

Abstract
Aberrant glycosylation has been implicated in various types of cancers and changes in glycosylation may be associated with signaling pathways during malignant transformation. Glycomic profiling of blood serum, in which cancer cell proteins or their fragments with altered glycosylation patterns are shed, could reveal the altered glycosylation. We performed glycomic profiling of serum from patients with no known disease (N=18), patients with high grade dysplasia (HGD, N=11) and Barrett’s (N=5), and patients with esophageal adenocarcinoma (EAC, N=50) in an attempt to delineate distinct differences in glycosylation between these groups. The relative intensities of 98 features were significantly different among the disease onsets; 26 of these correspond to known glycan structures. The changes in the relative intensities of three of the known glycan structures predicted esophageal adenocarcinoma with 94% sensitivity and better than 60% specificity as determined by receiver operating characteristic (ROC) analysis. We have demonstrated that comparative glycomic profiling of EAC reveals a subset of glycans that can be selected as candidate biomarkers. These markers can differentiate disease-free from HGD, disease-free from EAC, and HGD from EAC. The clinical utility of these glycan biomarkers requires further validation.
INTRODUCTION
The predominant subtypes of esophageal cancer are squamous cell carcinoma and adenocarcinoma. While the incidence of squamous cell carcinoma of the esophagus has remained largely unchanged, there has been a dramatic increase in the incidence of adenocarcinoma. Over the past two decades, the incidence of esophageal adenocarcinoma has increased in many countries, including the US, at a rate that exceeds that of any other malignancy 1. Currently, adenocarcinoma of the distal esophagus represents 60%–90% of all esophageal cancers 2. While surgical resection remains the mainstay of treatment, the poor overall survival with resection alone has led to the investigation of multiple modalities in addition to surgery, such as adjuvant or neo-adjuvant chemotherapy and radiation therapy, in an attempt to improve survival. Even with the best available treatment, fewer than 20% of patients with esophageal cancer can be expected to survive beyond 3 years 3. Several factors contribute to this poor survival, the most important of which is the fact that a majority of patients demonstrate locally advanced disease or metastatic disease at diagnosis. Furthermore, there is no non-invasive test to screen patients at risk for the development of esophageal cancer. Like other types of cancers, patients diagnosed with early stage esophageal cancer have a better prognosis. Therefore, the ability to diagnose and treat patients with esophageal adenocarcinoma at an earlier stage of disease will have a significant impact on patient survival.
It is now generally accepted that esophageal adenocarcinomas develop from a premalignant lesion of the esophagus referred to as Barrett’s esophagus (BE). Barrett’s esophagus is the metaplastic change of the normal squamous cell epithelium of the esophagus to a columnar cell epithelium. This change may occur over any length of the esophagus but is generally noted most commonly in the distal esophagus. The development of BE is strongly associated with longstanding gastroesophageal reflux. Although three subtypes of BE have been described, the specialized intestinal type is the one most closely associated with malignant transformation. The risk of esophageal adenocarcinoma in patients with BE is approximately 30–125 fold greater than that in the general population 4. It is widely held that patients with BE who eventually progress to adenocarcinoma do so by a gradual progression at the cellular level from a normal squamous cell to the metaplastic columnar cell that is synonymous with BE. The columnar cell may then undergo dysplastic transformation that can ultimately result in a malignant cell. The various pathologic states are clearly identifiable histologically and one or more of them may be observed in the same esophagus. However, the precise underlying molecular mechanisms by which such progression occurs and those BE patients at greatest risk for the development of adenocarcinoma have yet to be elucidated. Although BE with high grade dysplasia is considered a precursor to invasive adenocarcinoma, a better understanding of the pathophyisiology of Barrett’s adenocarcinoma may help to identify those patients at increased risk for malignant transformation. The identification of novel biomarkers involved in the progression and ultimate malignant transformation has the potential to result in the earlier diagnosis and, thus, the potential to improve the prognosis of patients with esophageal adenocarcinoma. Furthermore, such biomarkers may ultimately lead to the prevention of esophageal adenocarcinoma by identifying patients in whom intervention should be carried out in one of the premalignant states. Previously, we have shown that proteomic analysis of serum hold potential in identifying patients with esophageal adenocarcinoma 5.
Aberrant glycosylation has been implicated in different types of cancer, with numerous glycosyl epitopes known to constitute tumor-associated antigens 6–8. Also, it has been shown that molecular changes in glycosylation could be associated with the signaling pathways for the malignant transformation of cells 9. Since the correlation between certain structures of glycans and a clinical prognosis in cancer were first suggested over a decade ago 10, 11, the interest in structural studies of glycans and other related molecules on cellular surfaces has increased substantially. Moreover, cancerous cells with altered glycosylation of their surface proteins eventually shed such proteins or their fragments into the circulating fluids. Consequently, glycomic profiling of such fluids could reveal the altered glycosylation. There can be many biomarker candidates for cancer-related alterations due to the inherent structural complexity of blood glycoproteins. Glycomic profiling has been recently utilized to reveal glycan alterations as a result of the progression of different cancer diseases, including breast cancer 12–14, ovarian cancer 15, prostate cancer 16 and hepatocellular carcinoma 17.
In this study, we use our previously developed mass-spectrometric (MS) procedures 12, 18–20 in profiling of N-glycans derived from 10-μL serum sample aliquots. We have recently demonstrated the potential utility of these procedures in the diagnosis of breast 12, 14, prostate 16 and liver cancers 17. They are extended here to display statistically distinctive differences between disease-free samples and esophageal adenocarcinoma. Additionally, MS profiling allowed a distinction between the disease-free subjects and patients with HGD and Barrett’s esophagus. The study has involved quantitative recording and statistical comparisons of the glycomic profiles obtained from the blood sera of 18 disease free individuals, five individuals with Barrett’s esophagus, 11 individuals with HGD and 50 individuals with esophageal adenocarcinoma.
