

Abstract
Response regulator signaling proteins and phosphatases of the haloacid dehalogenase (HAD) superfamily share strikingly similar folds, active site geometries, and reaction chemistry. Proteins from both families catalyze the transfer of a phosphoryl group from a substrate to one of their own aspartyl residues, and subsequent hydrolysis of the phosphoprotein. Notable differences include an additional Asp that functions as an acid/base catalyst and an active site well-structured prior to phosphorylation in HAD phosphatases. Both features contribute to substantially faster reactions than for response regulators. To investigate mechanisms underlying the functional differences between response regulators and HAD phosphatases, we characterized five double mutants of the response regulator CheY designed to mimic HAD phosphatases. Each mutant contained the extra Asp paired with a phosphatase-inspired substitution to potentially position the Asp properly. Only CheY DR (Arg as anchor) exhibited enhanced rates of both autophosphorylation with phosphoramidate and autodephosphorylation compared to wild type CheY. Crystal structures of CheY DR complexed with MoO42− or WO42− revealed active site hydrogen-bonding networks similar to those in HAD·substrate complexes, with the extra Asp positioned for direct interaction with a leaving group (phosphorylation) or nucleophile (dephosphorylation). However, CheY DR reaction kinetics did not exhibit the pH sensitivities expected for acid/base catalysis. Biochemical analysis indicated CheY DR had an enhanced propensity to adopt the active conformation without phosphorylation, but a crystal structure revealed unphosphorylated CheY DR was not locked in the active conformation. Thus, the enhanced reactivity of CheY DR reflected partial acquisition of catalytic and structural features of HAD phosphatases.
Keywords: CheY, response regulator, haloacid dehalogenase, HAD phosphatase, molybdate, tungstate
Bacteria monitor the extracellular milieu, respond to metabolic conditions and effect virulence using two component signaling systems1, 2. Each two-component system is minimally comprised of a histidine kinase that detects the signal and a partner response regulator that executes an output response. The response regulator acts as an allosteric molecular switch that is turned on and off by the reversible phosphorylation of an aspartyl residue. Although histidine kinases and phosphatases often regulate rates of response regulator phosphorylation and dephosphorylation in vivo, the response regulator receiver domain contains the conserved catalytic residues necessary and sufficient for both reactions. The ability to self-catalyze phosphoryl chemistry allows for response regulator autophosphorylation using small molecule phosphodonors, such as acetyl phosphate (AcP: CH3CO2PO32−) or phosphoramidate (PAM; NH3+PO32−)3, 4, as well as subsequent hydrolytic autodephosphorylation5. Autophosphorylation occurs via in-line nucleophilic attack by the conserved Asp (D) on the phosphorus atom of the phosphodonor and autodephosphorylation progresses by a water molecule attacking the phosphorus within the phosphoaspartate (Figure 1B)6. Both substitution reactions occur with inversion of stereochemistry around the phosphorus atom. The proposed transition states for both reactions are believed to be stabilized by a conserved active site that includes a bound divalent cation [(coordinated by a conserved pair of acid residues (DD)], as well as threonine/serine (T) and lysine (K) residues (Figure 1A,B).
Figure 1.

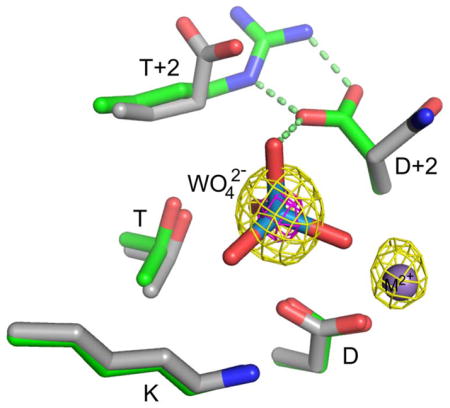
Similarity of active site geometries and reaction chemistry of response regulators and HAD phosphatases. (A) Stereo view of superimposed active sites of E. coli CheY (gray sticks, PDB entry 1FQW) and Bacteroides thetaiotaomicron VPI-5482 hexose phosphate phosphatase (HPP) (green sticks, PDB entry 2RBK). The divalent cations are shown as purple (CheY) or green (HPP) spheres. The BeF3− ion shown is present in the CheY structure. The VO42− that occupies the same location in HPP is not shown for clarity. D, DD, T, and K are defined in the text and are conserved in both families. Additionally, the residue at position D+2 is conserved as an Asp in HAD phosphatases. (B) Proposed trigonal bipyramidal transition states (black) for the phosphorylation (top) and dephosphorylation (bottom) substitution reactions of response regulators and HAD phosphatases. Conserved active site features involved in transition state stabilization that are shared by response regulators and HAD phosphatases (T, K, and Mg2+) are colored green. In addition, HAD phosphatases contain a conserved Asp at position D+2 (red) that acts sequentially as an acid (top) and base (bottom) catalyst and is anchored by the residue at position T+2 (red).
Phosphatases within the haloacid dehalogenase (HAD) superfamily share a highly similar active site and catalyze the same fundamental chemistry as response regulator receiver domains7–10. Like receiver domains, HAD phosphatases have a set of conserved active site residues located on the loops that follow β-strands within a domain that consists of an alternating pattern of β-strands and α-helices. Although the primary sequences of receiver domains and HAD phosphatases are related by circular permutation7, 10, the relative locations of the conserved D, K, T, DD, and divalent cation within receivers and HAD active sites are strikingly similar (Figure 1A). Directly analogous to the autophosphorylation/autodephosphorylation reactions of response regulators, HAD phosphatases catalyze dephosphorylation of their substrates via a two-step mechanism using a phosphoaspartate enzyme intermediate that is subsequently hydrolyzed (Figure 1B)11, 12. HAD phosphatases have a broad range of natural substrates, many of which are phosphomonoesters such as phosphorylated sugars9 and phosphoproteins13. Despite the similar active sites, HAD phosphatases typically exhibit rate constants ~102–104 fold greater than response regulators14. This difference highlights the different roles for the phosphorylated intermediate in the two families. HAD phosphatases evolved to catalyze dephosphorylation and typically only briefly exist as phosphorylated enzyme intermediates. In contrast, response regulators must persist in the phosphorylated (activated) form to facilitate signal transduction, and have dephosphorylation kinetics on the timescale of the processes they regulate15.
Two features of HAD phosphatases that likely contribute to their faster kinetics are an additional conserved Asp residue positioned two residues C-terminal to the site of phosphorylation (position D+2) and an active site so well structured to stabilize the transition state as to be capable of distorting the shape of substrate analogs11. The additional Asp acts alternately as a catalytic acid and base in the two phosphatase half reactions16–18 (Figure 1B) and is often anchored in position by a semi-conserved residue (often an Arg, Lys, Thr, Trp, or Tyr) at position T+2 (two residues C-terminal to the conserved Ser/Thr), which helps to maintain the active site structure11. In contrast, position D+2 is rarely an Asp in response regulators, accounting for less than 2% of response regulator sequences in a survey (Immormino and Bourret, unpublished) of ~14,000 sequences from the MiST2.119 database. In addition, response regulators are dynamic proteins that exhibit modest but functionally important conformational changes at the active site and allosterically linked surfaces. Residue T+2 lies on a particularly mobile loop20, 21. Despite these differences with HAD phosphatases, the residues at positions D+2 and T+2 in response regulators have been identified as playing roles in modulation of both autophosphorylation and autodephosphorylation reactions15, 22, 23. Furthermore, the D+2 and T+2 residues interact with each other in multiple X-ray crystal structures of response regulator receiver domains15, 24
In order to more fully understand the mechanistic similarities and differences between response regulators and HAD phosphatases, we explored the effect of placing an Asp at position D+2 and a partner residue at T+2 in the response regulator Escherichia coli CheY. CheY consists only of a receiver domain and has been used extensively as a model system to elucidate kinetic determinants that modulate receiver domain phosphotransfer reactions22, 23 as well as the allosteric conformational changes linked to phosphorylation21, 25. Of a set of five CheY double mutants designed to mimic the HAD active site, CheY DR (an Asp at D+2 and an Arg at T+2) uniquely exhibited enhanced rate constants for both autophosphorylation and autodephosphorylation. Structural and biochemical analysis indicated that the enhanced kinetics of CheY DR could be explained by partial acquisition of some properties of HAD phosphatases. Crystal structures of CheY DR complexed with MoO42− or WO42− revealed a CheY backbone in a fully activated conformation and an active site hydrogen bonding network between Asp at D+2 and the Arg at T+2 that positioned the Asp to interact with the leaving group atom of the phosphodonor (for autophosphorylation) or the attacking water molecule (for autodephosphorylation). However pH profiles of autocatalytic rate constants did not indicate an altered pKa for the Asp at D+2 as would be predicted for function as a true acid/base catalyst. CheY DR also exhibited an enhanced binding affinity for the target peptide FliM1–16 relative to wild type CheY, suggesting that a shift toward an activated (i.e. phosphorylated/FliM binding) global conformation provided a more complete active site mold as exhibited by HAD phosphatases.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis and Protein Purification
Genes encoding the CheY variants [referred to as CheY (D+2)(T+2), i.e. CheY DR, CheY DK, CheY DY, CheY DQ, and CheY DT) were generated by QuikChange mutagenesis (Agilent Technologies) of pKC126, a pET28a derived vector encoding wild type Escherichia coli CheY with an N-terminal thrombin-cleavable hexa-histidine tag. The substitutions at T+2 were chosen based on anchoring residues (Arg, Lys, Thr, Trp, Tyr) found in HAD phosphatase structures11. A Trp substitution at T+2 was not constructed because our assays of reaction kinetics depend on fluorescence of the unique Trp present at D+1 in wild type CheY. A Gln substitution at T+2 was tested because its size and chemical properties were potentially compatible with anchoring the Asp at D+2. After thrombin cleavage, an extra GlySerHis was present at the CheY N-terminus, which does not affect CheY autophosphorylation26 or autodephosphorylation27 kinetics. Variation at the N-terminus is not known to affect the structure of the active site, located on the opposite side of the receiver domain. Overexpression of the CheY DX variants was performed with BL21(DE3) cells containing the appropriate pKC1 plasmid. Briefly, 1 L lysogeny broth containing 30 μg/ml kanamycin was inoculated with 15 mL of overnight culture and grown to an OD600 of 1.0. The culture was then induced with 1 mM isopropyl-β-D-1-thiogalactopyranoside at room temperature overnight. Cell lysates (in 50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole) were purified by Ni-NTA agarose (Qiagen) chromatography. The eluted protein (in 50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 150 mM imidazole) was incubated with human α-thrombin (Haemolytic Technologies Inc.) at a 1:6000 (w/w; thrombin:CheY) ratio overnight at room temperature. A final purification step to separate CheY from thrombin and the cleaved tag was gel filtration (Superdex75 16/60, GE) in TMG buffer (25 mM Tris pH 7.5, 5 mM MgCl2, 10% v/v glycerol). Wild type CheY and the T+2 single mutants (CheY NK, NR, NA, NY, NQ) were expressed and purified as previously described28. Briefly, K0641recA containing pRS329 or mutant plasmids were grown to an OD600 of 1.0 and induced with 100 μg/mL β-indole acrylic acid at 37°C overnight. Cell lysates in TMG buffer were purified by dye-affinity chromatography (Affigel-Blue, Bio-Rad) followed by size-exclusion chromatography (Superdex 75, GE Healthcare). Concentrations for all purified CheY preparations were determined spectrophotometrically using an extinction coefficient at 280 nm of 0.727 (mg/mL)−1 cm−1 4.
