

Abstract
Objective:
To investigate the effects of NEFL Glu396Lys mutation on the expression and assembly of neurofilaments (NFs) in cutaneous nerve fibers of patients with Charcot-Marie-Tooth disease type 2E (CMT2E).
Methods:
A large family with CMT2E underwent clinical, electrophysiologic, and skin biopsy studies. Biopsies were processed by indirect immunofluorescence (IF), electron microscopy (EM), and Western blot analysis.
Results:
The clinical features demonstrated intrafamilial phenotypic variability, and the electrophysiologic findings revealed nerve conductions that were either slow or in the intermediate range. All patients had reduced or absent compound muscular action potential amplitudes. Skin biopsies showed axons labeled with the axonal markers protein gene product 9.5 and α-tubulin, but not with NFs. The results of Western blot analysis were consistent with those of IF, showing reduced or absent NFs and normal expression of α-tubulin. EM revealed clusters of regenerated fibers, in absence of myelin sheath abnormalities. Both IF and EM failed to show NF aggregates in dermal axons. The morphometric analysis showed a smaller axonal caliber in patients than in controls. The study of the nodal/paranodal architecture demonstrated that sodium channels and Caspr were correctly localized in patients with CMT2E.
Conclusions:
Decrease in NF abundance may be a pathologic marker of CMT2E. The lack of NF aggregates, consistent with prior studies, suggests that they occur proximally leading to subsequent alterations in the axonal cytoskeleton. The small axonal caliber, along with the normal molecular architecture of nodes and paranodes, explain the reduced velocities detected in patients with CMT2E. Our results also demonstrate that skin biopsy can provide evidence of pathologic and pathogenic abnormalities in patients with CMT2E.
Charcot-Marie-Tooth disease (CMT) is a group of peripheral neuropathies associated with mutations in more than 80 distinct genes. CMT is divided into different forms based on the pattern of inheritance and neurophysiology. Electrophysiologic studies allow for classification of CMT into demyelinating (CMT1) and axonal (CMT2) forms.1 The classification of CMT has been further divided into subtypes, identified by letters, as defined by the mutated gene.2 CMT2E refers to autosomal dominant CMT2 caused by mutations in the neurofilament light chain (NEFL) gene. Some patients with NEFL mutations have slow conduction velocities, in the “demyelinating” range, and are characterized as having CMT1F. Patients with CMT2E and CMT1F are clinically heterogeneous and the age at onset ranges from infancy, typically with severe impairment, to adulthood with milder impairment. Usually those with slowed conductions present early either in infancy with delayed motor milestones or in late childhood/early adolescence.3
Neurofilaments (NFs) are neuron-specific intermediate filaments that are necessary for maintaining the caliber of large-diameter axons in the peripheral nervous system. Axonal caliber is maintained by precise stoichiometric ratios of the 3 NF subunits: light (NF-L), medium (NF-M), and heavy (NF-H).4 NEFL mutations are distributed throughout the 3 functional domains of the NF-L (head, rod, and tail) and different mutations have been shown to lead to distinct pathologic effect.5,6 Previous studies in a transgenic mouse model carrying a Glu396Lys have demonstrated accumulation of NFs in the neuronal cell body, with decreased NFs in axons.6 Sural nerve biopsies from patients with the Glu396Lys have shown the presence of axons with a variable density of NFs.7,8 We have evaluated a large 4-generation family with the Glu396Lys mutation and investigated the expression and assembly of NFs in cutaneous nerve fibers.
METHODS
We evaluated 9 affected individuals from 2 generations of a large family with a clinical and genetic diagnosis of CMT2E (Glu396Lys) (figure 1). Clinical impairment was evaluated using the validated CMT Neuropathy Score version 2 (CMTNSv2).9 All patients but one underwent skin biopsies that were processed for indirect immunofluorescence (IF), electron microscopy (EM), or Western blot (WB) analysis. A control population of healthy age-matched controls was included for the morphologic and morphometric studies.
Figure 1. Pedigree of the family.

Standard protocol approvals, registrations, and patient consents.
The Institutional Review Board (Ethics) Committee approved the study, and a written informed consent was obtained from all participants in the study.
Skin biopsy: IF.
