

Abstract
A variety of heritable and acquired disorders is associated with excessive signaling by mutant or overstimulated GPCRs. Since any conceivable treatment of diseases caused by gain-of-function mutations requires gene transfer, one possible approach is functional compensation. Several structurally distinct forms of enhanced arrestins that bind phosphorylated and even non-phosphorylated active GPCRs with much higher affinity than parental wild-type proteins have the ability to dampen the signaling by hyperactive GPCR, pushing the balance closer to normal. In vivo this approach was so far tested only in rod photoreceptors deficient in rhodopsin phosphorylation, where enhanced arrestin improved the morphology and light sensitivity of rods, prolonged their survival, and accelerated photoresponse recovery. Considering that rods harbor the fastest, as well as the most demanding and sensitive GPCR-driven signaling cascade, even partial success of functional compensation of defect in rhodopsin phosphorylation by enhanced arrestin demonstrates the feasibility of this strategy and its therapeutic potential.
Keywords: Congenital disorders, Gain-of-function mutants, GPCRs, Arrestin activation, Enhanced arrestins, Functional compensation, Gene therapy, Protein-based therapeutics
1 Disorders Associated with Defects in GPCR Phosphorylation and Excessive Receptor Activity
Congenital disorders fall into two broad categories. Many are associated with loss-of-function mutations in particular genes. As a rule, these disorders are recessive, because normal wild-type (WT) protein encoded by the second allele can do the job. Rare cases of haplo-insufficiency are the only exception, where we need both functional alleles to produce necessary amounts of the protein. Thus, in most cases the disease develops only when both alleles carry loss-of-function mutations (i.e., the patient is compound heterozygous). Conceptually gene therapy of these disorders is quite straightforward: a gene encoding fully functional protein needs to be delivered. Even though technically this is not easy, recent success of three independent clinical trials where gene encoding functional RPE65 was delivered to patients with Leber's congenital amaurosis carrying loss-of-function mutations in this protein (Cideciyan et al. 2008; Hauswirth et al. 2008; Maguire et al. 2008; Bainbridge et al. 2008) demonstrate the feasibility of this type of gene therapy.
Diseases caused by gain-of-function mutations present much greater challenge. They are dominant, as the effect of one allele encoding hyperfunctional protein cannot be alleviated by the second perfectly normal allele. Most importantly, there are no good strategies to address this type of disorders. One possible approach is to deliver a ribozyme specifically designed to destroy mutant mRNA without touching the normal one. It must be very effective against the mutant form, as even very low expression of overactive protein is harmful (Chen et al. 1995). However, in case of many missense or frame-shift mutations, mutant and normal mRNAs differ only by a single nucleotide. This makes designing a ribozyme, which is very effective against the mutant form yet highly selective, so that it does not destroy virtually identical normal mRNA, next to impossible. The only alternative approach proposed so far is compensational gene therapy. The strategy here is to design a mutant version of a protein interacting with the one affected by disease-causing mutation, with functional characteristics changed in such a way that it will compensate for the excessive activity of inherited mutant. For example, if mutant G protein-coupled receptor (GPCR) signals too much, redesigned arrestin that quenches this signaling more effectively than WT form would normalize the signaling, shifting the balance in the cell closer to the norm.
Several human disorders are associated with excessive activity of GPCRs (Schöneberg et al. 2004). In some cases these are genetic, when one allele encodes a constitutively active receptor, or a form that cannot be shut off by the normal two-step mechanism employed by most GPCRs: phosphorylation of active receptor by G protein-coupled receptor kinases (GRKs), followed by arrestin binding to active phosphoreceptor. Several cases of retinitis pigmentosa (a form of retinal degeneration leading to complete blindness) are caused by mutations in rhodopsin that eliminate the sites phosphorylated in WT rhodopsin by GRK1 (rhodopsin kinase) (Apfelstedt-Sylla et al. 1993; Kim et al. 1993; Restagno et al. 1993). Upon activation by light these mutants effectively couple to visual G protein transducin, but their signaling cannot be quenched by GRK- and arrestin-mediated mechanism common in GPCR superfamily (Gurevich et al. 2011, 2012).
In other cases mutant receptors have GRK phosphorylation sites, but demonstrate higher than normal constitutive (ligand-independent) activity. Constitutively active mutant of PTH-PTHrP receptor causes Jansen-type metaphyseal chondrodysplasia (Schipani et al. 1995). Constitutively active mutants of TSH receptor cause toxic thyroid adenoma, multinodular toxic goiter, and autosomal dominant non-autoimmune hyperthyroidism (Paschke 1996; Khoo et al. 1999; Claus et al. 2005). Moreover, certain forms of cancer are caused by activating mutations in Gq-coupled GPCRs: ectopic expression of serotonin 1c receptor was shown to trigger malignant transformation (Julius et al. 1989), and Gq-coupled muscarinic receptors were found to act as agonist-dependent oncogenes (Gutkind et al. 1991). Interestingly, Gq-coupled angiotensin receptor was first cloned as mas oncogene before the true identity of this protein was discovered (Jackson et al. 1988).
The signaling by most GPCRs is turned off by a conserved two-step mechanism: first, active receptor is phosphorylated by specific GPCR kinases (GRKs) (Gurevich et al. 2012), whereupon arrestin specifically binds active phosphoreceptor (Gurevich and Gurevich 2004). Bound arrestin covers the cytoplasmic tip of the receptor, thereby blocking its coupling to G proteins by steric exclusion (Wilden 1995; Krupnick et al. 1997). The mutation in a GPCR can eliminate GRK phosphorylation sites (Apfelstedt-Sylla et al. 1993; Kim et al. 1993; Restagno et al. 1993; Chen et al. 1995), so that mutant receptor is normally activated by an appropriate stimulus, but cannot be turned off by GRKs and arrestins (Chen et al. 1995). In many other cases the receptor is perfectly normal, and its excessive activity is the result of genetic or acquired signaling defects upstream, e.g., abnormally high levels of its activating endogenous agonist.
Regardless whether the original error is genetic or acquired, in all these cases the net result is essentially the same: excessive receptor signaling that leads to imbalances that underlie the disease. Thus, an arrestin with enhanced ability to quench the signaling by overactive GPCR has a good chance to compensate and bring the signaling balance closer to normal.