Fluorescence Measurement of CheY Autophosphorylation and Autodephosphorylation Kinetics
With the exception of CheY DR and CheY DK autophosphorylation reactions, autophosphorylation and autodephosphorylation time courses were measured by continuous monitoring of CheY intrinsic tryptophan fluorescence on a Perkin-Elmer LS-50B spectrofluorimeter with an Applied Photophysics (Surrey, U.K.) RX2000 rapid mixer accessory (dead time = 8 ms) and Perkin-Elmer FL WINLAB V.1.1 software as previously described23, 30. The kinetics of CheY DR and CheY DK autophosphorylation were too fast to be measured using the LS-50B, so measurements were made using a Jobin Yvon FluoroLog 322 spectrofluorimeter (response time = 3 ms) equipped with a F-3009 μFlow stopped flow apparatus (dead time < 5 ms) and associated DATAMAX Version 2 software at the UNC Macromolecular Interactions Facility. In each case, the excitation and emission wavelengths were set to 295 nm and 346 nm respectively and samples were maintained at a constant temperature of 25°C with a circulating water bath. Data points were recorded at 20 msec intervals on the LS-50B and 10 msec intervals on the FluorLog 322.
For autophosphorylation experiments, equal volumes of 10 μM CheY and varying concentrations of phosphodonor- each in 100 mM HEPES, pH 7.0, 10 mM MgCl2 -were mixed. The phosphodonor was either phosphoramidate (PAM) with four final reaction concentrations ranging from 0.5–30 mM or acetyl phosphate (AcP) with four final reaction concentrations ranging from 10–50 mM. Because ionic strength affects CheY autophosphorylation kinetics31, 32, KCl was added to the phosphodonor solution to maintain a constant ionic strength of 300 mM (phosphodonor plus KCl). Time courses monitoring the accumulation of CheY-P were fit to a single exponential decay model to determine kobs. The data were further analyzed based on the following reaction scheme3, 32 (illustrated with PAM as phosphodonor):
Scheme 1.
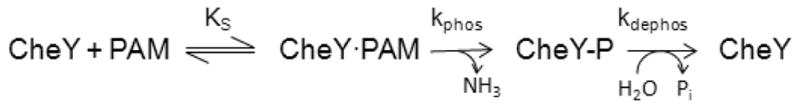
where KS is the equilibrium dissociation constant for formation of the noncovalent complex between CheY and PAM, kphos is the rate constant for phosphotransfer within the complex, and kdephos is the autodephosphorylation rate constant. kobs is a function of the kinetic parameters from scheme 1 as follows32:
| (eqn. 1) |
For each CheY variant, kobs was measured at four different phosphodonor concentrations. Plots of kobs versus phosphodonor concentration were linear in all cases and the slope gave the apparent bimolecular rate constant, kphos/KS.
Rate constants for CheY autodephosphorylation were determined using the pH jump method27, 32. Mutant or wild type CheY (10 μM in 10 mM HEPES pH 7.0, 20 mM MgCl2) was phosphorylated with PAM at ~five times the concentration of PAM required to phosphorylate half of the CheY at steady state. The sample was then mixed using the rapid mixing accessory with an equal volume of pH jump buffer (200 mM sodium carbonate, pH 10.2), which drastically retards the phosphorylation reaction, so that the observed time course for increase in fluorescence reflects the dephosphorylation reaction alone. Each time course was fit to a single exponential decay to determine the first order rate constant, kdephos. CheY autodephosphorylation rate constants determined by the pH jump method and by loss of radioactive phosphoryl groups agree within a factor of less than two27.
Rapid Dilution Assay to Determine the pH Dependence of Autodephosphorylation
The effect of pH on autodephosphorylation was tested with a rapid dilution assay. To isolate the dephosphorylation reaction, 100 μM CheY in autophosphorylation buffer was first phosphorylated with PAM (50 mM for wild type CheY, or 5 mM for CheY DR and CheY DY) in a 15 μL reaction volume. The reaction was then diluted 100-fold with 10 mM MgCl2 and 100 mM of one of the following buffers: sodium carbonate, pH 10.2; Tris, pH 8.9; Tris, pH 8.0; HEPES, pH 7.0; MES, pH 6.5; sodium cacodylate, pH 5.9; sodium citrate, pH 5; or sodium acetate, pH 4.5. The phosphorylation and dilution were performed in a 1.5 mL quartz fluorescence cuvette while monitoring Trp fluorescence. The observed pseudo first-order rate of autophosphorylation is proportional to the concentration of phosphodonor and thus is reduced by two orders of magnitude upon dilution. The observed change in fluorescence for these experiments (kobs, eqn. 1) following dilution is dominated by the kdephos term.
Autophosphorylation pH Profiles
The pH dependence of autophosphorylation was measured using a variation of the autophosphorylation assay described above. Briefly, equal volumes of 10 μM CheY and a phosphodonor solution of 5 mM PAM and 95 mM KCl were mixed using the rapid mixer accessory. Both CheY and phosphodonor were suspended in the set of pH buffers consisting of 10 mM MgCl2 and 100 mM buffer as described for the rapid dilution assay. The observed rate constant (kobs, eqn. 1) reflects contributions from both rate constants for autophosphorylation (kphos/KS) and autodephosphorylation (kdephos). To separate the effect of pH on kphos/KS, the kdephos value determined using the pH jump method was subtracted from kobs giving equation 2. To more readily compare the CheY mutants, kobs - kdephos was normalized to the value at pH 6.5 giving equation 3.
| (eqn. 2) |
| (eqn. 3) |
Enzyme Activity Towards Para-Nitro-Phenyl Phosphate
Wild type CheY or CheY DR (18.4 μM) was added to 200 mM para-nitro-phenyl phosphate (pNPP, Sigma) in a 1.0 mL cuvette and the optical density at 450 nm read continuously to monitor the release of para-nitrophenolate. The buffers (present at 100 mM) were sodium salts of HEPES (pH 7.0), cacodylate (pH 6.5), citrate (5.0), or acetate (4.5). All buffers contained 10 mM MgCl2. The positive controls were the HAD phosphatase E. coli YbiV33 or calf intestinal phosphatase.
Fluorescence Measurement of Binding Between CheY and FliM1–16
Binding affinities between the peptide MGDSILSQAEIDALLN, which corresponds to the amino terminal 16 residues of E. coli FliM (FliM1–16), and CheY were measured using tryptophan fluorescence as previously described34, 35. FliM1–16 (10 – 1000 μM final concentration) was titrated into 5 μM CheY in autodephosphorylation buffer (10 mM HEPES, pH 7.0, 20 mM MgCl2) and the resulting fluorescence quench was monitored. The measured intensities were corrected for dilution and Kd values were determined using Prism (GraphPad) to fit the data to a one-site binding model.
Crystallization, Data Collection, Structure Solution, and Refinement
Crystals of CheY DX mutants were grown by hanging drop vapor diffusion at room temperature. Crystals of the CheY·BeF3−·Mn2+ complexes were obtained by adding 20 mM MnCl2, 1 mM BeCl2, and 10 mM NaF to the CheY variants (at 4.3–8.9 mg/mL) prior to crystallization. Diffraction quality crystals were found in conditions36 ranging from 1.6–2.4 M ammonium sulfate, 5% (v/v) glycerol, 100 mM Tris, pH 7.5–8.25 with a drop ratio of 1:1 protein:reservoir buffer. Crystals of the CheY variants in the absence of BeF3− were obtained using 120–160 mM calcium acetate, 100 mM sodium cacodylate, pH 6.0, 20 mM MnCl2, and 28–31% (v/v) PEG8000 as the reservoir solution with a 1:1 drop ratio37. Crystals generally grew overnight or over the course of less than one week in various forms. For conditions with BeF3−, the main crystal type was rhombic bipyramidal, but several of the CheY variants also formed rod shaped crystals, which tended to diffract better.
CheY·BeF3−·Mn2+ and CheY·Mn2+ crystals were cryoprotected with glycerol as previously described15, or by serial transfers through buffers with increasing amounts of glycerol. Cryoprotected crystals were flash cooled in liquid nitrogen in preparation for data collection. X-ray diffraction images were collected at the SERCAT beamlines 22-BM and 22-ID at APS. Diffraction data were reduced and scaled using HKL200038 or XDS39.
Structures were also obtained for CheY DR bound to MoO42− or WO42−. The crystals were grown from conditions similar to the CheY·BeF3−·Mn2+ complexes (2.45–2.6 M ammonium sulfate, 5% (v/v) glycerol, 100 mM Tris, pH 7.5). Prior to setting up crystallization drops, 20 mM MnCl2, and either 2 mM ammonium molybdate or 10 mM sodium tungstate were added to CheY DR and incubated for 10 min. Single anomalous dispersion (SAD) data was collected at the LI absorption edge of tungsten (1.02631 Å) to 2.1 Å resolution for both complexes (Table 2). At this wavelength tungsten, molybdenum, and manganese have anomalous signals of 12.2 e−1, 1.3 e−1 and 1.4 e−1 respectively40, allowing for confident distinction of molybdate and tungstate from sulfate ions (0.25 e−1) present at up to 2.6 M in the crystallization buffer.
Table 2.