Three skin samples for IF were obtained by a 2.5-mm punch from the proximal phalanx of the II finger. Specimens were fixed in 4% paraformaldehyde for 30 minutes, washed in phosphate-buffered saline 3 times for 10 minutes, and then embedded in optimal cutting temperature medium in a plastic mold and gradually frozen. The tissues were then cut into 20-µm-thick sections and mounted on glass slides. These slides were then incubated with a panel of primary antibodies at 4°C overnight, reacted with secondary antibodies for 2 hours the following day, washed, dried, and coverslipped for examination with a Zeiss 710 confocal microscope. Primary and secondary antibody features, sources, and dilutions are listed in table e-1 on the Neurology® Web site at Neurology.org.
Fluorescence quantification.
Images were obtained on a Zeiss 710 confocal microscope using a 40× objective. All image parameters including pinhole size, detector gain, amplifier offset, amplifier gain, and laser intensity were first set for normal control tissue, and the same setting used for all samples imaged on a given day.10 Both control and patient sample images were obtained at the same day after the staining. For each sample, 5 nonoverlapping sections were acquired. Image analysis was performed using Image J software (NIH, Bethesda, MD). Fluorescence intensity was measured for both NFs and protein gene product (PGP) 9.5 at the same time, choosing an intensity threshold in the PGP channel so that only the axons were highlighted, corresponding to our regions of interest on both PGP and NF channel. Then we calculate the NF/PGP ratio to obtain a relative value of NF intensity over the PGP-stained axons and rule out a possible role of nerve fiber loss in reduced NF staining. Alpha-tubulin fluorescence intensity was evaluated as well.
Morphometric analysis.
Z-stack digital images of all myelinated fibers present in 5 PGP/myelin basic protein double-stained sections were acquired using confocal microscopy (Zeiss 710 confocal microscope). The axonal caliber was measured using Image J software (NIH). Four caliber measurements for each nerve fiber were obtained and a mean value was reported.11,12
Skin biopsy: EM.
Two skin samples for EM study were obtained by a 2.5-mm punch from the volar aspect of the forearm. Specimens were fixed in 2.5% glutaraldehyde solution overnight, then washed in phosphate-buffered saline 3 times for 10 minutes, osmicated for 1.5 hours, dehydrated, and embedded in Epon. The semithin sections (1-μm thickness) were stained with toluidine blue and examined under a light microscope. The ultrathin sections were contrasted with lead citrate and uranyl acetate and examined under EM (Transmission Electron Microscope, JEOL 1230, Peabody, MA).
Skin biopsy: WB.
Three skin samples for WB analysis were obtained by a 3.5-mm punch from the shoulder and immediately frozen in liquid nitrogen. Snap-frozen samples were then pulverized, dissolved in lysis buffer (RIPA [radioimmunoprecipitation assay] buffer, Sigma R0278, and proteinase inhibitors), kept on ice for at least 30 minutes, and centrifuged at 14,000g for 10 minutes at 4°C. The protein content of the supernatant was determined using a bovine serum albumin standard curve. Equal amounts of the protein were loaded on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels, and electroblotted onto polyvinyl difluoride membranes.13 The primary antibodies used are listed in table e-1. Horseradish peroxidase–conjugated secondary antibodies (1:5,000–1:20,000 dilution; Sigma) were detected using enhanced chemiluminescence (ECL) reagents (Amersham ECL Prime Western Blotting Detection Reagent, RPN 2232) with autoradiography film (Kodak Scientific Imaging Film, Blue XB).13 Results were normalized for α-tubulin and the control sample signal was used as the reference. Moreover, the signal density was calculated using Image J software (NIH), the ratio NF-L/α-tubulin was obtained for both patients and controls, and any value less than the control value was considered abnormal.
Statistical analysis.
Statistical analysis was performed using SigmaStat 3.0 for Windows (StataCorp LP, College Station, TX). Student t test for unpaired data and Mann–Whitney test (when analyzing nonparametric data) were used to compare skin biopsy findings from patients and controls. Spearman rank correlation test was used to correlate fluorescence intensity values with patients' disease severity determined by the CMTNSv2. A p value of <0.01 was considered significant.
RESULTS
Patients.
