expressed in healthy myocardium. 52 Animal models have demonstrated that increased circulating TNFα stimulate myocyte hypertrophy and remodeling.18 TNFα has negative inotropic effects by decreasing intracellular Ca2+ release.14,22 In addition, TNFα directly induces cardiomyocyte hypertrophy and apoptosis.8,53,54 Increased circulating TNFα is correlated with worsening HF, poor prognosis and sudden death.18,53
NLRP3 Inflammasome in Cardiac Remodeling
The work examining the inflammasome in cardiac remodeling and repair has primarily focused on fibroblasts, which comprise up to two-thirds of cells in cardiac tissue.14 The NLRP3 inflammasome in the myocardial fibroblast is the initial sensor of DAMPs after myocardial injury, and thus is primarily responsible for the initiation of the inflammatory response in injured cardiac tissue. Fibroblasts have been shown to activate the inflammasome via reactive oxygen species and K+ efflux after myocardial ischemic injury.14,55
Fibroblasts are able to withstand oxidative stress and thus are able to respond to hypoxia and initiate a hypoxic immune response. In the face of hypoxia, fibroblasts develop an inflammatory and fibrogeneic phenotype, leading to increased cytokines, inflammatory cell infiltration, myofibroblast transdifferentiation, and increased collagen production.14,55 Ischemic insult leads to ROS and K+ efflux, which activates the inflammasome in the fibroblast. The activated inflammasome produces IL-1β, which activates the initial immune response. The inflammatory fibroblast releases chemokines that recruit macrophages and neutrophils to the site of insult. These recruited leukocytes also contain inflammasomes that release IL-1β and IL-18, further propagating the inflammatory response after ischemic insult or injury. Thus, the cardiac fibroblasts play a central role in the initiation and enhancement of myocardial injury after ischemia.
NLRP3 plays a role in cardiac fibroblast differentiation through the NACHT domain, independent of inflammasome formation. In cardiac fibroblasts, NLRP3 was found to localize to mitochondria as a regulator of mitochondrial ROS (mROS) production and augment R-Smad signaling, ultimately leading to profibrotic gene expression.56
Using a calcineurin transgene mouse model of inflammatory and hypertrophic cardiomyopathy, Bracey et al.12 demonstrated that the inflammasome, via IL-1β activation, plays a role in pyroptosis during the development of HF and has direct effects on Ca2+ homeostasis, myocardial contractility, and excitation-contraction coupling. NLRP3 can also play a role in myocardial death in an inflammasome-independent manner; in cardiomyocytes, the NLRP3 inflammasome can lead to loss of myocardium by pyroptosis, independent of IL-1β.13
In a sample of patients with acute myocarditis, more inflammasome-containing leukocytes were found in myocardial tissue of persons with reduced EF (<40%) and those with more severe HF, as categorized by NYHA Class III and IV, than others.57 While this is merely associative, and does not demonstrate causality, the continued presence of increased inflammasome formation in injured and stressed myocardium suggests a mechanistic role in the pathogenesis and worsening of HF. Although the NRLP3 inflammasome is implicated in the development of HF, the mechanisms of adverse myocardial remodeling are not completely understood. Further work to elucidate the mechanisms of myocardial remodeling and repair in the pathogenesis of and worsening of HF are warranted.