2 The Mechanism of Arrestin Activation by Receptor-Attached Phosphates
Mammals express four arrestin subtypes (Hanson et al. 2006b). Two are specialized visual: arrestin-11 is expressed at very high levels in rod (Strissel et al. 2006; Hanson et al. 2007a; Song et al. 2011) and cone (Nikonov et al. 2008) photoreceptors, whereas arrestin-4 is cone specific (Craft et al. 1994; Nikonov et al. 2008). The two nonvisual subtypes, arrestin-2 and arrestin-3, are ubiquitously expressed and regulate signaling by hundreds of different GPCRs (Gurevich and Gurevich 2006b). Structurally, all four vertebrate arrestins are very similar (Hirsch et al. 1999; Han et al. 2001; Sutton et al. 2005; Zhan et al. 2011): they are elongated (long axis ~75A) two-domain molecules with relatively few contacts between domains, one of which is the interaction of the C-tail coming back from the C-domain and interacting with two elements in the N-domain, β-strand I and α-helix I (Fig. 1). Numerous studies showed that the residues that directly interact with receptors are localized on the concave sides of both domains (Gurevich and Benovic 1993; Ohguro et al. 1994; Gurevich et al. 1995; Pulvermuller et al. 2000a; Dinculescu et al. 2002; Vishnivetskiy et al. 2004; Hanson et al. 2006a; Hanson and Gurevich 2006; Vishnivetskiy et al. 2011; Gimenez et al. 2012c) (Fig. 2). As could be expected in a protein that preferentially binds phosphorylated GPCRs, arrestins have numerous positively charged phosphate-binding residues, all but one of which are conserved in the family: two lysines in β-strand I (Lys-14,15; Lys-10,11, Lys-11,12; and Lys-6,7 in arrestin-1, -2, -3, and -4, respectively), and two lysines and two arginines in β-strand X (Gurevich and Benovic 1995, 1997; Vishnivetskiy et al. 2000) (Fig. 2). Arg-18 in the loop between β-strands I and II (Fig. 2) is only present in the most phosphorylation-dependent family member, arrestin-1 (Sutton et al. 2005), whereas in others there are uncharged residues in equivalent position (Pro-14, Pro-15, and Ser-10 in arrestin-2, -3, and -4, respectively).
Fig. 1.
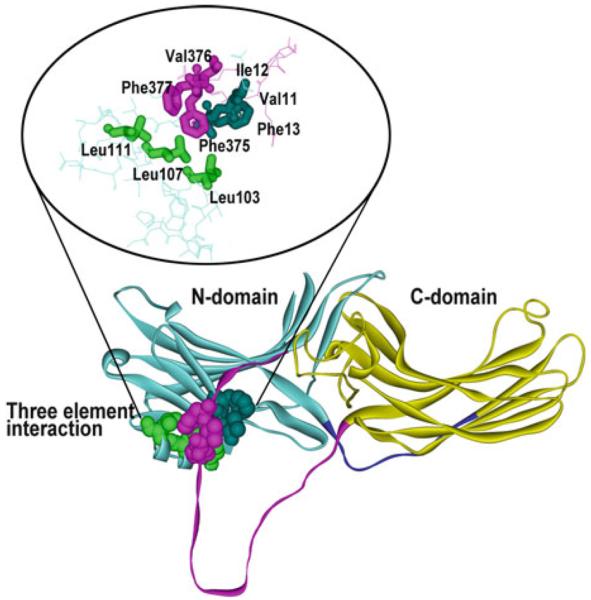
Three-element interaction. Arrestins are elongated molecules consisting of the N-domain (light blue), C-domain (yellow), connected by a 12-resiude hinge (dark blue), and the C-tail (magenta). One of the interactions stabilizing the basal arrestin conformation involves bulky hydrophobic residues in b-strand XX in the C-tail (Phe375, Val376, Phe377, magenta), which comes back and interacts with b-strand I (Val11, Ile12, Phe13, dark blue) and a-helix I (Leu103, Leu107, Leu111, green) in the N-domain. The structure of arrestin-1 [1CF1 (Hirsch et al. 1999)] where this arrangement was first discovered is shown, but this structural feature is conserved in all arrestins (Han et al. 2001; Sutton et al. 2005; Zhan et al. 2011). In all arrestins, destabilization of this interaction by triple alanine substitution of the hydrophobic residues in the C-tail (3A mutation) results in receptor binding-independent release of the C-tail and yields enhanced mutants that bind with higher affinity phosphorylated and non-phosphorylated active forms of their cognate receptors
Fig. 2.
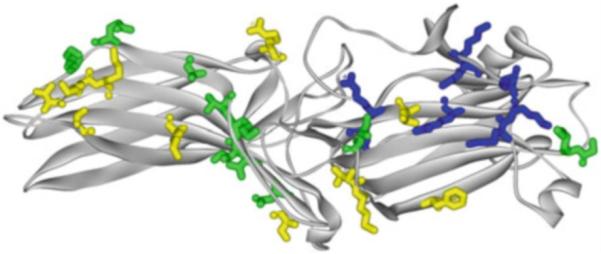
Receptor-binding residues in arrestins. Crystal structure of arrestin-1 [1CF1 (Hirsch et al. 1999)] viewed from the concave side of both domains. Side chains of receptor-binding residues shown as stick models, color coded, as follows: dark blue, positive charges engaged by receptor-attached phosphates (Lys14, Ly15, Arg18, Lys166, Lys167, Arg171, Arg175, Lys176) (Gurevich and Benovic 1995, 1997; Vishnivetskiy et al. 2000; Sutton et al. 2005); green, residues that affect receptor selectivity of arrestins (Gly54, Lys55, Ile72, Val244, Asn246, Ile256, Lys257, Thr258, Ala261, Gln265, Lys267) (Vishnivetskiy et al. 2011; Gimenez et al. 2012c); yellow, other receptor-binding residues identified by site-directed mutagenesis (Gurevich and Benovic 1997; Hanson and Gurevich 2006) or site-directed spin labeling and EPR of arrestin-1 (Hanson et al. 2006b) or arrestin-2 (Vishnivetskiy et al. 2011) (Val74, Met75, Phe85, Leu173, Lys232, Thr233, Lys235, Lys236, Arg288, Lys330, Thr344). Homologous residues in other arrestin subtypes play the same roles
In all arrestins an unusual (for a soluble protein) arrangement of five virtually solvent-excluded charged residues is found in the inter-domain interface (Fig. 3), which was termed the polar core (Hirsch et al. 1999). It includes two positive charges (Arg-175 and -382; Arg-169 and -393; Arg-170 and -392; Arg-165 and -370 in arrestin-1, -2, -3, and -4, respectively) and three negative charges (Asp-30, -296, and -303; Asp-26, -290, and -297; Asp-27, -291, and -298; Asp-22, -287, and -294 in arrestin-1, -2, -3, and -4, respectively). One of the positive charges, Arg175/169/170 in arrestin-1/2/3, directly binds receptor-attached phosphates (Gurevich and Benovic 1995, 1997). The neutralization or reversal of this charge by mutagenesis yields arrestin mutants that bind active non-phosphorylated forms of their cognate receptors with high affinity (Gurevich and Benovic 1995, 1997; Gray-Keller et al. 1997; Gurevich et al. 1997; Kovoor et al. 1999b; Vishnivetskiy et al. 1999; Celver et al. 2002a; Pan et al. 2003). The reversal of the negative charge of Asp296, which forms the salt bridge with Arg175, yields essentially the same enhancement of phosphorylation-independent binding to active receptors (Hirsch et al. 1999; Vishnivetskiy et al. 1999). Interestingly, simultaneous reversal of both charges, which restores the salt bridge in opposite configuration, fully restores strict dependence of arrestin binding on receptor phosphorylation (Vishnivetskiy et al. 1999). These results suggest that this salt bridge in the polar core is the main phosphate sensor in arrestins. Its disruption by negatively charged receptor-attached phosphates, which can occur regardless of the configuration of the bridge, turns arrestin “on,” allowing its transition into high-affinity receptor-binding state (Gurevich and Gurevich 2004). Breakup of this salt bridge by mutations essentially “tricks” arrestin into perceiving any active form of the receptor as phosphorylated. Obviously, purely ionic mechanism of the phosphate sensor action only requires the presence of spatially concentrated negative charge on the receptor and therefore is independent of the sequence context of phosphorylated serines and threonines. This simple mechanism of arrestin activation explained for the first time how two nonvisual arrestins in vertebrates (and only one in insects) can specifically bind active phosphorylated forms of hundreds of different GPCR subtypes (Gurevich and Gurevich 2006b).
