

Abstract
The mitochondrion is a vital intracellular organelle for retinal cell function and survival. There is growing confirmation to support an association between mitochondrial dysfunction and a number of retinal degenerations. Investigations have also unveiled mitochondrial genomic instability as one of the contributing factors for age-related retinal pathophysiology. This review highlights the role of mitochondrial dysfunction originating from oxidative stress in the etiology of retinal diseases including diabetic retinopathy, glaucoma and age-related macular degeneration (AMD). Moreover, mitochondrial DNA (mtDNA) damage associated with AMD due to susceptibility of mtDNA to oxidative damage and failure of mtDNA repair pathways is also highlighted in this review. The susceptibility of neural retina and retinal pigment epithelium (RPE) mitochondria to oxidative damage with ageing appears to be a major factor in retinal degeneration. It thus appears that the mitochondrion is a weak link in the antioxidant defenses of retinal cells. In addition, failure of mtDNA repair pathways can also specifically contribute towards pathogenesis of AMD. This review will further summarize the prospective role of mitochondria targeting therapeutic agents for the treatment of retinal disease. Mitochondria based drug targeting to diminish oxidative stress or promote repair of mtDNA damage may offer potential alternatives for the treatment of various retinal degenerative diseases.
Keywords: Mitochondria, Diabetic retinopathy, Glaucoma, Retinal degeneration, Age-related macular degeneration
INTRODUCTION
The mitochondrion is a critical organelle for cell function and survival. Its primary roles are adenosine triphosphate (ATP) production, control of cellular metabolism and regulation of apoptosis (programmed cell death). It consists of inner and outer membranes composed of phospholipid bilayers containing numerous integral proteins. Mitochondrial density varies among different cell types and is expressed abundantly in highly metabolic active cell types such as retinal pigment epithelium (RPE).1 Due to its critical functioning, mitochondrial dysfunctions may severely affect tissue homeostasis. Oxidative damage induced mitochondrial dysfunction has been proposed as a most prevalent ageing theory.1–4 Furthermore, involvement of mitochondrial DNA (mtDNA) damage and mitochondrial theory of ageing has been observed in age-related macular degeneration (AMD).5
Acute and chronic mitochondrial dysfunctioning is associated with a number of age-related degenerative diseases including Parkinson’s, Alzheimer’s and AMD. In this review, we will summarize recent developments in understanding the role of the mitochondrion as a weaker link in the antioxidant defenses of retinal cells and a potential contributor to the pathogenesis of retinal degenerations such as diabetic retinopathy, glaucoma and AMD. We will further highlight the mechanistic basis of mtDNA damage associated to AMD. Moreover, the potential role of mitochondria targeting agents for the treatment of retinal diseases will be outlined at the end of the review.
Diabetic Retinopathy
Diabetic retinopathy is the most prevalent microvascular complication of diabetes, and remains a leading cause of vision loss in many developed countries. The development of this microvascular disease occurs gradually and silently in as many as 50% of type I and 10% of type II diabetic patients within 15 years of diagnosis.6,7 Chronic hyperglycemia and other risk factors (hypertension, dyslipidaemia) are believed to trigger a cascade of biochemical and physiological changes that lead to microvascular damage and retinal dysfunction. The retinal manifestations of diabetes mellitus are broadly classified as either non-proliferative diabetic retinopathy (NPDR) or proliferative diabetic retinopathy (PDR). The progression of NPDR, which covers only intraretinal microvascular changes, starts from mild non-proliferative abnormalities (altered retinal vascular permeability) to moderate and severe NPDR (vascular closure). The eventual progression of PDR is characterized by new blood vessel formation and sometimes fibrous band proliferation on the retinal surface. Macular edema, characterized by retinal thickening from leaky blood vessels, can develop in both stages of retinopathy due to increased retinal vascular permeability which leads to fluid accumulation in the retina.8–10
Various hyperglycemia-induced metabolic abnormalities, including increased activity of the polyol pathway, advanced glycation end products (AGEs) and protein kinase C (PKC) activation are implicated in the progress of diabetic retinopathy. These metabolic pathways are considered to be interconnected and may mediate oxidative stress. Elevated oxidative stress plays a major role in the pathogenesis of diabetic complications.11,12 Oxidative stress is the accumulation of reactive oxygen species (ROS) beyond the capacity of a cell to defend, because of increased generation or impaired removal of ROS.13 The presence of a high content of polyunsaturated fatty acids, high oxygen uptake and glucose oxidation renders the retina more susceptible to oxidative stress relative than any other tissue.14 Critical events suggested in the pathogenesis of diabetic retinopathy are hyperglycemia, changes in the redox homeostasis and oxidative stress. Increased oxidative stress is reported in diabetic retina of animal models (diabetic and galactosemic rats).15 Elevated retinal levels of lipid peroxides, superoxide and hydrogen peroxide and down-regulation of the mRNA of the enzymes responsible for scavenging superoxide, superoxide dismutase (SOD), and glutathione reductase were reported in diabetic rat and mouse models.16–18 A decreased level of intracellular antioxidant (GSH) and impairment of antioxidant defense enzymes in the retina was also reported in diabetic rats.19–20 Furthermore, increased oxidative stress is also observed in bovine retinal endothelial cells (BREC) and pericytes incubated in high glucose medium and in other non-vascular retinal cells including transformed retinal Muller cells (rMC-1) and photoreceptors.16,21 These experiments clearly suggested an important role of oxidative stress in the development of retinopathy in diabetes. Moreover, animal studies have confirmed that oxidative stress not only contributes to the development of diabetic retinopathy but also offers resistance to reversal of the conditions after glycemic control.22 However, the mechanism by which oxidative stress can contribute to the pathogenesis of diabetic retinopathy remains to be elucidated.
