

Abstract
Clinical trials are the final links in the chains of knowledge and for determining the roles of therapeutic advances. Unfortunately, in an important sense they are the weakest links. This article describes two designs that are being explored today: platform trials and basket trials. Both are attempting to merge clinical research and clinical practice.
Keywords: Biomarker-driven clinical trials, Platform trials, Basket trials, Bayesian adaptive clinical trials
1.
Molecular biomarkers point the way to unraveling cancer. Biologists are cracking cancer codes at an unprecedented rate and are learning how biomarker profiles interact with therapy. Even so, future historians will view today's cancer biology as nascent. What is still unknown dwarfs the known.
Clinical trials are the final links in the chains of knowledge and for determining the roles of therapeutic advances. Unfortunately, in an important sense they are the weakest links. Moreover, as biology advances still further traditional clinical trials will increasingly limit progress. In the 1940s A. Bradford Hill designed and conducted a randomized clinical trial (RCT) of streptomycin in tuberculosis. That pioneering achievement turned medicine from anecdote and case study into legitimate science. RCTs have changed little in the last 70 years. Stretching the analogy with pioneering, the rocketships of modern biology culminate their final stage of delivery in a wagon train.
Traditional clinical trials are straightforward and purposely simple. Each trial addresses a single scientific question, one that barely scratches the surface in unraveling the nature of complex diseases such as cancer. Answering a single question is a baby step in the big picture of understanding and curing cancer. Actually, some steps are backwards because that they occupy resources that would better serve if allocated more wisely. Simple, one‐question trials were satisfactory or at least tolerable when we had a small number of therapies to investigate for a disease that was regarded to be homogeneous.
The problem is that cancer is many, many diseases. Our erstwhile taxonomy by organ of origin may have hindered progress as much as it helped. A lung cancer is more like other lung cancers than it is like breast cancer, say. But the heterogeneity of lung cancer is fundamental to understanding it. And modeling across organ sites is fundamental to curing it.
Coupled with the greater understanding of the drivers and backseat drivers of cancer is the burgeoning number of small molecules that have anti‐cancer activity. Combinations of these molecules make for an essentially infinite number of therapies—far more therapies than patients. Researchers cannot address more than a small proportion of them in clinical trials. We speak of false negatives and false positives, but both are dwarfed by false neutrals—therapies that have not been and may never be evaluated in clinical trials. Resources have been allocated traditionally to evaluating a small number of therapies, ensuring that they are neither false positives nor false negatives. Like a myopic gold miner we sift the dirt in our tiny claim while nearby mountain ranges harbor the real treasures.
In view of the huge number of and the complicated nature of pathways involved in cancer, the “gold in them thar hills” involves combinations and sequential therapies. Consider the space of single agents that have potential anti‐cancer activity, the “therapy space.” Its dimension is the number of possible single agents. Suppose 1015 just to be specific. Dose is a whole other set of dimensions, at least another 1015. Now consider the tumor/patient biomarkers that are (or may be) associated with cancer, perhaps 1010. They constitute the “patient space.” The ways in which the therapy space interacts with the patient space are hugely complicated, at least 1040. We have barely begun to learn which of the myriad of combinations of therapies are best for which patients.
Guardians of yesteryear cling to what they know and shun the new and the unknown. Tradition is a crutch, one that impedes progress. The advances in medicine made by RCTs confirm their value. But their number is too small. We must take the RCT to new levels. And we must consider a variety of approaches to clinical trials as the Brave New World unfolds. It is ironic that we take the same clinical trial approach to evaluate all manner of potentially amazing transformative experimental therapies and yet we don't experiment with the design of the clinical trial itself.
1. Experimenting with experiments
Traditional clinical trials focus on large populations. Clinical trials keep getting bigger, making therapies more expensive and delaying their availability to patients. The rub is that modern biology is slicing and dicing cancer into ever smaller subsets. Soon every cancer patient will have an ultra‐orphan disease. Traditional approaches to designing clinical trials are helpless and hopeless when evaluating therapies for diseases with an incidence of a few hundred patients per year. And yet this is the future of all cancer.
The Belmont Report of 1979 draws a clear distinction between clinical research and clinical practice, stressing that the former is “designed to test an hypothesis … and thereby to … contribute to generalizable knowledge.” (US Department of Health (1979)) “Generalizable knowledge” is that applicable to patients who have the disease and present after the trial. But the world has changed, and it continues to rapidly evolve. Traditional, large clinical trials take many years to design and run. And they can be straightjackets for clinical practice by providing answers to outdated questions. In the interim cancer biology will have asked many new questions, perhaps including which combinations of the experimental agents under investigation in the trial are appropriate for which patient subsets. These subsets will likely be defined by biomarkers that were unknown at the start of the trial.