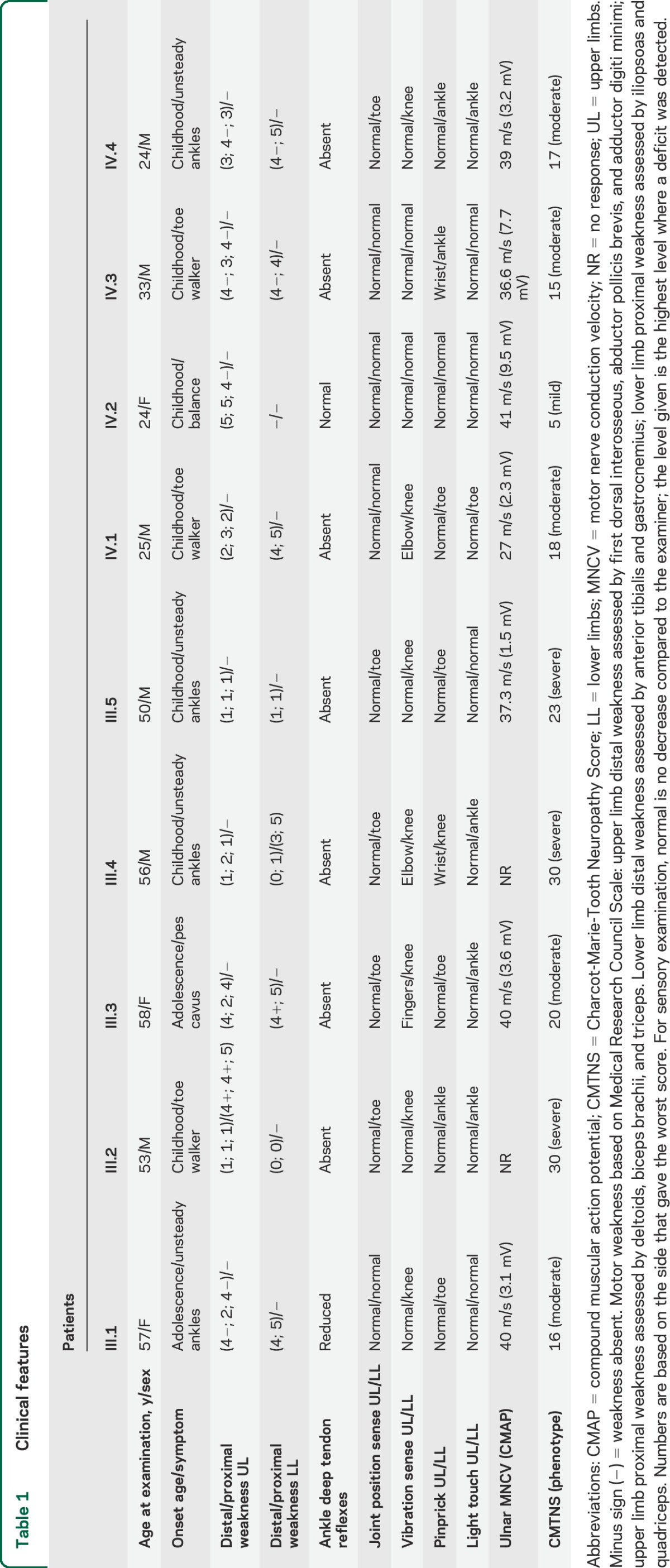
Disease severity, as determined by the CMTNSv2, ranged from mild to severe impairment (scores between 5 and 30). Electrophysiologic findings revealed either slow or intermediate motor nerve conduction velocities when a compound muscular action potential was recordable. Individual scores along with additional clinical information are provided in table 1.
Table 1.
Clinical features
Skin biopsy.
Cutaneous nerve fibers show a decrease in NF abundance.