Mitochondrial Dysfunction in Diabetic Retinopathy
Mitochondria are considered to be the prime endogenous source of superoxides, peroxynitrite, and hydroxyl radicals, and are also a target for damaging effect of oxidants, suggesting the existence of a vicious cycle of oxidative damage.23 Chronic overproduction of ROS in the retina results in abnormal mitochondrial functions in diabetes (Figure 1).13,14,24 Inhibition of antioxidant scavengers is one of the ROS-induced dysfunctions in mitochondria that may lead to enhanced sensitivity of retinal cells to oxidative stress. The isoform of SOD in the mitochondria (MnSOD) along with GSH may be suppressed in the diabetic and high glucose-cultured retinal mitochondria.25–27 Increased mitochondrial DNA damage has been reported in the diabetic retina of an animal model.28 ROS mediated damage to the mitochondrial lipid membrane enhances permeability of the mitochondrial membrane which represents another cellular dysfunction caused by ROS. Increased swelling of the mitochondria was observed in the retina of diabetic mice.26 It is widely accepted that apoptosis of retinal cells is the most common incident in diabetic retinopathy. Kowluru et al. have demonstrated that the release of cytochrome c from mitochondria to the cytoplasm and Bax translocation from the cytoplasm to mitochondria that could drive cell apoptosis were increased in retinal capillary cells in diabetes.29 Thus, it is evident that mitochondria play a crucial role in the development of retinopathy in diabetes, and oxidative stress can modulate mitochondrial cytochrome c release resulting in increased retinal capillary cell death.
FIGURE 1.

Hyperglycemia mediated mitochondrial dysfunctioning in diabetic retinopathy.
Diabetic Retinopathy Treatments Targeted to Mitochondrial Dysfunction
Diabetic retinopathy is a complex disease which is difficult to treat due to its multifactorial nature. An efficient therapy will probably be a combination of drugs targeting multiple pathways involved in the pathogenesis of diabetic retinopathy. Since the disease pathogenesis is partially attributed to mitochondrial dysfunction, treatment options should also consider restoring normal mitochondrial function. Oxidative stress is the significant instigator of hyperglycemia-induced mitochondrial damage. Therefore, treatment strategies to improve mitochondrial function by lowering oxidative stress may be an appropriate alternative.15,24 Sheu et al. has described a number of antioxidants which can be transported into mitochondria but only a few have been tested in diabetic retinopathy.30 Considering the role of superoxides in the development of diabetic retinopathy, SOD mimetics may represent a class of treatment to counter the effect of oxidative stress. MnSOD mimics are low-molecular weight, manganese containing, non-peptide molecules with similar function, and catalytic rates of native SOD enzymes. Kowluru et al.24,29 have shown that MnTBAP [manganese (III) meso-tetrakis (4-carboxypheny) porphyrin], a MnSOD mimic, can significantly inhibit glucose-induced decrease in GSH levels and translocation of cytochrome c from mitochondria and Bax into the mitochondria of retinal capillary cells. It can also inhibit apoptosis by suppressing caspase-3 activation.24,29
It is therefore essential to recognize treatment strategies that could inhibit superoxide production which might represent a possible direction for clinical research in diabetes. Although the mimetics appear to be very promising, it is not known whether any of these mitochondria-targeted treatments are beneficial in diabetic retinopathy. Furthermore, pharmacokinetics and route of administration also need to be addressed. Additional animal studies and clinical trials can resolve such issues.15,24
Glaucoma
Glaucoma is a neurodegenerative disease of the optic nerve characterized by the accelerated death of retinal ganglion cells (RGCs) and their axons which ultimately leads to progressive vision loss. Elevated intraocular pressure (IOP) and age are key risk factors for glaucoma. Reducing IOP is the only current treatment option available to retard glaucoma progression in clinical practice. However, control of IOP by itself may not be sufficient to arrest the progression of glaucoma and strategies that compliment IOP control for protecting the optic nerve are required.31,32
MITOC HONDRIA L DYSFUNCTION IN GLAUCOMA
Mitochondrial dysfunction is believed to contribute to the pathogenesis of a number of neurodegenerative disorders. Glaucoma has similar etiology to other neurodegenerative diseases, such as selective loss of a single neuronal cell population, age related incidence, and neuronal cell death. The dense distribution of mitochondria around the optic nerve head, the primary site of glaucomatous axonal injury, reflects a high requirement for ATP.31,32 Ju et al.33 have recently demonstrated the induction of mitochondrial fission, abnormal cristae depletion, and cellular ATP reduction in differentiated RGC-5 cells exposed to high hydrostatic pressure over 3 days. This observation suggests that elevated pressure, a major risk factor in glaucoma, may disrupt mitochondrial structure and function in RGCs, possibly leading to apoptosis (Figure 2).31,33 An association between glaucoma and mitochondrial dysfunction has also been suggested in a recent clinical study where a 20% reduction in mitochondrial respiratory function and an increase in mitochondrial DNA mutation were observed in peripheral blood of primary open-angle glaucoma patients relative to an age-matched control group.34 The mitochondrion is a key regulator of apoptosis and is implicated as the prime factor responsible for neuronal loss in neurodegenerative diseases. Evidence has also suggested that apoptosis is an important mechanism of cell death in glaucoma neurodegeneration. RGC apoptosis occurs in animal glaucoma models and in the retina of glaucoma patients.35,36 There is growing evidence to support involvement of mitochondria-mediated ROS-induced RGC injury. ROS generation is noticed in the retina and optic nerve of glaucoma animal models and in retinal ischemia reperfusion models.37,38 Furthermore, human studies have also confirmed the involvement of mitochondria-mediated oxidative stress in glaucoma due to up-regulation of various antioxidant proteins (serum auto-antibodies, glutathione S-transferase and iron-regulating proteins).39,40
FIGURE 2.