2. Clinical trials in the Brave New World
The focus of traditional clinical trial design is hypothesis testing, including control of type I error rate and statistical power. In a typical scenario, investigators propose a clinical trial comparing an experimental arm to a control. Assuming the primary endpoint is time to a particular type of negative event they might want to be able to detect a 25% reduction in hazard with 90% power and 5% two‐sided type I error rate. Suppose the median time to event in the control arm is expected to be 3 years, accrual rate is 4 patients per month, and minimum subsequent follow‐up is 3 years then they would calculate that about 650 patients would be required.
Such a trial would report results 16 years hence, when the trial's conclusion may be irrelevant. Nobody would run such a trial. Instead they would pretend that they expect a 65% reduction for which moderate sample size would give reasonable power. It's a joke. The punchline is that the same hypothesis testing approach is the status quo for both common conditions such as hyperlipidemia and rare diseases such as high risk relapsed Wilms' tumor. It may work for the former but it does not work without pretenses such as the above for the latter. The rub is that the latter is a glimpse into the future of all cancer research. The ability to run definitive clinical trials that address questions biologists are asking is dwindling, including for diseases that were common when they were categorized by organ of origin.
Future trials in cancer (and more generally) must explicitly consider disease prevalence. They must also consider the rapidity of advances in biology and the rate at which alternative therapies are being developed. This will mean smaller, shorter, and more focused trials. The new clinical trial paradigm should have the goal of delivering good medicine to patients who have or will have the narrowly defined disease in question. Randomization will continue to play a role, albeit one that is more refined, as I will describe. But hypothesis testing's role will be at most ancillary. Trial designs in the Brave New World will be radically different. No one knows what drug development and drug regulation will be like, but dramatic change is inevitable.
In later sections I will describe some trial designs and approaches that are being investigated in the face of a burgeoning number of patient subpopulations that are ever shrinking in size. Some of these designs may presage the future. Some are radically different from the usual approaches to designing clinical trials. But they are not different enough. All the designs I consider are anchored in the past in the sense that they take a standard hypothesis testing perspective.
3. A taxonomy of modern clinical trial designs
The terminology associated with modern oncology clinical trial designs has not been well established. In particular, the term “umbrella trial” has been used to mean a variety of types of trials but it has no specificity. For example, the “umbrella” may cover agents, biomarker subtypes, or both.
A design is determined by the questions being addressed. I offer a taxonomy of trial designs in Table 1. My definitions are not generally accepted. Combinations of design characteristics are possible. For example, an indication finder may move seamlessly from phase 2 to phase 3.
Table 1.
Categorizing modern oncology clinical trial designs.
| Name | Description |
|---|---|
| Platform trial | Evaluates many therapies in a particular disease or group of diseases. Therapies usually have different sponsors and may be combinations or sequences. |
| Standing trial | Platform trial in which therapies enter and leave over time. |
| Master protocol | A trial with multiple treatment options requiring separate protocols but under the same aegis. Informed consent is usually required for both the master protocol and the respective individual protocol. |
| Indication finder | Evaluates a particular therapy across multiple cancers that are defined by organ type, or across subtypes within a specific organ type. The goal is to determine which diseases or which biomarker subtypes are appropriate for further development. |
| Basket (or bucket) trial | Evaluates the effect of a particular targeted therapy on a particular genetic or molecular aberration across cancer organ types. Variant of indication finder but the therapy is not evaluated for its off‐target effects. |
| Umbrella trial | This term may be useless because it is used for very different designs by different researchers and reporters. I use it for platform trials (many therapies) that are indication finders for each therapy. |
| Adaptive trial | Trials in which unblinded data are monitored and used to determine the future course of the trial based on prospectively defined decision rules. |
| Seamless phasesa | A particular kind of adaptive trial that moves from one phase of drug development to another without pausing accrual. Decisions at the phase switch usually involve greater focus. Examples include dropping arms, dropping doses or schedules, dropping patient subsets, changing randomization proportions, estimating the sample size for the next phase, and there are many other possibilities. (I do not include changing primary endpoint for reasons indicated in the text.) |
FDA's February 2010 draft “Guidance for Industry, Adaptive Design Clinical Trials for Drugs and Biologics” <http://www.fda.gov/downloads/Drugs/Guidances/ucm201790.pdf> is focused on phase 3 trials. It avoids the term “seamless phase 2/3” because the term provides “no additional meaning beyond the term adaptive.”
Trials that address many questions can be complicated. For example, a standing platform trial might also seek to determine an appropriate indication for each therapy that is being considered. Moreover, any design can be made adaptive, making it both more efficient and better able to answer the questions accurately (Berry, 2012).
4. Biomarker Development and Adaptive Randomization
Historically, the development of biologic therapies has taken one of two very different tacks. The first is to treat all patients with a particular organ‐specific disease and determine or validate the therapy's indication retrospectively—perhaps with an analysis that is prospectively defined in the trial's protocol. The “indication” may be the therapy's maximally responsive subpopulation. The second tack is to limit the trial's eligibility to the patients whose tumors harbor the therapy's targeted genomic or molecular aberration.