Fig. 3.
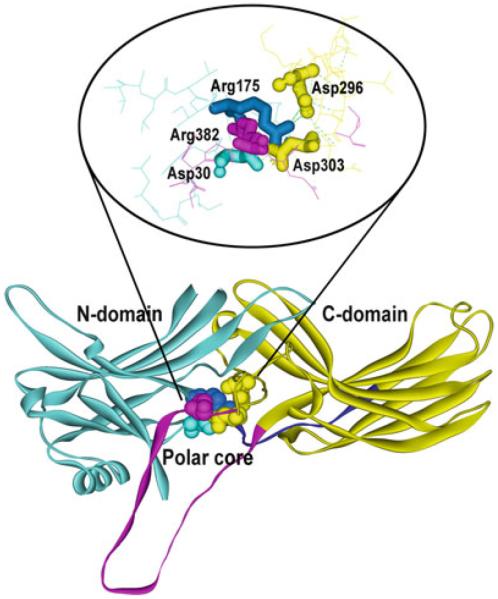
The polar core—key phosphate sensor in arrestins. Arrestins are elongated molecules consisting of the N-domain (light blue), C-domain (yellow), connected by a 12-resiude hinge (dark blue), and the C-tail (magenta). Basal arrestin conformation is also stabilized by the network of ionic interactions on the inter-domain interface among five residues: three negatively charged Asp30 (from the N-domain, light blue), Asp 296, and Asp303 (from the C-domain, yellow) and two positively charged, Arg175 (from the N-domain, dark blue) and Arg382 (from the C-tail, magenta). The enraged image on top is rotated to better show all five residues involved. The structure of arrestin-1 [1CF1 (Hirsch et al. 1999)] where the polar core was first discovered and got its name is shown, but this structural feature is conserved in all arrestins (Han et al. 2001; Sutton et al. 2005; Zhan et al. 2011). The polar core, particularly the salt bridge between Arg175 and Asp296, serves as the phosphate sensor: negatively charged receptor-attached phosphates break this salt bridge, which promotes arrestin transition into high-affinity receptor-binding state. The disruption of the polar core by mutations neutralizing or reversing the charge of Arg175 or Asp296 (or homologous residues in other arrestin subtypes) yields enhanced phosphorylation-independent mutants than bind with high-affinity active forms of their cognate receptors regardless of phosphorylation
It appears that by engaging the two lysines in the β-strand I receptor-attached phosphates also destabilize another key intramolecular “clasp” that holds arrestin in the basal conformation, three-element interaction of the C-tail, β-strand I, and α-helix I (Fig. 1) (Vishnivetskiy et al. 2000, 2010). Interestingly, these lysines are necessary for high-affinity binding of WT arrestin to active phosphoreceptor, but not for the binding of “pre-activated” mutants with either polar core or three-element interaction destabilized by mutations (Vishnivetskiy et al. 2000). Thus, it appears that highly exposed lysines on β-strand I “meet” receptor-attached phosphates first and then “guide” them to buried Arg-175 in the polar core, where they can destabilize this main phosphate sensor (Vishnivetskiy et al. 2000; Gurevich et al. 2011). Therefore, when the phosphate sensor is already turned “on” by mutagenesis, the job of the phosphates is done, making these “guiding” lysines in arrestin dispensable.
3 Construction of Enhanced Phosphorylation-Independent Arrestins
The main phosphate sensor, the polar core, is artificially turned “on” by mutations that disrupt the key salt bridge from either side, charge reversals of the Arg-175 and Asp-296 being virtually equipotent (Gurevich and Gurevich 2004). The fact that the disruption of the polar core, which supports the basal conformation in all arrestins (Hirsch et al. 1999; Han et al. 2001; Sutton et al. 2005; Zhan et al. 2011), pre-activates them, facilitating the binding even to non-phosphorylated receptors, is consistent with the idea that receptor binding is associated with a global conformational change in arrestin (Schleicher et al. 1989; Palczewski et al. 1991; Gurevich and Benovic 1993). It turns out that mutational disruption of the three-element interaction of the C-tail, β-strand I, and α-helix I, induces similar changes in the conformational equilibrium (Carter et al. 2005) and also greatly increases arrestin ability to bind active non-phosphorylated GPCRs (Gurevich 1998; Kovoor et al. 1999b; Celver et al. 2001, 2002a; Pan et al. 2003). One of the positive charges in the polar core, Arg-382/393/392 in arrestin-1/2/3, is localized in the C-tail near the three-element interaction. However, forcible detachment of the C-tail by substitution of three bulky hydrophobics anchoring it to the body of the molecule (Fig. 1) with alanines (3A mutation) was found to be a more potent activating mutation that elimination of the positive charge of Arg-382 or its equivalent in nonvisual arrestins (Gurevich 1998; Kovoor et al. 1999b; Celver et al. 2002a). Interestingly, the deletion of the C-tail beyond its point of contact with the β-strand I and α-helix I yields essentially the same level of phosphorylation-independent binding as its detachment by alanine substitution in all arrestins (Gurevich et al. 1997; Gurevich 1998; Celver et al. 2002a; Vishnivetskiy et al. 2013a, b, c).
Thus, there are at least three types of mutations capable of significantly enhancing arrestin binding to active non-phosphorylated receptors (1) charge reversals of either Arg or Asp forming key salt bridge in the polar core; (2) detachment of the C-tail by substituting three bulky hydrophobic residues with alanines; and (3) deletion of the distal C-tail just beyond these bulky hydrophobics engaged in the three-element interaction. All yield enhanced arrestins with fairly high affinity for active receptors regardless of their phosphorylation. One limitation is that to be potentially useful for compensational gene therapy, mutant protein has to be stable, preferably as stable as parental WT one. One would expect any mutation that “loosens up” the basal conformation to reduce protein stability to some extent. This issue was systematically addressed in mouse arrestin-1 (Song et al. 2009; Vishnivetskiy et al. 2013a, b). Unexpectedly, it turned out that some of the activating mutations are a lot more detrimental for arrestin-1 stability than others (Song et al. 2009; Vishnivetskiy et al. 2013a, b). Charge reversals in the polar core greatly destabilize arrestin, whereas 3A mutation and C-tail deletion are fairly well tolerated (Song et al. 2009). In fact, even 3A variants of mouse arrestin-1 with additional mutations on the receptor-binding surface that increase their affinity for non-phosphorylated light-activated rhodopsin (Rh*) are fairly stable (Vishnivetskiy et al. 2013b) paving the way to the testing of these more potent pre-activated mutants in vivo.