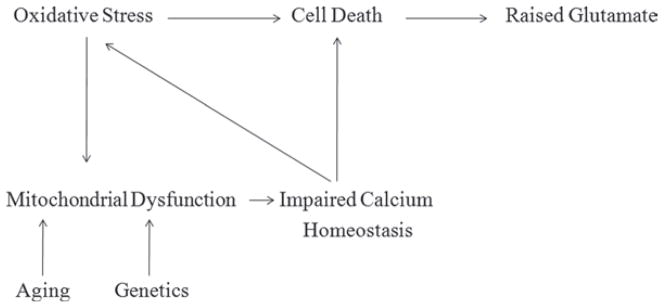
Involvement of mitochondrial dysfunctioning in glaucoma pathogenesis.
GLAUCOMA TREATM ENTS TARGETED TO MITOC HONDRIAL DYSFUNCTION
As a potential neuroprotective therapy, various antioxidants have been studied in neurodegenerative diseases with partial success. Advances have also been made in targeting antioxidants directly to the mitochondria, a major site of ROS production, by conjugating antioxidants to lipophilic cations.30,41 The mitochondria-targeted cationic plastoquinone derivative SkQ1 (10-(6′-plastoquinonyl) decyltriphenylphosphonium) has been investigated as a prospective tool for treating experimental glaucoma induced by serial injections of 2% hydroxypropyl methyl cellulose to the anterior segment of the rabbit eye. Once, daily drops of 5 μM SkQ1 caused a reduction in glaucomatous changes.42 SkQ1 appears to be a promising candidate for treating glaucoma. Involvement of mitochondria in glaucoma pathogenesis might therefore represent a new therapeutic target for protecting the optic nerve and preventing vision loss.
AGE-RELAT ED MACULAR DEGENERATION
AMD is a progressive neurodegenerative disease that primarily affects the central region of the retina (macula) and is a leading cause of blindness in the elderly. The presence of macroscopically visible soft drusen, with areas of hyper- or de-pigmentation are characteristic early symptoms of AMD, whereas atrophy of photoreceptors and RPE or choroidal neovascularization are evident during later stages of the disease.43–47
MITOC HONDRIA L DYSFUNCTION IN AMD
Strong evidence showing mitochondrial dysfunction in AMD has been reported. An association between AMD and a variant of mitochondrial protein (LOC387715/ARMS2 (age-related maculopathy susceptibility 2)) has been identified.48 In AMD genetic variants at two chromosomal loci, 1q32 and 10q26 confer major disease risks. A consensus from multiple studies is that the 1q32 and 10q26 region simultaneously harbors a first and second major genetic determinant of AMD susceptibility.49–52 Previous studies have identified chromosome 1q32 as harboring a susceptibility locus (complement factor H) for AMD which does not have any connection to the mitochondrion.49,50 However, signals at 10q26 overlap two nearby genes, LOC387715/ARMS2 (age-related maculopathy susceptibility 2) and HTRA1/PRSS11 (high-temperature requirement factor A1). The LOC387715/ARMS2 gene produces a protein of unknown function that localizes in the mitochondria, and polymorphisms in LOC387715/ARMS2 alter the risk of AMD by modulating the function of this gene.48,51–53 Furthermore, changes in the activity or regulation of LOC387715/ARMS2 are likely to be responsible for the impact on AMD disease susceptibility.53
Retinal pigment abnormalities and RPE atrophy similar to those present in the early AMD phenotypes are detected in 75% of individuals with the MELAS A3243G mitochondrial DNA mutation.54 mtDNA haplogroups which are associated with either increased or decreased prevalence of age-related maculopathy have been identified.55 So far there is no report available which confirms the association of similar mtDNA haplotypes with AMD. Based on this finding one can further explore the possible involvement of mtDNA haplogroups in AMD.