Mechanisms of Myocardial Epigenetic Regulation
The primary epigenetic mechanisms linked to the pathogenesis of HF are chromatin modifications, histone modifications, DNA methylation, and microRNAs (miRNA). These epigenetic mechanisms control key processes such as cardiomyocyte hypertrophy, fibrosis and cardiac failure. Several studies have focused on genome-wide mapping of global epigenetic differences between HF and healthy controls,58-60 which have led to the discovery of several mechanisms and pathways of myocardial epigenetic regulation in HF. Epigenetic changes in promoter regions of p53 response elements, such as complete demethylation of p21 or complete methylation of cyclin D1, are related to cell cycle arrest and have been implicated in the development of diabetic cardiomyopathy.61 Altered Dicer expression modulates cardiac function and cardiac electrophysiology through disturbed ion channel function, presumably through altered miRNA expression.61 Several genes related to angiogenesis, including PCAM1, are differentially methylated in HF, suggesting epigenetic regulation of the angiogenic process during HF pathogenesis.62 Changes in histone deacetylases, such as loss of HDAC1 and HDAC2 in cardiac tissue, lead to in arrhythmias and HF.63
Pathogenic cardiac remodeling leading to HF is characterized by a re-activation of fetal programming genes and a repression of adult genes.64 At the onset of HF, reprogramming of gene expression leads to changes in gene expression, including downregulation of α-MHC (major histocompatibility complex) and SERCA (sarcoplasmic reticulum Ca2+ ATPase) genes and reactivation of the fetal cardiac genes, ANF (atrial natriuretic factor) and BNP (B-type natriuretic peptide). This reprogramming causes structural and electrophysiological changes that lead to HF.65 In a model of pressure overload-induced HF, histone acetylation and chromatin modifications led to transcriptional reprogramming and fetal gene expression. These modifications resulted in the de novo synthesis of contractile and structural proteins, and ultimately cardiac hypertrophy.64
While epigenetic regulation of the myocardial changes implicated in the pathogenesis of HF have yet to be linked to inflammasome formation, increased circulating IL-18 had been shown to lead to epigenetic changes in the chromatin of ANF and MyHC (myosin heavy chain) genes. These chromatin changes are associated with cardiac hypertrophy via upregulation of β-MyHC and down regulation of α-MyHC, indicative of fetal reprogramming.66,67 Expression of a vital regulatory component of the NLRP3 inflammasome, ASC, is controlled through epigenetic modification via DNA methylation. ASC methylation is inversely correlated with ASC protein expression and is silenced by overexpression of DNA methyltransferase.36,37 Hypermethylation leads to an inactive state in which no ASC protein is expressed, while complete demethylation induces apoptosis via p53 and TRAK activation.36 Thus, inflammasome formation and activation may be reduced via increased ASC methylation. ASC expression increases with age,37 while moderate intensity aerobic exercise increases ASC methylation.24 However these exercise-induced epigenetic changes have not been correlated with circulating cytokines, such as IL-1β or IL-18, nor has ASC methylation been assessed in persons with HF.
Attempts to Modulate Inflammasome
Strategies to alter inflammasome function in mice, such as genetic knockouts of inflammasome proteins and interleukin-1β, binding circulating proteins (e.g., IL-1 Trap), IL-1 receptor antagonist (e.g., anakinra), and siRNA silencing, have demonstrated reduced ventricular remodeling in myocardial infarction-induced or ischemic models of HF. Preliminary studies using anakinra post ST-segment elevation myocardial infarction in humans reduced the incidence of subsequent HF,24,25 demonstrating that a reduction in IL-1β activity decreases ventricular remodeling. A two-week treatment with anakinra improved peak VO2 in persons with HFpEF in the absence of an exercise intervention.44 A recent trial of an intermediate substrate in the synthesis of glyburide (16673-34-0) demonstrated inhibition of NLRP3 in a mouse model of acute myocardial infarction.68 IL-18 binding protein (IL-18BP) has been shown to prevent systolic dysfunction in mice treated with plasma from decompensated HF patients.69 Use of IL-18BP in rheumatoid arthritis (RA) treatment demonstrated only mild to moderate adverse events.70 Anti-IL-18 antibodies are currently under investigation for treatment of type II diabetes mellitus, and may prove to be an effective therapy for HF. Because IL-1 family cytokines are important in innate immune processes, such as fever, treatment with IL-18 antibodies may be an important component of reducing deleterious inflammatory processes initiated by inflammasome activation with less immune suppression.