We performed immunohistochemistry on myelinated dermal nerves from patients and controls. Results from controls demonstrated many axons labeled by the axonal marker PGP 9.5 and with antibodies to NF-L (figure 2, A.a and A.b). Alternatively, axons from patients were not labeled with antibodies to NF-L but were labeled by PGP (figure 2, A.d and A.e). To measure the difference between controls and patients, we quantified the fluorescence intensity in the 2 groups. The analysis demonstrated a lower NF/PGP ratio in patients than in controls (p < 0.01) (figure 2B). There was no correlation between NF-L levels and clinical severity as measured by the CMTNSv2 (p = not significant). We then labeled axons with antibodies to α-tubulin to determine whether the transport of other molecules such as α-tubulin was affected by the mutation. Many axons in both patients and controls were labeled with α-tubulin and no difference in staining intensity was detectable between the 2 groups. In addition, many axons from patients but not controls were labeled with α-tubulin but not with NF-L (figure 2, A.c and A.f). We concluded that the decreased NF-L immunolabeling was not a consequence of axonal degeneration but rather was a specific consequence of the NF-L mutation. Results from WB analysis confirmed these data as NF-L expression was decreased in patients with a complete absence of NF-L band in 2 of them, while α-tubulin expression was normal (figure 2C). The ratio NF-L/α-tubulin was markedly reduced in patients (figure 2D).
Figure 2. Cutaneous nerve fibers from patients with CMT2E show a lack of NFs.

(A) Confocal images of dermal axons stained with antibodies against PGP 9.5 (red), NF-L (green), and MBP (blue) in control (A.a, A.b) and in patient (A.d, A.e) samples. A reduced or absent expression of NFs is observed in the patient compared with the control sample. Confocal images of dermal nerve fibers stained with antibodies against α-tubulin (red), NF-L (green), and PGP 9.5 (blue) in control (A.c) and in patient (A.f) samples. No difference in the expression of α-tubulin is found but a reduced NF labeling is still present in the patient compared with the control sample. Scale bar: 20 µm. (B) NF fluorescence intensity (expressed as the percentage of the value of NFs over the PGP intensity: NF/PGP ratio) between patients and controls. The ratio is significantly lower in each patient than in the controls (p < 0.01). (C) Western blot analysis of NF-L from skin of patients and controls, normalized for α-tubulin. NF-L expression is decreased in patients with a complete absence of NF-L band in 2 of them, while α-tubulin expression is normal. (D) NF-L/α-tubulin ratio is markedly reduced in patients compared with controls. αtub = α-tubulin; CMT2E = Charcot-Marie-Tooth disease type 2E; MBP = myelin basic protein; NF = neurofilament; NF-L = neurofilament light chain; PGP = protein gene product.
NF aggregates are not detected distally in the axon.
To investigate NF accumulation, we also evaluated dermal nerves from our patients by EM (figure 3A). No abnormalities of myelin sheathes were observed on either semithin or EM sections. Many Schwann cells without axons (bands of Büngner) were present, suggesting prior axonal degeneration. No aggregates were observed. Our results were similar to those previously reported in sural nerve biopsies from patients with CMT2E and Glu396Lys.7,8
Figure 3. Electron microscopy images of dermal nerves in CMT2E, and morphometric analysis of axonal caliber.

(A) Electron microscopy images of dermal axons in a patient with CMT2E. Arrowheads: myelinated axons; arrow: unmyelinated axons; asterisk: Schwann cells without axons (bands of Büngner). No neurofilament aggregates were detected. Scale bar: 2 µm. (B) Morphometric analysis showed smaller axonal calibers in patients than in controls (p < 0.01). CMT2E = Charcot-Marie-Tooth disease type 2E.
Because the aggregates were made up of phosphorylated NFs in the mutant mouse,6 we performed additional staining with antibodies to phosphorylated NFs at HIC, but again, we did not detect any aggregate formation (data not shown).
Furthermore, previous studies showed that the distribution of mitochondria was altered in cultured cells with aggregated NFs because of expression of CMT-causing NEFL mutants.14 In our patients, mitochondria were distributed normally into the axons and neither their shape nor their number was affected by NF disruption.
Patients with CMT2E have smaller axonal caliber than controls.
Prior studies have demonstrated a close correlation between the number of NFs and the diameter of myelinated axons in the peripheral nervous system.15,16 To determine whether the reduction of NF-L affected the caliber of axons in our patients, we performed a morphometric analysis on the dermal nerves from our patients and controls. Our results demonstrated smaller axonal calibers in patients compared with controls (p < 0.01) (figure 3B), providing further evidence of the NF role in the maintenance of the axonal diameter and supporting the feasibility of evaluating dermal nerves to address this question.
Nodal and paranodal regions are unaffected by the lack of axonal NFs.