In addition, the oxidative stress hypothesis of AMD proposes that cumulative oxidative damage to proteins, lipids, and DNA also leads to disease progression. Changes in selected redox proteins (an indicator of increased oxidative stress) and altered protein expression reflecting impaired mitochondrial biogenesis were found in human donor eyes with the progression of AMD.56,57 These findings led to the proposition that bioenergetic consequences of mtDNA derangements may be expressed in macular RPE as a maculopathy and contribute to the development of AMD. Evidence of mitochondrial dysfunction from human tissue examination and animal models has also been reported. Feher et al.58 revealed through morphometric studies a significant diminution in number and area of RPE mitochondria as well as loss of cristae and matrix density with age in AMD specimens. These decreases were significantly greater in AMD than in normal aging. This study has further confirmed that besides changes in mtDNA, alterations of mitochondrial membranes may also play a crucial role in the development of mitochondrial dysfunctions in AMD.58
Despite the evidence of mitochondrial dysfunction in AMD where emphasis is focused on the RPE, only a few studies have been focused the role of mtDNA damage and repair in the retina. Barreau et al. has identified mtDNA deletions in aged human retina.59 Increased mtDNA damage and decreased repair, along with reduced mitochondrial respiration in RPE and choroid of rodents are also observed with progression of ageing.60–62 Thus, mtDNA damage in RPE is associated with aging and may be a susceptibility factor in the development of AMD.
Cell culture studies have shown damage to mtDNA but not to nuclear DNA (nDNA) of human RPE cells when exposed to oxidizing or alkylating agents.63 Furthermore, nDNA damage repair appears to be rapid relative to mtDNA repair in the RPE, which appears to be slow and relatively inefficient. The lack of evidence for mtDNA repair in response to oxidative stress further suggests that the mitochondrion is a weaker link in the RPE cell’s defense against oxidative damage.5,64,65 A collective outcome of mtDNA damage will be the reduction in metabolic activity and/or an increased propensity for apoptosis. However, mtDNA repair capacity appears to be overwhelmed, resulting in diminished mtDNA repair which may play a significant role in the commencement of AMD.5,60 Further evidence has shown that human RPE cells treated with H2O2 resulted in mtDNA damage, which leads to compromised mitochondrial redox function due to impaired repair mechanism.5,63 This impaired repair mechanism clearly explains the susceptibility of mtDNA to oxidative damage in human RPE cells, together with the age-related decrease of the cellular antioxidant system.5
The susceptibility of RPE mtDNA to oxidative damage along with failure of mtDNA repair provides an intriguing and plausible mechanism for a mitochondria-based model of AMD and retinal degeneration. Figure 3 summarizes the possible mechanisms of AMD.1,5,66
FIGURE 3.

ROS-induced mtDNA damage based model for development of AMD (1,5,66).
AMD TREATM ENTS TARGETED TO MITOC HONDRIAL DYSFUNCTION
Treatment of AMD has always been a challenge for ophthalmologists. Therapeutic strategies targeted to the VEGF-signaling pathway has shown some success in the treatment of neovascular wet AMD, however, there are no proven medical treatments for the more common ‘dry’ AMD. Potential use of antioxidants for delaying later stage progression of AMD has been confirmed by the AREDS study.67 However, success of this study was likely limited by the choice of dietary antioxidants (which was later addressed in a new NEI funded clinical trial) and the subsequent realization that dietary antioxidants provide differential subcellular protection in the epithelial cells.68 In support of the therapeutic concept for retinal degeneration, a study by Jarrett et al. has further confirmed that dietary interventions can provide oxidative protection to hippocampal mtDNA and can lower ROS levels in rats.69 Reports have further proven that Bcl-2 overexpression, melatonin, ascorbic acid and glutathione-S-transferase can prevent mtDNA damage in cultured RPE.68,70–72 Several studies have also reported that SOD2 up-regulation, resveratrol, N-tertbutyl hydroxylamine, a-crystallin and l-carnitine have the ability to protect against mitochondrial dysfunction in the RPE.73–77 Whether these antioxidants simply act by reducing ROS levels or act by a more direct effect on mtDNA is yet to be fully elucidated.
An alteration of mitochondrial membranes in AMD has immediate clinical significance as several in vitro and in vivo studies showed that mitochondrial membranes may be a target for the treatment of mitochondrial dysfunctions. Compounds with specific affinity for mitochondrial membranes (mitochondriotropic) can restore mitochondrial functions.42 Alterations of mitochondrial membranes are accompanied by considerable accumulation of lipofuscin which may account for oxidative damage to the RPE.78 It may result in impaired lipid metabolism and apoptosis, characteristics of late AMD.79 Improvement in lipid metabolism in the RPE may be an innovative therapeutic approach for preventing AMD. As mitochondria and peroxisomes are implicated in both lipid biosynthesis and catabolism, Feher et al.80 suggested that the most effective dietary intervention may be achieved by targeting these organelles. These researchers developed a new metabolic approach by combining mitochondriotropic compounds including acetyl-l-carnitine (ALC), n-3 fatty acids, coenzyme Q10 and vitamin E for improvement of retinal function in early AMD. Clinical studies have confirmed this finding, since a combination of ALC, ω-3 fatty acids and coenzyme Q10, after an initial improvement, stabilized several visual functions in early AMD.80 Recently, these preliminary results were confirmed by a randomized, double-masked, placebo controlled, clinical trial that showed improvement in both visual functions and fundus alterations in early AMD.81 These results on restoration of mitochondrial function are certainly very promising for a new therapeutic approach for the treatment of AMD.
