

Abstract
The dopamine D3 receptor (D3R) is a target for developing medications to treat substance use disorders. D3R-selective compounds with high affinity and varying efficacies have been discovered, providing critical research tools for cell-based studies that have been translated to in vivo models of drug abuse. D3R antagonists and partial agonists have shown especially promising results in rodent models of relapse-like behavior, including stress-, drug-, and cue-induced reinstatement of drug seeking. However, to date, translation to human studies has been limited. Herein, we present an overview and illustrate some of the pitfalls and challenges of developing novel D3R-selective compounds toward clinical utility, especially for treatment of cocaine abuse. Future research and development of D3R-selective antagonists and partial agonists for substance abuse remains critically important but will also require further evaluation and development of translational animal models to determine the best time in the addiction cycle to target D3Rs for optimal therapeutic efficacy.
1. Introduction
A decade ago, we (A.H.N. and M.A.N.) wrote a Perspective entitled Dopamine D3 Receptor Partial Agonists and Antagonists as Potential Drug Abuse Therapeutics.(1) We posited that, as all drugs of abuse either directly or indirectly increase dopamine (DA) levels in the mesolimbic region of the brain, DA receptors were one obvious target for medication development. We highlighted the supporting literature up to that time, focusing on the DA D3 receptor (D3R), and further emphasized that despite tremendous progress in identifying mechanistic underpinnings of the psychoactive actions of addictive drugs, particularly for the psychostimulants such as cocaine and methamphetamine (METH), not a single pharmacological treatment had been approved by the FDA. As millions of people in the United States and worldwide suffer from psychostimulant abuse and addiction, the public health need to develop medications to treat these substance use disorders was and remains significant. Since 2005, more than 6000 papers have been published on basic, preclinical, and clinical cocaine abuse research in an effort to develop effective treatments. Nonetheless, despite our best efforts with hypothesis-driven investigations producing promising results in animal models of psychostimulant addiction, we have not yet succeeded in identifying a single medication that can meet FDA safety and efficacy requirements in this population of patients. The broader question is why have we failed? A narrower question, and the focus of this Perspective, relates directly to the development of medications that target the D3R. In this review, we once again set the stage for the D3R as a lead target for medication discovery for psychostimulant addiction and highlight the application of small molecule structure–activity relationships (SAR), with the aid of structure-based design using the D3R crystal structure,2−4 toward novel drug-like molecules. We then illustrate some of the challenges of moving our basic hypotheses through the rigors of drug development using our lead molecule 1 (PG648, Chart 1)5 as an example.
Chart 1. Chemical Structures of Highlighted D3R-Selective Partial Agonists and Antagonists.

All known addictive drugs enhance DA signaling within key corticolimbic circuits of the brain that control reward, emotion, cognition, and motivation. Although the molecular targets of addictive drugs vary widely,6 they all appear to directly or indirectly enhance DA signaling in the ventral striatum, particularly the nucleus accumbens,7,8 and activate neural circuitry that normally mediates reward responses to natural stimuli such as food and sex. Other dopaminergic pathways that project into the dorsal striatum and frontal cortex have also been identified as contributing to drug reward and may be especially affected in the progression to addiction.7,9 Specifically, psychostimulant drugs directly increase synaptic DA by altering the function of the DA transporter (DAT). DAT blockers (e.g., cocaine, methylphenidate) inhibit DA removal from the synaptic cleft; DAT substrates (e.g., amphetamines) have more complex actions and can induce nonvesicular DA release into the synapse.10 The commonality between these mechanistically distinct drugs of abuse is that rapid and profound increases in synaptic DA lead to stimulation of DA receptors, producing the stimulant and rewarding euphoric effects that can lead to abuse and addiction. What makes cocaine unique, though, is the rapid reversal of elevated DA, leading to compulsive drug taking.11,12 For example, cocaine and methylphenidate have similar regional distributions in the striatum, but the rapid pharmacokinetics of cocaine likely lead to its higher abuse potential.12
DA signaling is mediated by a family of G protein-coupled receptors (GPCRs). The five known DA receptors (D1R–D5R) are classified into two families on the basis of sequence similarity and second messenger activity. D1-like receptors (D1R and D5R) signal through Gαs and enhance the production of intracellular cyclic AMP (cAMP); D2-like receptors (D2R, D3R, and D4R) signal through Gαi/o and inhibit the production of intracellular cAMP.
In comparison to D2R pharmacology, D3R pharmacology has been historically more enigmatic. D3Rs have relatively small shifts in agonist binding affinity in response to guanyl nucleotides, which may indicate relatively poor receptor coupling to G proteins or, alternatively, that the receptor structure is relatively rigid and only modestly affected by G protein association; relatedly, D3R receptor high- and low-affinity states are reported to differ only 5–10-fold in heterologous systems.13 D3Rs do couple to G proteins in heterologous systems, but not exclusively to Gαi/o (some signaling through Gαq has been reported), and the adenylate cyclase V isozyme is required for agonist-mediated inhibition of cAMP production.13,14 Furthermore, recent evidence indicates that D3Rs likely form functional heteromers with D1Rs in the striatum.15,16 The functional consequences of this interaction in vivo have yet to be elucidated, but it may play an important role in a variety of neuropsychiatric disorders.17
The D3R has long been a target of interest in addiction pharmacotherapy due to its relatively focal localization within the ventral striatum and its enhanced expression in drug-exposed brains.1,18,19 Several research groups have discovered highly selective D3R antagonists, partial agonists, and full agonists using small molecule SAR (for recent reviews, see refs (20−22)) and more recently using the D3R crystal structure, computational methods, and molecular pharmacology.2,3,18,23,24 Many of these D3R-selective ligands have served as essential research tools for pharmacological investigations at the molecular, cellular, and behavioral levels.
Herein, we briefly discuss the history of D3R as a target for addiction treatment, including a preview of limited clinical studies. We discuss the viability of identifying a novel translational candidate for psychostimulant addiction, practical concerns for future development of D3R-targeted pharmacotherapies, and general obstacles to medication development for addiction. Translation of hypotheses based on preclinical findings has proven to be challenging due to the lack of clinically available, D3R-preferential compounds. One concern is that failure in the clinic of a single lead molecule could prematurely eliminate the D3R as a medication target for addiction pharmacotherapy.