Homologous mutations yield similarly enhanced phosphorylation-independent variants of nonvisual arrestins (Gurevich et al. 1997; Kovoor et al. 1999b; Celver et al. 2002a; Pan et al. 2003). These mutants effectively quench signaling by nonvisual GPCRs that are not phosphorylated either due to the loss of GRK sites or because GRKs are absent (Kovoor et al. 1999b; Celver et al. 2001, 2002a; Macey et al. 2006). Thus, enhanced nonvisual arrestins are perfectly ready for in vivo testing, but an additional serious issue needs to be addressed. Both nonvisual arrestins are promiscuous, comparably interacting with numerous GPCRs (Gurevich et al. 1995; Barak et al. 1997; Gimenez et al. 2012c). Virtually every cell in the body expresses 5–20 different GPCR subtypes. The expression of enhanced nonvisual arrestins will certainly suppress the signaling by hyperactive GPCR mutant. However, due to lack of receptor selectivity, enhanced versions of either arrestin-2 or -3 would dampen the signaling by all other perfectly normal GPCRs in the same cell, possibly doing more harm than good. Thus, practical use of enhanced nonvisual arrestins for gene therapy requires further work to significantly increase their receptor specificity (discussed in Chap. 8).
4 Compensational Approach to Gene Therapy
So far the ability of enhanced arrestin to compensate for defects in receptor phosphorylation was tested only in rod photoreceptors using a single model: rhodopsin kinase knockout (Song et al. 2009). Moreover, out of two stable enhanced mutants tested, mouse arrestin-1-3A and truncated arrestin-1-(1-377), only the former expressed at near-physiological levels in transgenic animals (Nair et al. 2005; Cleghorn et al. 2011; Song et al. 2011), and therefore only 3A mutant expressed at ~50 and ~220 % of WT arrestin-1, was actually tested for its compensational potential (Song et al. 2009). Considering that rods contain the fastest and the most sensitive GPCR-driven signaling cascade (Baylor et al. 1979; Gross and Burns 2010), which makes the visual system extremely demanding, this attempt was quite successful. Rod photoreceptors of rhodopsin kinase (GRK1) knockout mice (RKKO) rapidly lose their rhodopsin-containing signaling compartment, the outer segment, and then gradually degenerate (Chen et al. 1999; Song et al. 2009). The replacement of WT arrestin-1 in these rods with 3A mutant significantly improves their histological appearance and prolongs their survival (Song et al. 2009). Due to rod defect, RKKO animals demonstrate fairly low light sensitivity, effectively responding to brighter flashes that stimulate cones. In contrast, RKKO rods expressing 3A mutant at moderate level are functional and significantly more light sensitive (Song et al. 2009), again demonstrating certain level of compensation.
Since rhodopsin phosphorylation followed by arrestin-1 binding is critical for proper timing of the photoresponse (Xu et al. 1997; Chen et al. 1999; Mendez et al. 2000; Gross and Burns 2010), it was particularly important to test the effect of enhanced arrestin on the rate of photoresponse recovery. While WT rods rapidly restore sensitivity after moderately bright flashes, with time of half-recovery on the sub-second scale, RKKO rods recover extremely slow, with time of half-recovery ~18 s (Song et al. 2009). Good news is that the replacement of WT arrestin-1 with enhanced 3A mutant accelerated recovery about fivefold (Song et al. 2009), demonstrating that in principle compensational approach to gene therapy works even in the extremely demanding visual system. Bad news is that the recovery rate in “compensated” rods was still ~10-fold slower than in WT photoreceptors. Recent design of novel further enhanced mutants of arrestin-1 that bind to Rh* much better than the original 3A form and retain acceptable stability (Vishnivetskiy et al. 2013b) paves the way to testing the limits of this type of compensational gene therapy in rods.
It is entirely possible that the level of compensation achieved with 3A mutant would have been sufficient in any GPCR-driven signaling system that is less demanding than rod photoreceptors. In fact, in Xenopus oocytes mutants of arrestin-2 and -3 enhanced by homologous substitutions desensitize non-phosphorylated β2-adrenergic, μ- and δ-opioid receptors with virtually the same kinetics as WT arrestins in the presence of GRKs (Kovoor et al. 1999b; Celver et al. 2001, 2002a). However, enhanced versions of nonvisual arrestins would only become therapeutically usable when their receptor specificity is narrowed to groups of receptors or even individual GPCRs (discussed in Chap. 8).
In some cases the mutation in a GPCR reduces arrestin binding to other functional forms than active phosphorylated receptor. Gly90Asp mutation in rhodopsin generates constitutively active form that causes night blindness in humans by desensitizing rods even in the dark (Sieving et al. 1995). This mutation impedes rhodopsin regeneration by retinal, because introduced aspartic acid forms a salt bridge with Lys 296 where retinal attaches (Singhal et al. 2013), so that a significant fraction of it exists as opsin that can activate transducin (Sieving et al. 1995). Interestingly, whereas WT phosphorylated opsin is the second highest affinity target of WT arrestin-1 (Sommer et al. 2012; Zhuang et al. 2013), phospho-opsin form of G90D mutant shows reduced arrestin-1 binding (Singhal et al. 2013). The same pre-activated arrestin-1 mutants that bind Rh* much better than WT, 3A, and truncated (Gurevich 1998; Song et al. 2009), demonstrate essentially normal binding to G90D phospho-opsin (Vishnivetskiy et al. 2013c), suggesting that reengineered arrestins have a potential to compensate for this type of defect, as well.
Importantly, even in case of arrestin-1, which until recently was believed to interact only with rhodopsin, other functional characteristics need to be taken into account. It was recently shown that even though arrestin-1 binds clathrin adaptor AP2 with affinity that is ~30 times lower than that of arrestin-2, excessive AP2 binding by rhodopsin-associated arrestin-1 can induce rod death (Moaven et al. 2013). AP2-binding site is localized in the arrestin C terminus (Kim and Benovic 2002), and the replacement of WT arrestin-1 in rods with its truncated form lacking AP2-binding site was shown to protect photoreceptors expressing constitutively active rhodopsin (Moaven et al. 2013). Also, arrestin-1-3A, in addition to apparently beneficial ability to bind Rh* (Song et al. 2009), is impaired in self-association (Song et al. 2013) (see Chap. 11). Resulting excessive concentration of monomer in mouse line expressing high level of this mutant appears to induce photoreceptor death via yet another mechanism (Song et al. 2013) (see Chap. 16). Collectively, these data suggest that to effectively compensate for defects of rhodopsin phosphorylation without unwanted side effects, enhanced form of arrestin-1 should not be able to bind AP2 and should either robustly self-associate or should be expressed at a relatively low safe level (Song et al. 2009, 2013).
5 Excessive Desensitization of Pre-activated Receptor Mutants and Normal Receptors Can Underlie the Pathology
Interestingly, in some cases constitutively active GPCRs are constitutively desensitized via hyper-phosphorylation by GRKs and virtually permanent association with cognate arrestin. For example, certain constitutively active rhodopsin mutants were shown to be hyper-phosphorylated and associated with arrestin in vivo (Rim and Oprian 1995), suggesting that disease phenotype is just as likely to be caused by constitutive desensitization as by uncontrolled signaling. Another well-studied example is a naturally occurring R137H mutation in vasopressin receptor associated with familial nephrogenic diabetes insipidus. It was originally described as loss-of-function, but later mutant receptor was shown to be constitutively active, which leads to constitutive phosphorylation and arrestin binding in cells, resulting in receptor endocytosis and sequestration in the intracellular vesicles (Barak et al. 2001). Thus, this is another case where constitutive desensitization and internalization of overactive receptor gives an appearance of nonsignaling phenotype. It is entirely possible that some of the described loss-of-function GPCR mutations are in fact gain-of-function, but lead to constitutive desensitization in vivo.