The main translational focus of immune modulation in HF has been on cytokine proteins, however, clinical trials targeting pro-inflammatory cytokines, such as anti-TNFα therapy, have yet to demonstrate improvements in HF outcomes and, in some cases, have proven to be harmful.16,45 Efforts at immune modulation using disease-modifying agents of rheumatoid diseases (DMARDs), such as anakinra and methotrexate, have demonstrated improvements for chronic inflammatory diseases, such as rheumatoid arthritis, gout, and recently post-infarction HF.16 Potent anti-inflammatory therapy, anti-TNFα in particular, has a risk for infection.16 Treatment of gram-positive sepsis with anti-TNFα therapy had a higher mortality rate than those not receiving treatment.71 In contrast, treatment of IL-1 receptor antagonist for sepsis demonstrated treatment safety and dose-dependent efficacy with increased survival and decreased IL-6 and cytokine expression.72 While trials in RA with anakinra demonstrated increased risk of upper respiratory infections, it should be noted that participants also took other immune suppressing drugs, including NSAIDs, corticosteroids, and other DMARDs. In fact, those who took anakinra without corticosteroids had a lower infection rate than participants not taking corticosteroids.
A large anti-cytokine study, The Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS, www.thecantos.org), is currently examining if IL-1β inhibition via canakinumab, an IL-1β neutralizing monoclonal antibody, leads to decreased rates of recurrent MI, stroke, and cardiovascular death in persons with coronary artery disease and increased CRP. This study will enroll 17,500 participants who are post-AMI and have persistently elevated CRP.73 Preliminary analysis of 556 participants examined the relationship between inflammation and atherosclerosis, and found that treatment with canakinumab decreased CRP over placebo, with no group differences in clinical adverse events.74 This study may reveal a new approach to preventing ischemic HF through dampening the effects of inflammasome activation.
Anti-cytokine therapy may decrease cytokine levels below the physiologic levels needed for myocardial repair. Alternative methods, such as epigenetic regulation of inflammatory response and targeting upstream components of the signaling cascade have been proposed16 however no trials in humans have been reported to date. The NLRP3 inflammasome has been shown to contribute to the development of HF after myocardial infarction.5 ASC is a key component of the NLRP3 inflammasome and is necessary for caspase-1 mediated activation of IL-1β and IL-18. Inflammation in HF can be regulated by ASC methylation (Figure 2). This pathway is important to our understanding of the pathological processes behind myocardial remodeling in HF as our current understanding does not distinguish the individual contributions of the cytokines involved.
Figure 2.
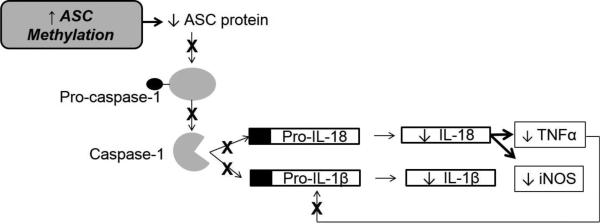
Proposed Pathway of Epigenetic Regulation of the Inflammasome in Heart Failure
Future Directions
Historically anti-inflammatory therapies, e.g. NSAIDs and TNFα inhibitors infliximab and etanercept were not beneficial in HF.8,44 While recent trials with other DMARDs, e.g. anakinra, have demonstrated prevention of remodeling post infarction, the over benefit of anti-inflammatory therapy in HF is unknown.24,25 Anti-inflammatory therapy can lead to compromised host defense or amplification of inflammatory processes due to the many redundancies.16 Further exploration of drug therapy that targets the inflammasome is needed. One new drug, 16673-34-0 was found to inhibit inflammasome formation post infarction in a mouse model.68
It is possible that epigenetic regulation of ASC activation in HF could dampen adverse inflammatory processes without disrupting cellular and tissue homeostasis. The involvement of the inflammasome components in various pathways makes finding specific inflammasome targets a challenge as it may lead to unintended consequences. For example, by suppressing NALP3 function, it may be possible to also remove an important intracellular surveyor of danger signals. Some degree of inflammation is necessary for proper healing after insult or injury. The study by Nakajima et al.31 demonstrated an increase in ASC methylation in older healthy adults after a moderate intensity aerobic exercise program. This study found that ASC methylation decreases with age and that these changes can be modified with moderate intensity aerobic exercise. While this study did not examine inflammatory markers related to levels of ASC methylation, regular aerobic exercise may prove to be an effective non-pharmacological modulator of inflammasome activation in HF.