The first tack enables assessing whether the therapy's effect is indeed specific to its target, whether it has off‐target effects, and whether the marker assay used appropriately identifies the target. This information is valuable but its cost in terms of time and resources is great.
The second tack will miss off‐target effects but is more economical. In theory, if the therapy is ineffective against its target then it is unlikely to be effective against tumors harboring aberrations that are not specifically targeted. And off‐target effects can be assessed later for any therapy that is effective against its target. A problem is that there may be an inordinate delay before this information becomes available. An example is trastuzumab for breast cancer. The NSABP is conducting a trial involving 3260 adjuvant breast cancer patients whose tumor HER2 expression is “low.” The trial started in 2011 and is due to report in 2017. <https://clinicaltrials.gov/ct2/show/NCT01275677> This will be 19 years after the drug's FDA approval in HER2‐high metastatic breast cancer and 11 years after its approval in HER2‐high adjuvant disease.
Adaptive randomization is a compromise between these two extremes, one that can achieve the benefits of both with relatively small additional cost in terms of resources. Namely, the trial can have broad eligibility criteria with non‐responsive patient subgroups dropped as the trial proceeds. In a platform trial that includes many experimental therapies, a particular therapy's non‐responsive subgroup is not dropped from the trial but is gradually assigned to other therapies.
5. Platform trials
A simple device for greatly increasing the efficiency of traditional RCTs is to compare multiple experimental arms against the same control arm in a single “platform trial.” Having two experimental arms and one control in a single trial reduces total sample size by 25% in comparison with two two‐armed trials. For a trial with a greater number of experimental arms the savings approaches 50%. Not all experimental arms need be included in the randomization scheme at the start of the trial but instead arms can be added as they become available: a “standing trial.” Indeed the “trial” may be an unending screening process.
Even greater efficiency can be achieved by including a range of types of patients while adaptively identifying and/or confirming which patient/biomarker subsets—if any—benefit from which of the therapies: an “indication finder.” Another efficiency is to modify the randomization probabilities based on the data accumulating in the trial to increase the probability of assigning better performing therapies within patient subtype: an “adaptive design.” This has the effect of moving better performing therapies through the trial faster. Finally, since primary clinical endpoint information may be delayed, statistical models of tumor burden over time can enable more informed adaptive decisions about therapeutic benefits, again depending on patient subtype.
A prototypic example of the above description I‐SPY 2. (Berry, 2012; I‐SPY 2 Trial, 2015; ClinicalTrials, 2015; Barker et al., 2009) It is a phase 2 adaptively randomized “umbrella trial” in high‐risk neoadjuvant breast cancer. Specifically, it is a standing platform trial that seeks to identify each therapy's indication, or biomarker “signature.” This is subset of patients who benefit from the therapy. The primary endpoint in I‐SPY 2 is pathologic complete response (pCR), an endpoint that the U.S. FDA has come to accept for accelerated approval in neoadjuvant breast cancer. (fda, 2015; Driving Biomedical Innovation, October 2011) Moreover, to get an early read regarding the likelihood that each patient will be a pCR on her assigned therapy, we use an adaptive statistical longitudinal model of tumor burden based on MRI volume measurements at 3 weeks and 12 weeks after the start of therapy.
I‐SPY 2 considers experimental therapies in primary disease. It bucks the tradition of first using experimental therapies in metastatic disease and then, if the therapy is successful, evaluate it in primary disease. The problem with the traditional approach is that it misses effective agents that prevent metastases but have no or little effect once metastases have occurred.
Table 2 shows the 8 disease subtypes considered in I‐SPY 2. These are defined by tumor hormone‐receptor status (estrogen‐ or progesterone‐receptor positive versus neither), HER2‐positive versus not, and MammaPrint® high‐plus versus other. Table 2 also shows the 10 signatures (or indications) considered for the therapies in the trial. These are biologically plausible combinations of the 8 basic subtypes.
Table 2.
There are 8 biomarker subtypes in I‐SPY 2. These are defined by hormone‐receptor (HR), HER2, and MammaPrint® (MP) statuses. There are 10 biomarker signatures. These are combinations of the 8 subtypes as shown. Signature prevalences are estimated from I‐SPY 1, (Esserman et al., 2012) which like I‐SPY 2 focused on high‐risk primary breast cancer treated neoadjuvantly.
| Biomarker signature | Subtypes of breast cancer: HR, HER2, MP | Estimated prevalence | |||||||
|---|---|---|---|---|---|---|---|---|---|
| +++ | ++− | +−+ | +−− | −++ | −+− | −−+ | −−− | ||
| 1: All | X | X | X | X | X | X | X | X | 100% |
| 2: HR+ | X | X | X | X | 49% | ||||
| 3: HR− | X | X | X | X | 51% | ||||
| 4: HER2+ | X | X | X | X | 37% | ||||
| 5: HER2− | X | X | X | X | 63% | ||||
| 6: MP+ | X | X | X | X | 48% | ||||
| 7: −−a | X | X | 34% | ||||||
| 8: −+a | X | X | 17% | ||||||
| 9: ++a | X | X | 20% | ||||||
| 10: +−a | X | X | 29% | ||||||
Indicates either MP+ or MP−.