Another well-established case where excessive desensitization plays critical role in pathology is congestive heart failure. Phenotypically, failing heart does not adequately respond to adrenergic stimulation, which is easily explained by decreased density of β-adrenergic receptors (Bristow et al. 1982). It has been shown that the expression of GRK2 is elevated in human failing heart and animal models of heart failure (Ungerer et al. 1993). High GRK2 activity results in excessive phosphorylation of β-adrenergic receptors and corresponding reduction in their responsiveness to sympathetic neurotransmitter norepinephrine and the adrenal hormone epinephrine. Importantly, the reduction of GRK2 activity toward β-adrenergic receptors by underexpression in hemizygous GRK2+/− mice or by transgenic expression of GRK2 C terminus that competes with endogenous GRK2 for G protein βγ subunits, thereby suppressing its recruitment to the plasma membrane where adrenergic receptors reside, restores receptor sensitivity to catecholamines, and improves heart function (Rockman et al. 1998a, b; Akhter et al. 1999).
6 Enhanced Arrestins Protect Receptor from Excessive Phosphorylation and Prevent its Downregulation
By virtue of binding to active non-phosphorylated receptors, enhanced phosphorylation-independent arrestin mutants compete with GRKs. Enhanced arrestin-2 mutant with disrupted polar core (R169E) was shown to suppress the phosphorylation of purified β2-adrenergic receptor by pure GRK2 in vitro, as well as in living cells (Pan et al. 2003). Interestingly, the expression of either arrestin-2-R169E or arrestin-2-3A (another enhanced mutant where the C-tail is detached) in cells was shown to prevent downregulation of β2-adrenergic receptor upon prolonged agonist stimulation (Pan et al. 2003). It was shown that in response to an agonist β2-adrenergic receptor is internalized equally rapidly in cells expressing WT arrestin-2 or enhanced mutant. The main difference was found to be in the rate of receptor recycling back to the plasma membrane, which was many times faster in mutant-expressing cells (Pan et al. 2003).
The mechanisms of GPCR cycling provide the simplest explanation of these findings. Under normal circumstances arrestin binding to the active phosphorylated receptor induces the release of the arrestin C-tail (Hanson et al. 2006b; Vishnivetskiy et al. 2010; Kim et al. 2012). This greatly increases the availability of clathrin and AP2-binding sites localized in this element of nonvisual arrestins (Goodman et al. 1996; Laporte et al. 1999; Kim and Benovic 2002), facilitating the recruitment of the arrestin–receptor complex to the coated pits. Internalized receptor is transported to endosomes, where its extracellular surface with bound agonist faces the lumen with relatively low pH. It is generally believed that this induces the release of bound agonist, which promotes receptor transition back to inactive state. Inactive phosphorylated receptors demonstrate 30–50 % lower arrestin binding than active phosphorylated forms (Gurevich et al. 1993, 1995; Kovoor et al. 1999a; Celver et al. 2002b), suggesting that receptor inactivation facilitates the release of arrestin. Since bound arrestin shields receptor-attached phosphates (Palczewski et al. 1989), arrestin dissociation is necessary to allow receptor dephosphorylation by cytoplasmic protein phosphatases, whereupon it becomes recycling competent (Morrison et al. 1996; Hsieh et al. 1999). The situation changes dramatically when the complex of enhanced arrestin with non-phosphorylated receptor is internalized. In this case receptor deactivation reduces arrestin binding manifold, rather than by a mere 30–50 % (Kovoor et al. 1999a; Celver et al. 2002b; Pan et al. 2003), suggesting that arrestin release would be much faster. Importantly, unphosphorylated and therefore fully recycling-competent receptor emerges immediately upon arrestin dissociation. These two factors acting together explain extremely rapid recycling of internalized β2-adrenergic receptor in cells expressing enhanced mutants (Pan et al. 2003). It appears that as it zooms through endosomes and back to the plasma membrane, receptor does not spend enough time in sorting endosomes to be diverted to lysosomes and degraded, which would explain how enhanced mutants protect the receptor from downregulation.
These data suggest that enhanced mutants of nonvisual arrestins can serve yet another purpose: protect the receptor from excessive phosphorylation and facilitate its recycling, which apparently prevents downregulation of the receptor. It appears that in situations associated with excessive phosphorylation and loss of the receptor, like congestive heart failure, this is likely to be beneficial. The ability of phosphorylation-independent arrestin mutants to protect heart function in conditions causing its failure needs to be tested experimentally.
7 Therapeutic Potential of Enhanced Visual and Nonvisual Arrestins
The success of the first proof-of-concept experiments in highly demanding visual system, where enhanced arrestin-1 partially compensated for the lack of rhodopsin phosphorylation, improving photoreceptor health, survival, and functional performance (Song et al. 2009) demonstrates the feasibility of compensational gene therapy and its potential. However, neither morphology of “compensated” rods nor the rate of the recovery of their photoresponse was fully normalized: photoreceptors in WT animals outperformed compensated rods (Song et al. 2009). Thus, the challenge in this system is to construct more effective enhanced forms of arrestin-1, with much higher ability to bind non-phosphorylated light-activated rhodopsin and shut off its signaling. Several recent developments will facilitate progress in this direction. These include the analysis of rhodopsin binding-induced conformational changes in arrestin-1 using intramolecular distance measurements by pulse EPR (Kim et al. 2012), the identification of arrestin-1 elements engaged by different functional forms of rhodopsin using solution NMR (Zhuang et al. 2013), as well as the structures of truncated forms of arrestin-1 (Kim et al. 2013) and arrestin-2 (Shukla et al. 2013) that reveal the direction of the conformational changes in the process of arrestin activation by cognate receptors. Continuing improvements in engineered arrestin-1 mutants with phosphorylation-independent binding to active rhodopsin (Vishnivetskiy et al. 2013b) suggest that this goal is attainable. Next, the ability of new and improved enhanced mutant to compensate for impaired rhodopsin phosphorylation must be tested in different models of defective rhodopsin phosphorylation. In addition to GRK1 (rhodopsin kinase) knockout mice, where the previous mutant was tested (Song et al. 2009), compensation potential of new mutants should be tested in mice expressing rhodopsin without GRK1 phosphorylation sites, as well as those expressing rhodopsin with only one or two remaining sites (Mendez et al. 2000), which are insufficient for high-affinity arrestin-1 binding (Vishnivetskiy et al. 2007) in vitro and rapid rhodopsin shutoff in vivo (Mendez et al. 2000). Another unanswered question is whether these enhanced mutants should retain their ability to self-associate (see Chap. 11) or should be made constitutively monomeric (Hanson et al. 2008; Kim et al. 2011), since only arrestin-1 monomer can bind rhodopsin (Hanson et al. 2007b). Recent study showed that reduced self-association can be combined with enhanced phosphorylation-independent binding to active rhodopsin (Vishnivetskiy et al. 2013b). Optimal expression level of enhanced arrestin-1 is another issue that needs to be solved: previous experiments showed that the line expressing moderate (~50 % of WT) levels of enhanced arrestin-1 shows much better compensation that the line expressing it at more than twice WT level (Song et al. 2009). Moreover, progressive death of photoreceptors was documented in higher expressing line (Song et al. 2009). Since similarly high expression of WT arrestin-1 does not adversely affect photoreceptors (Song et al. 2011), this detrimental effect appears to be connected with mutation-induced changes in the molecule. Mutant-induced rod death was apparently associated with its impaired self-association that yields excessive concentration of arrestin-1 monomer (Song et al. 2013). Human homologue of the most promising form of enhanced mouse arrestin-1 that emerges from these experiments, expressed at optimal level, will be a good candidate to test for actual gene therapy in human patients. Thus, the challenge in the visual system, where it is clear that arrestin-1 subtype specifically regulates rhodopsin signaling, is advanced engineering of a more powerful phosphorylation-independent mutant and precise determination of the range of expression levels that ensure safety and functional efficiency.