Over the past decade, we have discovered many D3R-selective ligands with varying efficacies as research tools that have high affinity (Ki < 10 nM) and D3R selectivity (>100-fold over D2R).5,25,26 One of our lead compounds, N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide (1), is a high-affinity D3R-selective antagonist first reported in 2009.5 Computational studies following the publication of the D3R crystal structure identified key interactions within the orthosteric (DA) binding site and the 2,3-diCl-phenylpiperazine moiety of 1, as well as a secondary binding pocket that together likely contribute to the 4-phenylpiperazine class of compounds’ affinity, subtype selectivity, and efficacy.2,27 Further studies determined that D2R–D3R subtype selectivity of 1 and related compounds depended critically on a divergent glycine residue in extracellular loop 1.23 Given the success of earlier and structurally related compounds 2 (NGB2904),28,293 (PG01037),30 and 4 (SB277011A)31 in animal models of addiction, compound 1’s favorable receptor selectivity profile made it an enticing preclinical candidate. Hence, we highlight the development and preclinical evaluation of 1 as an instructive example of a D3R-selective antagonist that ultimately will not make it to the clinic; identifying pitfalls in the medications development process and areas of need in behavioral models of addiction.
2. Rationale: D3R as a Pharmacotherapeutic Target for Psychostimulant Addiction
As a substantial cause of morbidity and mortality, drug addiction exerts major sociological and financial costs on individuals and society at large. A recent estimate of $500 billion in economic costs to the U.S. alone has been used to inspire further support and development of Addiction Medicine Programs.32 However, while there are presently pharmacotherapeutic options to treat dependence on nicotine,33 alcohol,34 and opiates,35,36 there are currently no FDA-approved medications for psychostimulant addiction. This deficit in our medical armamentarium significantly reduces what clinicians can do for psychostimulant abusers, which underscores the public health need to develop medications for treatment.
It is well-established that prolonged exposure to psychostimulant drugs leads to adaptive neurobiological changes.37 Key observations that encouraged the development of D3R-specific compounds for psychostimulant addiction treatment came from human postmortem studies showing elevated expression of D3Rs following acute or chronic exposure to cocaine.38−40 The phenomenon of increased D3R expression has been confirmed and extended to other drugs, including METH and alcohol, via animal and human studies using various molecular biology techniques and PET imaging.41−46 As the focus of this Perspective is targeting D3R for psychostimulant addiction treatment, we encourage interested readers to consult other excellent reviews for discussions of the pharmacotherapeutic potential of D3R-selective compounds for substance use disorders associated with other classes of addictive drugs, including ethanol and opiates.41,47−50
Since the cloning of the D3R gene in 1990,51 several key differences have been identified between D3R and the closely related D2R (sharing 78% sequence identity in the transmembrane and binding domains). D2R expression in the human brain is widespread but most prominent in the dorsal striatum;52 D3R expression is overall lower than D2R, and the distribution of striatal D3R is more limited to ventral regions, particularly the nucleus accumbens.48,52,53 A recent autoradiography study in postmortem human brain tissue reported that D3R expression is greater and more widespread than has previously been appreciated, including moderate dorsal striatal expression (although still lower density than D2R) and appreciable expression in extrastriatal regions such as the thalamus.54 The recent development of newer radioligands, such as 5 ([3H]LS-3-134 in Chart 1), with greater D3R selectivity over D2R may further resolve the longstanding difficulty in differentiating D2R and D3R receptor densities in neuronal tissues.55
The localization of D3Rs within the ventral striatum suggests the receptor may play an important role in the rewarding effects of drugs and control of motivational behaviors. The relatively low D3R density in the dorsal striatum, compared to D2R, suggests that ligands sufficiently selective for D3R over D2R may also avoid the undesirable motor coordination and extrapyramidal side effects associated with nonselective D2-like antagonists (e.g., antipsychotics), commonly attributed to D2R blockade.56,57 Moreover, a recent meta-analysis of randomized placebo-controlled trials with a variety of neuroleptics showed that these clinically available D2R/D3R antagonists were no more efficacious than placebo in improving abstinence or reducing craving for cocaine or METH, further suggesting that nonselective D2-like antagonists do not have clinical utility in this patient population.58 However, cariprazine (6, RGH-188, Chart 1), a partial agonist approximately 10-fold D3R-preferential, is in clinical development as an antipsychotic agent (for review, see ref (59)) and has shown potential utility in preclinical models of cocaine abuse.60
While the major goal of addiction pharmacotherapy development is to block drug seeking, there has been recent interest in also focusing on cognitive deficits induced by long-term drug abuse.61 In one of the earlier studies, Laszy et al.62 used a water labyrinth test to assess spatial memory in rats and found that cognitive impairments induced by scopolamine were reversed by D3R antagonists. More recently, Mugnaini et al.63 reported that the D3R antagonist 7 (GSK598,809 in Chart 1) partially attenuated an attentional bias, as assessed with a Stroop Task, in abstinent smokers. In a review on D3R and cognition, Nakajima et al.64 concluded that D3R blockade enhances cognitive function, whereas agonists at D3R appear to impair cognition. Recent preclinical studies suggest that the mechanism by which D3R blockade improves cognition involves the facilitation of both cholinergic and DA transmission in the frontal cortex.62,65−67 D3R may also influence cognition by modulating CREB signaling in the hippocampus68 as well as through glutamatergic–D3R interactions.69 The relationship between D3R and cognition is an area of research that requires much additional work. While Nakajima et al.64 reported reversal in “compromised” animals, it remains to be determined whether selective D3R antagonists and/or partial agonists can improve cognition in subjects with cocaine- or METH-induced cognitive disruptions.
2.1. Designing D3R-Selective Antagonists and Partial Agonists
The initial success of the partial agonist 8 (BP897 in Chart 1) in a rodent model of cocaine addiction70 spurred many laboratories in both academia and pharma to develop new drug-like molecules with D3R selectivity. SAR studies published by a number of research groups have been reviewed previously.1,3,21,71−74 Recently reported analogues in the 4-phenylpiperazine template include functionalization of the butyl linking chain5,25,26,75−77 and elaboration of both the terminal aryl amide and phenylpiperazine or head group.24 Recently reported D3R-selective compounds can be seen in Chart 2. These compounds typically follow and expand upon the SAR already established for D3R-selective antagonists or partial agonists and, for those that have been tested in vivo, have similar behavioral profiles in animal models of psychostimulant abuse. A novel approach that inserted a cyclobutyl group in the linking chain and replaced the phenylpiperazine head group with tranylcypromine resulted in a recently reported novel series of D3R-selective antagonists.78
Chart 2. Recently Reported D3R-Preferential Ligands with Novel Structural Templates.