The numbers of patients assigned to each therapy both within tumor subtypes and overall are determined by the trial results. The totality of results determine when a therapy graduates from the trial or drops for futility. These results are used to find a Bayesian predictive probability of success in a future phase 3 clinical trial (Berry, 2006). In particular, a therapy graduates from I‐SPY 2 if the predictive probability that it will show statistical superiority over control in an equally randomized 300‐patient phase 3 trial restricted to its graduating signature(s) is at least 85%. Upon graduation accrual stops in all patient subtypes. Stopping accrual is easy to effect by setting all the therapy's randomization probabilities to 0. This happens silently with only selected individuals at the therapy's owner being informed. Announcements of graduations to the trial's investigators and the general public occur after all the patients assigned to the therapy and the concurrently randomized control patients have had surgery, which is approximately 6 months after graduation. Similarly, accrual stops for futility if the predictive probability of statistical superiority in a future trial is less than 10% for all 10 signatures.
Figure 1 is a flow chart showing the adaptive process used in I‐SPY 2.
Figure 1.
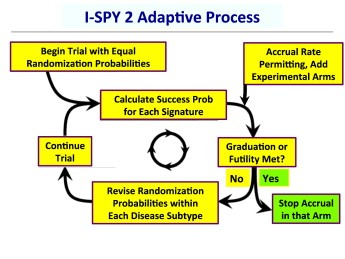
Flow chart showing the I‐SPY 2 process. “Success prob” is the Bayesian predictive probability of statistical significance in a confirmatory 300‐patient phase 3 confirmatory trial in the therapy's graduating signature.
As of this writing I‐SPY 2 has evaluated or is still in the process of evaluating 8 experimental therapies from 6 different pharmaceutical companies, and more therapies are in the queue. Two therapies have graduated to phase 3: veliparib/carboplatin with a signature of triple‐negative disease, (Rugo et al., 2013; Helwick, 2014) which is Signature 37 in Table 2, and neratinib in hormone‐receptor‐negative/HER2‐positive disease, (Park et al., 2014; Neratinib graduates, 2015) which is Signature #8 in Table 2.
Standard therapy in I‐SPY 2 is 12 weeks of paclitaxel followed by 4 cycles of doxorubicin/cyclophosphamide. Patients with HER2‐positive tumors receive trastuzumab in addition to paclitaxel. The neratinib arm was similar except that neratinib was given in addition to paclitaxel irrespective of HER2‐status and in lieu of trastuzumab for patients with HER2‐positive tumors.
An example of the point in section “Biomarker Development and Adaptive Randomization” above is that the neratinib arm performed well in comparison with control in HER2‐positive subsets but not very well in HER2‐negative subsets. The adaptive randomization algorithm “learned” to focus neratinib on HER2‐positives. In the latter part of its tenure in the trial the randomization probability of neratinib became 0 for both HER2‐negative, MP‐negative subsets. When neratinib graduated it had been assigned to a total of 115 patients. As a consequence of the algorithm focusing on neratinib for patients with HER2‐positive tumors, especially those with HER2‐positive/HR‐negative tumors, 65 (57%) of these 115 were HER2‐positive. In contrast, of the 78 controls that had been concurrently randomized versus neratinib only 22/78 (28%) were HER2‐positive (Park et al., 2014). This focus on HER2‐positives had the benefit of providing more and faster information about neratinib for those subsets of patients where it was effective and it had an obvious benefit for patients with HER2‐negative tumors who were not assigned to that arm.
The I‐SPY 2 concept is being applied in other cancers and in other diseases as well, including Alzheimer's disease, diabetes, acute respiratory infections, anti‐infective agents, autoimmune diseases, and Parkinson's disease (Mullard, 2014; Goldman et al., 2015).
An early platform trial in cancer, and perhaps the first umbrella trial, was BATTLE (Kim et al., 2011; Berry et al., 2012). It employed a master protocol and individual protocols for the 4 treatment arms. It randomized 255 lung cancer patients at M.D. Anderson Cancer Center from 2006 to 2009. BATTLE shared some characteristics with I‐SPY 2. In particular, both trials had Bayesian designs, both used adaptive randomization, and both sought to determine effective therapies in biomarker‐defined disease subsets. There were differences as well, perhaps most noteworthy was that BATTLE did not have a control arm (Rubin et al., 2011). However, it did compare the various treatments within the 5 predefined biomarker subsets.