Other GPCR-driven signaling systems are not as sensitive as rod photoreceptors and demonstrate much slower shutoff kinetics (Carman and Benovic 1998; Violin et al. 2008). Phosphorylation-independent mutants of nonvisual arrestin-2 and -3 block G protein coupling of β2-adrenergic, μ-, and δ-opioid receptors yielding desensitization in the absence of receptor phosphorylation with essentially the same time course that WT arrestins yield in the presence of GRKs (Kovoor et al. 1999a; Celver et al. 2001, 2002b). Thus, it appears that there is no need to increase their efficiency, although homologues of some mutations on the receptor-binding surface of arrestin-1 with increased ability to bind non-phosphorylated receptors (Hanson and Gurevich 2006; Vishnivetskiy et al. 2013a, b) might further improve the performance of enhanced nonvisual arrestins. However, the main challenge with nonvisual arrestins is not efficiency, but receptor specificity. Both WT arrestin-2 and -3 are fairly promiscuous, binding comparably to numerous GPCRs (Gurevich et al. 1995; Barak et al. 1997), even though relative contribution of receptor-attached phosphates to arrestin binding varies widely in different cases (Gimenez et al. 2012a). Since most cells express a variety of receptors, only one of which would be mutant in each particular patient, the expression of enhanced versions of WT nonvisual arrestins would likely dampen the signaling by all GPCRs present in the same cell, instead of selectively suppressing faulty signaling by the mutant. Thus, to make enhanced nonvisual arrestins suitable for therapeutic purposes, it is imperative to increase their receptor specificity.
Receptor binding to any arrestin engages fairly large surface, covering most of the concave sides of both arrestin domains (Gurevich and Benovic 1993; Ohguro et al. 1994; Pulvermuller et al. 2000b; Hanson et al. 2006b; Hanson and Gurevich 2006; Vishnivetskiy et al. 2011). However, element swapping between arrestin-1 and -2 showed that only part of this extensive surface plays a role in receptor preference (Vishnivetskiy et al. 2004), and subsequent mutagenesis identified surprisingly few residues on it that define receptor specificity (Vishnivetskiy et al. 2011). These results, along with the fact that very few different amino acids occupied each of the key positions in arrestins from a variety of animal species from Caenorhabditis elegans to mammals (Gurevich and Gurevich 2006a), allowed the construction of a limited number of variants with point mutations in distinct receptor-discriminator sites (Gimenez et al. 2012b). Interestingly, out of the first 12 mutations tested on a set of 5 different GPCRs, 11 significantly affected receptor preference (Gimenez et al. 2012b). These results demonstrate that the construction of nonvisual arrestins with narrow receptor specificity is feasible (Chap. 8). Considering that the binding of phosphorylation-independent mutants to non-phosphorylated receptors tends to be more subtype specific (Kovoor et al. 1999a; Celver et al. 2002b), it is likely that enhanced versions of selective arrestins will retain narrow receptor specificity of parental mutants, although this still needs to be tested experimentally. If this turns out to be the case, enhanced receptor-specific variants of arrestin-2 and -3 would be ready for in vivo testing of their ability to selectively suppress the signaling of only one of many GPCRs expressed in the same cell.
The ability of phosphorylation-independent arrestin-2 mutants to protect the receptor from excessive phosphorylation and downregulation was so far only demonstrated in cultured cells (Pan et al. 2003). If combining enhancing mutations with narrow receptor specificity proves feasible, these mutants should be tested for their ability to prevent phosphorylation and loss of an individual GPCR subtype among several in the same cell. The success of these experiments will provide reasons for testing the ability of receptor-specific versions of phosphorylation-independent nonvisual arrestins to protect β-adrenergic receptors in mouse models of heart failure and to improve heart function in these conditions. If these mutants work in living mice, as expected, this will pave the way for their therapeutic use for treating human patients with failing heart.
Footnotes
Different systems of arrestin names are used in the field and in this book. We use systematic names of arrestin proteins: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons its gene is called “arrestin 3” in the HUGO database).
References
- Akhter SA, Eckhart AD, Rockman HA, Shotwell K, Lefkowitz RJ, Koch WJ. In vivo inhibition of elevated myocardial beta-adrenergic receptor kinase activity in hybrid transgenic mice restores normal beta-adrenergic signaling and function. Circulation. 1999;100:648–653. doi: 10.1161/01.cir.100.6.648. [DOI] [PubMed] [Google Scholar]
- Apfelstedt-Sylla E, Kunisch M, Horn M, Ruther K, Gerding H, Gal A, Zrenner E. Ocular findings in a family with autosomal dominant retinitis pigmentosa and a frameshift mutation altering the carboxyl terminal sequence of rhodopsin. Br J Ophthalmol. 1993;77:495–501. doi: 10.1136/bjo.77.8.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- Barak LS, Ferguson SS, Zhang J, Caron MG. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- Barak LS, Oakley RH, Laporte SA, Caron MG. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc Natl Acad Sci USA. 2001;98:93–98. doi: 10.1073/pnas.011303698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol. 1979;288:613–634. [PMC free article] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Luni K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- Carter JM, Gurevich VV, Prossnitz ER, Engen JR. Conformational differences between arrestin2 and pre-activated mutants as revealed by hydrogen exchange mass spectrometry. J Mol Biol. 2005;351:865–878. doi: 10.1016/j.jmb.2005.06.048. [DOI] [PubMed] [Google Scholar]
- Celver J, Lowe J, Kovoor A, Gurevich VV, Chavkin C. Threonine 180 is requred for G protein-coupled receptor kinase 3 and b-arrestin mediated desensitization of the m-opioid receptor in Xenopus oocytes. J Biol Chem. 2001;276:4894–4900. doi: 10.1074/jbc.M007437200. [DOI] [PubMed] [Google Scholar]
- Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, Pang JJ, Sumaroka A, Windsor EA, Wilson JM, Flotte TR, Fishman GA, Heon E, Stone EM, Byrne BJ, Jacobson SG, Hauswirth WW. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus M, Maier J, Paschke R, Kujat C, Stumvoll M, Führer D. Novel thyrotropin receptor germline mutation (Ile568Val) in a Saxonian family with hereditary nonautoimmune hyper-thyroidism. Thyroid. 2005;15:1089–1094. doi: 10.1089/thy.2005.15.1089. [DOI] [PubMed] [Google Scholar]
- Cleghorn WM, Tsakem EL, Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Progressive reduction of its expression in rods reveals two pools of arrestin-1 in the outer segment with different roles in photoresponse recovery. PLoS One. 2011;6:e22797. doi: 10.1371/journal.pone.0022797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft CM, Whitmore DH, Wiechmann AF. Cone arrestin identified by targeting expression of a functional family. J Biol Chem. 1994;269:4613–4619. [PubMed] [Google Scholar]