Summary of data collection and refinement statistics for CheY DR complexed with MoO42− or WO42−.
| Diffraction Data | CheY DR + MoO42−, Mn2+ | CheY DR + WO42−, Mn2+ |
|---|---|---|
| Diffraction Data Statistics | ||
|
| ||
| PDB code | 3RVR | 3RVS |
| Source | APS 22-BM | APS 22-BM |
| Space Group | P212121 | P212121 |
| a, b, c (Å) | 53.51, 53.63, 161.84 | 53.56, 53.62, 162.82 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 1.02631 | 1.02631 |
| Resolution (Å)a | 50-2.10 | 50-2.10 |
| (Last Shell) (Å) | 2.15-2.10 | 2.15-2.10 |
| Unique Reflections | 27965 | 52812 |
| Completeness (Last Shell) (%) | 99.8 (100) | 99.9 (100) |
| Average I/σI (Last Shell) | 20.2 (5.3) | 15.3 (4.3) |
| Redundancy (Last Shell) | 7.0 (6.7) | 11.2 (11.4) |
| Rsymb (Last Shell) (%) | 7.2 (38.4) | 11.3 (60.3) |
| Refinement Statistics | ||
| Refinement Package | PHENIX 1.7_650 | PHENIX 1.7_650 |
| Resolution range (Å) | 19.73-2.10 | 19.31-2.10 |
| Reflections | 27715 | 52807 |
| Non-solvent atomsc | 1995 | 1986 |
| Solvent and hetero-atomsc | 385 | 378 |
| Molecules in Asymmetric Unit | 2 | 2 |
| Rms deviation from ideality | ||
| Bond lengths (Å) | 0.011 | 0.010 |
| Bond angles (°) | 1.288 | 1.328 |
| Rd value (%) | 17.1 | 14.8 |
| Rd free (%) | 18.8 | 18.4 |
Resolution limit was defined as the highest resolution shell where the average I/σI was >2.
Rmerge = ΣhklΣi|Ii(hkl) - <I(hkl)> |/ΣhklΣII(hkl).
Non-hydrogen atoms, alternate atoms are counted once.
R = Σ | Fo - Fc |/ΣFo. ~5% of reflections were used to calculate Rfree.
Initial phases for the CheY crystals were obtained by molecular replacement using PDB entry 1FQW41 (CheY·BeF3−·Mg2+ with CheY in the activated conformation) or 3CHY42 (apo-CheY with CheY in an unactivated conformation) as the search models. Initial models were improved by iterative rounds of model building in Coot43 and structure refinement with PHENIX44. Prior to deposition, the models were validated using MolProbity45. Tables 2, 3, and S1 contain summaries of diffraction data and refinement statistics. For structural comparisons, Cα RMSD (root-mean-square deviation) values were determined using the align command in PyMOL (PyMOL Molecular Graphics System, Version 1.4, Schrödinger, LLC.).
Table 3.
Summary of data collection and refinement statistics for CheY•BeF3−•Mn2+ complexes.
| Diffraction Data | CheY DR + BeF3−, Mn2+ | CheY DK + BeF3−, Mn2+ | CheY DY + BeF3−, Mn2+ | CheY DQ + BeF3−, Mn2+ |
|---|---|---|---|---|
| Diffraction Data Statistics | ||||
|
| ||||
| PDB code | 3RVL | 3RVP | 3RVN | 3RVJ |
| Source | APS 22-BM | APS 22-ID | APS 22-BM | APS 22-BM |
| Space Group | P21221 | P212121 | P212121 | P212121 |
| a, b, c (Å) | 37.63, 72.44, 107.28 | 53.49, 53.63, 160.42 | 53.44, 53.61, 161.31 | 53.58, 53.70, 160.66 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.97933 | 1.00882 | 0.97933 | 1.0 |
| Resolution (Å)a | 50-1.55 | 50-2.40 | 50-2.25 | 50-2.10 |
| (Last Shell) (Å) | 1.58-1.55 | 2.44-2.40 | 2.29-2.25 | 2.14-2.10 |
| Unique Reflections | 43539 | 18144 | 22709 | 26982 |
| Completeness (Last Shell) (%) | 99.3 (96.5) | 96.9 (70.7) | 99.8 (99.9) | 96.3 (91.9) |
| Average I/σI (Last Shell) | 18.8 (3.0) | 22.3 (2.6) | 12.8 (2.7) | 14.4 (2.4) |
| Redundancy (Last Shell) | 5.6 (4.0) | 6.4 (3.1) | 5.0 (4.4) | 6.2 (4.7) |
| Rsymb (Last Shell) (%) | 9.4 (40.3) | 7.9 (33.7) | 13.9 (53.1) | 11.0 (62.4) |
| Refinement Statistics | ||||
| Refinement Package | PHENIX 1.7_650 | PHENIX 1.7_650 | PHENIX 1.7_650 | PHENIX 1.7_650 |
| Resolution range (Å) | 25.92-1.55 | 44.58-2.40 | 27.6-2.25 | 27.6-2.10 |
| Reflections | 42133 | 17615 | 21750 | 26051 |
| Non-solvent atomsc | 1930 | 1990 | 1996 | 1986 |
| Solvent and hetero-atomsc | 453 | 288 | 323 | 325 |
| Molecules in Asymmetric Unit | 2 | 2 | 2 | 2 |
| Rms deviation from ideality | ||||
| Bond lengths (Å) | 0.015 | 0.007 | 0.007 | 0.007 |
| Bond angles (°) | 1.588 | 1.058 | 1.110 | 1.095 |
| Rd value (%) | 15.6 | 17.3 | 18.6 | 17.7 |
| Rd free (%) | 18.2 | 21.5 | 22.5 | 20.1 |
Resolution limit was defined as the highest resolution shell where the average I/σI was >2.
Rmerge = ΣhklΣi|Ii(hkl) - <I(hkl)>|/ΣhklΣII(hkl).
Non-hydrogen atoms, alternate atoms are counted once.
R = Σ|Fo - Fc|/ΣFo. ~5% of reflections were used to calculate Rfree.
RESULTS
A CheY HAD Mimic Exhibited Enhanced Rates of Both Autophosphorylation and Autodephosphorylation
The faster reaction kinetics observed for HAD phosphatases relative to response regulators are due in part to an additional conserved active site Asp at position D+2 [two residues from the phosphorylatable Asp (D)]. The Asp at D+2 is often held in place via hydrogen bonding or salt bridge interactions with a residue at position T+211, thus orienting the Asp side chain for direct interaction with the leaving group (during formation of the Asp-P intermediate) or with the water oxygen (during hydrolysis of the Asp-P intermediate) (Figure 1B). To assess the impact of the presence of an Asp at D+2 in response regulators with various potential anchoring residues at T+2, we constructed a set of five CheY DX proteins where D represents the substitution of an Asp at position D+2 (Asn59 in wild type CheY) and X is the substitution of either Arg, Gln, Lys, Thr or Tyr, at position T+2 (Glu89 in wild type CheY). For CheY, the two steps that correspond to the HAD phosphatase half reactions (autophosphorylation and autodephosphorylation, respectively) can be monitored independently and with high precision using stopped-flow fluorescence and exploiting the difference in tryptophan emission for phosphorylated and unphosphorylated CheY.
Measured bimolecular rate constants (kphos/Ks) for autophosphorylation of CheY DX double mutants with the phosphodonors acetyl phosphate (AcP) and phosphoramidate (PAM) as well as autodephosphorylation rate constants (kdephos) are listed in Table 1. Because the goal of the current study was to assess the extent to which the CheY scaffold can kinetically and mechanistically mimic an HAD phosphatase by utilizing an aspartate at position D+2 as an additional catalytic residue, the rate constants were compared to those of the analogous CheY single mutants with an Asn (the wild type residue) at position D+2 and the same residue at T+2, reported in a previous study23. For autophosphorylation with AcP, the five CheY DX variants for which comparison was possible gave reduced rate constants (between ~7–26 fold) relative to the matched CheY NX single mutant, indicating that the Asp at D+2 actually had detrimental effects on kinetics, opposite the expected result for successful mimicry of catalysis by HAD phosphatases. This result is consistent with the previously observed deleterious effect of net negative charge of D+2/T+2 side chains for CheY autophosphorylation with AcP23, which likely masked any potential specific catalytic function of the Asp at D+2.
Table 1.
Autophosphorylation and autodephosphorylation rate constants of CheY variants.
| CheY designationa | Amino acid at position | kphos/KS (M−1s−1)b
|
kdephos (min−1)c | ||
|---|---|---|---|---|---|
| D+2 | T+2 | Phosphoramidate | Acetyl phosphate | ||
| DX set (HAD mimics) | |||||
| DR | D | R | 820 ± 26 | 3.9 ± 0.4 | 20 ± 1.7 |
| DK | D | K | 170 ± 3 | 5.7 ± 0.5 | 2.8 ± 0.06 |
| DQ | D | Q | 13 ± 0.2 | 1.5 ± 0.05 | 4.8 ± 0.03 |
| DY | D | Y | 130 ± 10 | 0.5 ± 0.05 | 0.7 ± 0.05 |
| DT | D | T | 4.1 ± 0.05 | 0.3 ± 0.1 | 1.7 ± 0.02 |
| DE | D | E | 3.7 ± 0.55d | < 0.2d | 5.5 ± 0.4e |
| NX set | |||||
| NR | N | R | 93 ± 18d | 62 ± 5.5d | 0.69 ± 0.04e |
| NK | N | K | 60 ± 7.3d | 42 ± 0.9d | 1.2 ± 0.3e |
| NQ | N | Q | 19 ± 3d | 36 ± 1.8d | 3.9f |
| NY | N | Y | 240 ± 34d | 13 ± 1.9d | 0.26 ± 0.01e |
| NA | N | A | 10 ± 0.8d | 18 ± 2.0d | 1.5f |
| wild type | N | E | 9.2 ± 0.9 | 10 ± 0.7 | 3.2 ± 0.2 |
CheY variants are referred to in the text by ‘CheY’ followed by the single letter codes for the amino acids at positions D+2 and T+2 (CheY residues 59 and 89).
Mean and standard deviation of autophosphorylation rate constants (n=3). See Experimental Procedures or footnote d where indicated.
Mean and standard deviation of autodephosphorylation rate constants (n=3). See Experimental Procedures or footnotes e or f where indicated.
Thomas et al.23
Thomas et al.22
Silversmith et al.79
In contrast to the AcP results, there was significant variation in the rate constants for the CheY DX variants relative to the CheY NX mutants for autophosphorylation with PAM (Table 1). We previously found that rate constant variation for CheY autophosphorylation with zwitterionic PAM correlates with the nonpolar surface area of D+2/T+2 side chains23 and the rate constants for the CheY NX set largely reflects these differences. Three of the five CheY HAD mimics (CheY DY, DQ, and DK) gave PAM autophosphorylation rate constants that were within three-fold of the corresponding CheY NX variant, indicating that the Asp at D+2 functioned similarly to the Asn. CheY DT exhibited an autophosphorylation rate constant for PAM that was less than that of any measured CheY NX protein23, again suggesting no special contribution from the Asp at D+2. The CheY NT mutant was not available for direct comparison, and CheY DT was not characterized further. CheY DR, however, exhibited a PAM autophosphorylation rate constant that was about nine-fold greater than CheY NR (and ~90-fold greater than wild type CheY), suggesting that the negative charge of the Asp at D+2 functioned in catalysis. Furthermore, CheY DR was also the only CheY DX variant with a substantially enhanced autodephosphorylation rate constant. Whereas CheY DY, DQ, and DK all gave autodephosphorylation rate constants within ~three-fold of the CheY NX analog, CheY DR gave an autodephosphorylation rate constant ~30-times that of CheY NR (and ~six-fold greater than wild type CheY). Thus CheY DR is the most probable candidate for a CheY HAD mimic in which the Asp at D+2 plays a catalytic role that is dependent on the acidic side chain.