Many of the newer analogues have capitalized on variations of the 4-phenylpiperazine template, but some interesting and diverse molecules have also been discovered. Notably, the evolution of SAR in the Glaxo program, led by Micheli and colleagues, abandoned the arylamide terminus for a heteroaryltriazole, as seen in 7.20,79 This modification resulted in high affinity and selectivity for the D3R target but also metabolic stability and low hERG channel activity, predicting that this compound would be bioavailable and safe in humans. Other groups have replaced the 4-phenylpiperazine with the tetrahydrobenzothiazole of pramipexole to give full80 or partial81 D3R agonists with both high affinity and subtype selectivity. Of note, the groups led by Dutta and Reith have also reported tetrahydrobenzothiazole analogues, but they have replaced the arylamide of the more classic D3R ligands with this functional group and retained an arylpiperazine or an arylamide piperazine to give a novel set of D3R-selective agonists.82 In this series, it is interesting to contemplate which end of these molecules binds to the orthosteric (DA) binding site and whether the other end binds to the secondary binding pocket believed to be occupied by the aryl amide of (R)-12 or a different site within the D3R protein to produce a full agonist profile. A similar hybridization approach that replaced the 4-phenylpiperazine with a 5-aminohydropyrazolopyridine group also resulted in a very interesting series of highly potent D3R agonists that were reported to demonstrate functional bias,83 a hot topic in the D2-like receptor drug discovery arena (for review, see refs (84 and 85)) but beyond the scope of the present review. Gmeiner and colleagues are avidly following these leads and discovering other templates that display functionally biased profiles.86,87 The field is still in its infancy in terms of understanding the pharmacological and behavioral consequences of D3R-mediated biased agonism. Nevertheless, the combination of small molecule SAR and the D3R crystal structure with computational modeling will undoubtedly produce structurally and pharmacologically variant tools in the near future with which to probe these questions.3
2.2. Translating D3R-Selective Antagonists and Partial Agonists to the Clinic
To date, very few D3R-selective or preferential antagonists or partial agonists have been tested in clinical trials for substance use disorders. Thus far, 7 appears to be the most clinically investigated compound based on publications in the literature and perusal of clinicaltrials.gov (e.g., ClinicalTrials.gov identifiers NCT00437632, NCT01188967, NCT00793468, NCT00605241, and NCT01039454).
Briefly, after validating D3R occupancy in a PET study using [11C]PHNO53 and evaluating its pharmacokinetics,887 was tested in several clinical studies. In overweight and obese subjects, 7 reduced approach bias89 and attentional bias to palatable food cues90 but did not alter fMRI brain responses to food images.91 In tobacco smokers, 7 transiently alleviated craving in smokers after overnight abstinence, although smoking increased.63 Although 7 had some qualified successes in these clinical populations, and many other D3R-selective antagonists and partial agonists have been successful in preclinical models of cocaine or METH abuse, it is unclear at this time whether or not 7 will be evaluated in this patient population.
One concern that has emerged is the potential for 7 and other D3R antagonists to increase blood pressure, especially if taken in combination with stimulants (Nate Appel and Jane Acri, National Institute on Drug Abuse, personal communication), as D3Rs reside in the kidney and contribute to DA-mediated regulation of blood pressure.92−95 Cocaine’s cardiovascular effects have been investigated, and increases in both blood pressure and heart rate have been well-documented.96,97 Indeed, cardiovascular toxicity is likely the main contributor to medical complications and overdose. Both central and peripheral mediation of increased blood pressure upon acute and chronic administration of cocaine have been investigated with varying results depending on species, dose of cocaine, and acute versus chronic administration.96 The concern, of course, is that if subjects relapse to cocaine taking, blood pressure elevations due to cocaine exposure could be exacerbated in the presence of a D3R antagonist. Human studies investigating the effects of the D2R/D3R partial agonist aripiprazole on cocaine self-administration, drug discrimination, and cardiovascular effects reported a small, acute increase in blood pressure that was not sustained after repeated cocaine administration.98,99 Further investigation with D3R-selective drugs is required in order to clearly determine whether D3R-selective partial agonists or antagonists are likely to exhibit this potential side effect. For example, in ongoing preclinical studies using nonhuman primates implanted with indwelling telemetry devices, administration of cocaine increases blood pressure and heart rate; these effects are either unaltered or enhanced by D3R antagonists, but they appear to be attenuated by D3R partial agonists (M.A. Nader, A.H. Newman, unpublished results). Also, it is critically important to determine whether this potential cardiovascular risk is mediated via D3R or is an off-target effect of the drug molecule: the latter might be addressed with appropriate structural modifications of the lead compound(s). Hence, continued evaluation of structurally diverse molecules with differing off-target profiles remains a priority to further validate the D3R as a target for medication development, especially for psychostimulant abuse. Of course, substance use disorders that by themselves do not alter blood pressure or heart rate may be completely treatable with D3R antagonists or partial agonists, without concern for cardiovascular safety. In addition, increases in blood pressure can be monitored and treated, if necessary, with clinically available drugs.
2.3. Repurposing Buspirone for Treatment of Cocaine Abuse
In response to the interest in investigating D3R antagonists as potential pharmacotherapies for cocaine abuse, the clinically available anxiolytic buspirone was evaluated in several preclinical addiction models and in a clinical trial with cocaine abusers. While by no means D3R-selective, Bergman et al.100 reported that buspirone had higher affinity at the D3R compared to the D2R. When tested in monkeys self-administering cocaine, they found that buspirone decreased cocaine-maintained behavior to a larger degree than food-reinforced behavior. Mello et al.,101 using rhesus monkeys that self-administered cocaine 4 times per day, found that continuous infusions of buspirone also decreased cocaine self-administration to a greater degree than food-reinforced responding. These findings supported the use of buspirone as a pharmacotherapy for cocaine addiction. Unfortunately, the positive results from these preclinical studies did not extend to clinical trials. Winhusen et al.102 reported negative effects of buspirone in cocaine abusers and, in fact, found that buspirone increased cocaine use in women. However, it should be noted that this study had a remarkable placebo effect: subjects that were not administered buspirone dramatically reduced their cocaine use. Further, it is not clear that the single tested dose (60 mg) of buspirone was adequate to achieve high occupancy of D3R for a sufficient duration of time to be effective.102 Supporting this assumption is a recent study in primates measuring D3/D2R occupancy of buspirone using PET imaging.103 It was found that the low dose of oral buspirone (1.0 mg/kg) tested, approximately the same dose tested in the Winhusen et al. study, exhibited minimal D3R occupancy (<20%). In contrast, 80% sustained D3R occupancy was achieved by a dose that was 3-fold higher (3.0 mg/kg) and was well-tolerated. These data indicate the importance of using receptor occupancy as a guideline for therapeutic efficacy in that high and sustained levels are most likely needed for the successful treatment of addiction.
On the basis of these clinical findings with buspirone, one may (prematurely) conclude that the D3R and, more specifically, D3R antagonists are not viable targets for psychostimulant addiction. We would caution against such conclusions for two reasons: First, buspirone has very complex pharmacology, with known effects at 5-HT1A, D2R, D3R, and D4Rs, as well as active metabolites that interact with α2-adrenergic receptors.100,104,105 Second, the choice of drug reinforcement schedules used in the preclinical evaluations of buspirone may not have been ideal to detect D3R-mediated antiaddiction effects.