MATERIALS AND METHODS
Materials
The endoglycosidase, Peptide-N-Glycosidase F (PNGase F; EC 3.5.1.52), isolated from E. coli, used for deglycosylation, was obtained from Cape Cod Company (East Falmouth, MA). Trifluoroethanol (TFE), 2,5-dihydroxybenzoic acid (DHB), and sodium hydroxide were purchased from Aldrich (Milwaukee, WI). Chloroform, iodomethane and sodium chloride were received from EM Science (Gibbstown, NJ). Ammonium bicarbonate was received from Mallinckrodt Chemical Company (Paris, KY). Acetonitrile (ACN) was purchased from Fisher Scientific (Fair Lawn, NJ). All other common chemicals of analytical-grade quality were purchased from Sigma (St. Louis, MO).
Serum samples and clinical diagnosis
The study was approved by the Indiana University institutional review board. Serum samples from patients with documented Barrett’s esophagus (N=5), high-grade dysplasia (HGD, N=11) and esophageal adenocarcinoma (N=50) were collected. Serum samples from 18 healthy volunteers was obtained and used as control. Clinical data related to the samples used in this study, including age, sex and diseases stage, are summarized in Supplementary Table 1. It is important to use age-matched control in this kind of study. Venous blood samples were taken in the morning’s fasting state, being collected with the minimal stasis in evacuated tubes. After at least 30 min, but all within 2 hrs, the tubes were centrifuged at 20 °C for 12 min at 1200 g, while the sera were stored frozen in plastic vials at −80 °C until the time of glycomic analyses. Glycomic profiling of 10-μl blood serum aliquots was conducted according to our recently published procedures 14, 16 which are described in detail next.
Release of N-glycans from glycoproteins
A 10-μl aliquot of human blood serum was lyophilized and then resuspended in 100 μl of 25 mM ammonium bicarbonate. Next, human blood serum N-glycans were enzymatically released using PNGase F according to our previously published procedure 20. A 5 mU aliquot of PNGase F was added to the reaction mixture and incubated overnight (18–22 hrs) at 37 °C.
Solid-phase extraction of enzymatically released N-glycans
The volume of enzymatically released glycans was adjusted to 1 ml by adding deionized water. Samples were then applied to both C18 Sep-Pak® cartridges (Waters, Milford, MA) and activated charcoal cartridges (Harvard Apparatus, Holliston, MA). The use of C18 Sep-Pak® cartridges is necessary to isolate the glycans from peptides and proteins, which would otherwise interfere with trapping on the activated charcoal cartridges. The reaction mixture was first applied to C18 Sep-Pak® cartridge that had been preconditioned with ethanol and deionized water according to the manufacturer’s recommendation. The reaction mixture was circulated through the C18 Sep-Pak® cartridge 5-times prior to washing with water. Peptides and O-linked glycopeptides were retained on the C18 Sep-Pak® cartridge, while the released glycans were collected as eluents. Next, the C18 Sep-Pak® cartridge was washed with 1 ml of deionized water. The combined eluents containing the released N-glycans were then passed over activated charcoal microcolumns. The columns were preconditioned with 1 ml of ACN and 1 ml of 0.1% trifluroacetic acid (TFA) aqueous solution, as recommended by the manufacturer. After applying the sample, the microcolumn was washed with 1 ml of 0.1% TFA aqueous solution. The samples were then eluted with a 1-ml aliquot of 50% ACN aqueous solution containing 0.1% TFA. Finally, the purified glycans were evaporated to dryness using vacuum CentriVap Concentrator (Labconco Corporation, Kansas City, MO) prior to solid-phase permethylation.
Solid-phase permethylation
Permethylation of enzymatically released and solid-phase purified N-glycans was accomplished utilizing our recently published solid-phase permethylation technique 18, 19. This approach involves packing of sodium hydroxide beads in peek tubes (1 mm i.d.; Polymicro Technologies, Phoenix, AZ), permitting complete derivatization. Tubes, nuts and ferrules from Upchurch Scientific (Oak Harbor, WA) were employed for assembling the sodium hydroxide capillary reactor. Sodium hydroxide powder was suspended in ACN for packing. A 100-μl syringe from Hamilton (Reno, NV) and a syringe pump from KD Scientific, Inc. (Holliston, MA) were employed for introducing the sample into the reactor. Sodium hydroxide reactor was first conditioned with 60 μl of dimethyl sulfoxide (DMSO) at a 5 μl/min flow rate. Samples were resuspended in DMSO and mixed with methyl iodide solution containing traces of deionized water. Typically, released and purified N-glycans were resuspended in a 50-μl aliquot of DMSO, to which 0.3 μl of water and 22 μl methyl iodide were added. This permethylation procedure has been shown to minimize oxidative degradation, peeling reactions as well as to avoid the need of excessive clean-up 18, 19. Samples were infused through the reactor at a slow flow rate of 2 μl/min, as previously described 18, 19. The reactor was then washed with 230 μl ACN (flow rate: 5 μl/min). All eluents were combined, and the permethylated N-glycans were finally extracted using 200 μl chloroform and washed repeatedly (3 times) with 200 μl of water prior to drying.
MALDI-TOF MS instrumentation
Permethylated glycans were resuspended in 2 μl of (50:50) methanol:water solution. A 0.5-μl aliquot of the sample was then spotted directly on the MALDI plate and mixed with the equal volume of DHB-matrix prepared by suspending 10 mg of DHB in 1 ml of (50:50) water:methanol solution, containing 1 mM sodium acetate. The inclusion of sodium acetate is to promote a nearly complete sodium adduct formation in MALDI-MS. The MALDI plate was then dried under vacuum to ensure uniform crystallization. Mass spectra were acquired using the Applied Biosystems 4800 MALDI TOF/TOF Analyzer (Applied Biosystems Inc., Framingham, MA). This instrument is equipped with Nd:YAG laser with 355-nm wavelength. MALDI-spectra were recorded solely in the positive-ion mode, since permethylation eliminates the negative charge normally associated with sialylated glycans. External calibration was attained using a mixture of permethylated N-glycans derived from standard glycoproteins, including ribonuclease B, alpha acid glycoprotein and fetuin. This mixture includes high-mannose glycans and sialylated and fucosylated complex glycans, thus allowing mass accuracy of 50 ppm or better.