Accelerated reaction rates for CheY DR are not due to acid/base catalysis
One possible mechanism to account for the accelerated reaction rates for both CheY DR autophosphorylation with PAM and autodephosphorylation (Table 1) could be that the Asp at position D+2 acts as an acid/base catalyst, as in the HAD phosphatases (Figure 1B). Acidic residues (Asp or Glu) that act as acid/base catalysts typically have pKa values near neutral46 so that appreciable amounts of both the protonated and unprotonated forms are present. If the Asp at D+2 in CheY DR acts as a catalytic base in autodephosphorylation, then pH profiles for CheY DR autodephosphorylation might be expected to exhibit a transition towards lower rate constants in the mildly acidic pH range, in contrast to wild type CheY, for which autodephosphorylation rate constants are essentially pH independent over a broad pH range4. However, the autodephosphorylation rate constants for CheY DR, CheY DY, and wild type CheY were essentially constant over a wide pH range (4.5–10.2) (Figure 2A). Thus, none of the three tested CheY variants contain a residue with pKa > ~4.5 that contributes to catalysis in its deprotonated state, as would be expected for a base catalyst.
Figure 2.

pH sensitivities of autodephosphorylation and autophosphorylation rate constants. (A) Autodephosphorylation rate constants (mean and standard deviation, n≥3) are plotted for wild type CheY at 25 °C (squares), CheY DY at 25 °C (triangles), and CheY DR at 15 °C (inverted triangles). The rate constants were measured with fluorescence using the rapid dilution method. Horizontal lines indicate the autodephosphorylation rate constants measured by the pH jump method (pH 10.2). (B) Rate constants (mean and standard deviation, n 3) for autophosphorylation with PAM normalized to the rate at pH 6.5 for wild type CheY (red), CheY DR (green), CheY DK (orange), CheY DY (yellow), and CheY DQ (blue).
We also considered the possibility that acid catalysis could account for the accelerated rate of CheY DR autophosphorylation with PAM (Table 1). However, at pH 7.0 (the pH at which the rate constant was measured), PAM exists as a zwitterion with a protonated nitrogen (+1) and two oxygen monoanions (−2). Thus, for PAM, the proton is already present on the leaving group atom and acid catalysis by the enzyme is not required. PAM loses the nitrogen proton with a pKa of ~8.0 and wild type CheY reacts with PAM only under pH conditions where the nitrogen is protonated4. We investigated the possibility that CheY DR autophosphorylation may exhibit a broader pH range than wild type CheY, which could reflect acid catalysis by a residue with pKa >~7. However, the normalized pH dependences of PAM autophosphorylation for wild type CheY and CheY DR (as well as CheY DK, DY, and DQ; Figure 2B) were virtually superimposable despite large differences in absolute rates. All of the pH profiles had a transition that correlated with the pKa of PAM47, thus giving no positive evidence for acid catalysis by CheY DR.
Finally, we tested the ability of CheY DR to hydrolyze para-nitro-phenyl phosphate (pNPP). Many HAD phosphatases readily react with pNPP14, a phosphomonoester whose rate of hydrolysis would be expected to benefit greatly from acid catalysis. However, CheY DR exhibited no detectable reactivity towards pNPP over the pH range 4.5–7.5 (data not shown). Although a negative result, taken together with the pH profile results (Figure 2), we conclude that it is unlikely that the Asp at D+2 in CheY DR acted as a true acid/base catalyst in the phosphorylation and dephosphorylation reactions.
Structures of CheY DR Complexed with WO42− or MoO42−
Without evidence for acid/base catalysis, structural analysis was used to gain insight into the mechanism by which CheY DR uniquely enhanced rates of both CheY autophosphorylation and autodephosphorylation. We obtained high quality crystals of CheY DR complexed with Mn2+ and the phosphate analogs molybdate (MoO42−) or tungstate (WO42−). The CheY DR·MoO42−·Mn2+ and CheY DR·WO42−·Mn2+ structures, both solved to 2.1 Å resolution (Table 2), represent the first structures of which we are aware of receiver domains complexed with these ions. The presence of MoO42− or WO42− was confirmed by calculation of anomalous difference Fourier maps using phases calculated from the molecular replacement solution. As seen in Figures 3 and S1, the >4 σ peak for molybdenum and a >15 σ peak for tungsten indicate that the ions are bound at the active site.
Figure 3.
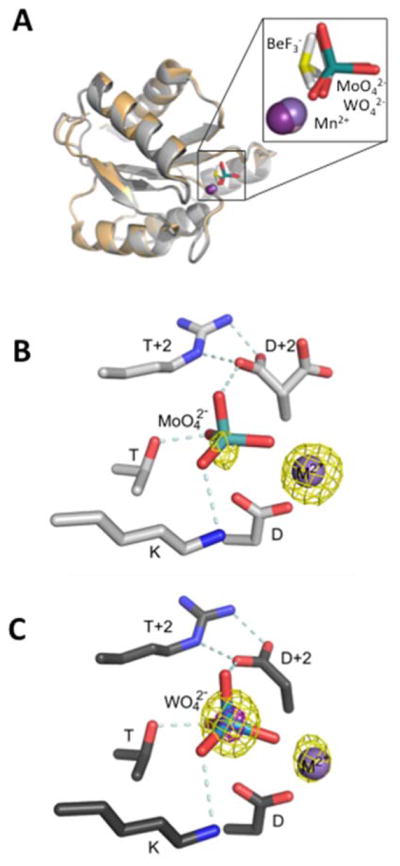
X-ray crystal structures of CheY DR with bound MoO42− or WO42−. (A) Overlay of CheY DR·MoO42− ·Mn2+ (light gray ribbon), CheY DR·WO42− ·Mn2+ (dark gray ribbon), and wild type CheY·BeF3− ·Mn2+ (PDB entry 1FQW, orange ribbon). Mn2+ ions are represented with purple spheres and the MoO42−, WO42−, or BeF3− ions are shown as sticks with Mo and W (teal), Be (yellow), and F (white). The inset is a zoomed version of the active site ligands. (B) and (C) Close-up views of the active sites of CheY DR·MoO42− ·Mn2+ (B) and CheY DR·WO42− ·Mn2+ (C) using the same coloring as panel (A). In both structures, the Asp at D+2 is oriented to form a divalent salt bridge with the Arg at T+2. Additionally, the Asp at D+2 forms a hydrogen bond with the apical oxygen of the bound molybdate or tungstate. In panel B the anomalous difference Fourier map (yellow mesh) is contoured at 4σ and has peaks that roughly correspond with the positions of the Mn and Mo atoms. Similarly, in panel C, peaks are observed at 5σ (yellow mesh) for Mn and W, and additionally at 15σ (magenta mesh) for W. The relative peak heights and atom identities for the anomalous scattering atoms are consistent with expectations for the X-ray wavelength used [LI absorption edge of tungsten (1.02631 Å)]. Hydrogen bonding and electrostatic interactions are represented by light blue dashed lines and Mn2+ is represented as a purple sphere.
The CheY DR co-crystal structures were analyzed for both global backbone conformation and details of the active site. Like all receiver domains, CheY is an allosteric protein and exhibits both ‘unactivated’ and ‘activated’ global conformations for signal transduction, with the activated signaling conformation dominating in the phosphorylated state. The conserved differences between unactivated and activated receiver domain conformations are evident in comparison of structures of the unphosphorylated domain with structures of models of the phosphorylated protein, most often using the phosphoryl analog BeF3− 41. The phosphoprotein intermediate is generally too labile to isolate in crystals. In both the MoO42−and WO42− structures, the CheY backbone and relevant side chains displayed all the hallmarks of a fully activated receiver domain including rotameric switching of residues Y106 and T87 and a ~4 Å shift of the β4α4 loop. The global similarity (Figure 3A) was reflected in Cα RMSD values of 0.182 and 0.217 Å in comparison of CheY·BeF3−·Mn2+ (PDB entry 1FQW) to CheY DR·MoO42−·Mn2+ and CheY DR·WO42−·Mn2+, respectively. In contrast, the Cα RMSD values for comparison of CheY DR·MoO42−·Mn2+, CheY DR·WO42−·Mn2+, and CheY·BeF3−·Mn2+ to the unactive CheY•Mg 2+ structure (PDB entry 2CHE) were 0.874, 0.882, and 0.841 Å, respectively.
Close-up views of the active sites show that both the MoO42− and WO42− anions retained an essentially tetrahedral geometry (Figure 3, Figure S1 for 2Fo-Fc density maps). In both structures, three of the four oxygen atoms from the anion essentially overlay positions occupied by the three fluorine atoms in structures of CheY•BeF3−•Mn2+ (Figure 3A, inset) and the three oxygen atoms make identical interactions with protein atoms as the fluorines in the BeF3− structure. However, the stereochemistry of the MoO42− or WO42− is inverted relative to the BeF3−, with the fourth oxygen atom projecting out towards solvent (Figure 3A, inset). This arrangement suggests that these structures are models of substrate binding to CheY prior to phosphorylation or models of the dephosphorylated protein before dissociation of the phosphate product. In this context, the fourth oxygen atom would be in the location of the leaving group atom during autophosphorylation or the water oxygen during autodephosphorylation.
Further examination of the active site revealed a network of interactions involving the MoO42− or WO42− anion and the D+2/T+2 side chains. One of the key active site interactions in HAD phosphatases is the interaction between the Asp at D+2 and a polar residue at T+2, which positions the Asp appropriately for catalysis and stabilizes the active site structure11. In wild type CheY, the Asn at D+2 is oriented away from the Glu at T+2 and away from the active site. A simple rotamer switch, observed in some structures of mutant CheY15, 48, however, allows the D+2 Asn residue to extend toward T+2 and the active site. In both the CheY DR·MoO42−·Mn2+ and CheY DR·WO42−·Mn2+ structures, the side chain of the Asp at D+2 assumes the rotamer directed towards the active site and forms a bidentate salt bridge with the Arg at T+2 (Figure 3B,C). Both the nature of the Asp (D+2)/Arg (T+2) interactions and the consequent positioning of the Asp at D+2 are highly similar to interactions seen in HAD phosphatases.
The adoption of the HAD phosphatase-like positioning of the Asp at D+2 in CheY DR puts the side chain carboxylate within hydrogen bonding distance of the apical oxygen of the bound molybdate or tungstate anion (Figure 3B,C). Similar hydrogen bonding interactions could occur between the carboxylate side chain and the leaving group atom of a small molecule phosphodonor during phosphorylation, or an attacking water molecule during dephosphorylation, both of which would occupy nearly the same position in space as the apical oxygen atom. Thus the orientation and novel interactions observed for D+2 and T+2 in the CheY DR co-crystal complexes with MoO42− and WO42− likely contribute to the accelerated autophosphorylation and autodephosphorylation rates of CheY DR.