A previous study of nodal architecture in NF-deficient mice showed normal placement, organization, and physical dimensions of nodes of Ranvier.17 Our results demonstrated that sodium channels and Caspr are correctly localized in patients with CMT2E (figure 4), confirming that, despite the axonal NF deficiency, the molecular architecture of nodal and paranodal regions is not impaired in CMT2E. This observation supports the hypothesis that NFs, as cytoskeletal components of the internodal domain of axons, are not required to position nodal and paranodal molecules.
Figure 4. Nodal/paranodal architecture of dermal nerves.

Confocal images of dermal axons stained with antibodies against Caspr (red), sodium channels (green), and MBP (blue) show normal architecture of nodal and paranodal regions in both control (A.a, A.b) and patient (A.c, A.d) samples. Panel A.d and the 2 small left corner figures in A.a and A.c are 3-dimensional images of single or few nodes of Ranvier at higher magnification. MBP = myelin basic protein.
DISCUSSION
Our results demonstrated a reduced expression of NFs and smaller axonal diameter in cutaneous nerve fibers of patients with CMT2E. In addition, our findings did not show NF aggregates in the axons, suggesting that their proximal localization in the cell body may lead to NF disruption distally. The cytoskeletal perturbation is specific for NFs and does not affect other components involved in the axonal transport and scaffold, and in nodal/paranodal architecture.
NEFL-linked CMT2E mutations are distributed throughout the 3 functional domains and different mutations have been shown to cause distinct pathologic effect.5,6 The mutation carried by our patients that leads to a substitution of glutamic acid to lysine at position 396 (Glu396Lys) occurs in a highly conserved motive at the end of the rod domain, and several families with the same mutation as well as a transgenic mouse model have been reported in literature.5–7,18,19 The axons of the mutant mouse had fewer NFs than those of the wild-type, and accordingly, sural nerve biopsies from patients have shown the presence of axons either devoid of or with a variable density of NFs. Our results from both immunohistochemistry and WB analysis are in keeping with these data, demonstrating reduced levels of NFs in cutaneous nerve fibers. We think that a decrease in NF abundance in peripheral nerves might be a pathologic marker of CMT2E and independent from the axonal loss. Specifically, the Glu396Lys mutation, as occurs in a protein domain involved in the dimerization process, may affect the ability of NFs to assemble properly into filaments, disrupting the NF network and reducing the transport of NFs into the axons. The loss of axonal NFs distally may be a consequence of their accumulation proximally in both nerve fiber and cell body (as shown in transgenic models and iPSC-derived motor neurons from a patient with CMT2E20), leading to the formation of aggregates and to subsequent alterations in the axonal cytoskeleton. The lack of NF aggregates distally supports this hypothesis. Of note, Fabrizi et al.8 reported the presence of giant axons with NF accumulation in sural nerve biopsies from 4 patients with CMT2E carrying 3 different NEFL mutations, but not in their patient with the Glu396Lys mutation. These findings confirm that different mutations behave differently as far as disruption of NF organization is concerned, and that abnormalities of NF organization may be a marker of CMT2E. The possible alteration of axonal transport due to NF disruption does not involve other molecules that have a role in this process. Indeed, we did not find either an increase or a defect in the expression of acetylated α-tubulin as respectively shown in NEFL knockout mice and in other models of axonal CMT.21,22
A linear relationship between conduction velocity and fiber diameter is well known.23 Reduction in axon diameter decreases velocity, largely because the longitudinal resistance is higher. The most essential function of NFs is regulating the axonal caliber and thereby the conduction velocity. NFs are therefore particularly abundant in neurons with large-diameter axons where fast impulse conduction velocities are crucial for proper functioning.24 Within the skin, NF is expressed by both Aδ and Aβ fibers, which are fast-conducting nerve fibers, but not by C fibers that conduct more slowly.25 In transgenic mice, the loss of NF-L resulted in axons with reduced diameters and slowed conduction velocities.21,26 Morphometric analysis of sural nerve biopsies from patients with CMT2E disclosed decreased axonal diameters in myelinated fibers.7,8 Our morphometric data demonstrated that reduced axonal diameters can also be detected in a less invasive way in cutaneous nerve fibers. These smaller axonal calibers can explain the slow conduction velocities observed in CMT2E, supporting the notion that conduction velocity can be slow even in the absence of demyelination or abnormalities of nodes and paranodes, since no significant myelin sheath, nodal, or paranodal abnormalities were detected in our patients. Although the decreased axonal caliber observed in our patient might be the consequence of the loss of large-diameter fibers. Indeed, mice in which NF-L has been deleted (NF-L −/−) showed a unimodal distribution of axon calibers with the large-sized myelinated axons being shifted to the small-size axons, and a reduction in the large-diameter fibers with loss of the bimodal spectrum have been reported in patients with CMT2E as well.7,8,21 We believe that both phenomena occur in CMT2E and, along with the normal molecular architecture of nodes and paranodes, explain the reduced velocities detected in patients with CMT2E.