CONCLUSION
Mitochondria are attractive targets for drug-delivery because of their roles in cellular energy metabolism, programmed (apoptotic) cell death, and cell signaling. This review highlights the importance of mitochondria derived oxidative stress and to some extent mtDNA damage in the etiology of various retinal diseases (diabetic retinopathy, glaucoma and AMD). substantial efforts must be devoted to developing strategies to target small and large therapeutic molecules to mitochondria. A number of possibilities exist for mitochondrial targeted drug delivery such as (i) therapeutic drug targeting based on negative inner membrane potential of mitochondria, (ii) mitochondrial based enzyme catalyze drug release from prodrugs, and (iii) mitochondrial localized transporter or receptor mediated prodrug delivery (Figure 4).82–86 Continued research in this exciting area will undoubtedly provide a greater understanding of how oxidative stress and deficiencies of the mtDNA contribute to the pathogenesis of neurodegenerative retinal diseases. Future therapeutic strategies should target mitochondria with the ultimate goal of blocking or retarding the effects of chronic mitochondrial dysfunction.
FIGURE 4.
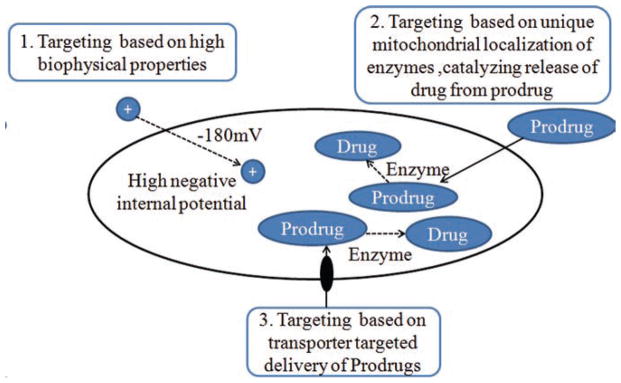
Strategies for targeted delivery of drugs to Mitochondria.
Acknowledgments
This work has been supported by NIH grants RO1 EY 09171-16 and RO1 EY 10659-14.
Footnotes
Declaration of interest: The authors report no conflicts of interest.
References
- 1.Jarrett SG, Lin H, Godley BF, Boulton ME. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog Retin Eye Res. 2008;27:596–607. doi: 10.1016/j.preteyeres.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev. 2008;129:383–390. doi: 10.1016/j.mad.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Golden TR, Melov S. Mitochondrial DNA mutations, oxidative stress, and aging. Mech Ageing Dev. 2001;122:1577–1589. doi: 10.1016/s0047-6374(01)00288-3. [DOI] [PubMed] [Google Scholar]
- 4.Sastre J, Pallardó FV, Viña J. Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life. 2000;49:427–435. doi: 10.1080/152165400410281. [DOI] [PubMed] [Google Scholar]
- 5.Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403. doi: 10.1016/s0014-4835(03)00023-x. [DOI] [PubMed] [Google Scholar]
- 6.Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 7.Aylward GW. Progressive changes in diabetics and their management. Eye (Lond) 2005;19:1115–1118. doi: 10.1038/sj.eye.6701969. [DOI] [PubMed] [Google Scholar]
- 8.Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. 2010;376:124–136. doi: 10.1016/S0140-6736(09)62124-3. [DOI] [PubMed] [Google Scholar]
- 9.Fong DS, Aiello L, Gardner TW, et al. American Diabetes Association. Retinopathy in diabetes. Diabetes Care. 2004;27 (Suppl 1):S84–S87. doi: 10.2337/diacare.27.2007.s84. [DOI] [PubMed] [Google Scholar]
- 10.Meyerle CB, Chew EY, Ferris FL. Nonproliferative diabetic retinopathy. In: Duh E, editor. Diabetic Retinopathy. Humana press; 2008. pp. 3–6. [Google Scholar]
- 11.Xia P, Inoguchi T, Kern TS, Engerman RL, Oates PJ, King GL. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes. 1994;43:1122–1129. doi: 10.2337/diab.43.9.1122. [DOI] [PubMed] [Google Scholar]
- 12.Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001;50:1938–1942. doi: 10.2337/diabetes.50.8.1938. [DOI] [PubMed] [Google Scholar]
- 13.Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp Diabetes Res. 2007;2007:43603. doi: 10.1155/2007/43603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson RE, Rapp LM, Wiegand RD. Lipid peroxidation and retinal degeneration. Curr Eye Res. 1984;3:223–227. doi: 10.3109/02713688408997203. [DOI] [PubMed] [Google Scholar]
- 15.Chan PS, Kowluru RA. Role of retinal mitochondria in the development of diabetic retinopathy. Expert Rev of Ophthalmol. 2007;2:237–247. [Google Scholar]
- 16.Du Y, Miller CM, Kern TS. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic Biol Med. 2003;35:1491–1499. doi: 10.1016/j.freeradbiomed.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 17.Ellis EA, Guberski DL, Somogyi-Mann M, Grant MB. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med. 2000;28:91–101. doi: 10.1016/s0891-5849(99)00216-6. [DOI] [PubMed] [Google Scholar]
- 18.Li W, Yanoff M, Jian B, He Z. Altered mRNA levels of antioxidant enzymes in pre-apoptotic pericytes from human diabetic retinas. Cell Mol Biol (Noisy-le-grand) 1999;45:59–66. [PubMed] [Google Scholar]