One of the interesting aspects of the behavioral pharmacology of D3R compounds is the importance of the schedules of reinforcement used in self-administration studies (see refs (1 and 48) for review). In the preclinical models used by Bergman et al.100 and Mello et al.,101 there were no competing reinforcers when cocaine self-administration was studied, and this may be critical to understanding how medications, such as buspirone, can decrease one type of behavior (in their case, food-maintained responding) to a smaller degree than cocaine self-administration. More complex models, such as food–drug choice paradigms, measure reinforcing strength (efficacy) with the goal of examining treatments that decrease cocaine self-administration and reallocate responding from cocaine to food. In a recent study, John et al.106 found that 5 day treatment with buspirone increased cocaine choice, a finding consistent with the Winhusen et al.102 clinical study. Clearly, the animal models used to evaluate D3R compounds on cocaine self-administration need to be more thoughtfully analyzed in order to achieve translation of preclinical findings to clinical success. For example, in a study using socially housed monkeys, acute buspirone administration decreased cocaine choice relative to food in dominant monkeys but not subordinate animals, suggesting a possible interaction between environmental variables and efficacy of buspirone.107 Importantly, though, it is our premise that using food–drug choice paradigms and the study of a range of D3R compounds (see below) will allow for (1) a better understanding of the role of D3Rs in cocaine abuse and (2) the identification of potential pharmacotherapies based on the D3R. Thus, buspirone should not be considered a representative D3R antagonist, and it is our perspective that this single clinical trial should not deter further research toward developing a D3R-selective antagonist or partial agonist for substance use disorders. It should, however, be noted that treatment-emergent adverse events reported in this study did not include increases in blood pressure in the subjects receiving buspirone.102
3. Recent Development of Novel D3R-Selective Compounds as in Vivo Tools
On the basis of the preclinical promise of early leads, our group and others have focused efforts on optimizing D3R affinity and selectivity as well as physical properties (e.g., cLogP, tPSA, metabolic stability, etc.) to improve their utility as in vivo tools. As drug abuse is a human behavior, we ultimately must develop tools that are stable in vivo, penetrate the blood–brain barrier, and selectively engage D3R. We must also be able to readily scale up the synthesis of these molecules for behavioral studies in rodent and nonhuman primate models, the most translational of which require chronic dosing. Like other medicinal chemists, we have optimized lead compounds, using structure–activity relationships and, more recently, the D3R crystal structure, to design novel molecules. These efforts have led to the identification of several new lead molecules, including (±)-, R-, and S-1, 3, and others including 17 (CJB090),10818 (PG619),7519 (GCC3-09),5 and 20 (BAK2-66),26 to highlight a few (Chart 3). These compounds have been evaluated in cell-based binding assays along with compounds reported in the literature to obtain side-by-side data comparisons across the D2-like family of receptors.
Chart 3. Behaviorally Tested D3R-Preferential Ligands.

3.1. Relative Binding Affinity and D2-Like Receptor Subtype Selectivity of Dopaminergic Compounds
The binding affinities of representative D2-like compounds, in Table 1, were obtained from human D2Rs, D3Rs, and D4Rs expressed in HEK293 cells. Using membranes from these cells and the antagonist radioligand [3H]N-methylspiperone, direct comparisons can be made of these structurally divergent compounds under identical experimental conditions (see Supporting Information for methods).
Table 1. D2-Like Binding Affinity and Subtype Selectivity of Selected Ligands Using [3H]N-Methylspiperone.
| D2R | D3R | D4R | subtype
selectivity |
|||
|---|---|---|---|---|---|---|
| compound | Ki ± SEM (nM)a | Ki ± SEM (nM)a | Ki ± SEM (nM)a | D2/D3 | D4/D3 | cLogP |
| N-methylspiperone78 | 0.133 ± 0.00885 | 0.265 ± 0.00752 | 0.461 ± 0.00789 | 0.50 | 1.7 | |
| eticlopride78 | 0.0860 ± 0.000951 | 0.134 ± 0.00437 | 46.4 ± 6.94 | 0.64 | 346 | |
| raclopride78 | 12.7 ± 1.21 | 13.4 ± 0.695 | 14 700 ± 357 | 0.95 | 1100 | |
| butaclamol78 | 2.58 ± 0.473 | 6.39 ± 0.584 | 229 ± 57.0 | 0.40 | 36 | |
| aripiprazole | 0.343 ± 0.0899 | 1.00 ± 0.0250 | 57.1 ± 16.2 | 0.34 | 57 | |
| (±)-1 (PG648)76 | 746 ± 123 | 1.88 ± 0.112 | 2600 ± 660 | 397 | 1380 | 4.8 |
| (R)-176 | 295 ± 65.0 | 0.528 ± 0.0910 | 3980 ± 882 | 559 | 7540 | 4.8 |
| (S)-176 | 786 ± 160 | 3.89 ± 0.365 | 1900 ± 491 | 202 | 489 | 4.8 |
| 2 (NGB2904) | 54.7 ± 5.20 | 0.233 ± 0.0089 | 3670 ± 626 | 235 | 15 800 | 6.7 |
| 3 (PG01037) | 74.0 ± 16.6 | 0.316 ± 0.0284 | 550 ± 28.3 | 234 | 1740 | 5.5 |
| 7 (GSK598,809) | 2110 ± 485 | 3.15 ± 0.335 | 35 600 ± 8,880 | 670 | 11 300 | 2.6 |
| 18 (PG619)78 | 1090 ± 20.6 | 6.70 ± 0.768 | 4470 ± 993 | 163 | 667 | 3.3 |
| 19 (GCC3-09) | 217 ± 35.6 | 0.920 ± 0.102 | 2040 ± 677 | 236 | 2210 | 4.9 |
| 20 (BAK2-66)76 | 956 ± 273 | 10.3 ± 1.98 | NT | 93 | 5.3 | |
| 21 (YQA14) | 46.9 ± 7.36 | 3.04 ± 0.369 | 2370 ± 223 | 15 | 780 | 3.7 |
Ki values determined by competitive inhibition of [3H]N-methylpsiperone binding in membranes harvested from HEK 293 cells stably expressing hD2R, hD3R, or hD4R. Detailed methods have been described previously76,78 and are in the Supporting Information. NT, not tested. References indicate previously published binding data using identical methods.