Residues at Positions D+2 and T+2 Do Not Interact in the Activated Conformations of the Other CheY HAD Mimics
To assess whether the details of the active site hydrogen bonding network were unique to CheY DR, co-crystal structures of other CheY HAD mimics (CheY DQ, DY, and DK) complexed with Mn2+ and BeF3− were solved to between 1.55 and 2.4 Å resolution (Table 3). In all of the co-crystal structures, the CheY backbone was quite similar to that of activated wild type CheY (PDB entry 1FQW)41, with Cα RMSDs of 0.263 Å, CheY DK; 0.260 Å, CheY DY; and 0.255 Å, CheY DQ. In contrast to CheY DR·MoO42−·Mn2+ and CheY DR·WO42−·Mn2+, the Asp at D+2 is oriented away from the active site pocket and is not anchored by the polar residue at position T+2 in CheY DQ, CheY DK, and CheY DY complexed with BeF3− and Mn2+ (Figure 4B, C, D). We also solved the crystal structure of CheY DR complexed with Mn2+ and BeF3−. Interestingly, the CheY DR·BeF3−·Mn2+ complex crystallized in the orthorhombic space group P21221 rather than the more common P212121 space group observed previously for many BeF3− bound CheY co-crystal structures, likely accounting for the slightly higher RMSD (Cα RMSD of 0.633 Å) relative to wild type CheY·BeF3−·Mn2+ (PDB entry 1FQW). With the P21221 packing environment, the Asp at D+2 formed a salt bridge with Arg19 from helix α1 of a symmetry related molecule and not with the Arg at T+2 within the same monomer (Figure 4F). However, the CheY DR Asp residue at position D+2 was still oriented in the appropriate position for catalysis (Figure 4E).
Figure 4.
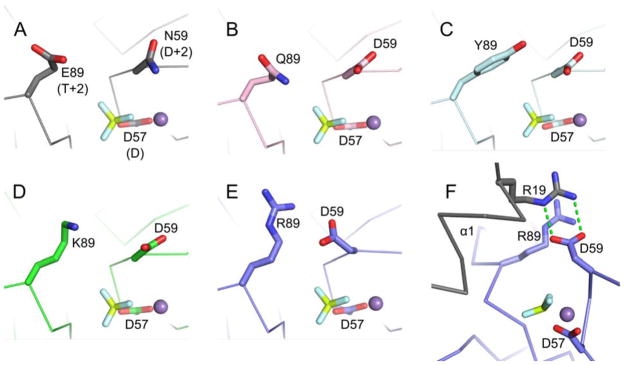
Positioning of residues D+2 and T+2 in CheYs complexed with BeF3− and Mn2+. In wild type CheY (A, PDB entry 1FQW), the side chain of Asn59 at position D+2 is directed away from Glu89 at position T+2 and is not positioned to interact with the leaving group atom (in autophosphorylation) or the nucleophilic water molecule (in autodephosphorylation). Similar orientations are observed for the Asp at D+2 in CheY DQ·BeF3− ·Mn2+ (B, pink carbons), CheY DY·BeF3− ·Mn2+ (C, cyan carbons), and CheY DK·BeF3− ·Mn2+ (D, green carbons). The active site of CheY DR·BeF3− ·Mn2+ (E/F, slate carbons) showed a reorientation of the Asp at D+2 toward the Arg at 89 and in position to aid catalysis. The Asp at D+2 in the CheY DR structure does not hydrogen bond with the Arg at T+2 as seen in the MO42− and WO42− structures, but instead interacts with Arg19 from helix α1 (grey) of a symmetry related molecule (F). In all panels, the BeF3− is shown as yellow and white sticks and the M2+ as a purple sphere.
Enhanced Binding of FliM1–16 to CheY DR Indicates a More Activated Conformational Landscape
The bidendate salt bridge between the Asp at D+2 and the Arg at T+2 observed in the CheY DR·MoO42−·Mn2+ and CheY DR·WO42−·Mn2+ complexes, reminiscent of similar hydrogen bonding networks seen in the active sites of HAD phosphatases, raised the possibility that the CheY DR mutant had an enhanced propensity to adopt the activated conformation. To assess this possibility, we measured binding affinities for the N-terminal peptide of the flagellar motor protein FliM (FliM1–16) using tryptophan fluorescence49, 50 (Table 4). A population of CheY shifted toward a fully activated signaling conformation - either by phosphorylation or mutation - exhibits tighter binding to FliM1–1649, 50. FliM1–16 binds to a CheY surface distinct from the active site, so FliM1–16 binding affinity serves as a measure of CheY conformation independent of active site modification. The measured Kd for wild type CheY (370 μM) compares reasonably to a previously published measurement from our lab (280 μM)34. Notably, CheY DR exhibited a binding constant (Kd of 65 μM) that was nearly six-fold tighter than wild type CheY and three-fold tighter than CheY NR. The Kd of 65 μM is also less than three-fold weaker than reported values for FliM1–16 binding to phosphorylated wild type CheY (Kd = 26–27 μM) 34, 49, believed to be largely in an activated conformation. The increased affinity of CheY DR for FliM1–16 indicates that CheY DR has an enhanced propensity to acquire an activated conformation in the absence of phosphorylation. In turn, the enhanced propensity might be due to stabilization of the activated conformation of CheY DR by the interactions between the Asp at D+2 and the Arg at T+2 observed in the crystal structures.
Table 4.
Equilibrium dissociation constants for CheY binding to FliM1–16 peptide.
| CheY designation | Amino acid at position | Kd FliM1–16(μM)a | |
|---|---|---|---|
| D+2 | T+2 | ||
| DX set (HAD mimics) | |||
| DR | D | R | 65 ± 5 |
| DK | D | K | 120 ± 8 |
| DY | D | Y | 370 ± 30 |
| DQ | D | Q | 380 ± 50 |
| NX set | |||
| NR | N | R | 180 ± 9 |
| NK | N | K | 230 ± 40 |
| NY | N | Y | 160 ± 10 |
| wild type | N | E | 370 ± 20 |
Mean and standard deviation, n=3.
The highly structured active site “scaffold” or “mold” of HAD phosphatases is noted as one of the contributors to their fast catalytic rates11. Likewise, the shift to an activated conformation could have important implications regarding enhancement of autophosphorylation rates for CheY DR. Such models are consistent with earlier studies on the effect of switching the conformational state of CheY by binding the FliM1–16 peptide or the P2 domain of CheA30. A 160-fold increase in CheY autophosphorylation rate constant was observed for the activated versus unactivated state30, perhaps due to more optimal locations of conserved active site residues51. Thus, in addition to the positioning of the Asp at D+2, the enhanced autophosphorylation kinetics of CheY DR also likely have contributions from a shift toward the activated conformation. Though more modest, several of the other CheY DX and NX mutants - CheY DK, NR, NK, and NY - also exhibited enhanced binding affinities for FliM1–16 relative to wild type CheY (Table 4). Thus, shift in relative activation also likely contribute to the enhanced PAM autophosphorylation rate constants for these CheYs (Table 1).
A shift to an activated conformation could potentially be evident from crystal structures of CheY solved in the absence of a phosphate or phosphoryl group analog. We solved four crystal structures to investigate this possibility. However, only the unactivated conformation was observed in crystal structures of CheY DR, DK, DY, or DQ complexed with Mn2+ in the absence of BeF3− (Table S1). This suggests that the conditions used for crystallization preferentially stabilize the unactive conformation. Furthermore, the observation of CheY DR in the unactive conformation reveals that despite more facile transitions implied by the FliM1–16 binding data, CheY DR is still conformationally dynamic, and unlike the HAD phosphatases does not constitutively maintain a complete active site mold.
DISCUSSION
Meticulous regulation of the rates of both phosphorylation and dephosphorylation of response regulator signaling proteins is essential for effective signal transduction. Here we extend our use of CheY as a model system to elucidate mechanisms of response regulator rate modulation by assessing mechanistic similarities to a family of enzymes (HAD phosphatases) that catalyze nearly identical chemistry using a highly similar active site. Structural and biochemical characterization of a set of CheY double mutants designed to mimic the more efficient HAD phosphatases revealed that one of the engineered CheYs – CheY DR – acquired some properties of the HADs that resulted in enhanced reaction rate constants, although CheY DR appeared to fall short of bona fide acid/base catalysis or an active site preformed to fit the transition state. The results highlight the role of an active site hydrogen bonding network that allows direct interaction with the leaving group/nucleophile atom in rate enhancement of both autophosphorylation and dephosphorylation of response regulators. Additionally, the results provided insight into how the propensity to adopt the activated global conformation positively modulates response regulator catalysis.
Anchoring of the Asp at D+2 by the Residue at T+2 Enhances Catalysis in Both HAD Enzymes and CheY DR
The importance of ‘pinning’ the Asp at D+2 in position to enable function as an acid/base catalyst in HAD phosphatases is supported by a large body of structural and functional analysis11, 52–54. Pinning almost exclusively occurs through hydrogen bonding interactions between the side chains of the Asp at D+2 and the residue at position T+2, usually an Arg, Lys, Thr, Trp, or Tyr11. In the one HAD phosphatase of which we are aware that has been directly tested experimentally, substitution of the anchoring residue at T+2 (an arginine) results in a loss of hexose phosphate phosphatase catalytic activity similar in magnitude to substitution of the catalytic Asp at D+211.
Structures of CheY DR bound to the substrate/product analogs WO42− or MO42− revealed an active site configuration with features of a HAD phosphatase. The geometries of the bidentate salt bridge between the Asp and Arg side chains in CheY DR mimicked the Asp-Arg interactions in multiple HAD structures and placed the Asp at D+2 in the rotameric form (Figure 3B,C) to hydrogen bond with the apical oxygen atom of WO42−/MO42−, which occupied the space expected for the nucleophilic water oxygen or leaving group nitrogen. The unique interactions between the D+2 and T+2 side chains observed in CheY DR but absent from other CheY DX variants (Figure 4) correlate with the highest rate constants for catalysis of both PAM autophosphorylation and autodephosphorylation (Table 1).
Hydrogen Bonding Interactions Versus Acid/Base Catalysis as Mechanisms of Catalysis
Despite the structural evidence that correlated the anchoring of Asp at D+2 with enhanced catalysis, our pH profile data indicated that CheY DR was unlikely to act as a true acid/base catalyst. The rate constants for CheY DR and DY autodephosphorylation were pH independent (Figure 2A), ruling out base catalysis. The autophosphorylation experiment to assess acid catalysis (Figure 2B) was less direct, because the PAM phosphodonor can acquire the proton required for reaction without assistance from CheY. However, the pH dependence of the bimolecular autophosphorylation rate constant was indistinguishable for all five CheY variants tested, indicating that the Asp at D+2 (absent from wild type CheY) likely does not act as an acid catalyst in CheY DR. The more compelling evidence that Asp59 is not a base catalyst in CheY DR also implies that the Asp at D+2 does not act as a dual acid/base catalyst in CheY DR as it does in HAD phosphatases.