Data evaluation
The obtained MALDI-MS data were further processed using DataExplorer 4.0 (Applied Biosystems, Framingham, MA) to generate ASCII files listing m/z values and intensities. An in-house developed software tool (PeakCalc 2.0) was then used to extract the intensities of N-glycans. Principal component analysis (PCA) was performed using MarkerView (ABI, Framingham, MA), allowing the visualization of multivariate information. Supervised PCA methods were employed, using a prior knowledge of the sample groups as healthy vs. diseased. MS data were weighted using the base-e logarithm of the peak intensities. The peak intensities were also scaled using pareto option in which each value is subtracted by the average and divided by the square root of the standard deviation. This option is suitable for MS data, since it prevents intense peaks from completely dominating the PCA process, thus allowing any peak with good signal-to-noise ratio to contribute.
We also used Receiver Operating Characteristics (ROC) curve analysis using AccuROC 2.5 software for Windows (Accumetric Corporation, Montreal, Canada) to assess the sensitivity and selectivity of the potential diagnostic variables. ROC curve is defined as a plot of test sensitivity, in its y-axis versus its specificity or false-positive rate as the x-axis. This type of statistical analysis is an effective method of evaluating the quality or performance of diagnostic tests, as has been widely used in radiology to evaluate performance of many tests 21.
The intensity values of 421 features were collected from 84 individuals. Among them, 18 were healthy controls, five had Barrett’s esophagus, 11 suffered from HGD, and the other 50 were diagnosed with esophageal adenocarcinoma. As the primary goal was to compare these groups in terms of the relative intensity values for each feature (chemical identity of a glycan). Since the number of Barrett’s esophagus samples were very limited (N=5), we combined Barrett’s esophagus and HGD groups for statistical comaprsion. It has been widely accepted that the total intensity for each individual varies, thus the relative intensity is utilized for comparison throughout the following statistical analysis.
Exploratory data analyses indicated that the relative intensity of the original data violated the normality and constant variance assumptions. After logarithm transformation of the relative intensity, normality was improved but still not achieved. As a result, a non-parametric method, Kruskal-Wallis test 22, was employed to test the equality of the medians. Due to the large number of tests, Bonferroni method was used to correct for multiple comparisons with overall Type I error rate controlled at 0.05. The statistical significance of each feature was determined by comparing the p-value from Kruskal-Wallis test with 1.1876e-004. If the statistical significance was reached, comparisons of the relative intensities between each pair of the two groups were performed using the Wilcoxon rank-sum test 23.
Results from the Kruskal-Wallis test indicated that the relative intensities of 98 features were significantly different among the three groups of which 26 features corresponded to known glycan structures. Further pairwise comparisons using Wilcoxon rank-sum test showed that only 8 features with known structures are significantly different in all three pairwise comparisons, i.e., the comparisons between esophageal adenocarcinoma and healthy controls, between esophageal adenocarcinoma and Barrett’s+HGD, between healthy control and Barrett’s+HGD. The other 18 features with known structures reach the statistical significance in two out of the three comparisons. On the basis of these results, a subset of features can be selected as candidate biomarkers to differentiate all three groups or to differentiate one group from the other two. The p-values from Wilcoxon rank-sum tests for the significant features with known glycan strcutures are summarized in Tables 1–3. All statistical analyses were carried out using R version 2.5.0 and Matlab version 7.4.0 (the MathWorks).
Table 1.
Area under the curve (AUC) values from ROC analysis and p-values of Wilcoxon rank sum test for N-glycans derived from human blood serum of sex- and age-matched control and esophageal cancer patients. Highlighted rows are the N-glycans which received a highly accurate ROC analysis AUC values (AUC > 0.8) and low p-values of Wilcoxon rank sum test, while others are N-glycans which received a moderately accurate ROC analysis AUC values (0.7<AUC<0.8) and low p-values of Wilcoxon rank sum test. Values listed in red indicate a decrease in the relative intensity of N-glycans derived from esophageal adenocarcinoma samples.
| Control vs. Cancer | |||||
|---|---|---|---|---|---|
| Glycan [M+Na]+ Theoretical | Glycan [M+Na]+ Observed | ppm | Structure | P-value | ROC AUC |
| 2244.13 | 2244.12 | −2 |
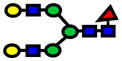
|
2.60E-09 | 0.99±0.01 |
| 2040.03 | 2040.03 | 1 |
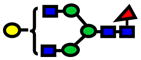
|
2.10E-07 | 0.95±0.03 |
| 2227.11 | 2227.12 | 3 |
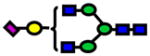
|
1.81E-08 | 0.90±0.04 |
| 2605.30 | 2605.30 | 2 |
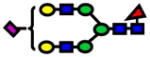
|
4.67E-05 | 0.88±0.05 |
| 2070.04 | 2070.03 | −3 |
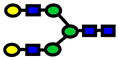
|
4.13E-07 | 0.87±0.06 |
| 3950.96 | 3950.92 | −10 |
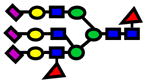
|
6.70E-05 | 0.86±0.06 |
| 1906.96 | 1906.96 | −2 |
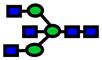
|
1.10E-05 | 0.84±0.06 |
| 2635.99 | 2635.96 | −13 |
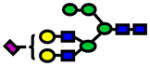
|
1.96E-08 | 0.82±0.06 |
| 1590.80 | 1590.80 | −1 |
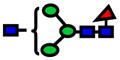
|
4.45E-07 | 0.79±0.08 |
| 1794.90 | 1794.90 | −1 |
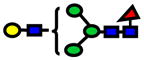
|
1.17E-05 | 0.79±0.08 |
| 2274.14 | 2274.14 | 4 |
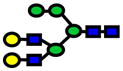
|
8.43E-06 | 0.77±0.08 |
| 2214.11 | 2214.10 | −7 |
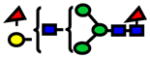
|
2.22E-05 | 0.76±0.07 |
| 2029.01 | 2029.02 | 6 |
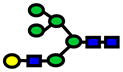
|
1.67E-07 | 0.76±0.07 |
| 1620.81 | 1620.81 | −2 |
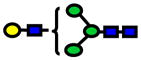
|
2.49E-06 | 0.75±0.07 |
| 1764.89 | 1764.86 | −15 |
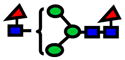
|
0.000151645 | 0.74±0.08 |
| 2431.21 | 2431.21 | 1 |
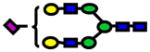
|
0.006978279 | 0.74±0.08 |
Table 3.