Table 3 gives a partial list of platform trials in oncology. The trials have different goals. Most are phase 2 trials. Lung‐MAP is a seamless phase 2/3 trial in squamous cell lung cancer. (LUNG‐MAP, January 2015; Lung‐MAP, January 2015) It is similar to BATTLE in considering 5 biomarker subsets including one “other” category, although the biomarkers and agents are different from those in BATTLE. The targeted agents are from 4 different pharmaceutical companies. Patients in the “other” category have none of the mutations under consideration and are randomized to an anti‐PD‐L1 agent provided by a 5th company.
Table 3.
Partial list of oncology platform clinical trials that are active, completed, or under development, showing characteristics described in the text.
| Characteristics | Trials | |||||||
|---|---|---|---|---|---|---|---|---|
| I‐SPY 2a | BATTLEb | Lung‐MAPc | UK Matrixd | NCI‐MATCHe | NCI‐MPACTf | IMPACT 2g | MICATh | |
| Screen all patients for markers | X | X | X | X | X | X | X | X |
| Master protocol | X | X | X | X | X | X | ||
| Drugs from many companies | X | X | X | X | X | X | X | X |
| Treats all comers | X | X | X | X | ||||
| Combination therapies | X | X | ? | X | X | |||
| Sequential therapies | X | |||||||
| Regimens enter & leave trial | X | X | X | |||||
| Learn off‐target effects | X | X | X | |||||
| Pair regimens with biomarkers | X | X | X | |||||
| Common control arm | X | X | X | |||||
| Adaptive randomization | X | X | X | X | ||||
| Adaptive sample size | X | X | X | |||||
| Early “curable” disease | X | |||||||
| Registration endpoint | X | X | X | X | ? | |||
| Longitudinal disease modeling | X | X | ||||||
| Seamless phases 2/3 | X | |||||||
Berry, 2012; I‐SPY 2 Trial, 2015; ClinicalTrials, 2015; Barker et al., 2009.
Kim et al., 2011; Berry et al., 2012.
LUNG‐MAP, January 2015; Lung‐MAP, January 2015.
Cancer Research UK Science, 2015.
NCI‐MATCH, January 2015.
NCI‐MPACT, January 2015.
IMPACT 2, January 2015.
MICAT, January 2015.
Lung‐MAP is an experiment in using biomarkers and targeted therapies. Its master protocol is a device for triaging patients into 4 or 5 substudies with separate protocols. The individual substudies have traditional designs, with distinct control groups. There is no relationship between the different trials except for the triaging aspect.
In each of the substudies that make up Lung‐MAP the phase‐2 endpoint is PFS and the phase‐3 endpoint is OS. Patients accrued in the phase‐2 portion also contribute to OS in phase 3. This is especially apt because a “phase‐2 patient” has longer follow‐up time and so contributes more information regarding OS than does a patient accrued during phase 3. However, the design does not model the relationship between PFS and OS. Lung‐MAP is not adaptive except that a predetermined cutpoint of the PFS hazard ratio is used at a predetermined time for deciding whether to shift into phase 3. The trial's performance and efficiency could be improved, for example, by modeling OS as the sum of PFS and SPP (survival post‐progression) depending on treatment arm and sizing the phase‐3 portion of the trial based on the PFS/OS results observed during the trial, an adaptive characteristic (Broglio and Berry, 2009).
Lung‐MAP is not adaptive and there are no cross‐substudy comparisons. Its substudies have different control groups (although most have the same treatment, docetaxel). Its approach may not be sustainable. For example, when the trial was just getting started in March 2015, the FDA approved the drug nivolumab based on a trial in the same population of patients as Lung‐MAP's in which the drug had demonstrated a substantial improvement in overall survival in comparison with docetaxel. As of this writing the trial's steering committee is working to salvage the trial but its future is uncertain.
The National Lung Matrix Trial in late stage non‐small‐cell lung cancer is a collaboration of Cancer Research UK, UK National Health Service, and pharmaceutical companies AstraZeneca and Pfizer. It too is a device for triaging into independent cohorts but at an earlier phase of development than Lung‐MAP. The goal of this uncontrolled “signal‐finding trial” is to evaluate experimental targeted agents for tumors with specific genetic or molecular aberrations. The number of therapies is variously reported to be between 8 and 14, the latter including 12 from AstraZeneca and 2 from Pfizer (Torjesen, 2014; Harrison, 2014). An agent that “shows promise” in 15–30 patients will lead to a separate, larger trial. Specifics of the UK Matrix design are not publicly available.
MICAT is the Melanoma International Consortium for Adaptive Trials. As indicated in Table 3, a highlight of the trial is its evaluation of sequential use of therapies as well as therapies in combination. The trial is under development and is not yet accruing patients.