Additional evidence against acid catalysis in CheY DR can be inferred from examination of autophosphorylation with AcP. AcP does not require protonation to react with CheY, but acid catalysis would be expected to accelerate the reaction. For the Asp at D+2 to serve as an acid catalyst, the pKa should be shifted to result in protonation near neutral pH. However, all of the CheY DX variants tested had substantially slower rate constants for reaction with AcP than their CheY NX counterparts (Table 1). Because negative charge at D+2 and T+2 retards CheY autophosphorylation with AcP23, we can infer that the Asp at D+2 is likely unprotonated. Furthermore, exploratory experiments revealed no effect of pH on autophosphorylation of CheY DR with 100 mM AcP across the range pH 4.7 to 8.0 (data not shown), again suggesting the Asp at D+2 in CheY DR is not protonated and therefore does not act as an acid catalyst.
The rate enhancements of autophosphorylation with PAM and autodephosphorylation observed for CheY DR compared to CheY NR (Table 1) therefore must be due to effects of the oriented Asp side chain other than acid/base catalysis. For dephosphorylation, this could be positioning the water molecule for in-line nucleophilic attack. Positioning the water molecule as a mechanism to enhance response regulator dephosphorylation is the strategy used by multiple families of response regulator phosphatases. The CheX55, CheZ56, Rap57, and His KA_3 sensor kinase58 families of response regulator phosphatases all function by inserting a side chain (Gln, Asn, or Thr) into the active site of the response regulator to hydrogen bond with (and position) the attacking water molecule. For autophosphorylation by PAM, direct interaction between the carboxylate oxygen on the Asp at D+2 and the cationic nitrogen on PAM would be expected to enhance PAM binding through positive electrostatic interactions (salt bridge formation). This stabilizing interaction would be expected to persist in the transition state where the nitrogen atom carries a partial positive charge.
The proposed introduction of hydrogen bonding interactions between the Asp at D+2 and nucleophilic/leaving group atoms as the mechanism of CheY DR rate enhancement (as well as the absence of acid/base catalysis) is consistent with the magnitude of reaction rate enhancements exhibited by CheY DR, which were ~101–102 greater than CheY NR or wild type CheY (Table 1). Response regulator phosphatases, which function by positioning the water, generally provide about one to two orders of magnitude increase in the rate dephosphorylation over that catalyzed by a response regulator in the absence of its phosphatase55, 56. There is also evidence from HAD phosphatases that orienting hydrogen bonds increase catalytic rates ~101–102 fold8, 59, 60. In contrast, the involvement of a bona fide catalytic acid/base typically increases rates by ~102–104 fold61.
What structural feature(s) present in HAD phosphatases but absent in CheY DR might account for the lack of acid/base catalysis for CheY DR? One structural difference is the HAD cap domain8, which, when closed over the active site, yields a relatively hydrophobic pocket. This hydrophobic environment facilitates the shift in pKa towards neutral for the catalytic Asp, allowing for acid/base catalysis46. Even the so-called “capless” HAD phosphatases, which typically dephosphorylate macromolecular substrates, benefit from a hydrophobic active site pocket created by the HAD/substrate interface62, 63. In contrast, CheY has a surface exposed active site and lacks a capping domain. CheY T+2 mutants that increase the nonpolar surface area at the active site do enhance autophosphorylation23, but are apparently not sufficient to functionally mimic a capping domain.
Global Conformation as a Kinetic Determinant for CheY DX
The active site for many HAD enzymes has been likened to a preformed mold where the catalytic residues are arranged similarly throughout the catalytic cycle11. In contrast, response regulators are allosteric proteins with activated and unactivated global conformational states that are in dynamic equilibrium21, 41. For unphosphorylated response regulators, the equilibrium lies towards the unactive conformation. However, ‘pushing’ the conformational equilibrium towards the activated conformation by mutation35, relief of inhibitory interactions with the output domain64, or providing a binding partner30, 65 can accelerate autophosphorylation more than 100-fold30, 64. Although the mechanistic basis for the rate enhancement is not fully understood, the structure of unphosphorylated CheY bound to FliM1–1651 (which has many but not all of the features of a fully activated conformation) shows that the conserved Thr87 has shifted closer to the other catalytic residues and thus may provide a more complete active site mold, reminiscent of the HAD enzymes.
Our observation that unphosphorylated CheY DR exhibited enhanced binding to FliM1–16, the peptide target of phosphorylated CheY, provided compelling evidence of a shift to a conformation that is more active for both autophosphorylation and output signaling. FliM binds to the β4α4 surface on CheY, a surface distal from the active site whose topology is changed as a result of phosphorylation. The six-fold enhancement of FliM1–16 binding by CheY DR (Kd ~65 μM) relative to wild type CheY (Kd ~370 μM) is considerable compared to the ~10–14-fold30, 49 enhancement in FliM1–16 binding upon CheY phosphorylation. However, it is clear that CheY DR, while shifted more towards an activated conformation than wild type CheY, is not ‘locked’ in the activated conformation. The structure of CheY DR•Mn2+ (no substrate, product, or phosphoryl group analog present) displayed an unactivated conformation. Although the backbone is in the fully activated conformation in the CheY DR complexes with MoO42− and WO42−, the presence of the substrate/product analog may tip the delicate energy balance between conformational states towards the activated conformation. Though more modest than CheY DR, several of the other CheY DX and NX mutants also displayed enhanced FliM binding affinities relative to wild type CheY (Table 4). Thus, these CheY mutants may also have conformational equilibria shifted towards a more activated conformation as a contribution to their enhanced autophosphorylation. For example, CheY DK and CheY NY, exhibited both enhanced autophosphorylation rate constants with PAM and modest (~two-fold) enhanced FliM binding relative to wild type CheY. It is tempting to speculate that the observed dual hydrogen bonding interactions between Asp at D+2 and Arg T+2 (Figure 3B,C) may contribute energy to the conformational shift of CheY DR. In the absence of a substrate/product analog, the same additional interactions could lower the energy of the activated conformation. Conformation is expected to have less impact on the control of autodephosphorylation rates for response regulators because the reactant (CheY-P) is already in an activated conformation.
Structural Relationships Between Response Regulators and HAD Enzymes
The hallmarks of the CheY DR MoO42− and WO42− structures presented here were that 1) the CheY backbone was in the fully activated conformation and 2) the activated conformation enabled interactions between conserved active site features and the MoO42−/WO42− that directly mimicked those seen with HAD complexes. Together, these properties provide direct evidence in support of the notion that the nature of the protein:substrate or protein:product complexes in response regulators is highly similar to that of HAD phosphatases.
Structures of HAD phosphatases containing a phosphate or a phosphate analog in the active site have served as informative models of enzyme:substrate complexes (e.g. Ref. 16) and it is notable that such structures constitute a significant fraction of the total HAD structures in current public databases. In our survey of 285 HAD structures (compiled from PDB searches for Pfam groups PF00702, PF03767, PF12689, and PF08645), we found 71 that contained inorganic phosphate, a phosphate analog, or a phosphate-containing substrate properly oriented in the HAD active site mold. In each of these structures the HAD phosphate analog structures show conserved interactions between three phosphate oxygen atoms (or equivalent) and the bound divalent metal ion, the conserved Thr, and Lys. These interactions are preserved in structures of many of the same HAD phosphatases complexed with planar molecules that model the transition state, and thus are likely to stay intact throughout the reaction cycle.
In contrast, in a survey of 255 receiver domain structures (excluding the structures presented in this report), we identified only seven PDB entries (four different response regulators) with HPO42− (phosphate) or HPO42− analog within the active site, even though many of the structures were solved using crystals containing high concentrations of SO42−, a phosphate analog. Strikingly, in all seven of these structures (PDB entries 3GWG, 3H1G, 3H1F, 3OO0, 3DGE, 1I3C, 1JLK)66–68, like the CheY DR·MoO42− and CheY DR·WO42− structures reported here, the receiver domain was in an activated global conformation. Of the seven entries, four also contained a divalent metal ion and in three of these four (PDB entries 3GWG, 3H1G, and 3OO0), there are direct interactions between the oxygen atoms of the phosphoryl analog (sulfate in all cases) and the M2+, Lys, and Thr, as observed in HAD phosphatase:substrate structures. To the best of our knowledge, no response regulator structures have been determined with an actual small molecule substrate bound to the active site. The paucity of structures for response regulators with a bound active site phosphate/phosphate analog is likely because unphosphorylated response regulators are dominated by the unactivated conformation and only very weakly bind phosphate. In the same vein, the correlation between the presence of the activated conformation and a bound phosphate/phosphate analog is consistent with the notion that the activated conformation of response regulators is the reactive molecule for phosphorylation26, 30, 64, 65 and further, that the interactions between active site residues and substrate/product molecules are highly similar to those in HAD phosphatases. Response regulator proteins such as CheY DR that form crystallizable complexes with phosphate analogs may serve as good candidates for which to attempt co-crystallization with substrate analogs or with planar transition state analogs, as has already been achieved for multiple HAD phosphatases.
Enzyme Families With Functionally Tuned Reaction Mechanisms
Receiver domains and HAD enzymes are distantly related and share structural similarities69. However, the reactions catalyzed by receiver domains and HAD phosphatases have diverged kinetically to fulfill different purposes. HAD phosphatases, with the conserved Asp at D+2 providing acid/base catalysis, the anchoring residue at T+2, and a structured active site, support rapid hydrolysis of the phosphoryl group both from the substrate and the phospho-enzyme intermediate. Receiver domains, lacking the catalytic Asp at D+2 and undergoing allosteric conformational changes, support a relatively stable phospho-enzyme intermediate for signal transduction. Our attempt to endow a receiver domain with HAD-like properties by engineering specific residues at positions D+2 and T+2 was partially successful.
A potentially analogous circumstance is provided by the GHKL protein superfamily, composed of gyrase-like topoisomerases, Hsp90 chaperones, histidine kinases, and MutL DNA repair enzymes70. Each constituent family contains a similar ATP-binding domain, which supports ATP hydrolysis in three (gyrases, Hsp90s, MutLs) of the four families. In each case, ATP hydrolysis is coupled to protein conformational changes associated with interactions with their macromolecular substrates71, 72. In contrast, in histidine kinases, the γ-phosphoryl group of ATP is transferred to a His residue instead of water. The phospho-His residue then fulfills a signal transduction function, serving as the phosphodonor for receiver domains. The absence of ATPase activity in the histidine kinases is hypothesized to be due to lack of a conserved Glu that serves as a catalytic base, as well as lack of a positively charged Lys or Arg that interacts with the γ-phosphoryl group of ATP70, 73, 74. Nevertheless, GHKL ATP-binding domains are sufficiently similar that radicicol, a drug that inhibits the ATPase activities of Hsp90s and topoisomerases75, also inhibits the autophosphorylation activity of at least two histidine kinases76, 77.