Area under the curve (AUC) values from ROC analysis and p-values of Wilcoxon rank sum test for N-glycans derived from human blood serum of esophageal adenocarcinoma patients and patients with high-grade dysplasia (HGD).
| Cancer vs. HGD | |||||
|---|---|---|---|---|---|
| Glycan [M+Na]+ Theoretical | Glycan [M+Na]+ Observed | ppm | Structure | P-value | ROC AUC |
| 1865.94 | 1865.92 | −9 |
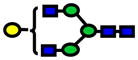
|
1.0E-05 | 0.83±0.09 |
| 2274.14 | 2274.14 | 4 |
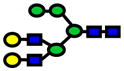
|
2.0E-05 | 0.83±0.09 |
| 2070.04 | 2070.03 | −3 |
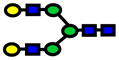
|
2.0E-04 | 0.83±0.08 |
| 3084.54 | 3084.47 | −21 |
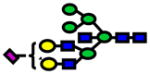
|
2.0E-05 | 0.82±0.09 |
| 2519.26 | 2519.22 | −16 |
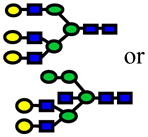
|
8.0E-06 | 0.79±0.1 |
| 2227.11 | 2227.12 | 3 |
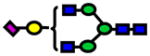
|
3.0E-03 | 0.79±0.09 |
| 2938.48 | 2938.43 | −15 |
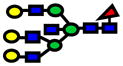
|
1.0E-06 | 0.79±0.07 |
| 1794.90 | 1794.89 | −3 |
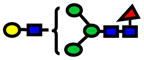
|
2.0E-05 | 0.76±0.1 |
| 1661.84 | 1661.79 | −26 |
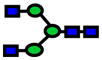
|
1.0E-05 | 0.76±0.08 |
| 3142.58 | 3142.51 | −20 |
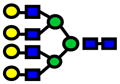
|
1.0E-05 | 0.73±0.10 |
| 1590.80 | 1590.80 | −1 |
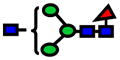
|
5.0E-05 | 0.73±0.1 |
| 1620.81 | 1620.81 | −2 |
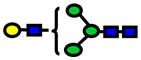
|
9.0E-06 | 0.73±0.09 |
| 2874.44 | 2874.36 | −25 |
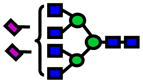
|
7.0E-04 | 0.72±0.1 |
| 1764.89 | 1764.86 | −15 |
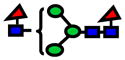
|
8.0E-06 | 0.72±0.09 |
| 3131.56 | 3131.43 | −42 |
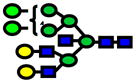
|
8.0E-05 | 0.71±0.09 |
The range of values throughout this study was expressed as a standard error of the mean (SEM) value, which accounts for a sample size. Standard deviation is the most common measure of statistical dispersion, measuring how spread-out the values in a data set appear (e.g., due to limitations in measurement reproducibility). However, when working with biological samples, any observed variation might be intrinsic to the phenomenon that distinct members of a population differ greatly (biochemical individuality). Consequently, the standard error (SE), or SEM, signifies an estimate of the standard deviation of the sampling distribution of means, based on the data from one or more random samples. SEM then accounts for the number of real samples, implicating their biodiversity in the evaluation process.
RESULTS
Glycomic profiles derived from human sera
We have previously demonstrated that comparative glycomic profiling allows quantitative distinction between the glycan structures derived from the sera of healthy individuals and cancer patients 14, 16, 17. Here, the same approach has been utilized to distinguish esophageal adenocarcinoma, Barrett’s esophagus, HGD esophagus from disease free subjects. It also allows the distinction between the different disease onsets. The profiles of permethylated N-glycans derived from 10 μl of serum volumes were recorded for the m/z range of 1500–5000 using MALDI-MS. The profiles generally appeared as different between the four sample sets.
The repeatability of mass spectrometry for glycomic analysis has been recently addressed in a pilot study conducted by the Human Proteome Organisation Human Disease Glycomics/Proteome Initiative (HUPO HGPI) and included 20 different laboratories including ours 33. The study involved comparing the MS analyses of N-linked glycans of standard samples acquired in 20 laboratories 24. The study concluded that, in general, MALDI/TOF MS of permethylated oligosaccharide mixtures carried out in six laboratories yielded good quantitation, and the results could be correlated to those obtained through liquid chromatography of the reductive amination derivatives of glycans. The study also revealed the high reproducibility of the MS analysis of permethylated glycans.
Principal component analysis (PCA) of measured spectra
For an informative chemometric analysis of the acquired glycomic profiles, PCA was employed, as it is commonly used in microarray research for cluster analysis. PCA is designed to capture a variance in the given data sets in terms of their principal components, meaning a set of variables which defines a projection encapsulating the maximum amount of variation in a dataset. It is orthogonal (and, therefore, uncorrelated) to the previous principal component 25, 26. PCA is a chemometric tool which is commonly employed to establish the differences among sample sets.
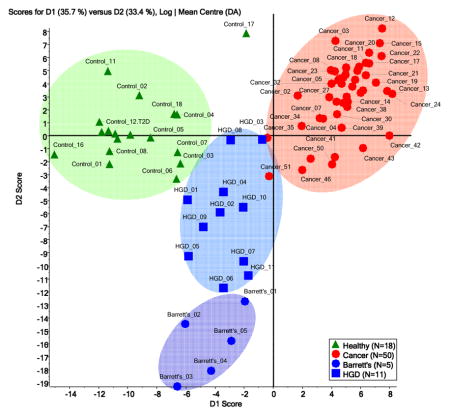
A plot of the scores of principal component one and two for the samples of the four disease onsets is illustrated in Figure 1. The four sets of samples received, to a various degrees, distinguishable first principal component (PC1) scores. Consequently, the four sets clustered in a manner representative of the glycomic profile differences for the four onsets (Figure 1). However, samples from patients with Barrett’s esophagus were distinguishable from those with HGD esophagus only in the second principle component, suggesting very limited differences between the two onsets. According to PCA clustering, there is a vast distinction between disease-free samples and those with esophageal adenocarcinoma. This difference is less distinct in the case of samples from patients with HGD and Barrett’s esophagus. It is also suggested by PCA of the glycomic profiles that glycomic changes associated with HGD and Barrett’s esophagus are intermediate, relative to the alteration shown by esophageal adenocarcinoma samples.