A two-week treatment with anakinra, an IL-1 receptor antagonist, improved peak exercise oxygen consumption in HF in the absence of an exercise intervention.44 Thus, aerobic capacity in HF may be related to increase circulating inflammatory cytokines and aerobic exercise reduces inflammatory cytokines. But the effect of aerobic exercise on ASC methylation and IL-18 in HF has not been previously examined. Aerobic exercise-induced ASC methylation may be a non-pharmacologic method of inflammasome modulation leading to decreased inflammation and improved outcomes in HF.
Conclusion
Attenuating HF disease progression by way of dampening inflammatory process is an ongoing area of intervention research with much promise. Considering the poor prognosis for HF patients, identification of novel inflammatory pathways and effectively intervening to slow HF progression is an important goal for HF research. Anti-inflammatory drug therapy can lead to compromised host defense or further amplification of inflammatory processes due to the many redundancies and compensatory responses built into this complicated defense system. Targeting specific inflammatory pathways, such as NLRP3 activation, may provide a more precise approach to reducing deleterious inflammation in HF while leaving some innate host defense intact. Further research on targeting the NLRP3 inflammasome in HF patients is warranted.
Acknowledgments
Funding Sources:
Effort was funded for B. Butts through the National Institutes of Health National Institute of Nursing Research grant number T2NR012715 (PI-S. Dunbar) and Heart Failure Society of America Nursing Research Grant (PI-B. Butts).
References
- 1.Eslick GD, Thampan BV, Nalos M, McLean AS, Sluyter R. Circulating interleukin-18 concentrations and a loss-of-function P2×7 polymorphism in heart failure. 2008 doi: 10.1016/j.ijcard.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 2.Van Tassell B, Varma A, Salloum FN, et al. Interleukin-1 trap attenuates cardiac remodeling after experimental acute myocardial infarction in mice. J. Cardiovasc. Pharmacol. 2010;22:117–122. doi: 10.1097/FJC.0b013e3181c87e53. [DOI] [PubMed] [Google Scholar]
- 3.Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933–944. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 4.Yancy CW, Jessup M, Bozkurt B, et al. ACCF/AHA guideline for the management of heart failure. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 20132013 doi: 10.1016/j.jacc.2013.05.019. epub ahead of print. doi:doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 5.Abbate A. The heart on fire: Inflammasome and cardiomyopathy. Exp. Physiol. 2013;98(2):385. doi: 10.1113/expphysiol.2012.069021. [DOI] [PubMed] [Google Scholar]
- 6.Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J. Am. Coll. Cardiol. 1992;20(1):248–254. doi: 10.1016/0735-1097(92)90167-l. [DOI] [PubMed] [Google Scholar]
- 7.Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J. Card. Fail. 1996;2(3):243–249. doi: 10.1016/s1071-9164(96)80047-9. [DOI] [PubMed] [Google Scholar]
- 8.Chung ES, Packer M, Lo KH, Adedigbo A, Fasanmade, Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of Infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure. Results of the Anti-TNF Therapy Against Cognestive Heart Failure (ATTACH) Trial. Circulation. 2003;107:3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 9.Hofmann U, Frantz S. How can we cure a heart “in flame”? A translational view on inflammation in heart failure. Basic Res. Cardiol. 2013;108:356–375. doi: 10.1007/s00395-013-0356-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Menyar AA. Cytokines and myocardial dysfunction: state of the art. J. Card. Fail. 2008;14(1):61–74. doi: 10.1016/j.cardfail.2007.09.006. doi:10.1016/j.cardfail.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 11.von Haehling S, Schefold JC, Lainscak M, Doehner W, Anker SD. Inflammatory biomarkers in heart failure revisited: Much more than innocent bystanders. Heart Failure Clinician. 2009;5:549–560. doi: 10.1016/j.hfc.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Bracey NA, Beck PL, Muruve DA, et al. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp. Physiol. 2013;98(2):462–472. doi: 10.1113/expphysiol.2012.068338. [DOI] [PubMed] [Google Scholar]