Table 1 shows the experimentally derived binding dissociation constants (Ki) for a number of dopaminergic compounds with varying selectivities for D3R over D2R and D4R. Of note, N-methylspiperone, eticlopride, raclopride, and butaclamol are well-known D2-like antagonists, demonstrating high binding affinities across the D2-family of receptors, with the notable exceptions of butaclamol and especially raclopride at D4R. Aripiprazole is a clinically available D2-like partial agonist, marketed as Abilify, used in the treatment of schizophrenia and mood disorders. It binds with high affinity to both D2R and D3R and has been shown to decrease cocaine self-administration and attenuate reinstatement in laboratory animals.109−112 Although mixed results have emerged from human studies,98,99,113 a recent phase II study using chronic treatment with lower doses reported that aripiprazole decreased cocaine craving.114
Because identical binding conditions were used, the data presented in Table 1 permit direct binding affinity comparisons across compound classes. Compound 7 was the most subtype-selective compound in this set, with 670-fold D3R selectivity over D2R. Notably, binding for 21 (YQA14) in this system did not show two-site binding kinetics at D3R as reported previously.115 The Ki determined in this analysis (3.04 nM) is consistent with the Ki-Low reported previously (2.11 nM); coupled with a higher D2R affinity in this analysis than in the previous report (46.9 vs 335 nM),115 compound 21 appears to be only moderately preferential for D3R binding over D2R when compared to others in this class of D3R ligands and points to the limitations of using cell-based binding affinities to predict D3R selectivities and potencies in vivo. Nevertheless, radioligand binding is the first line of testing in most drug discovery programs and is useful as long as compounds are evaluated side-by side under the same assay conditions. As noted in our previous Perspective, cell-based functional assay data remain difficult to interpret from an SAR standpoint;1 hence, binding data remain our primary source of SAR, despite these limitations.
Compound 2 is one of the first D3R-selective antagonists to be reported116 and has served as an important preclinical tool. Early reports demonstrated that 2 significantly lowered the break point in rats trained to self-administer cocaine under a progressive-ratio (PR) schedule of reinforcement.29 PR schedules require an increase in number of responses following injections of cocaine and the last ratio completed (the break point) is thought to be a measure of reinforcing strength.117 Compound 2 also inhibited cocaine-induced enhancement of brain stimulation reward threshold while neither maintaining self-administration nor altering brain-reward thresholds on its own. In addition, 2 blocked both cue- and cocaine-induced reinstatement of cocaine seeking, a preclinical model of relapse.28,29,118 Compound 2, along with 4 and 8, was also evaluated in METH-enhanced brain stimulation reward in rats. Notably, the two antagonists (2 and 4) effectively attenuated METH-enhanced brain stimulation reward and did not affect brain stimulation reward on their own; the partial agonist 8 attenuated METH-enhanced brain stimulation reward, but a high dose inhibited brain stimulation reward itself.119 Hence, 2, along with 4 and 8, demonstrated potential for this class of ligands to be developed toward medications to treat cocaine and METH abuse. Although none of these agents would be translated to the clinic, all of them served as templates for the design of new analogues. While 4 was the precursor to 7,192 served as the starting point from which our early lead compounds 3 and 17 were derived.25,108,120 Compound 17 was the first partial agonist in our series that was evaluated in nonhuman primates and compared to 2 in two models of cocaine abuse.121 Interestingly, 17, but not 2, attenuated cocaine’s discriminative stimulus effects and decreased both cocaine- and food-maintained responding in monkeys that were trained on a second-order schedule of reinforcement.121 However, in a separate study with squirrel monkeys, 17 failed to attenuate cocaine self-administration or cocaine-induced reinstatement of extinguished cocaine-seeking behavior.122 As elaborated by Achat-Mendes et al.,122 there are several possible reasons for these discrepant results including the different schedule parameters, the different frequencies of cocaine injection per session (maximally 10 in the Achat-Mendes et al. study122 and 2 in the Martelle et al. study121), and the different species used (squirrel monkeys vs rhesus monkeys). Another possible explanation could be the cocaine history of the subjects, which has been shown to influence the behavioral effects of D3R compounds.123−125
Both 17 and the structurally related but more D3R-selective antagonist 3 reduced PR METH self-administration in rats with a history of long access (6 h per day, 6 days per week) to METH.12617, in contrast to 3, also reduced PR METH self-administration in rats with a history of short-access to METH (1 h per day, 3 days per week).126 It is unclear at this time whether or not differences in pharmacokinetics or pharmacodynamics can explain these subtle differences in efficacy across species and models, although pharmacokinetic and metabolism data suggested that 3, like 2, had suboptimal bioavailability.127 Overall, coupled with many other reports in the literature of similar findings with compounds such as 4, 7, and newer generation analogues, these data supported further optimization and development of D3R-selective antagonists or partial agonists. Recent reports from the Neisewander lab highlight novel 4-phenylpiperazines, exemplified by 22 (Chart 3), each varying in their subtype selectivity and degree of partial agonism at D3R, that reduce cocaine self-administration.128−130
Compound 17 and a newer-generation partial agonist, 18,75 exhibited interesting effects in rhesus monkeys with a history of cocaine self-administration in comparison to their drug-naïve counterparts.123 As part of our efforts to identify D3R-based behaviors in vivo, a model of D3R-induced yawning in rats131,132 was modified, in which yawning could be produced in rhesus monkeys upon administration of the D3/D2R agonist quinpirole. In cocaine-naïve animals, quinpirole induces robust yawning, but the partial agonists 17 and 18 failed to do so.121,123 However, in monkeys with a history of cocaine self-administration, both 17 and 18 induced yawning similar in magnitude to quinpirole.123 We reasoned that increased sensitivity to yawning might occur in the monkeys with a cocaine history, as increases in D3R densities had been reported in both human cocaine fatalities38 as well as in cocaine-exposed rodents.133 Nevertheless, in that study, 18 was unable to attenuate cocaine self-administration under a fixed-ratio (FR) 30 schedule of reinforcement. Unlike quinpirole, however, 18 did not reinstate cocaine seeking in these monkeys but rather attenuated cocaine-induced reinstatement.123
In an effort to continue to optimize D3R affinity, selectivity, and bioavailability, modifications of these lead molecules have led to newer generation compounds such as 19 and 1 (Chart 1).5 Both of these compounds have high affinity (Ki 1–2 nM) for D3R and are highly selective over D2R and other off-target receptors.5 Of note, (±)-1 was evaluated in 64 radioligand/enzyme assays through the NIDA Addiction Treatment Discovery Program. Other than the reported binding affinities at D1R (Ki = 4630 nM) and 5-HT1A receptors (Ki = 104 nM),5 (±)-1 did not produce >50% inhibition of binding at any of the receptors evaluated at a concentration of 100 nM and only “hit” a few at 10 μM [e.g., α1 adrenergic, α2 adrenergic, histamine H1, sodium channel site 2, cholecystokinin 1 (CCK1), and neurokinin 2 (NK2)]. In addition, the IC50 for (R)-1 at the hERG channel was 0.38 μM, as determined using the PatchXpress assay134 through the NIMH Psychoactive Drug Screening Program (PDSP; http://pdsp.med.unc.edu). Considering its Ki at D3R is ∼2 nM, (±)-1 was considered to be highly selective for the D3R, and, despite its marginally acceptable cLogP value of 4.8, we chose it as our lead candidate for further development.