Figure 1.

Principal component analysis (PCA) scores plot for the mass spectra of permethylated N-glycans derived from blood sera of disease-free subjects (N=18) and patients with esophageal adenocarcinoma (N=50), HGD esophagus (N=11) and Barrett’s esophagus (N=5).
Changes in the relative intensities of fucosylated and sialylated structures
Comparing the relative intensities of all fucosylated structures or sialylated structures as a sum of all individual relative intensities is yet another means to monitor changes in glycosylation patterns. Here, a decrease in the total fucosylation of N-glycans derived from samples collected from esophageal adenocarcinoma was observed (Figure 2a). This decrease was statistically significant with a p-value less than 0.005. The decrease was also observed for samples collected from patients with HGD and Barrett’s esophagus; however, such decreases were not statistically significant. As suggested by PCA plot (Figure 1), the glycomic changes associated with HGD and Barrett’s esophagus samples are expected to be intermediate to those associated with esophageal adenocarcinoma. This decrease in fucosylation as a result of disease progression is in stark contrast to what we had previously observed in breast 14 and prostate cancers 16. On the other hand, no significant change in sialylation was detected among the different disease onsets (Figure 2b).
Figure 2.
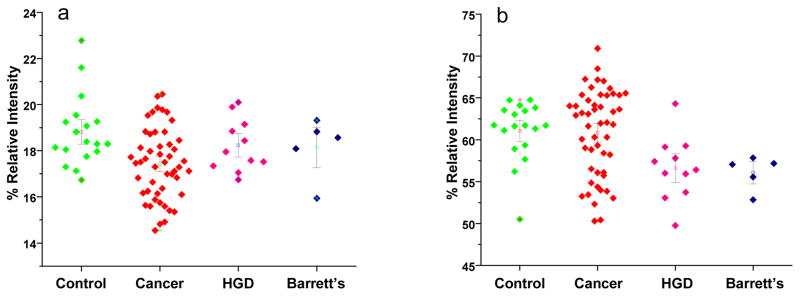
Dot-plots reflecting the overall changes in total fucosylation (a) and total sialylation (b) among the different disease onsets.
ROC analysis and Wilcoxon rank sum test
A statistical evaluation of changes in the intensities associated with all known N-glycans that were observed in the MALDI mass spectrum was further performed to validate the aforementioned differences for the individual glycans, thus testing their potential use in diagnosis of the different disease onsets. Such statistical evaluations were performed using two independent approaches: Wilcoxon rank sum test and ROC curve analyses. The changes in relative intensities were evaluated using Wilcoxon rank sum test. In this case, a statistically significant difference in the relative intensity of a glycan structure between two states has been associated with a low p-value. Generally, a change associated with a p-value lower than 0.01 is considered to be significant, suggesting a 1% probability that the difference is not valid.
The ROC analysis is used in test situations where the diagnostic test yields numerical results that can be compared to an independent diagnosis, confirming either the presence or absence of a disease 27, 28. The software, that was designed for medical use, operates on two parameters: sensitivity and specificity. AUC is a combined measure of sensitivity and specificity and, consequently, the overall performance of a diagnostic test, which can be interpreted as the average value of sensitivity for all possible values of specificity. It can take on any value between 0 and 1. The closer AUC is to 1, the better the overall diagnostic performance of this test, while a test with an AUC value of 1 is the one that is perfectly accurate. A test is considered to be highly accurate for the AUC values of 0.9 or higher, while a moderately accurate test demonstrates AUC value between 0.7–0.89. An AUC value lower than 0.7 suggests an inaccurate test 27, 28.
The AUC values and their corresponding Wilcoxon rank sum test p-values for the glycan structures with a moderate to highly accurate ROC analysis are listed in Tables 1–3. The relative intensities of sixteen N-glycan structures were statistically different among disease free subjects and esophageal adenocarcinoma patients as suggested by Wilcoxon rank sum test p-values (Table 1). According to the AUC values of ROC analysis, three of these glycanss predict esophageal adenocarcinoma with high accuracy, while the other structures could be useful to predict adenocarcinoma with a moderate accuracy. The N-glycan at m/z value 2244 predicts esophageal adenocarcinoma with 94% sensitivity and specificity as suggested by ROC analysis, while the N-glycan at m/z value 2040 predicts the disease at 94% sensitivity and 86% specificity. The relative intensities of 14 N-glycans were lower in esophageal adenocarcinoma patients, while those of two N-glycans were higher. Dot plots representing the relative intensities observed for some of the structures listed in Table 1 are illustrated in Figure 3. Eight of the structures listed in Table 1 are fucosylated, while five are acidic glycans featuring sialic acid residues.
Figure 3.

Dot-plots comparing the average relative intensities of some of the N-glycans listed in Table 1. Symbols:
 , N-acetylglucosamine;
, N-acetylglucosamine;
 , mannose;
, mannose;
 , galactose;
, galactose;
 , fucose;
, fucose;
 , N-acetylneuraminic acid.
, N-acetylneuraminic acid.
Additionally, the relative intensities of sixteen N-glycan structures were statistically different between the disease-free subjects and the patients with HGD esophagus as suggested by Wilcoxon rank sum test p-values (Table 2). According to the AUC values of ROC analysis, all of the structures listed in Table 2 predict HGD esophagus with moderate accuracy. On the other hand, the relative intensities of 15 N-glycan structures were statistically different between esophageal adenocarcinoma patients and those with HGD esophagus, as suggested by Wilcoxon rank sum test p-values (Table 3). According to the AUC values of ROC analysis, all of the structures listed in Table 3 predict HGD esophagus with moderate accuracy. The dot plots representing the relative intensities observed for some of the structures, listed in Tables 2 and 3, are illustrated in Figures 4 and 5, respectively. Six of the glycan structures listed in Table 2 are fucosylated, while seven are sialylated. Only four glycan structures listed in Table 3 are fucosylated, while only three are sialylated.