To the best of our knowledge, restoration of ATPase activity in histidine kinases by introduction of the catalytic Glu found in GHL family members has not been attempted. However, engineering a monovalent cation binding site from a K family member into a GHL protein has been achieved78. When an Ala adjacent to the MutL cation binding site was replaced with a Pro found in kinases, the coordination of the ion in MutL changed (two ligands lost and two gained) to display the same coordination found in the kinase that inspired the Pro substitution. However, the binding site in the kinase is specific for potassium, whereas the mutant MutL binding site with the same coordination pattern is specific for sodium. As with our attempt to engineer HAD phosphatase properties into a receiver domain, transplanting a key amino acid onto a related protein backbone endowed the recipient with some, but not all, of the properties of the “donor” protein.
Supplementary Material
Acknowledgments
Funding Source Statement:
This work was supported by National Institutes of Health Grant R01GM050860 awarded to R.B.B. Chrystal Starbird was supported by National Institutes of Health Grant R25 GM089569. Financial support for use of beamlines 22-BM and 22-ID at Advanced Photon Source, Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-3 and the Southeast Regional Collaborative Access Team (SER-CAT).
We thank Karen Allen for very helpful discussions about HAD enzymes and Ash Tripathy at the Macromolecular Interaction Facility at UNC for help with stop-flow fluorescence measurements. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- D
the conserved phosphorylated aspartate shared by response regulators and HAD phosphatases
- T
the conserved threonine/serine shared by response regulators and HAD phosphatases
- K
the conserved lysine shared by response regulators and HAD phosphatases
- DD
the conserved acid residues that coordinate the Mg2+ shared by response regulators and HAD phosphatases
- D+2 or T+2
the position in the primary sequence of response regulators or HAD phosphatases that is two residues carboxyl-terminal to D or T, respectively
- CheY DX
E. coli CheY double mutant with an Asp substituted for CheY Asn59 at position D+2 and X representing the residue substituted for CheY Glu89 at position T+2. AcP, acetyl phosphate
- PAM
phosphoramidate
Footnotes
Coordinates from the X-ray structures were deposited in the Protein Data Bank as PDB entries 3RVJ, 3RVK, 3RVL, 3RVM, 3RVN, 3RVO, 3RVP, 3RVQ, 3RVR, and 3RVS.
SUPPORTING INFORMATION AVAILABLE
Supporting information includes Table S1, which contains a summary of data collection and refinement statistics for CheY DR·Mn2+, CheY DK·Mn2+, CheY DY·Mn2+, and CheY DQ·Mn2+; and Figure S1, which contains electron density maps in the active site regions of CheY DR complexed with Mn2+ and MoO42− or WO42−. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bourret RB, Silversmith RE. Two-component signal transduction. Curr Opin Microbiol. 2010;13:113–115. doi: 10.1016/j.mib.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capra EJ, Laub MT. Evolution of two-component signal transduction systems. Annu Rev Microbiol. 2012;66:325–347. doi: 10.1146/annurev-micro-092611-150039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukat GS, McCleary WR, Stock AM, Stock JB. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc Natl Acad Sci U S A. 1992;89:718–722. doi: 10.1073/pnas.89.2.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silversmith RE, Appleby JL, Bourret RB. Catalytic mechanism of phosphorylation and dephosphorylation of CheY: kinetic characterization of imidazole phosphates as phosphodonors and the role of acid catalysis. Biochemistry. 1997;36:14965–14974. doi: 10.1021/bi9715573. [DOI] [PubMed] [Google Scholar]
- 5.Hess JF, Bourret RB, Oosawa K, Matsumura P, Simon MI. Protein phosphorylation and bacterial chemotaxis. Cold Spring Harb Symp Quant Biol. 1988;53(Pt 1):41–48. doi: 10.1101/sqb.1988.053.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Wolanin PM, Webre DJ, Stock JB. Mechanism of phosphatase activity in the chemotaxis response regulator CheY. Biochemistry. 2003;42:14075–14082. doi: 10.1021/bi034883t. [DOI] [PubMed] [Google Scholar]
- 7.Cho H, Wang W, Kim R, Yokota H, Damo S, Kim SH, Wemmer D, Kustu S, Yan D. BeF3− acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a BeF3− complex with phosphoserine phosphatase. Proc Natl Acad Sci U S A. 2001;98:8525–8530. doi: 10.1073/pnas.131213698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen KN, Dunaway-Mariano D. Phosphoryl group transfer: evolution of a catalytic scaffold. Trends Biochem Sci. 2004;29:495–503. doi: 10.1016/j.tibs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Lu Z, Dunaway-Mariano D, Allen KN. HAD superfamily phosphotransferase substrate diversification: structure and function analysis of HAD subclass IIB sugar phosphatase BT4131. Biochemistry. 2005;44:8684–8696. doi: 10.1021/bi050009j. [DOI] [PubMed] [Google Scholar]
- 10.Ridder IS, Dijkstra BW. Identification of the Mg2+-binding site in the P-type ATPase and phosphatase members of the HAD (haloacid dehalogenase) superfamily by structural similarity to the response regulator protein CheY. Biochem J. 1999;339(Pt 2):223–226. [PMC free article] [PubMed] [Google Scholar]
- 11.Lu Z, Dunaway-Mariano D, Allen KN. The catalytic scaffold of the haloalkanoic acid dehalogenase enzyme superfamily acts as a mold for the trigonal bipyramidal transition state. Proc Natl Acad Sci U S A. 2008;105:5687–5692. doi: 10.1073/pnas.0710800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collet JF, Stroobant V, Van Schaftingen E. Mechanistic studies of phosphoserine phosphatase, an enzyme related to P-type ATPases. J Biol Chem. 1999;274:33985–33990. doi: 10.1074/jbc.274.48.33985. [DOI] [PubMed] [Google Scholar]
- 13.Seifried A, Schultz J, Gohla A. Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J. 2012;280:549–571. doi: 10.1111/j.1742-4658.2012.08633.x. [DOI] [PubMed] [Google Scholar]
- 14.Kuznetsova E, Proudfoot M, Gonzalez CF, Brown G, Omelchenko MV, Borozan I, Carmel L, Wolf YI, Mori H, Savchenko AV, Arrowsmith CH, Koonin EV, Edwards AM, Yakunin AF. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J Biol Chem. 2006;281:36149–36161. doi: 10.1074/jbc.M605449200. [DOI] [PubMed] [Google Scholar]
- 15.Pazy Y, Wollish AC, Thomas SA, Miller PJ, Collins EJ, Bourret RB, Silversmith RE. Matching biochemical reaction kinetics to the timescales of life: structural determinants that influence the autodephosphorylation rate of response regulator proteins. J Mol Biol. 2009;392:1205–1220. doi: 10.1016/j.jmb.2009.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang W, Cho HS, Kim R, Jancarik J, Yokota H, Nguyen HH, Grigoriev IV, Wemmer DE, Kim SH. Structural characterization of the reaction pathway in phosphoserine phosphatase: crystallographic “snapshots” of intermediate states. J Mol Biol. 2002;319:421–431. doi: 10.1016/S0022-2836(02)00324-8. [DOI] [PubMed] [Google Scholar]
- 17.Dai J, Finci L, Zhang C, Lahiri S, Zhang G, Peisach E, Allen KN, Dunaway-Mariano D. Analysis of the structural determinants underlying discrimination between substrate and solvent in beta-phosphoglucomutase catalysis. Biochemistry. 2009;48:1984–1995. doi: 10.1021/bi801653r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang M, Liu J, Kim Y, Dixon JE, Pfaff SL, Gill GN, Noel JP, Zhang Y. Structural and functional analysis of the phosphoryl transfer reaction mediated by the human small C-terminal domain phosphatase, Scp1. Protein Sci. 2010;19:974–986. doi: 10.1002/pro.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ulrich LE, Zhulin IB. The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 2010;38:D401–D407. doi: 10.1093/nar/gkp940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho HS, Lee SY, Yan D, Pan X, Parkinson JS, Kustu S, Wemmer DE, Pelton JG. NMR structure of activated CheY. J Mol Biol. 2000;297:543–551. doi: 10.1006/jmbi.2000.3595. [DOI] [PubMed] [Google Scholar]
- 21.McDonald LR, Boyer JA, Lee AL. Segmental motions, not a two-state concerted switch, underlie allostery in CheY. Structure. 2012;20:1363–1373. doi: 10.1016/j.str.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas SA, Brewster JA, Bourret RB. Two variable active site residues modulate response regulator phosphoryl group stability. Mol Microbiol. 2008;69:453–465. doi: 10.1111/j.1365-2958.2008.06296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas SA, Immormino RM, Bourret RB, Silversmith RE. Nonconserved active site residues modulate CheY autophosphorylation kinetics and phosphodonor preference. Biochemistry. 2013;52:2262–2273. doi: 10.1021/bi301654m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gouet P, Fabry B, Guillet V, Birck C, Mourey L, Kahn D, Samama JP. Structural transitions in the FixJ receiver domain. Structure. 1999;7:1517–1526. doi: 10.1016/s0969-2126(00)88342-2. [DOI] [PubMed] [Google Scholar]
- 25.Formaneck MS, Ma L, Cui Q. Reconciling the “old” and “new” views of protein allostery: a molecular simulation study of chemotaxis Y protein (CheY) Proteins. 2006;63:846–867. doi: 10.1002/prot.20893. [DOI] [PubMed] [Google Scholar]
- 26.Creager-Allen RL, Silversmith RE, Bourret RB. A link between autophosphorylation and dimerization of the PhoB response regulator. J Biol Chem. 2013;288:21755–21769. doi: 10.1074/jbc.M113.471763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bourret RB, Thomas SA, Page SC, Creager-Allen RL, Moore AM, Silversmith RE. Measurement of response regulator autodephosphorylation rates spanning six orders of magnitude. Methods Enzymol. 2010;471:89–114. doi: 10.1016/S0076-6879(10)71006-5. [DOI] [PubMed] [Google Scholar]
- 28.Hess JF, Bourret RB, Simon MI. Phosphorylation assays for proteins of the two-component regulatory system controlling chemotaxis in Escherichia coli. Methods Enzymol. 1991;200:188–204. doi: 10.1016/0076-6879(91)00139-n. [DOI] [PubMed] [Google Scholar]