- 13.Mezzaroma E, Toldoa S, Farkasb D, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. PNAS. 2011;108(49):19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 15.Nakayama H, Otsu K. Translation of hemodynamic stress to sterile inflammation in the heart. Trends in endocrinology and metabolism: TEM. 2013 Nov;24(11):546–553. doi: 10.1016/j.tem.2013.06.004. doi:10.1016/j.tem.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;336:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otani H. The role of nitric oxide in myocardial repair and remodeling. Antioxidant & Redox Signaling. 2009;11(8):1913–1928. doi: 10.1089/ars.2009.2453. [DOI] [PubMed] [Google Scholar]
- 18.Elahi M, Asopa S, Matata B. NO-cGMP and TNF-α counter regulatory system in blood: Understanding the mechanisms leading to myocardial dysfunction and failure. Biochim. Biophys. Acta. 2010;1772:5–14. doi: 10.1016/j.bbadis.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Vaduganathan M, Greene SJ, Butler J, et al. The immunological axis in heart failure: importance of the leukocyte differential. Heart Fail Rev. 2013 Nov;18(6):835–845. doi: 10.1007/s10741-012-9352-9. doi:10.1007/s10741-012-9352-9. [DOI] [PubMed] [Google Scholar]
- 20.Campos JC, Gomes KMS, Ferreira JCB. Impact of exercise training on redox signaling in cardiovascular diseases. Food Chem. Toxicol. 2013;62:107–119. doi: 10.1016/j.fct.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 21.Glezeva N, Baugh JA. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Failure Reviews. 2013 doi: 10.1007/s10741-013-9405-8. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 22.Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: A sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 23.Kanneganti TD, M L, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Abbate A, Kontos MC, Grizzard JD, et al. Interleukin-1 blockade with Anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] pilot study). Am. J. Cardiol. 2010;105:1371–1377. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 25.Abbate A, Van Tassell BW, Biondi-Zoccai G, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am. J. Cardiol. 2013;111(10):1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroder K, Tschopp J. The inflammasome. Cell Adhes. Commun. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 27.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. doi:10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 28.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nat. Biotechnol. 2012;481:278–286. doi: 10.1038/nature10759. doi:10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 29.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014 doi: 10.1111/nyas.12458. doi:10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi M. NLRP3 inflammasome as a novel player in myocardial infarction. International Heart Journal. 2014;55:101–105. doi: 10.1536/ihj.13-388. [DOI] [PubMed] [Google Scholar]
- 31.Nakajima K, Takeoka M, Mori M, et al. Exercise effects on methylation of ASC gene. Int. J. Sports Med. 2010;31:671–375. doi: 10.1055/s-0029-1246140. [DOI] [PubMed] [Google Scholar]
- 32.Okamura H, Tsutsui H, Kashiwamura S-I, Yoshimoto T, Nakanishi K. Interleukin-18: A novel cytokinethat augments both innate and acquired immunity. Adv. Immunol. 1998;70:281–312. doi: 10.1016/s0065-2776(08)60389-2. [DOI] [PubMed] [Google Scholar]
- 33.Fedak PWM, Verma S, Weisel RD, Li R-K. Cardiac remodeling and failure. From molecules to Man (part 1). Cardiovascular Pathology. 2005;14:1–11. doi: 10.1016/j.carpath.2004.12.002. doi:10.1016/j.carpath.2001.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-κB activating pattern recoginition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. doi:10.4049/jimmunol.901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathwayson ROS production? Nature Reviews Immunology. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 36.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. doi:10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 37.Martinon F, Mayor A, Tschopp J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. doi:10.1146/annurev.immunol.021905.132715. [DOI] [PubMed] [Google Scholar]