- 19.Kowluru RA, Kern TS, Engerman RL. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. IV. Antioxidant defense system. Free Radic Biol Med. 1997;22:587–592. doi: 10.1016/s0891-5849(96)00347-4. [DOI] [PubMed] [Google Scholar]
- 20.Heath H, Rutter AC, Beck TC. Changes in the ascorbic acid and glutathione content of the retinae and adrenals from alloxan-diabetic rats. Vision Res. 1962;2:431–437. [Google Scholar]
- 21.Colantuoni A, Longoni B, Marchiafava PL. Retinal photoreceptors of Syrian hamsters undergo oxidative stress during streptozotocin-induced diabetes. Diabetologia. 2002;45:121–124. doi: 10.1007/s125-002-8252-0. [DOI] [PubMed] [Google Scholar]
- 22.Kowluru RA. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes. 2003;52:818–823. doi: 10.2337/diabetes.52.3.818. [DOI] [PubMed] [Google Scholar]
- 23.Green DR, Amarante-Mendes GP. The point of no return: mitochondria, caspases, and the commitment to cell death. Results Probl Cell Differ. 1998;24:45–61. doi: 10.1007/978-3-540-69185-3_3. [DOI] [PubMed] [Google Scholar]
- 24.Kowluru RA. Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal. 2005;7:1581–1587. doi: 10.1089/ars.2005.7.1581. [DOI] [PubMed] [Google Scholar]
- 25.Kowluru RA, Kowluru V, Xiong Y, Ho YS. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med. 2006;41:1191–1196. doi: 10.1016/j.freeradbiomed.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Kanwar M, Chan PS, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007;48:3805–3811. doi: 10.1167/iovs.06-1280. [DOI] [PubMed] [Google Scholar]
- 27.Kowluru RA, Atasi L, Ho YS. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2006;47:1594–1599. doi: 10.1167/iovs.05-1276. [DOI] [PubMed] [Google Scholar]
- 28.Madsen-Bouterse SA, Mohammad G, Kanwar M, Kowluru RA. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal. 2010;13:797–805. doi: 10.1089/ars.2009.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003;44:5327–5334. doi: 10.1167/iovs.03-0353. [DOI] [PubMed] [Google Scholar]
- 30.Sheu SS, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim Biophys Acta. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Kong GY, Van Bergen NJ, Trounce IA, Crowston JG. Mitochondrial dysfunction and glaucoma. J Glaucoma. 2009;18:93–100. doi: 10.1097/IJG.0b013e318181284f. [DOI] [PubMed] [Google Scholar]
- 32.Barron MJ, Griffiths P, Turnbull DM, Bates D, Nichols P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br J Ophthalmol. 2004;88:286–290. doi: 10.1136/bjo.2003.027664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ju WK, Liu Q, Kim KY, et al. Elevated hydrostatic pressure triggers mitochondrial fission and decreases cellular ATP in differentiated RGC-5 cells. Invest Ophthalmol Vis Sci. 2007;48:2145–2151. doi: 10.1167/iovs.06-0573. [DOI] [PubMed] [Google Scholar]
- 34.Abu-Amero KK, Morales J, Bosley TM. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2006;47:2533–2541. doi: 10.1167/iovs.05-1639. [DOI] [PubMed] [Google Scholar]
- 35.Calandrella N, Scarsella G, Pescosolido N, Risuleo G. Degenerative and apoptotic events at retinal and optic nerve level after experimental induction of ocular hypertension. Mol Cell Biochem. 2007;301:155–163. doi: 10.1007/s11010-006-9407-0. [DOI] [PubMed] [Google Scholar]
- 36.Kerrigan LA, Zack DJ, Quigley HA, Smith SD, Pease ME. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 1997;115:1031–1035. doi: 10.1001/archopht.1997.01100160201010. [DOI] [PubMed] [Google Scholar]
- 37.Moreno MC, Campanelli J, Sande P, Sánez DA, Keller Sarmiento MI, Rosenstein RE. Retinal oxidative stress induced by high intraocular pressure. Free Radic Biol Med. 2004;37:803–812. doi: 10.1016/j.freeradbiomed.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Bonne C, Muller A, Villain M. Free radicals in retinal ischemia. Gen Pharmacol. 1998;30:275–280. doi: 10.1016/s0306-3623(97)00357-1. [DOI] [PubMed] [Google Scholar]
- 39.Tezel G, Seigel GM, Wax MB. Autoantibodies to small heat shock proteins in glaucoma. Invest Ophthalmol Vis Sci. 1998;39:2277–2287. [PubMed] [Google Scholar]
- 40.Farkas RH, Chowers I, Hackam AS, et al. Increased expression of iron-regulating genes in monkey and human glaucoma. Invest Ophthalmol Vis Sci. 2004;45:1410–1417. doi: 10.1167/iovs.03-0872. [DOI] [PubMed] [Google Scholar]
- 41.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 42.Neroev VV, Archipova MM, Bakeeva LE, et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 4. Age-related eye disease. SkQ1 returns vision to blind animals. Biochemistry Mosc. 2008;73:1317–1328. doi: 10.1134/s0006297908120043. [DOI] [PubMed] [Google Scholar]