The addition of a 3-OH group in the linking chain of these molecules creates a chiral center; therefore, we synthesized the R- and S-enantiomers of 1. In radioligand binding competition studies, we observed a small but significant enantioselectivity at D3R, but not at D2R, with the R-enantiomer having higher D3R affinity.5 From a structural point of view, this was significant and gave insight on differential binding interactions at the receptor protein level that were further explored.5,27 To determine if behavioral effects were also enantioselective, we improved the enantioselective synthesis,76 evaluated the microsomal metabolism and pharmacokinetics of the racemate, and then tested the racemate and the R- and S-enantiomers in behavioral models of cocaine and METH abuse.
3.2. Further Development of Lead Compound 1
3.2.1. Pharmacokinetics and Metabolism Studies of (±)-1 in Mice
Pharmacokinetic and metabolism studies were performed in mice. The plasma concentration–time profile of (±)-1 after i.v. and p.o. dosing of 10 mg/kg is shown in Figure 1. A summary of the plasma pharmacokinetic parameters is listed in Table 2. Absorption for oral (±)-1 was slightly delayed, peaking 2 h after administration. The peak plasma concentration (Cmax) for (±)-1 was 7369 ng/mL following i.v. dosing compared to 522 ng/mL following p.o. dosing; comparing oral versus intravenous AUC parameters gives a low/moderate average absolute bioavailability fraction (F%) of 16.2%. The plasma half-life (t1/2) of i.v. (±)-1 was approximately 75 min. (±)-1 showed excellent brain penetrability following both p.o. and i.v. dosing, as shown in Table 3. The brain-to-plasma ratio ranged from 6- to 20-fold for (±)-1.
Figure 1.

Concentration profiles for 10 mg/kg (±)-1 in mouse plasma following i.v. (○) or p.o. (■) administration. Data are presented as mean ± SEM; n = 3 mice per time point.
Table 2. Noncompartmental PK Parameters for 10 mg/kg (±)-1 in Mouse Plasma Following i.v. or p.o. Administration.
| Cmaxa | Tmaxa | AUClasta | (AUCinf)a | Ke | t1/2a | F% | |
|---|---|---|---|---|---|---|---|
| (ng/mL) | (h) | (h·ng/mL) | (h·μg/mL) | (h) | |||
| plasma (i.v.) | 7369 | 0.08 | 7971 | 8895 | 0.55 | 1.241 | |
| plasma (p.o.) | 522 | 2 | 1299 | NCb | NCb | NCb | 16.2% |
Data are presented as mean; n = 3 mice per time point.
NC, not calculated.
Table 3. Brain Concentrations of (±)-1 in Mouse Following i.v. or p.o. Dosing at 10 mg/kg.
| (±)-1 | ||
|---|---|---|
| time (h) | i.v. conc. ng/g (SD) | p.o. conc. ng/g (SD) |
| 0.5 | 28 928 (3716) | 2979 (893) |
| 2 | 17 798 (3292) | 9231 (3478) |
We investigated the metabolism of (±)-1, and these data are presented in Table 4. (±)-1 was found to be very stable in mouse plasma over a period of 60 min, with 90% of the parent compound remaining. (±)-1 was, however, susceptible to phase I and phase II hepatic metabolism. In mouse liver microsomal incubations in the presence of NADPH, which measures phase I metabolism, 21% of the parent (±)-1 remained after 60 min. Notably, microsomal stability in rats was increased to 37% of the parent (±)-1 remaining after 60 min, under the same assay conditions, suggesting higher stability in this species. In mouse microsomes fortified with UDPGA, which measures phase II metabolism, 55% of the parent (±)-1 remained after 60 min of incubation. No metabolism was observed in microsomes without the cofactors, showing their specificity to CYP- and UGT-dependent instability, respectively.
Table 4. Metabolic Stability Results for (±)-1 in Mouse Plasma and Liver Microsomes Compared to Positive Control Compounds Procaine (Plasma), Testosterone (Phase I), and 4-Methyl Umbelliferone (Phase II).
| (±)-1 |
positive
control compounds |
||||||
|---|---|---|---|---|---|---|---|
| time | plasma | phase I | phase II | negative control | procaine | testosterone | 4-methyl umbelliferone |
| 0 | 100% | 100% | 100% | 100% | 100% | 100% | 100% |
| 15 | 108% | 97% | 108% | ||||
| 30 | 103% | 34% | 64% | 103% | |||
| 60 | 90% | 21% | 55% | 90% | 9.70% | 0% | 3.10% |
3.2.2. Effects of i.p. (±)-, (R)-, and (S)-1 on PR Responding for METH in Rats
Concurrent with the pharmacokinetic studies described above, (±)-, (R)-, and (S)-1 were evaluated in a rat yawning model131 to verify target engagement and determine an effective dose range for further behavioral evaluation. Although mouse yawning studies were initially attempted, it was discovered that D3R agonists do not induce yawning in mice, so that approach was abandoned. (±)-, (R)-, and (S)-1 each attenuated 7-OH DPAT-induced yawning in rats, as demonstrated by a rightward shift in the ascending limb of the dose–response curve; however, no clear evidence of enantioselectivity was observed (J.L. Katz and A.H. Newman, unpublished results). Further, this model proved to be difficult to use as an in vivo screen for numerous reasons, including tolerance to the D3R-agonist effect on yawning and significant variation across subjects. It also required a large quantity of test drug to obtain full dose–response curves. Hence, we discontinued testing compounds in the rat yawning model as an in vivo diagnostic and instead used binding affinities as a guide to dose ranges evaluated in the subsequent rodent studies.