Our study extends the pathology of CMT2E and provides clues to the pathogenesis, supporting the feasibility of skin biopsy to investigate its mechanisms. A better understanding of the pathomechanism underlying each single NEFL mutation can lead to identifying a clear genotype–phenotype correlation.
Supplementary Material
GLOSSARY
- CMT
Charcot-Marie-Tooth disease
- CMT2E
Charcot-Marie-Tooth disease type 2E
- CMTNSv2
Charcot-Marie-Tooth Neuropathy Score version 2
- EM
electron microscopy
- IF
immunofluorescence
- NEFL
neurofilament, light polypeptide
- NF
neurofilament
- PGP
protein gene product
- WB
Western blot
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
C. Pisciotta: study concept and design, data acquisition, data analysis, statistical analysis, drafting the manuscript. Y. Bai: data acquisition, data analysis. K. Brennan: data acquisition, data analysis. X. Wu: data acquisition, data analysis. T. Grider: data acquisition, data analysis. S. Feely: data acquisition, data analysis. S. Wang: data acquisition, data analysis. S. Moore: interpretation of the data. C. Siskind: data analysis. M. Gonzalez: data acquisition, data analysis. S. Zuchner: data acquisition, data analysis. M.E. Shy: study concept and design, study supervision, interpretation of the data, revising the manuscript.
STUDY FUNDING
This study was supported by the Inherited Neuropathy Consortium–Rare Disease Clinical Research Consortium (INC-RDCRC U54NS065712), NINDS/ORDR, NCATS, the Muscular Dystrophy Association (MDA), and the Charcot-Marie-Tooth Association (CMTA).
DISCLOSURE
C. Pisciotta, Y. Bai, K. Brennan, X. Wu, T. Grider, S. Feely, and S. Wang report no disclosures relevant to the manuscript. S. Moore reports NIH funding for Wellstone Muscular Dystrophy Cooperative Research Center (U54NS053672) and has contracts with Sarepta Therapeutics. C. Siskind, M. Gonzalez, S. Zuchner, and M. Shy report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Manganelli F, Tozza S, Pisciotta C, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes in a Southern Italy population. J Peripher Nerv Syst 2014;19:292–298. [DOI] [PubMed] [Google Scholar]
- 2.Pisciotta C, Shy ME. The inherited peripheral neuropathies. In: Louis ED, Mayer SA, Rowland LP. Merritt's Neurology, 13th ed Philadelphia: Wolters Kluwer; 2015. [Google Scholar]
- 3.Miltenberger-Miltenyi G, Janecke AR, Wanschitz JV, et al. Clinical and electrophysiological features in Charcot-Marie-Tooth disease with mutations in the NEFL gene. Arch Neurol 2007;64:966–970. [DOI] [PubMed] [Google Scholar]
- 4.Griffin JW, Hoke A. The control of axonal caliber. In: Dyck PJ, Thomas PK, editors. Peripheral Neuropathy, 4th ed Philadelphia: Elsevier; 2005:433–436. [Google Scholar]
- 5.Perez-Olle R, Jones ST, Liem RK. Phenotypic analysis of neurofilament light gene mutations linked to Charcot-Marie-Tooth disease in cell culture models. Hum Mol Genet 2004;13:2207–2220. [DOI] [PubMed] [Google Scholar]
- 6.Shen H, Barry DM, Dale JM, Garcia VB, Calcutt NA, Garcia ML. Muscle pathology without severe nerve pathology in a new mouse model of Charcot-Marie-Tooth disease type 2E. Hum Mol Genet 2011;20:2535–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Züchner S, Vorgerd M, Sindern E, Schröder JM. The novel neurofilament light (NEFL) mutation Glu397Lys is associated with a clinically and morphologically heterogeneous type of Charcot-Marie-Tooth neuropathy. Neuromuscul Disord 2004;14:147–157. [DOI] [PubMed] [Google Scholar]