- 43.Fine SL, Berger JW, Maguire MG, Ho AC. Age-related macular degeneration. N Engl J Med. 2000;342:483–492. doi: 10.1056/NEJM200002173420707. [DOI] [PubMed] [Google Scholar]
- 44.Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol. 1976;60:324–341. doi: 10.1136/bjo.60.5.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young RW. Pathophysiology of age-related macular degeneration. Surv Ophthalmol. 1987;31:291–306. doi: 10.1016/0039-6257(87)90115-9. [DOI] [PubMed] [Google Scholar]
- 46.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 47.Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32. [PMC free article] [PubMed] [Google Scholar]
- 48.Fritsche LG, Loenhardt T, Janssen A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40:892–896. doi: 10.1038/ng.170. [DOI] [PubMed] [Google Scholar]
- 49.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 50.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/ CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt S, Hauser MA, Scott WK, et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006;78:852–864. doi: 10.1086/503822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rivera A, Fisher SA, Fritsche LG, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14:3227–3236. doi: 10.1093/hmg/ddi353. [DOI] [PubMed] [Google Scholar]
- 53.Kanda A, Chen W, Othman M, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci USA. 2007;104:16227–16232. doi: 10.1073/pnas.0703933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones M, Mitchell P, Wang JJ, Sue C. MELAS A3243G mitochondrial DNA mutation and age related maculopathy. Am J Ophthalmol. 2004;138:1051–1053. doi: 10.1016/j.ajo.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 55.Jones MM, Manwaring N, Wang JJ, Rochtchina E, Mitchell P, Sue CM. Mitochondrial DNA haplogroups and age-related maculopathy. Arch Ophthalmol. 2007;125:1235–1240. doi: 10.1001/archopht.125.9.1235. [DOI] [PubMed] [Google Scholar]
- 56.Decanini A, Nordgaard CL, Feng X, Ferrington DA, Olsen TW. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2007;143:607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nordgaard CL, Berg KM, Kapphahn RJ, et al. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:815–822. doi: 10.1167/iovs.05-0976. [DOI] [PubMed] [Google Scholar]
- 58.Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. 2006;27:983–993. doi: 10.1016/j.neurobiolaging.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 59.Barreau E, Brossas JY, Courtois Y, Tréton JA. Accumulation of mitochondrial DNA deletions in human retina during aging. Invest Ophthalmol Vis Sci. 1996;37:384–391. [PubMed] [Google Scholar]
- 60.Wang AL, Lukas TJ, Yuan M, Neufeld AH. Increased mitochondrial DNA damage and down-regulation of DNA repair enzymes in aged rodent retinal pigment epithelium and choroid. Mol Vis. 2008;14:644–651. [PMC free article] [PubMed] [Google Scholar]
- 61.Godley BF, Xu H, Havey A, Xhong X, Lin H, Boulton ME. Mitochondrial DNA repair capacity decreases with progression of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:ARVO E-abstract. [Google Scholar]
- 62.Xu H, Zhong X, Lin H, Godley BF, Boulton ME. Endogenous ROS production increases and mitochondrial redox function decreases in the RPE as a function of age and stage of AMD. Invest Ophthalmol Vis Sci. 2008;49:ARVO E-abstract. [Google Scholar]
- 63.Ballinger SW, Van Houten B, Jin GF, Conklin CA, Godley BF. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp Eye Res. 1999;68:765–772. doi: 10.1006/exer.1998.0661. [DOI] [PubMed] [Google Scholar]
- 64.Jarrett SG, Boulton ME. Antioxidant up-regulation and increased nuclear DNA protection play key roles in adaptation to oxidative stress in epithelial cells. Free Radic Biol Med. 2005;38:1382–1391. doi: 10.1016/j.freeradbiomed.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 65.Jarrett SG, Boulton ME. Poly(ADP-ribose) polymerase offers protection against oxidative and alkylation damage to the nuclear and mitochondrial genomes of the retinal pigment epithelium. Ophthalmic Res. 2007;39:213–223. doi: 10.1159/000104683. [DOI] [PubMed] [Google Scholar]
- 66.Graziewicz MA, Day BJ, Copeland WC. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 2002;30:2817–2824. doi: 10.1093/nar/gkf392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.AREDS. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, β carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 2001. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jarrett SG, Cuenco J, Boulton M. Dietary antioxidants provide differential subcellular protection in epithelial cells. Redox Rep. 2006;11:144–152. doi: 10.1179/135100006X116646. [DOI] [PubMed] [Google Scholar]