Using methods described previously,135,1361 was evaluated for its effects in two rat models of drug-taking and drug-seeking behavior. Figure 2A shows METH self-administration in rats responding under a PR schedule of reinforcement, comparing baseline performance against pretreatments with vehicle or the enantiomers of 1; Figure 2B presents these data normalized to each individual animal’s baseline performance. Separate one-way repeated-measures ANOVAs were conducted on number of injections, which represents PR break points (i.e., the final ratio completed). Compound 1 significantly and dose-dependently reduced METH injections [(±)-1: F2,18: 19.36, p < 0.0001, Dunnett’s test: vehicle vs 3.0 mg/kg p > 0.05, vehicle vs 10 mg/kg p < 0.001; (R)-1: F2,18: 11.15, p < 0.001, Dunnett’s test: vehicle vs 3.0 mg/kg p < 0.01, vehicle vs 10 mg/kg p < 0.01; (S)-1: F2,18: 15.19, p < 0.0001, Dunnett’s test: vehicle vs 3.0 mg/kg p > 0.05, vehicle vs 10 mg/kg p < 0.001]. Similarly, one-way repeated-measures ANOVA revealed that 1 significantly reduced PR break points when normalized to baseline responding [(±)-1: F2,18: 16.67, p < 0.001, Dunnett’s test: vehicle vs 3.0 mg/kg p > 0.05, vehicle vs 10 mg/kg p < 0.001; (R)-1: F2,18: 13.16, p = 0.003, Dunnett’s test: vehicle vs 3.0 mg/kg p < 0.001, vehicle vs 10 mg/kg p < 0.001; (S)-1: F2,18: 25.16, p < 0.001, Dunnett’s test: vehicle vs 3.0 mg/kg p < 0.01, vehicle vs 10 mg/kg p < 0.001]. Although (R)-1 and (S)-1 vary in their in vitro affinity and selectivity for D3R (Table 1), the doses studied did not indicate a clear differential in vivo potency between enantiomers. Hence, only (±)-1 was tested further.
Figure 2.

Effects of i.p. vehicle or 1 (racemic and enantiomers) on PR self-administration of METH. Data are presented as mean ± SEM; * p < 0.05, ** p < 0.01, *** p < 0.001. Statistics in panel A refer to number of METH injections (right axis) compared to vehicle; statistics in panel B refer to break point values (last ratio completed), normalized to baseline responding, compared to vehicle.
PR drug self-administration paradigms are often used to evaluate the reinforcing efficacy of drugs of abuse.137−139 Because the response requirement to receive the next injection increases continuously, the break point (the last completed ratio) becomes a repeatable measure of the relative reinforcing value of a particular commodity (including drug dose). The PR procedure has been used in a variety of human research efforts, including some clinical populations, but has not been well-evaluated in measuring the therapeutic outcomes of pharmacological interventions in applied settings.140 However, recent findings that intranasal cocaine or intravenous opioids are effective reinforcers under PR schedules in humans141−143 suggest potential future translational utility in measuring the therapeutic efficacy of novel antiaddiction medications. Consistent with the results of other D3R antagonists, 1 effectively reduced the number of METH injections and the break points of treated rats. These findings suggest that 1 lowered the reinforcing efficacy of METH, providing further evidence that D3R signaling contributes to the rewarding value of psychostimulant drugs.
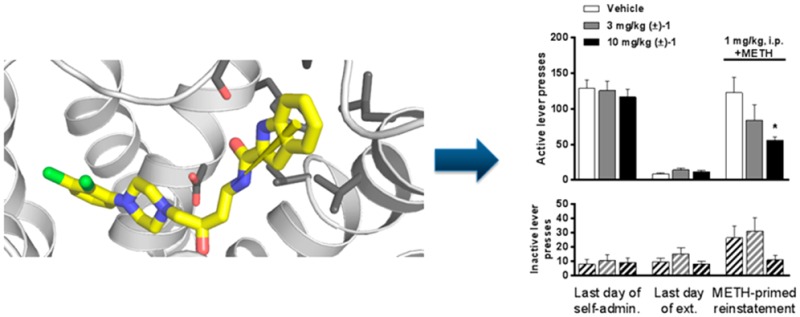
3.2.3. Effects of i.p. (±)-1 on Drug-Primed Reinstatement Responding for METH in Rats
Figure 3A illustrates the acquisition and extinction of METH self-administration. Acquisition of self-administration began with a high dose (0.1 mg/kg/inj) of METH using an FR 1 schedule of reinforcement over five daily training sessions, followed by seven daily training sessions with a lower dose (0.05 mg/kg/inj) using an FR 2 schedule. This format allowed rapid acquisition of drug taking with robust levels of lever pressing. Following seven daily extinction-training sessions, in which lever presses resulted in no drug delivery or cue presentation, the effect of (±)-1 on METH-primed reinstatement was tested (Figure 3B). One-way ANOVA revealed that (±)-1 significantly reduced METH-primed reinstatement responding on the active lever [F2,31: 3.817, p = 0.033, Dunnett’s test: vehicle vs 3.0 mg/kg p > 0.05, vehicle vs 10 mg/kg p < 0.05] but did not significantly alter inactive lever responding [F2,31: 2.12, p > 0.13, Dunnett’s test: vehicle vs 3.0 mg/kg p > 0.05, vehicle vs 10 mg/kg p > 0.05].
Figure 3.

(A) Acquisition of METH self-administration and extinction of self-administration. (B) Effects of i.p. vehicle or (±)-1 on METH-primed reinstatement of METH-seeking behavior. Data are presented as mean ± SEM; * p < 0.05 compared to vehicle.
Reinstatement of drug-seeking behavior, via stressors, drug-associated cues or contexts, or acute exposure to the self-administered drug or related drugs, is a widely used model of drug craving and relapse (for review, see refs (144−146); see also ref (147)). As with previous D3R-selective antagonists and partial agonists (e.g., 2,1183,30 and 4(31)), (±)-1 showed a dose-dependent reduction in METH-primed reinstatement responding. These results add to the growing evidence that D3R signaling plays an important role in the neurocircuitry that drives relapse to drug taking and that D3R-selective compounds could be useful therapeutics in the prevention of relapse. However, it should be noted here that assessing a medication that may prevent relapse requires that the subject abstain from drug taking for a period of time. If the D3R antagonists do not curb drug taking, then abstinence may be very difficult to achieve and hence clinical assessment for this therapeutic benefit will be challenging. It has been proposed that a “Rosetta Stone approach” be taken to developing drugs for addiction, wherein the addiction cycle is taken into account for a pharmacological strategy.148 In the case of D3R antagonists, additional treatment strategies may need to be in place to help subjects attain abstinence before administering D3R-selective therapeutics to prevent relapse.
3.2.4. Effects of Acute Intravenous (±)-1 on Food–Cocaine Choice in Cynomolgus Monkeys
In addition to the rodent model studies, male cynomolgus monkeys were trained to self-administer cocaine under a concurrent schedule in which food was the alternative reinforcer. Under these experimental conditions, complete cocaine self-administration dose–response curves were determined each session (see ref (112) and Supporting Information for methods). As shown in Figure 4, in all but one monkey, (±)-1 either shifted the cocaine choice curve to the left (4 subjects) or had no effect (2 subjects). In one monkey (C-6529), a decrease in choice of the highest cocaine dose was observed. When group data were analyzed, there was a main effect of cocaine dose (F4,30 = 81.8, p < 0.0001) but no main effect of drug pretreatment and no interaction. Posthoc multiple comparisons indicated a significant increase in cocaine choice when 0.01 mg/kg cocaine was available as the alternative to food. There were no significant effects of the highest dose tested (5.6 mg/kg) (±)-1 on any of the dependent variables of secondary interest (Table 5).