- 38.Bracey NA, Gershkovich B, Chun J, et al. Mitochondrial NLRP3 induces reactive oxygen species to promote Smad signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014 doi: 10.1074/jbc.M114.550624. doi:0.1074/jbc.M114.550624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.PvWilson S, Cassel SL. Inflammasome-mediated autoinflammatory disorders. Postgrad. Med. J. 2010;122(5):125–133. doi: 10.3810/pgm.2010.09.2209. doi:10.3810/pgm.2010.09.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taniguchi S, Sagara J. Regulatory molecules involved in inflammasome formation with special reference to a key mediator protein, ASC. Seminars in Immunopathology. 2007;29:231–238. doi: 10.1007/s00281-007-0082-3. [DOI] [PubMed] [Google Scholar]
- 41.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 42.Dinarello CA. A clinical perspective of IL-1b as the gatekeeper of inflammation. Eur. J. Immunol. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 43.Van Tassell B, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1b induces a reversible cardiomyopathy in the mouse. Inflamm. Res. 2013;62:637–640. doi: 10.1007/s00011-013-0625-0. [DOI] [PubMed] [Google Scholar]
- 44.Van Tassell B, Arena RA, Toldo S, et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS ONE. 2012;7(3):e33438. doi: 10.1371/journal.pone.0033438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase. 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1β. Proc. Natl. Acad. Sci. U. S. A. 2001;98(5):2871–2876. doi: 10.1073/pnas.041611398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abbate A, Van Tassell B, Seropian IM, et al. Interleukin-1b modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. European Journal of Heart Failure. 2010;12:319–322. doi: 10.1093/eurjhf/hfq017. [DOI] [PubMed] [Google Scholar]
- 47.Dinarello CA, Simon A, van der Meer J. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature Reviews Drug Discovery. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mallat Z, Heymes C, Corbaz A, et al. Evidence for altered interleukin (IL)-18 pathway in human heart failure. The FASEB Journal. 2004 doi: 10.1096/fj.04-2426fje. [DOI] [PubMed] [Google Scholar]
- 49.Yamaoka-Tojo M, Tojo T, Inomata T, Machida Y, Osada K, Izumi T. Circulating levels of interleukin. 18 reflect etiologies of heart failure: Th1/TH2 cytokine imbalance exaggerates the pathophysiology of advanced heart failure. J. Card. Fail. 2002;8(1):21–27. doi: 10.1054/jcaf.2002.31628. [DOI] [PubMed] [Google Scholar]
- 50.Ueno N, Kashiwamura S-i, Ueda H, et al. Role of interleukin. 18 in nitric oxide production and pancreatic damage during acute pancreatitis. Shock. 2005;24(6):564–570. doi: 10.1097/01.shk.0000184285.57375.bc. [DOI] [PubMed] [Google Scholar]
- 51.Umar S, van der Laarse A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic and failing heart. Mol. Cell. Biochem. 2010;333:191–201. doi: 10.1007/s11010-009-0219-x. [DOI] [PubMed] [Google Scholar]
- 52.Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N. Proinflammatory cytokines in heart failure: double-edged swords. Heart Failure Reviews. 2010;15:543–562. doi: 10.1007/s10741-010-9168-4. [DOI] [PubMed] [Google Scholar]
- 53.Bozkurt B, Torre-Amione G, Warren MS, et al. Results of targeted antitumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation. 2001;103:1044–1047. doi: 10.1161/01.cir.103.8.1044. [DOI] [PubMed] [Google Scholar]
- 54.Burkard T, Pfister O, Rickli H, et al. Prognostic impact of systemic inflammatory diseases in elderly patients with congestive heart failure. QJM. 2013 doi: 10.1093/qjmed/hct205. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 55.Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim. Biophys. Acta. 2013 Apr;1833(4):945–953. doi: 10.1016/j.bbamcr.2012.08.023. doi:10.1016/j.bbamcr.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bracey NA, Gershkovich B, Chun J, et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014 Jul 11;289(28):19571–19584. doi: 10.1074/jbc.M114.550624. doi:10.1074/jbc.M114.550624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toldo S, Kannan H, Bussani R, et al. Formation of the inflammasome in acute myocarditis. Int. J. Cardiol. 2014 Feb 15;171(3):e119–121. doi: 10.1016/j.ijcard.2013.12.137. doi:10.1016/j.ijcard.2013.12.137. [DOI] [PubMed] [Google Scholar]