NCI‐MATCH uses Next Generation Sequencing of solid tumors and lymphoma to identify patients with molecular aberrations who have progressed on at least one therapy. Patients are excluded if they have an aberration and tumor type for which a targetable agent has been FDA‐approved. Just as for Lung‐MAP and UK Matrix the MATCH master protocol involves triaging, with no borrowing across sub‐protocols. There will be up to 25 sub‐protocols, each of which will be traditional phase 2 single‐arm assessments with approximately 30 patients irrespective of the organ types of the tumors. The reason for question mark in Table 3 is that combinations are possible but there are no specific plans as yet for such therapies. The primary endpoint of each sub‐protocol is overall response rate, with power to distinguish between 25% and 5% (NCI‐MATCH, January 2015; Abrams et al., 2014). So each sub‐protocol is a basket trial in which all patients are regarded to be exchangeable across tumor types, and in particular as having the same underlying response rate irrespective of tumor type. (The next section deals with basket trials and does not make this highly questionable assumption.)
MPACT is an ongoing “pilot trial” considering four targeted therapies within three molecular pathways. The objective is to test the targeting strategy. The co‐primary endpoints are response rate (CR + PR) and PFS (NCI‐MATCH, January 2015). The goal is to have 180 patients overall, with 2:1 stratified randomization within molecular pathway, favoring targeted therapy.
IMPACT 2 is a single‐institution trial that was motivated by a non‐randomized comparison of phase‐1 patients of any tumor type treated with targeted agents whose cancers harbored the targets with matching patients who were not treated with a targeted agent. (Tsimberidou et al., 2012) IMPACT 2 employs randomization to targeted versus non‐targeted therapy in a phase‐1 setting similar to NCI‐MPACT, but with a much broader range of what may be targetable, similar to NCI‐MATCH. The sample size is 200 patients, with the first 100 assigned using balanced randomization. Subsequent patients will be assigned using adaptive randomization within the subtypes of patients defined by biomarker type and tumor type. The analysis and the adaptive features of the design uses Bayesian hierarchical modeling, an extension of the modeling presented in the next section.
6. Basket trials
As indicated in Table 1, basket trials evaluate the effects of a particular targeted therapy on a particular genetic or molecular aberration across organ types. Even though the patients have the same biomarker, the sensitivity of their diseases may depend on the tumor site. Moreover, different tumor types may have different historical response rates because standard therapies and their effectiveness differ by disease.
Simply pooling trial results across tumor types as in NCI‐MATCH is not reasonable. For example, two trials with identical results by tumor type could have very different overall response rates because they included different proportions of the various tumor types.
In the other extreme, it makes no sense to regard two different tumor types that have the same molecular aberration as being completely distinct entities. The two tumor types may respond similarly to targeted therapy. Suppose one tumor type has a 15% historical response rate and a second type has a 30% historical rate. Suppose there are 9 (60%) responders of 15 patients in the first type and 6 (30%) responders of 20 patients in the second type. The sample sizes are small. But doubling both historical rates is more suggestive of an improvement in both types than when viewing the treatment effects in the two types separately. Put another way, the confidence intervals of treatment effects in both tumor types should be narrow to account for the similarity of treatment effects in the two tumor types.
In another type of example, suppose three tumor types with the same historical rate of 20%, suppose there are, respectively, 12, 8, and 8 responders out of 20 patients of each type. Given the small sample sizes the observations 60%, 40%, and 40% are not very different, and the three true rates are probably closer together than the observed rates. The first one might be 55%, say, and the other two might be 43%.
This kind of estimation is called “shrinkage” or “regression to the mean.” It has a surprisingly powerful characteristic. In the 1950s Charles Stein showed that when there are at least 3 groups shrinkage estimates improve the naïve approach of no shrinkage regardless of the true state of nature (Stein, 1956; James and Stein, 1961). In particular, the usual (no shrinkage) estimator of group means has greater mean squared error regardless of the true values. Expressed colloquially, if someone goes through life and never shrinks their estimates, there are a countless number of people who do shrink and whose estimates are ordained to be better overall regardless of the underlying state of nature.
The shrinkage approach has led to Bayesian hierarchical modeling in which there is “partial borrowing” of results in the various groups, and where the amount of borrowing is greater across groups that have similar results. This method has been shown to enable building adaptive “indication‐finder” clinical trials that are efficient in terms of saving resources while at same time providing better estimates (Berry et al., 2013). The method applies for randomized trials and other types of endpoints, including times to an event.
Figure 2 shows hypothetical results for a clustering hierarchical borrowing method that is being used in industry basket trials. It exhibits regression toward the overall mean as well as toward the cluster means for positives and negatives separately.
Figure 2.
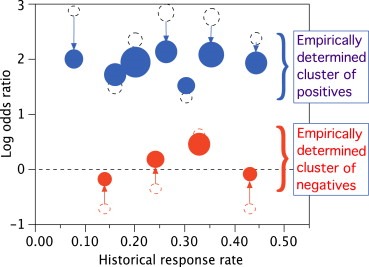
Open dashed circles show raw response rates for 11 tumor types. Solid circles show estimates adjusted for hierarchical borrowing from other tumor types and accounting for clustering. The area of each open circle is proportional to the actual sample size, which varies by a factor of greater than 2 across the tumor types. The area of each solid circle is proportional to the “equivalent sample size,” which is increased to demonstrate the greater precision of estimation that is effected by hierarchical borrowing. Raw estimates for tumor types with smaller sample sizes, further from the overall mean, and further from the cluster mean are regressed further. Tumor types with raw estimates further from the cluster mean borrow less and therefore have less increase in precision. (Results are hypothetical and effects are exaggerated to demonstrate the methodology.)