- 69.Jarrett SG, Milder JB, Liang LP, Patel M. The ketogenic diet increases mitochondrial glutathione levels. J Neurochem. 2008;106:1044–1051. doi: 10.1111/j.1471-4159.2008.05460.x. [DOI] [PubMed] [Google Scholar]
- 70.Godley BF, Jin GF, Guo YS, Hurst JS. Bcl-2 overexpression increases survival in human retinal pigment epithelial cells exposed to H(2)O(2) Exp Eye Res. 2002;74:663–669. doi: 10.1006/exer.2001.1146. [DOI] [PubMed] [Google Scholar]
- 71.Liang FQ, Green L, Wang C, Alssadi R, Godley BF. Melatonin protects human retinal pigment epithelial (RPE) cells against oxidative stress. Exp Eye Res. 2004;78:1069–1075. doi: 10.1016/j.exer.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 72.Liang FQ, Alssadi R, Morehead P, Awasthi YC, Godley BF. Enhanced expression of glutathione-S-transferase A1-1 protects against oxidative stress in human retinal pigment epithelial cells. Exp Eye Res. 2005;80:113–119. doi: 10.1016/j.exer.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 73.Kasahara E, Lin LR, Ho YS, Reddy VN. SOD2 protects against oxidation-induced apoptosis in mouse retinal pigment epithelium: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005;46:3426–3434. doi: 10.1167/iovs.05-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.King RE, Kent KD, Bomser JA. Resveratrol reduces oxidation and proliferation of human retinal pigment epithelial cells via extracellular signal-regulated kinase inhibition. Chem Biol Interact. 2005;151:143–149. doi: 10.1016/j.cbi.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 75.Voloboueva LA, Killilea DW, Atamna H, Ames BN. N-tertbutyl hydroxylamine, a mitochondrial antioxidant, protects human retinal pigment epithelial cells from iron overload: relevance to macular degeneration. FASEB J. 2007;21:4077–4086. doi: 10.1096/fj.07-8396com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yaung J, Jin M, Barron E, et al. alpha-Crystallin distribution in retinal pigment epithelium and effect of gene knockouts on sensitivity to oxidative stress. Mol Vis. 2007;13:566–577. [PMC free article] [PubMed] [Google Scholar]
- 77.Shamsi FA, Chaudhry IA, Boulton ME, Al-Rajhi AA. L-carnitine protects human retinal pigment epithelial cells from oxidative damage. Curr Eye Res. 2007;32:575–584. doi: 10.1080/02713680701363833. [DOI] [PubMed] [Google Scholar]
- 78.Kennedy CJ, Rakoczy PE, Constable IJ. Lipofuscin of the retinal pigment epithelium: a review. Eye (Lond) 1995;9 (Pt 6):763–771. doi: 10.1038/eye.1995.192. [DOI] [PubMed] [Google Scholar]
- 79.Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- 80.Feher J, Papale A, Mannino G, Gualdi L, Balacco Gabrieli C. Mitotropic compounds for the treatment of age-related macular degeneration. The metabolic approach and a pilot study. Ophthalmologica. 2003;217:351–357. doi: 10.1159/000071351. [DOI] [PubMed] [Google Scholar]
- 81.Feher J, Kovacs B, Kovacs I, Schveoller M, Papale A, Balacco Gabrieli C. Improvement of visual functions and fundus alterations in early age-related macular degeneration treated with a combination of acetyl-L-carnitine, n-3 fatty acids, and coenzyme Q10. Ophthalmologica. 2005;219:154–166. doi: 10.1159/000085248. [DOI] [PubMed] [Google Scholar]
- 82.Yousif LF, Stewart KM, Kelley SO. Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem. 2009;10:1939–1950. doi: 10.1002/cbic.200900185. [DOI] [PubMed] [Google Scholar]
- 83.Armstrong JS. Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br J Pharmacol. 2007;151:1154–1165. doi: 10.1038/sj.bjp.0707288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mukhopadhyay A, Weiner H. Delivery of drugs and macromolecules to mitochondria. Adv Drug Deliv Rev. 2007;59:729–738. doi: 10.1016/j.addr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamada Y, Harashima H. Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases. Adv Drug Deliv Rev. 2008;60:1439–1462. doi: 10.1016/j.addr.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 86.Barot M, Gokulgandhi MR, Haghnegahdar M, Dalvi P, Mitra AK. Effect of emergence of fluoroquinolone resistance on intrinsic expression of P-glycoprotein phenotype in corneal epithelial cells. J Ocul Pharmacol Ther. 2011 Aug 10; doi: 10.1089/jop.2011.0076. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]