- 58.Haas J, Frese KS, Park YJ, et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO molecular medicine. 2013 Mar;5(3):413–429. doi: 10.1002/emmm.201201553. doi:10.1002/emmm.201201553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaneda R, Takada S, Yamashita Y, et al. Genome-wide histone methylation profile for heart failure. Genes Cells. 2009 Jan;14(1):69–77. doi: 10.1111/j.1365-2443.2008.01252.x. doi:10.1111/j.1365-2443.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- 60.Koczor CA, Lee EK, Torres RA, et al. Detection of differentially methylated gene promoters in failing and nonfailing human left ventricle myocardium using computation analysis. Physiological genomics. 2013 Jul 15;45(14):597–605. doi: 10.1152/physiolgenomics.00013.2013. doi:10.1152/physiolgenomics.00013.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asrih M, Steffens S. Emerging role of epigenetics and miRNA in diabetic cardiomyopathy. Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology. 2013 Mar-Apr;22(2):117–125. doi: 10.1016/j.carpath.2012.07.004. doi:10.1016/j.carpath.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 62.Movassagh M, Choy MK, Goddard M, Bennett MR, Down TA, Foo RS. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS One. 2010;5(1):e8564. doi: 10.1371/journal.pone.0008564. doi:10.1371/journal.pone.0008564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Montgomery RL, Davis CA, Potthoff MJ, et al. Histone deacetylases. 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007 Jul 15;21(14):1790–1802. doi: 10.1101/gad.1563807. doi:10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Angrisano T, Schiattarella GG, Keller S, et al. Epigenetic switch at atp2a2 and myh7 gene promoters in pressure overload-induced heart failure. PLoS One. 2014;9(9):e106024. doi: 10.1371/journal.pone.0106024. doi:10.1371/journal.pone.0106024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duygu B, Poels EM, da Costa Martins PA. Genetics and epigenetics of arrhythmia and heart failure. Frontiers in genetics. 2013;4:219. doi: 10.3389/fgene.2013.00219. doi:10.3389/fgene.2013.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pandya K, Smithies O. beta-MyHC and cardiac hypertrophy: size does matter. Circ. Res. 2011 Sep 2;109(6):609–610. doi: 10.1161/CIRCRESAHA.111.252619. doi:10.1161/circresaha.111.252619. [DOI] [PubMed] [Google Scholar]
- 67.Majumdar G, Johnson IM, Kale S, Raghow R. Epigenetic regulation of cardiac muscle-specific genes in H9c2 cells by Interleukin-18 and histone deacetylase inhibitor m carboxycinnamic acid bis-hydroxamide. Mol. Cell. Biochem. 2008 May;312(1-2):47–60. doi: 10.1007/s11010-008-9720-x. doi:10.1007/s11010-008-9720-x. [DOI] [PubMed] [Google Scholar]
- 68.Marchetti C, Chojnacki J, Toldo S, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014;63:316–322. doi: 10.1097/FJC.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toldo S, Mezzaroma E, O'Brien L, et al. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. American journal of physiology. Heart and circulatory physiology. 2014 Apr 1;306(7):H1025–1031. doi: 10.1152/ajpheart.00795.2013. doi:10.1152/ajpheart.00795.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O'Brien LC, Mezzaroma E, Van Tassell BW, et al. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol. Med. 2014;20(1):221–229. doi: 10.2119/molmed.2014.00034. doi:10.2119/molmed.2014.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fisher CJ, Jr., Agosti JM, Opal SM, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N. Engl. J. Med. 1996 Jun 27;334(26):1697–1702. doi: 10.1056/NEJM199606273342603. doi:10.1056/nejm199606273342603. [DOI] [PubMed] [Google Scholar]
- 72.Fisher CJ, Jr., Slotman GJ, Opal SM, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit. Care Med. 1994 Jan;22(1):12–21. doi: 10.1097/00003246-199401000-00008. [DOI] [PubMed] [Google Scholar]
- 73.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am. Heart J. 2011 Oct;162(4):597–605. doi: 10.1016/j.ahj.2011.06.012. doi:10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 74.Ridker PM, Howard CP, Walter V, et al. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 2012 Dec 4;126(23):2739–2748. doi: 10.1161/CIRCULATIONAHA.112.122556. doi:10.1161/circulationaha.112.122556. [DOI] [PubMed] [Google Scholar]