Table 2.
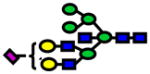
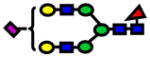
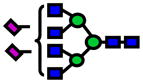
Area under the curve (AUC) values from ROC analysis and p-values of Wilcoxon rank sum test for N-glycans derived from human blood serum of sex- and age-matched control and patients with high-grade dysplasia (HGD). Values listed in red indicate a decrease in the relative intensity of N-glycans derived from HGD samples.
| Control vs. HGD | |||||
|---|---|---|---|---|---|
| Glycan [M+Na]+ Theoretical | Glycan [M+Na]+ Observed | ppm | Structure | P-value | ROC AUC |
| 3084.54 | 3084.41 | 41 |
|
1.0E-05 | 0.89±0.07 |
| 2605.30 | 2605.31 | 3 |
|
4.0E-06 | 0.87±0.07 |
| 2874.44 | 2874.38 | −18 |
|
1.0E-05 | 0.87±0.07 |
| 2938.48 | 2938.43 | −15 |
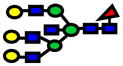
|
3.0E-07 | 0.85±0.08 |
| 3142.58 | 3142.59 | 5 |
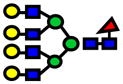
|
9.0E-06 | 0.85±0.07 |
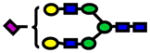
| 2244.13 | 244.13 | 1 |
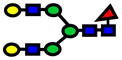
|
1.0E-05 | 0.85±0.07 |
| 4675.32 | 4675.29 | −25 |
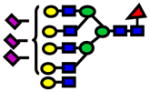
|
8.0E-04 | 0.85±0.07 |
| 1865.94 | 1865.91 | −15 |
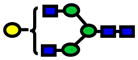
|
3.0E-03 | 0.84±0.09 |
| 3131.56 | 3131.51 | −16 |
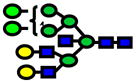
|
4.0E-06 | 0.84±0.08 |
| 2519.26 | 2519.24 | −10 |
|
4.0E-05 | 0.84±0.07 |
| 2227.11 | 2227.12 | 4 |
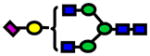
|
3.0E-05 | 0.80±0.1 |
| 1906.96 | 1906.95 | −7 |
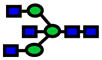
|
4.0E-03 | 0.79±0.08 |
| 2431.21 | 2431.23 | 7 |
|
1.0E-05 | 0.77±0.09 |
| 2040.03 | 2040.03 | 3 |
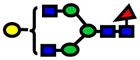
|
2.0E-05 | 0.77±0.09 |
| 2966.47 | 2966.36 | −37 |
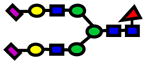
|
3.0E-05 | 0.77±0.09 |
| 1661.84 | 1661.78 | −35 |
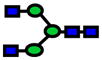
|
4.0E-04 | 0.77±0.09 |
Figure 4.

Dot-plots comparing the average relative intensities of some of the N-glycans listed in Table 2 which are significantly altered as a result of the progression of esophageal adenocarcinoma. Symbols: as in Figure 3
Figure 5.
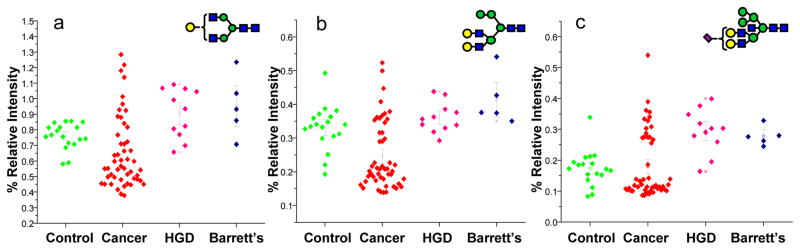
Dot-plots comparing the average relative intensities of some of the N-glycans listed in Table 3 which are significantly altered as a result of HGD esophagus. Symbols: as in Figure 3
The spectra of difucosylated/trisialylated triantennary N-glycan observed in disease-free sample (green trace) and esophageal adenocarcinoma sample (red trace) are depicted in Figure 6a. The spectra of core-fucosylated/biantennary N-glycan observed in disease-free sample (green trace) and esophageal adenocarcinoma sample (red trace) are depicted in Figure 6b. Intensity comparisons of a hybrid N-glycan which demonstrated highest AUC value of ROC analysis between disease-free samples and HGD esophagus samples, is shown in Figure 6c. The intensity of the biantennary N-glycan with one less galactose residue is substantially higher in the glycomic profile of patients with HGD esophagus when compared to that of esophageal adenocarcinoma patients (Figure 6d).
Figure 6.

MALDI mass spectra illustrating the difference in the intensity of N-glycans derived from human blood serum of (a, b) a disease-free subject (green trace) vs. patient with esophageal adenocarcinoma (red trace); (c) a disease free subject (green trace) vs. patient with HGD esophagus (pink trace) and (d) a patient with HGD esophagus (pink trace) vs. that with esophageal adenocarcinoma (red trace). Symbols: as in Figure 3
DISCUSSION
Small volumes (10 μl) of unfractionated human serum were used to generate MALDI-MS profiles of permethylated glycans, which were derived from the samples representative of disease-free subjects and three esophageal disease onsets, including esophageal adenocarcinoma as well as HGD and Barrett’s esophagus. The data provide some preliminary insight into the mechanisms of altered glycosylation and point to the structural changes of glycoproteins at the onset and during the course of cancer development. At this point, the study is not concerned with changes in the proteins bearing the aforementioned glycan structures. No information pertaining to the states of proteins which endured such glycosylation changes has been obtained. The potential of employing the glycomic approach as a diagnostic and prognostic tool has thus far been demonstrated here for esophageal disease as it has been previously demonstrated for breast14, prostate 16 and liver cancers 17.