7. Discussion, with an eye on the future
The designs of clinical trials in oncology are changing. But the fundamental approach and the trial design criteria are traditional. This is good and bad. We must build on what we have learned. But we cannot stop learning and building ever better trials, and trials suited to the changing landscape of disease and possible therapy. Clinical trialists must work to meet the challenges of modern cancer biology even though it's not clear how to best do that. The answer lies in experimenting with different approaches.
This article describes two designs that are being explored today: platform trials and basket trials. Both are attempting to merge clinical research and clinical practice. But neither is the final solution, and there probably is not a final solution. We must learn to synthesize evidence from all sources, including biological knowledge as well as information from randomized trials, preclinical studies, and patient databases. Such synthesis will be essential as clinical trials become smaller than today's by orders of magnitude. Large clinical trials in narrowly defined diseases are impossible. And all cancers are becoming increasingly narrowly defined.
No one understands the regulatory model in the Brave New World. And no one understands the corresponding business model for pharmaceutical companies. All that is clear is that both will be different from today's. The statistical design of clinical trials will also be different. Type I and type II error probabilities will fade out of existence and be replaced with an explicit goal of delivering effective therapy for patients who have the disease. The distinction between clinical practice and clinical trials will become even more blurred than it is now.
Berry Donald A., (2015), The Brave New World of clinical cancer research: Adaptive biomarker‐driven trials integrating clinical practice with clinical research, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.02.011.
This is a contribution to the special issue edited by Drs. Ringborg, Mendelsohn and Schilsky.
References
- Abrams, J. , Conley, B. , Mooney, 2014. National Cancer Institute's precision medicine initiatives for the new National Clinical Trials Network. Am. Soc. Clin. Oncol. Educ. Book. 71–76. [DOI] [PubMed] [Google Scholar]
- Barker, A.D. , Sigman, C.C. , Kelloff, G.J. , 2009. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin. Pharmacol. Ther. 86, 97–100. [DOI] [PubMed] [Google Scholar]
- Berry, D.A. , 2006. A guide to drug discovery: Bayesian clinical trials. Nat. Rev. Drug Discov. 5, 27–36. 10.1038/nrd1927 [DOI] [PubMed] [Google Scholar]
- Berry, D.A. , 2012. Adaptive clinical trials in oncology. Nat. Rev. Clin. Oncol. 9, 199–207. [DOI] [PubMed] [Google Scholar]
- Berry, D.A. , Herbst, R.S. , Rubin, E.H. , 2012. Design strategies for personalized therapy trials. Clin. Cancer Res. 18, 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry, S.M. , Broglio, K.R. , Groshen, S. , Berry, D.A. , 2013. Bayesian hierarchical modeling of patient subpopulations: efficient designs of phase II oncology clinical trials. Clin. Trials. 10, 720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broglio, K.R. , Berry, D.A. , 2009. Detecting an overall survival benefit that is derived from progression-free survival. J. Natl. Cancer Inst. 101, 1642–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01042379 (accessed 1 January 2015).
- Driving Biomedical Innovation: Initiatives to Improve Products for Patients. October 2011. http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UCM274464.pdf (accessed 1 January 2015).
- Esserman, L.J. , Berry, D.A. , DeMichele, A. , 2012. Pathologic complete response predicts recurrence-free survival more effectively by cancer subset: results from the I-SPY 1 TRIAL—CALGB 150007/150012, ACRIN 6657. J. Clin. Oncol. 30, 3242–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, M. , Seigneuret, N. , Eichler, H.-G. , 2015. The innovative medicines initiative: an engine for regulatory science. Nat. Rev. Drug Discov. 14, 1–2. 10.1038/nrd4520 [DOI] [PubMed] [Google Scholar]
- Harrison, C. , 2014. Trial watch: lung cancer trial aims to bolster personalized medicine. Nat. Rev. Drug Discov. 13, (407) 10.1038/nrd4341 http://www.nature.com/nrd/journal/v13/n6/full/nrd4341.html?message-global=remove (accessed 1 January 2015) [DOI] [PubMed] [Google Scholar]
- Helwick, C. , March 1, 2014. Innovative I-SPY 2 trial yields first results in triple-negative breast cancer. ASCO Post. 5, (4) [Google Scholar]
- http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm305501.pdf (accessed 1 January 2015).
- I-SPY 2 Trial, http://ispy2.org (accessed 1 January 2015).