- Dinculescu A, McDowell JH, Amici SA, Dugger DR, Richards N, Hargrave PA, Smith WC. Insertional mutagenesis and immunochemical analysis of visual arrestin interaction with rhodopsin. J Biol Chem. 2002;277:11703–11708. doi: 10.1074/jbc.M111833200. [DOI] [PubMed] [Google Scholar]
- Gimenez LE, Kook S, Vishnivetskiy SA, Ahmed MR, Gurevich EV, Gurevich VV. Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. J Biol Chem. 2012a;287:9028–9040. doi: 10.1074/jbc.M111.311803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Vishnivetskiy SA, Baameur F, Gurevich VV. Enhancing receptor specificity of non-visual arrestins by targeting receptor-discriminator residues. J Biol Chem. 2012b;287(35):29495–29505. doi: 10.1074/jbc.M112.366674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Vishnivetskiy SA, Baameur F, Gurevich VV. Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J Biol Chem. 2012c;287:29495–29505. doi: 10.1074/jbc.M112.366674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Gray-Keller MP, Detwiler PB, Benovic JL, Gurevich VV. Arrestin with a single amino acid sustitution quenches light-activated rhodopsin in a phosphorylation-independent fasion. Biochemistry. 1997;36:7058–7063. doi: 10.1021/bi963110k. [DOI] [PubMed] [Google Scholar]
- Gross OP, Burns ME. Control of rhodopsin's active lifetime by arrestin-1 expression in mammalian rods. J Neurosci. 2010;30:3450–3457. doi: 10.1523/JNEUROSCI.5391-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation-recignition region of arrestin. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Mechanism of phosphorylation-recognition by visual arrestin and the transition of arrestin into a high affinity binding state. Mol Pharmacol. 1997;51:161–169. doi: 10.1124/mol.51.1.161. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Gurevich VV. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006a;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol Ther. 2006b;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J Biol Chem. 1993;268:16879–16882. [PubMed] [Google Scholar]
- Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Hanson SM, Song X, Vishnivetskiy SA, Gurevich EV. The functional cycle of visual arrestins in photoreceptor cells. Prog Retin Eye Res. 2011;30:405–430. doi: 10.1016/j.preteyeres.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutkind JS, Novotny EA, Brann MR, Robbins KC. Muscarinic acetylcholine receptor subtypes as agonist-dependent oncogenes. Proc Natl Acad Sci USA. 1991;88:4703–4707. doi: 10.1073/pnas.88.11.4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- Hanson SM, Gurevich VV. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006a;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci USA. 2006b;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Gurevich EV, Vishnivetskiy SA, Ahmed MR, Song X, Gurevich VV. Each rhodopsin molecule binds its own arrestin. Proc Natl Acad Sci USA. 2007a;104:3125–3128. doi: 10.1073/pnas.0610886104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Arshavsky VY, Klug CS, Hubbell WL, Gurevich VV. Structure and function of the visual arrestin oligomer. EMBO J. 2007b;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Dawson ES, Francis DJ, Van Eps N, Klug CS, Hubbell WL, Meiler J, Gurevich VV. A model for the solution structure of the rod arrestin tetramer. Structure. 2008;16:924–934. doi: 10.1016/j.str.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, Jacobson SG. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Jackson TR, Blair LA, Marshall J, Goedert M, Hanley MR. The mas oncogene encodes an angiotensin receptor. Nature. 1988;335:437–440. doi: 10.1038/335437a0. [DOI] [PubMed] [Google Scholar]
- Julius D, Livelli TJ, Jessell TM, Axel R. Ectopic expression of the serotonin 1c receptor and the triggering of malignant transformation. Science. 1989;244:1057–1062. doi: 10.1126/science.2727693. [DOI] [PubMed] [Google Scholar]
- Khoo DH, Parma J, Rajasoorya C, Ho SC, Vassart G. A germline mutation of the thyrotropin receptor gene associated with thyrotoxicosis and mitral valve prolapse in a Chinese family. J Clin Endocrinol Metab. 1999;84:1459–1462. doi: 10.1210/jcem.84.4.5620. [DOI] [PubMed] [Google Scholar]
- Kim YM, Benovic JL. Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J Biol Chem. 2002;277:30760–30768. doi: 10.1074/jbc.M204528200. [DOI] [PubMed] [Google Scholar]
- Kim RY, al-Maghtheh M, Fitzke FW, Arden GB, Jay M, Bhattacharya SS, Bird AC. Dominant retinitis pigmentosa associated with two rhodopsin gene mutations. Leu-40-Arg and an insertion disrupting the 5'-splice junction of exon 5. Arch Ophthalmol. 1993;111:1518–1524. doi: 10.1001/archopht.1993.01090110084030. [DOI] [PubMed] [Google Scholar]
- Kim M, Hanson SM, Vishnivetskiy SA, Song X, Cleghorn WM, Hubbell WL, Gurevich VV. Robust self-association is a common feature of mammalian visual arrestin-1. Biochemistry. 2011;50:2235–2242. doi: 10.1021/bi1018607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Vishnivetskiy SA, Van Eps N, Alexander NS, Cleghorn WM, Zhan X, Hanson SM, Morizumi T, Ernst OP, Meiler J, Gurevich VV, Hubbell WL. Conformation of receptor-bound visual arrestin. Proc Natl Acad Sci USA. 2012;109:18407–18412. doi: 10.1073/pnas.1216304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Hofmann KP, Ernst OP, Scheerer P, Choe HW, Sommer ME. Crystal structure of pre-activated arrestin p44. Nature. 2013;497:142–146. doi: 10.1038/nature12133. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Gurevich VV, Benovic JL. Mechanism of quenching of phototransduction. Binding competition between arrestin and transducin for phosphorhodopsin. J Biol Chem. 1997;272:18125–18131. doi: 10.1074/jbc.272.29.18125. [DOI] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SG, Caron MG, Barak LS. The 2-adrenergic receptor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey TA, Lowe JD, Chavkin C. Mu opioid receptor activation of ERK1/2 is GRK3 and arrestin dependent in striatal neurons. J Biol Chem. 2006;281:34515–34524. doi: 10.1074/jbc.M604278200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh ENJ, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell'Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Moaven H, Koike Y, Jao CC, Gurevich VV, Langen R, Chen J. Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc Natl Acad Sci USA. 2013;110:9463–9468. doi: 10.1073/pnas.1301126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison KJ, Moore RH, Carsrud ND, Trial J, Millman EE, Tuvim M, Clark RB, Barber R, Dickey BF, Knoll BJ. Repetitive endocytosis and recycling of the beta 2-adrenergic receptor during agonist-induced steady state redistribution. Mol Pharmacol. 1996;50:692–699. [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov SS, Brown BM, Davis JA, Zuniga FI, Bragin A, Pugh ENJ, Craft CM. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron. 2008;59:462–474. doi: 10.1016/j.neuron.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohguro H, Palczewski K, Walsh KA, Johnson RS. Topographic study of arrestin using differential chemical modifications and hydrogen/deuterium exchange. Protein Sci. 1994;3:2428–2434. doi: 10.1002/pro.5560031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, McDowell H, Jakes S, Ingebritsen TS, Hargrave PA. Regulation of rhodopsin dephosphorylation by arrestin. J Biol Chem. 1989;264:15770–15773. [PubMed] [Google Scholar]