Alterations in the relative intensities of over 134 known N-glycans were initially determined in this work, however, only 26 structures demonstrated statistically significant changes as a result of disease progression. This is also in agreement with the general knowledge that aberrant glycosylation is commonly associated with cancer 6–8. It is also observed in this study that aberrant glycosylation associated with esophagus diseases such as HGD and Barrett’s, which commonly transform to esophagus adenocarcinoma, is intermediate and less pronounced relative to that observed in the case of cancer state. This is suggested by an intermediate decrease in fucosylation and the alterations associated with the different N-glycans that appeared to change as a result of disease progression.
Although an increase in fucosylation has been implicated in many cancers including prostate carcinoma LNPaC cells 29, pancreatic cancer 30, colorectal cancer 31, human leucocyte cancer 32, hepatocarcinomas 33, and renal carcinomas 34, we observed a fucosylation decrease in the case of esophageal adenocarcinoma. On the other hand, no significant alteration in the sialylation of N-glycans was observed in this study. Although an impaired sialylation pattern has been suggested in different types of cancer 35–38 through previous studies, our results do not implicate its importance in the case of N-glycans. This does not rule out its significance in association with O-glycans which were not investigated thus far. The results described here suggest that glycomic mapping is disease-specific, thus validating its potential use as a diagnostic tool.
Only four of the N-glycans listed in Table 1 demonstrated a statistically significant change in our previous prostate cancer glycomic profiling 16. However, they all demonstrated an opposite trend to what is observed here. These N-glycans were observed at m/z values of 2244.1, 2070.0, 1907.0 and 2431.2. On the other hand, only four of the N-glycans listed in Table 1 also demonstrated a statistically significant changed in our breast cancer glycomic profiling 14, of which one was also significantly changes in prostate cancer. These N-glycans were observed at m/z values of 2040.0, 2605.3, 3950.9, and 1907.0. Their changes were similar to the changes observed in this study except for the N-glycan observed at 2040.0 which demonstrated an opposite trend. Finally, the N-glycans demonstrating statistically significant changes in the case of our liver cancer glycomic profiling, were different from the ones listed in Table 1. Accordingly, glycomic changes observed for the four different cancers which we have studied thus far appear to be different, suggesting the specific nature of the observed changes. The changes in the glycan structures we have reported thus far 14, 16, 17, and those reported here, are demonstrating a very limited, but statistically distinguishable overlap. The analytical approach that we have developed and we are using here appears to be cancer-specific. This being said, thorough validation of this statement is needed and is currently being pursued.
The principal component analysis of the four groups of samples analyzed in this study demonstrates a very distinct clustering with a significant difference between disease-free and esophageal adenocarcinoma samples. In fact, it has resulted in a complete separation between these two groups of data. The differences between disease-free subjects and those with disease precursor conditions were intermediate in magnitude. This is in agreement with the fact that HGD and Barrett’s are intermediate between disease-free and cancer conditions. The manner in which the data cluster suggest a possibility of using serum glycomic profiling to effectively diagnose the different esophagus related-diseases.
CONCLUSIONS
We have demonstrated here the diagnostic potential of MS-based glycomic profiles to yet another type of cancer. This was again achieved with only a 10-μl volume of human serum. Glycomic profiles from disease-free subjects and patients with different esophageal diseases (esophageal adenocarcinoma, HGD and Barrett’s esophagus) demonstrated statistically significant changes, which were based on the results of the ROC analysis and Wilcoxon rank sum test. All statistical analyses confirmed differences in the N-glycosylation patterns among the four different conditions. On of the potential N-glycan biomarkers predicts esophageal adenocarcinom with 94% sensitivity and 94% specificity. The bioanalytical information acquired here provides some insight into the mechanisms of aberrant glycosylation and points to the structural changes of glycoproteins. Such methodologies may form the basis for development of new pre-screening method to aid early cancer detection through the determination of glycan-specific biomarkers. Comparing the results of the four studies we have thus far conducted 14, 16, 17 in different cancers suggests the specific nature of this methodological approach.
Supplementary Material
Acknowledgments
This work was primarily supported by grant No. CA128535-01 from the National Cancer Institute, U.S. Department of Health and Human Services. Additional support was provided by the Indiana Metabolomics and Cytomics Initiative (METACyt) and by NIH/NCRR – National Center for Glycomics and Glycoproteomics (NCGG), grant No - RR018942.
Footnotes
Supporting information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Jemal A, Murray T, Samuels A, Ghafour A, Wood E, Thun MJ. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Devasa SS, Blot WJ, Fraumeni JF. Cancer Res. 1998;83:2049–2053. [PubMed] [Google Scholar]