- IMPACT 2: Randomized Study Evaluating Molecular Profiling and Targeted Agents in Metastatic Cancer. https://www.clinicaltrials.gov/ct2/show/NCT02152254 (accessed 1 January 2015).
- James, W. , Stein, C. , 1961. Estimation with quadratic loss. Proc. Fourth Berkeley Symp. Math. Statist Prob. 1, 361–379. [Google Scholar]
- Kim, E.S. , Herbst, R.S. , Wistuba, 2011. The BATTLE Trial: personalizing therapy for lung cancer. Cancer Discov. 1, OF42–OF51. 10.1158/2159-8274.CD-10-0010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUNG-MAP. A groundbreaking collaborative approach to clinical trials. http://www.lung-map.org (accessed 1 January 2015).
- Lung-MAP: S1400 Biomarker-Targeted Second-Line Therapy in Treating Patients With Recurrent Stage IIIB-IV Squamous Cell Lung Cancer. https://clinicaltrials.gov/ct2/show/NCT02154490 (accessed 1 January 2015).
- MICAT. International Activities of NCI-Designated Cancer Centers. http://www.cancer.gov/aboutnci/organization/global-health/resources/International-Activities-of-NCI-Designated-Cancer-Centers.pdf (accessed 1 January 2015).
- Mullard, A. , 2014. Multicompany trials adapt to disciplines beyond cancer. Nat. Med. 20, 3 10.1038/nm0114-3 [DOI] [PubMed] [Google Scholar]
- NCI-MPACT: Molecular Profiling-Based Assignment of Cancer Therapy for Patients With Advanced Solid Tumors. https://clinicaltrials.gov/ct2/show/NCT01827384 (accessed 1 January 2015).
- Neratinib graduates to I-SPY 3. Cancer Discovery, OnlineFirst April 7, 2014;http://dx.doi.org/10.1158/2159-8290.CD-NB2014-055; http://cancerdiscovery.aacrjournals.org/content/early/2014/04/04/2159-8290.CD-NB2014-055.full (accessed 1 January 2015).
- NCI Molecular Analysis for Therapy Choice Program (MATCH) & Pediatric MATCH. http://www.cancer.gov/clinicaltrials/noteworthy-trials/match (accessed 1 January 2015).
- Park, J.W. , Liu, M.C. , Yee, D. , 2014. Neratinib plus standard neoadjuvant therapy for high-risk breast cancer: efficacy results from the I-SPY 2 TRIAL. Proceedings of the 105th Annual Meeting of the American Association for Cancer Research; 2014 Apr 5–9; San Diego, CA. Philadelphia (PA). AACR; Abstract nr CT227 http://www.abstractsonline.com/Plan/ViewAbstract.aspx?mID=3404&sKey=ed5341ec-ef13-493e-8844-e14be322679c&cKey=d9b44ad7-1673-4ec9-b360-11254fbb92be&mKey=6ffe1446-a164-476a-92e7-c26446874d93 (accessed 1 January 2015) [Google Scholar]
- Rubin, E.H. , Anderson, K.M. , Gause, C.K. , 2011. The BATTLE trial: a bold step toward improving the efficiency of biomarker-based drug development. Cancer Discov. 1, 17–20. 10.1158/2159-8274.CD-11-0036 [DOI] [PubMed] [Google Scholar]
- Rugo HS, Olopade O, DeMichele A, et al. Veliparib/carboplatin plus standard neoadjuvant therapy for high-risk breast cancer: First efficacy results from the I-SPY 2 trial. 2013, San Antonio Breast Cancer Symposium. Abstract S5–02. Presented December 13, 2013. http://sabcs.org/Documents/2013SABCSProgramWeb.pdf (accessed 1 January 2015).
- Stein, C. , 1956. Inadmissibility of the usual estimator for the mean of a multivariate normal distribution. Proc. Third Berkeley Symp. Math. Statist Prob. 1, 197–206. [Google Scholar]
- Stratified medicine and the lung cancer ‘Matrix’ trial – part of a cancer care revolution. Cancer Research UK Science Blog. http://scienceblog.cancerresearchuk.org/2014/04/17/stratified-medicine-and-the-lung-cancer-matrix-trial-part-of-a-cancer-care-revolution/(accessed 1 January 2015).
- Torjesen, I. , 2014. Large personalised medicine trial in lung cancer heralds new research partnership. BMJ. 348, g2837 http://www.bmj.com/content/348/bmj.g2837 (accessed 1 January 2015) [DOI] [PubMed] [Google Scholar]
- Tsimberidou, A. , Iskander, N.G. , Hong, D.S. , 2012. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center Initiative. Clin. Cancer Res. 18, 6373–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Department of Health, Education, and Welfare. The Belmont Report: Ethical principles and guidelines for the protection of human subjects of research, hhs.gov/ohrp/humansubjects/guidance/belmont.html (1979, accessed 1 January 2015). [PubMed]