- Palczewski K, Pulvermuller A, Buczylko J, Hofmann KP. Phosphorylated rhodopsin and heparin induce similar conformational changes in arrestin. J Biol Chem. 1991;266:18649–18654. [PubMed] [Google Scholar]
- Pan L, Gurevich EV, Gurevich VV. The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]
- Paschke R. Constitutively activating TSH receptor mutations as the cause of toxic thyroid adenoma, multinodular toxic goiter and autosomal dominant non autoimmune hyperthyroidism. Exp Clin Endocrinol Diabetes. 1996;104:129–132. doi: 10.1055/s-0029-1211720. [DOI] [PubMed] [Google Scholar]
- Pulvermuller A, Schroder K, Fischer T, Hofmann KP. Interactions of metarhodopsin II. Arrestin peptides compete with arrestin and transducin. J Biol Chem. 2000;275:37679–37685. doi: 10.1074/jbc.M006776200. [DOI] [PubMed] [Google Scholar]
- Restagno G, Maghtheh M, Bhattacharya S, Ferrone M, Garnerone S, Samuelly R, Carbonara A. A large deletion at the 3' end of the rhodopsin gene in an Italian family with a diffuse form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1993;2:207–208. doi: 10.1093/hmg/2.2.207. [DOI] [PubMed] [Google Scholar]
- Rim J, Oprian DD. Constitutive activation of opsin: interaction of mutants with rhodopsin kinase and arrestin. Biochemistry. 1995;34:11938–11945. doi: 10.1021/bi00037a035. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross JJ, Lefkowitz RJ, Koch WJ. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA. 1998a;95:7000–7005. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Choi DJ, Akhter SA, Jaber M, Giros B, Lefkowitz RJ, Caron MG, Koch WJ. Control of myocardial contractile function by the level of beta-adrenergic receptor kinase 1 in gene-targeted mice. J Biol Chem. 1998b;273:18180–18184. doi: 10.1074/jbc.273.29.18180. [DOI] [PubMed] [Google Scholar]
- Schipani E, Kruse K, Jüppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- Schleicher A, Kuhn H, Hofmann KP. Kinetics, binding constant, and activation energy of the 48-kDa protein-rhodopsin complex by extra-metarhodopsin II. Biochemistry. 1989;28:1770–1775. doi: 10.1021/bi00430a052. [DOI] [PubMed] [Google Scholar]
- Schöneberg T, Schulz A, Biebermann H, Hermsdorf T, Römpler H, Sangkuhl K. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol Ther. 2004;104:173–206. doi: 10.1016/j.pharmthera.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, Paduch M, Tripathi-Shukla P, Koide A, Koide S, Weis WI, Kossiakoff AA, Kobilka BK, Lefkowitz RJ. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieving PA, Richards JE, Naarendorp F, Bingham EL, Scott K, Alpern M. Dark-light: model for nightblindness from the human rhodopsin Gly-90-Asp mutation. Proc Natl Acad Sci USA. 1995;92:880–884. doi: 10.1073/pnas.92.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A, Ostermaier MK, Vishnivetskiy SA, Panneels V, Homan KT, Tesmer JJ, Veprintsev D, Deupi X, Gurevich VV, Schertler GF, Standfuss J. Insights into congenital night blindness based on the structure of G90D rhodopsin. EMBO Rep. 2013;14:520–526. doi: 10.1038/embor.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer ME, Hofmann KP, Heck M. Distinct loops in arrestin differentially regulate ligand binding within the GPCR opsin. Nat Commun. 2012;3:995. doi: 10.1038/ncomms2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, Gurevich EV, Burns ME, Gurevich VV. Enhanced arrestin facilitates recovery and protects rod photoreceptors deficient in rhodopsin phosphorylation. Curr Biol. 2009;19:700–705. doi: 10.1016/j.cub.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Arrestin-1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience. 2011;174:37–49. doi: 10.1016/j.neuroscience.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Seo J, Baameur F, Vishnivetskiy SA, Chen Q, Kook S, Kim M, Brooks EK, Altenbach C, Hong Y, Hanson SM, Palazzo MC, Chen J, Hubbell WL, Gurevich EV, Gurevich VV. Rapid degeneration of rod photoreceptors expressing self-association-deficient arrestin-1 mutant. Cell Signal. 2013;25:2613–2624. doi: 10.1016/j.cellsig.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal structure of cone arrestin at 2.3Å: evolution of receptor specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Expression of β-arrestins and β-adrenergic receptor kinases in the failing human heart. Circulation. 1993;87:454–463. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- Violin JD, DiPilato LM, Yildirim N, Elston TC, Zhang J, Lefkowitz RJ. beta2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J Biol Chem. 2008;283:2949–2961. doi: 10.1074/jbc.M707009200. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez MG, Gurevich VV. An additional phosphate-binding element in arrestin molecule. Implications for the mechanism of arrestin activation. J Biol Chem. 2000;275:41049–41057. doi: 10.1074/jbc.M007159200. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV. Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem. 2007;282:32075–32083. doi: 10.1074/jbc.M706057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Francis DJ, Van Eps N, Kim M, Hanson SM, Klug CS, Hubbell WL, Gurevich VV. The role of arrestin alpha-helix I in receptor binding. J Mol Biol. 2010;395:42–54. doi: 10.1016/j.jmb.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV. Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem. 2011;286:24288–24299. doi: 10.1074/jbc.M110.213835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Baameur F, Findley KR, Gurevich VV. Critical role of the central 139-loop in stability and binding selectivity of arrestin-1. J Biol Chem. 2013a;288:11741–11750. doi: 10.1074/jbc.M113.450031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Chen Q, Palazzo MC, Brooks EK, Altenbach C, Iverson TM, Hubbell WL, Gurevich VV. Engineering visual arrestin-1 with special functional characteristics. J Biol Chem. 2013b;288:11741–11750. doi: 10.1074/jbc.M112.445437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Ostermaierm MK, Singhal A, Panneels V, Homan KT, Glukhova A, Sligar SG, Tesmer JJ, Schertler GF, Standfuss J, Gurevich VV. Constitutively active rhodopsin mutants causing night blindness are effectively phosphorylated by GRKs but differ in arrestin-1 binding. Cell Signal. 2013c;25(11):2155–2162. doi: 10.1016/j.cellsig.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilden U. Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry. 1995;34:1446–1454. doi: 10.1021/bi00004a040. [DOI] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- Zhan X, Gimenez LE, Gurevich VV, Spiller BW. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol. 2011;406:467–478. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang T, Chen Q, Cho M-K, Vishnivetskiy SA, Iverson TI, Gurevich VV, Hubbell WL. Involvement of Distinct Arrestin-1 Elements in Binding to Different Functional Forms of Rhodopsin. Proc Natl Acad Sci USA. 2013;110:942–947. doi: 10.1073/pnas.1215176110. [DOI] [PMC free article] [PubMed] [Google Scholar]