- 8.Fabrizi GM, Cavallaro T, Angiari C, et al. Charcot-Marie-Tooth disease type 2E, a disorder of the cytoskeleton. Brain 2007;130:394–403. [DOI] [PubMed] [Google Scholar]
- 9.Murphy SM, Herrmann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2011;16:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor LE, Kaminoh YJ, Rodesch CK, Flanigan KM. Quantification of dystrophin immunofluorescence in dystrophinopathy muscle specimens. Neuropathol Appl Neurobiol 2012;38:591–601. [DOI] [PubMed] [Google Scholar]
- 11.Nolano M, Provitera V, Crisci C, et al. Quantification of myelinated endings and mechanoreceptors in human digital skin. Ann Neurol 2003;54:197–205. [DOI] [PubMed] [Google Scholar]
- 12.Provitera V, Nolano M, Pagano A, Caporaso G, Stancanelli A, Santoro L. Myelinated nerve endings in human skin. Muscle Nerve 2007;35:767–775. [DOI] [PubMed] [Google Scholar]
- 13.Patzkó A, Bai Y, Saporta MA, et al. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain 2012;135:3551–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tradewell ML, Durham HD, Mushynski WE, Gentil BJ. Mitochondrial and axonal abnormalities precede disruption of the neurofilament network in a model of Charcot-Marie-Tooth disease type 2E and are prevented by heat shock proteins in a mutant-specific fashion. J Neuropathol Exp Neurol 2009;68:642–652. [DOI] [PubMed] [Google Scholar]
- 15.Griffin JW, Watson DF. Axonal transport in neurological disease. Ann Neurol 1988;23:3–13. [DOI] [PubMed] [Google Scholar]
- 16.Hoffman PN, Griffin JW, Price DL. Control of axonal caliber by neurofilament transport. J Cell Biol 1984;99:705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perrot R, Lonchampt P, Peterson AC, Eyer J. Axonal neurofilaments control multiple fiber properties but do not influence structure or spacing of nodes of Ranvier. J Neurosci 2007;27:9573–9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elbracht M, Senderek J, Schara U, et al. Clinical and morphological variability of the E396K mutation in the neurofilament light chain gene in patients with Charcot-Marie-Tooth disease type 2E. Clin Neuropathol 2014;33:335–343. [DOI] [PubMed] [Google Scholar]
- 19.Abe A, Numakura C, Saito K, et al. Neurofilament light chain polypeptide gene mutations in Charcot-Marie-Tooth disease: nonsense mutation probably causes a recessive phenotype. J Hum Genet 2009;54:94–97. [DOI] [PubMed] [Google Scholar]
- 20.Saporta MA, Dang V, Volfson D, et al. Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp Neurol 2015;263:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Q, Couillard-Després S, Julien JP. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol 1997;148:299–316. [DOI] [PubMed] [Google Scholar]
- 22.d'Ydewalle C, Krishnan J, Chiheb DM, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 2011;17:968–974. [DOI] [PubMed] [Google Scholar]
- 23.Hursh JB. Conduction velocity and diameter of nerve fibres. Am J Physiol 1939;127:131–139. [Google Scholar]
- 24.Al-Chalabi A, Miller CC. Neurofilaments and neurological disease. Bioessays 2003;25:346–355. [DOI] [PubMed] [Google Scholar]
- 25.Reinisch CM, Tschachler E. The dimensions and characteristics of the subepidermal nerve plexus in human skin-terminal Schwann cells constitute a substantial cell population within the superficial dermis. J Dermatol Sci 2012;65:162–169. [DOI] [PubMed] [Google Scholar]
- 26.Sakaguchi T, Okada M, Kitamura T, Kawasaki K. Reduced diameter and conduction velocity of myelinated fibers in the sciatic nerve of a neurofilament-deficient mutant quail. Neurosci Lett 1993;153:65–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.