- 3.Fiorica F, DiBona D, Schepis F, Licata A, Shaheid L, Venturi A, Falchi AM, Craxi A, Camma C. Gut. 2004;53:925–930. doi: 10.1136/gut.2003.025080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blot WJ, Devasa SS, Faumeni JFJ. JAMA. 1993;270:1320. [PubMed] [Google Scholar]
- 5.Hammoud Z, Dobrolecki LE, Kesler KA, Rahmani E, Rieger K, Malkas LH, Hickey RJ. Ann Thorac Surg. 2007;84:384–392. doi: 10.1016/j.athoracsur.2007.03.088. [DOI] [PubMed] [Google Scholar]
- 6.Baldus SE, Wienand JR, Werner JP, Landsberg S, Drebber U, Hanisch FG, Dienes HP. Int J Oncol. 2005;27:1289–1297. [PubMed] [Google Scholar]
- 7.Dwek MV, Lacey HA, Leathem A. J Clin Chim Acta. 1998;271:191–202. doi: 10.1016/s0009-8981(97)00258-1. [DOI] [PubMed] [Google Scholar]
- 8.Handerson T, Camp R, Harigopal M, Rimm D, Pawelek J. Clin Cancer Res. 2005;11:2969–2973. doi: 10.1158/1078-0432.CCR-04-2211. [DOI] [PubMed] [Google Scholar]
- 9.Alper J. Science. 2003;301:159–160. doi: 10.1126/science.301.5630.159. [DOI] [PubMed] [Google Scholar]
- 10.Hakomori S. Cancer Res. 1996;56:5309–5318. [PubMed] [Google Scholar]
- 11.Kobata A. Glycoconj J. 1998;15:323–331. doi: 10.1023/a:1006961532182. [DOI] [PubMed] [Google Scholar]
- 12.Goetz JA, Mechref Y, Kang P, Jeng M-H, Novotny MV. Glycoconj J. 2008 doi: 10.1007/s10719-008-9170-4. in press. [DOI] [PubMed] [Google Scholar]
- 13.Kirmiz C, Li B, An HJ, Clowers BH, Chew HK, Lam KS, Ferrige A, Alecio R, Borowsky AD, Sulaimon S, Lebrilla CB, Miyamoto S. Mol Cell Proteomics. 2007;6:43–55. doi: 10.1074/mcp.M600171-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Kyselova Z, Mechref Y, Kang P, Goetz JA, Dobrolecki LE, Sledge G, Schnaper L, Hickey RJ, Malkas LH, Novotny MV. Clin Chem. 2008;54:1166–1175. doi: 10.1373/clinchem.2007.087148. [DOI] [PubMed] [Google Scholar]
- 15.An HJ, Miyamoto S, Lancaster KS, Kirmiz C, Li B, Lam KS, Leiserowitz GS, Lebrilla CB. J Proteome Res. 2006;5:1626–1635. doi: 10.1021/pr060010k. [DOI] [PubMed] [Google Scholar]
- 16.Kyselova Z, Mechref Y, Al Bataineh MM, Dobrolecki LE, Hickey RJ, Vinson J, Sweeney CJ, Novotny MV. J Proteome Res. 2007;6:1822–1832. doi: 10.1021/pr060664t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldman R, Ressom H, Varghese R, Goldman L, Bascug G, Loffredo C, Abdel-Hamid M, Gouda I, Abo-Elkhir S, Kyselova Z, Mechref Y, Novotny MV. Clin Cancer Res. 2008 doi: 10.1158/1078-0432.CCR-07-5261. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang P, Mechref Y, Novotny MV. Rapid Commun Mass Spectrom. 2008;22:721–734. doi: 10.1002/rcm.3395. [DOI] [PubMed] [Google Scholar]
- 20.Mechref Y, Novotny MV. Anal Chem. 1998;70:455–463. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- 21.Park SH, Goo JM, Jo CH. Korean J Radiology. 2004;5:11–18. doi: 10.3348/kjr.2004.5.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kruskal WH, Wallis WA. J Am Statist Asso. 1952;47:583–621. [Google Scholar]
- 23.Wilcoxon F. Biometrics Bulletin. 1945;1:80–83. [Google Scholar]
- 24.Wada Y, Azadi P, Costello CE, Dell A, Dwek RA, Geyer H, Geyer R, Kakehi K, Karlsson NG, Kato K, Kawasaki N, Khoo K-H, Kim S, Kondo A, Lattova E, Mechref Y, Miyoshi E, Nakamura K, Narimatsu H, Novotny MV, Packer NH, Perreault H, Peter-Katalinic J, Pohlentz G, Reinhold VN, Rudd PM, Suzuki A, NT Glycobiology. 2007;17:411–422. doi: 10.1093/glycob/cwl086. [DOI] [PubMed] [Google Scholar]
- 25.Hotelling H. J Educat Psych. 1933;24:417–441. [Google Scholar]
- 26.Musumarra G, Barresi V, Condorelli DF, Scire S. Biol Chem. 2003;384:321–327. doi: 10.1515/BC.2003.037. [DOI] [PubMed] [Google Scholar]
- 27.Hanley JA, McNeil BJ. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- 28.Pepe MS, Cai T, Longton G. Biometrics Bulletin. 2006;62:221–229. doi: 10.1111/j.1541-0420.2005.00420.x. [DOI] [PubMed] [Google Scholar]
- 29.Chandrasekaran EV, Chawda R, Locke RD, Piskorz CF, Matta KL. Glycobiology. 2002;2:153–162. doi: 10.1093/glycob/12.3.153. [DOI] [PubMed] [Google Scholar]
- 30.Mas E, Pasqualini E, Caillol N, El Battari A, Crotte C, Lombardo D, Sadoulet MO. Glycobiology. 1998;8:605–613. doi: 10.1093/glycob/8.6.605. [DOI] [PubMed] [Google Scholar]
- 31.Izawa M, Kumamoto K, Mitsuoka C, Kanamori C, Kanamori A, Ohmori K, Ishida H, Nakamura S, Kurata-Miura K, Sasaki K, Nishi T, Kannagi R. Cancer Res. 2000;60:1410–1416. [PubMed] [Google Scholar]
- 32.Mitsuoka C, Ohmori K, Kimura N, Kanamori A, Komba S, Ishida H, Kiso M, Kannagi R. Proc Natl Acad Sci USA. 1999;96:1597–1602. doi: 10.1073/pnas.96.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu F, Zhang Y, Zhang XY, Chen HL. J Cancer Res Clin Oncol. 2002;128:189–196. doi: 10.1007/s00432-001-0314-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saito S, Yamashita S, Endoh M, Yamato T, Hoshi S, Ohyama C, Watanabe R, Ito A, Satoh M, Wada T, Paulson JC, Arai Y, Miyagi T. Oncol Rep. 2002;9:1251–1255. [PubMed] [Google Scholar]
- 35.Chandrasekaran EV, Xue J, Xia J, Chawda R, Piskorz C, Locke RD, Neelamegham S, Matta KL. Biochemistry. 2005;44:15619–15635. doi: 10.1021/bi050246m. [DOI] [PubMed] [Google Scholar]
- 36.Peracaula R, Tabares GRL, Harvey DJ, Dwek RA, Rudd PM, de Llorens R. Glycobiology. 2003;13:457–470. doi: 10.1093/glycob/cwg041. [DOI] [PubMed] [Google Scholar]
- 37.von Der Ohe M, Wheeler SF, Wuhrer M, Harvey DJ, Liedtke S, Muhlenhoff M, Gerardy-Schahn R, Geyer H, Dwek RA, Geyer R, Wing DR, Schachner M. Glycobiology. 2002;12:47–63. doi: 10.1093/glycob/12.1.47. [DOI] [PubMed] [Google Scholar]
- 38.Wuhrer M, Geyer H, von der Ohe M, Gerardy-Schahn R, Schachner M, Geyer R. Biochemie. 2003;85:207–218. doi: 10.1016/s0300-9084(03)00043-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.