- 29.Boesch KC, Silversmith RE, Bourret RB. Isolation and characterization of nonchemotactic CheZ mutants of Escherichia coli. J Bacteriol. 2000;182:3544–3552. doi: 10.1128/jb.182.12.3544-3552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuster M, Silversmith RE, Bourret RB. Conformational coupling in the chemotaxis response regulator CheY. Proc Natl Acad Sci U S A. 2001;98:6003–6008. doi: 10.1073/pnas.101571298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Da Re SS, Deville-Bonne D, Tolstykh T, Veron M, Stock JB. Kinetics of CheY phosphorylation by small molecule phosphodonors. FEBS Lett. 1999;457:323–326. doi: 10.1016/s0014-5793(99)01057-1. [DOI] [PubMed] [Google Scholar]
- 32.Mayover TL, Halkides CJ, Stewart RC. Kinetic characterization of CheY phosphorylation reactions: comparison of P-CheA and small-molecule phosphodonors. Biochemistry. 1999;38:2259–2271. doi: 10.1021/bi981707p. [DOI] [PubMed] [Google Scholar]
- 33.Roberts A, Lee SY, McCullagh E, Silversmith RE, Wemmer DE. YbiV from Escherichia coli K12 is a HAD phosphatase. Proteins. 2005;58:790–801. doi: 10.1002/prot.20267. [DOI] [PubMed] [Google Scholar]
- 34.Schuster M, Zhao R, Bourret RB, Collins EJ. Correlated switch binding and signaling in bacterial chemotaxis. J Biol Chem. 2000;275:19752–19758. doi: 10.1074/jbc.M909908199. [DOI] [PubMed] [Google Scholar]
- 35.Smith JG, Latiolais JA, Guanga GP, Pennington JD, Silversmith RE, Bourret RB. A search for amino acid substitutions that universally activate response regulators. Mol Microbiol. 2004;51:887–901. doi: 10.1046/j.1365-2958.2003.03882.x. [DOI] [PubMed] [Google Scholar]
- 36.Volz K, Beman J, Matsumura P. Crystallization and preliminary characterization of CheY, a chemotaxis control protein from Escherichia coli. J Biol Chem. 1986;261:4723–4725. [PubMed] [Google Scholar]
- 37.Zhu X, Rebello J, Matsumura P, Volz K. Crystal structures of CheY mutants Y106W and T87I/Y106W. CheY activation correlates with movement of residue 106. J Biol Chem. 1997;272:5000–5006. doi: 10.1074/jbc.272.8.5000. [DOI] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 39.Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brennan S, Cowan PL. A suite of programs for calculating X-Ray absorption, reflection, and diffraction performance for a variety of materials at arbitrary wavelengths. Rev Sci Instr. 1992;63:850–853. [Google Scholar]
- 41.Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE. Crystal structure of activated CheY. Comparison with other activated receiver domains. J Biol Chem. 2001;276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 42.Volz K, Matsumura P. Crystal structure of Escherichia coli CheY refined at 1.7-A resolution. J Biol Chem. 1991;266:15511–15119. doi: 10.2210/pdb3chy/pdb. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris TK, Turner GJ. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- 47.Perrin DD. Ionization Constants of Inorganic Acids and Bases in Aqueous Solution. 2. Pergamon Press; Oxford: 1982. [Google Scholar]
- 48.Silversmith RE, Guanga GP, Betts L, Chu C, Zhao R, Bourret RB. CheZ-mediated dephosphorylation of the Escherichia coli chemotaxis response regulator CheY: role for CheY glutamate 89. J Bacteriol. 2003;185:1495–1502. doi: 10.1128/JB.185.5.1495-1502.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McEvoy MM, Bren A, Eisenbach M, Dahlquist FW. Identification of the binding interfaces on CheY for two of its targets, the phosphatase CheZ and the flagellar switch protein, FliM. J Mol Biol. 1999;289:1423–1433. doi: 10.1006/jmbi.1999.2830. [DOI] [PubMed] [Google Scholar]
- 50.Shukla D, Zhu XY, Matsumura P. Flagellar motor-switch binding face of CheY and the biochemical basis of suppression by CheY mutants that compensate for motor-switch defects in Escherichia coli. J Biol Chem. 1998;273:23993–23999. doi: 10.1074/jbc.273.37.23993. [DOI] [PubMed] [Google Scholar]
- 51.Dyer CM, Dahlquist FW. Switched or not?: the structure of unphosphorylated CheY bound to the N terminus of FliM. J Bacteriol. 2006;188:7354–7363. doi: 10.1128/JB.00637-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang G, Dai J, Wang L, Dunaway-Mariano D, Tremblay LW, Allen KN. Catalytic cycling in beta-phosphoglucomutase: a kinetic and structural analysis. Biochemistry. 2005;44:9404–9416. doi: 10.1021/bi050558p. [DOI] [PubMed] [Google Scholar]
- 53.Nguyen HH, Wang L, Huang H, Peisach E, Dunaway-Mariano D, Allen KN. Structural determinants of substrate recognition in the HAD superfamily member D-glycero-D-manno-heptose-1,7-bisphosphate phosphatase (GmhB) Biochemistry. 2010;49:1082–1092. doi: 10.1021/bi902019q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. Caught in the act: the structure of phosphorylated beta-phosphoglucomutase from Lactococcus lactis. Biochemistry. 2002;41:8351–8359. doi: 10.1021/bi0202373. [DOI] [PubMed] [Google Scholar]
- 55.Pazy Y, Motaleb MA, Guarnieri MT, Charon NW, Zhao R, Silversmith RE. Identical phosphatase mechanisms achieved through distinct modes of binding phosphoprotein substrate. Proc Natl Acad Sci U S A. 2010;107:1924–1929. doi: 10.1073/pnas.0911185107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao R, Collins EJ, Bourret RB, Silversmith RE. Structure and catalytic mechanism of the E. coli chemotaxis phosphatase CheZ. Nat Struct Biol. 2002;9:570–575. doi: 10.1038/nsb816. [DOI] [PubMed] [Google Scholar]
- 57.Parashar V, Mirouze N, Dubnau DA, Neiditch MB. Structural basis of response regulator dephosphorylation by Rap phosphatases. PLoS Biol. 2011;9:e1000589. doi: 10.1371/journal.pbio.1000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huynh TN, Noriega CE, Stewart V. Conserved mechanism for sensor phosphatase control of two-component signaling revealed in the nitrate sensor NarX. Proc Natl Acad Sci U S A. 2010;107:21140–21145. doi: 10.1073/pnas.1013081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sorensen TL, Dupont Y, Vilsen B, Andersen JP. Fast kinetic analysis of conformational changes in mutants of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 2000;275:5400–5408. doi: 10.1074/jbc.275.8.5400. [DOI] [PubMed] [Google Scholar]
- 60.Clausen JD, McIntosh DB, Woolley DG, Andersen JP. Importance of Thr-353 of the conserved phosphorylation loop of the sarcoplasmic reticulum Ca2+-ATPase in MgATP binding and catalytic activity. J Biol Chem. 2001;276:35741–35750. doi: 10.1074/jbc.M105434200. [DOI] [PubMed] [Google Scholar]
- 61.Fersht A. Structure and Mechanism in Protein Science. W.H. Freeman and Co; 1999. [Google Scholar]
- 62.Peisach E, Wang L, Burroughs AM, Aravind L, Dunaway-Mariano D, Allen KN. The X-ray crystallographic structure and activity analysis of a Pseudomonas-specific subfamily of the HAD enzyme superfamily evidences a novel biochemical function. Proteins. 2008;70:197–207. doi: 10.1002/prot.21583. [DOI] [PubMed] [Google Scholar]
- 63.Galburt EA, Pelletier J, Wilson G, Stoddard BL. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure. 2002;10:1249–1260. doi: 10.1016/s0969-2126(02)00835-3. [DOI] [PubMed] [Google Scholar]
- 64.Barbieri CM, Mack TR, Robinson VL, Miller MT, Stock AM. Regulation of response regulator autophosphorylation through interdomain contacts. J Biol Chem. 2010;285:32325–32335. doi: 10.1074/jbc.M110.157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ames SK, Frankema N, Kenney LJ. C-terminal DNA binding stimulates N-terminal phosphorylation of the outer membrane protein regulator OmpR from Escherichia coli. Proc Natl Acad Sci U S A. 1999;96:11792–11797. doi: 10.1073/pnas.96.21.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Casino P, Rubio V, Marina A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell. 2009;139:325–336. doi: 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 67.Im YJ, Rho SH, Park CM, Yang SS, Kang JG, Lee JY, Song PS, Eom SH. Crystal structure of a cyanobacterial phytochrome response regulator. Protein Sci. 2002;11:614–624. doi: 10.1110/ps.39102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lam KH, Ling TK, Au SW. Crystal structure of activated CheY1 from Helicobacter pylori. J Bacteriol. 2010;192:2324–2334. doi: 10.1128/JB.00603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol. 2006;361:1003–1034. doi: 10.1016/j.jmb.2006.06.049. [DOI] [PubMed] [Google Scholar]
- 70.Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 71.Corbett KD, Berger JM. Structural dissection of ATP turnover in the prototypical GHL ATPase TopoVI. Structure. 2005;13:873–882. doi: 10.1016/j.str.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 72.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 73.Cunningham CN, Southworth DR, Krukenberg KA, Agard DA. The conserved arginine 380 of Hsp90 is not a catalytic residue, but stabilizes the closed conformation required for ATP hydrolysis. Protein Sci. 2012;21:1162–1171. doi: 10.1002/pro.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilke KE, Carlson EE. All signals lost. Sci Transl Med. 2013;5:203ps212. doi: 10.1126/scitranslmed.3006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corbett KD, Berger JM. Structural basis for topoisomerase VI inhibition by the anti-Hsp90 drug radicicol. Nucleic Acids Res. 2006;34:4269–4277. doi: 10.1093/nar/gkl567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Besant PG, Lasker MV, Bui CD, Turck CW. Inhibition of branched-chain alpha-keto acid dehydrogenase kinase and Sln1 yeast histidine kinase by the antifungal antibiotic radicicol. Mol Pharmacol. 2002;62:289–296. doi: 10.1124/mol.62.2.289. [DOI] [PubMed] [Google Scholar]
- 77.Guarnieri MT, Zhang L, Shen J, Zhao R. The Hsp90 inhibitor radicicol interacts with the ATP-binding pocket of bacterial sensor kinase PhoQ. J Mol Biol. 2008;379:82–93. doi: 10.1016/j.jmb.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 78.Hu X, Machius M, Yang W. Monovalent cation dependence and preference of GHKL ATPases and kinases. FEBS Lett. 2003;544:268–273. doi: 10.1016/s0014-5793(03)00519-2. [DOI] [PubMed] [Google Scholar]
- 79.Silversmith RE, Smith JG, Guanga GP, Les JT, Bourret RB. Alteration of a nonconserved active site residue in the chemotaxis response regulator CheY affects phosphorylation and interaction with CheZ. J Biol Chem. 2001;276:18478–18484. doi: 10.1074/jbc.M011418200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.