Figure 4.

Effects of i.v. vehicle or (±)-1 on cocaine choice in seven monkeys individually and as a group (lower right panel). Ordinates, percent of total reinforcers earned that resulted in cocaine injections. Abscissae, dose of cocaine (mg/kg) available as an alternative to a food pellet. Data in lower right panel represent mean ± SEM.
Table 5. Secondary Dependent Variables in Food–Drug Choice Studies after Administration of Vehicle or (±)1.
| cynomolgus
monkeys | ||
|---|---|---|
| vehicle | 5.6 mg/kg (±)-1 | |
| food reinforcers | 30.0 ± 1.9 | 25.5 ± 2.4 |
| cocaine injections | 13.4 ± 1.3 | 15.5 ± 2.2 |
| total reinforcers | 43.4 ± 2.0 | 40.3 ± 1.7 |
| intake (mg/kg) | 0.77 ± 0.11 | 0.63 ± 0.11 |
| choice ED50 | 0.023 ± 0.006 | 0.012 ± 0.002 |
| rhesus
monkeys | ||
|---|---|---|
| baseline | 3.0 mg/kg (±)-1 | |
| food reinforcers | 19.9 ± 2.3 | 14.4 ± 2.8 |
| METH injections | 14.8 ± 2.8 | 15.2 ± 1.9 |
| total reinforcers | 34.7 ± 0.8 | 29.7 ± 4.5 |
| intake (mg/kg) | 1.08 ± 0.11 | 1.08 ± 0.23 |
| choice ED50 | 0.024 ± 0.011 | 0.011 ± 0.003 |
Although (±)-1 failed to reduce cocaine choice and increase food choice, which would be a result more congruent with a potential therapeutic, there are several testable hypotheses generated from these results. Both concurrent food–drug choice and PR responding are considered to be models of reinforcing strength.149 However, the effects of 1 are different in these two models, suggesting that they are measuring different aspects of self-administration (see ref (150) for an example). One could hypothesize that drugs that share discriminative stimulus effects with cocaine would shift the cocaine choice dose–response curve to the left, as seen with 1. However, in preliminary findings from monkeys trained to discriminate cocaine, 1 does not substitute for cocaine (M.A. Nader and A.H. Newman, unpublished results). Several investigators have shown that D2/D3R antagonists can shift the cocaine choice dose–response curve to the left (e.g., refs (112, 151, and 152)). It is also possible that the differences noted are due to drug and/or species differences (see ref (153)) or to differences in drug histories between the rat PR study and the monkey food–cocaine choice study. On the basis of our experience and the literature, D3R antagonists are most effective in models of relapse, and perhaps this is the clinical end point that should be singly targeted with this class of drugs. Nevertheless, from a basic research standpoint, additional behavioral models of addiction must be explored to fully elucidate the role of D3R in the development of addiction. Further, as described previously, neurobiological changes that occur upon chronic exposure to psychostimulant drugs and also during abstinence must also be quantified to better ascertain optimal timing of administration of D3R antagonists or partial agonists for potential therapeutic benefit. One obvious limitation to the present study is that 5.6 mg/kg was the highest dose that could be solubilized and administered intravenously to these subjects. It is certainly possible that the level of D3R occupancy required to observe behavioral effects was not achieved or sustained and future D3R occupancy determination, potentially using PET imaging or another biomarker, will need to be assessed.
3.2.5. Effects of Acute or Repeated Intravenous (±)-1 on Food–METH Choice in Rhesus Monkeys
In order to more fully model the clinical situation in which chronic drug treatments are likely to be employed, a follow-up study was conducted in rhesus monkeys that self-administered METH in which (±)-1 was administered for 5 consecutive days. In two of three monkeys, 5 day treatment with 3.0 mg/kg (±)-1 (i.v.) produced a leftward shift of the METH choice curve (Figure 5). Effects of acute treatment had a more pronounced effect in one monkey (R-1690), suggesting tolerance developed in this subject. When grouped data were analyzed, there were no statistically significant effects of 3.0 mg/kg (±)-1 on any of the dependent variables of secondary interest (Table 5). In addition to the possibility of suboptimal D3R occupancy, pharmacokinetic studies were conducted only in mice. Clearly, the pharmacokinetics and metabolism of 1 in monkeys may differ. These experimental sessions are approximately 2 h in duration, and drug self-administration doses are presented in ascending order. If the half-life of 1 is short in monkeys, then it is possible that there is insufficient drug blocking D3Rs later in the session when higher cocaine doses are available.
Figure 5.

Effects of acute and chronic administration of (±)-1 (i.v.) on METH choice in three rhesus monkeys (mean ± SEM of days 3–5 of daily treatment). Ordinates, percent of total reinforcers earned that resulted in METH injections. Abscissae, dose of METH (mg/kg) available as an alternative to a food pellet.
An additional possibility is that the elevated DA from cocaine or METH is displacing 1 and that D3R antagonists are less effective in models of self-administration compared to reinstatement models or under conditions in which treatment occurs in the absence of psychostimulant availability. Indeed, a PET imaging study in rhesus monkeys with [18F]LS-3-134 suggested a high level of competition between this D3R-selective PET imaging agent and endogenous dopamine in the absence of a psychostimulant drugs.154 Thus, in the presence of psychostimulants, competing DA levels may render D3R antagonists ineffective. Future research is clearly needed to better understand the conditions under which D3R-selective compounds are effective in nonhuman primate models of psychostimulant addiction and which of these models is reliably translatable to human psychostimulant abusers.
4. Discussion
In radioligand competition binding experiments using [3H]N-methylspiperone and membranes from HEK293 cells expressing hD2R, hD3R, or hD4R, (±)-1 exhibits nearly 400- and 1300-fold D3R selectivity over D2R and D4R, respectively. This is more pronounced when considering the enantioselectivity of D3R for the R-enantiomer.5 The in vitro D3R selectivity of 1, particularly (R)-1, was an improvement over previous generations of 2,3-dichlorophenylpiperazinebutylarylcarboxamides (e.g., 3).25,75 As such, 1 and its enantiomers were some of the most D3R-selective ligands in our library and thus the tools we chose to test in vivo.
To evaluate the in vivo stability of 1, plasma pharmacokinetics (PK) was studied in mice, revealing plasma stability 0–1.5 h following i.v. administration of 1. Additionally, 1 was orally bioavailable with good brain penetration. Compound 1 was actively metabolized by liver microsomes, but it showed a terminal half-life of approximately 1–1.5 h following i.v. administration. It should be noted that metabolism and PK studies were done in mice and thus these parameters may be different across species. Clearly, PK studies need to be extended to nonhuman primates and brain imaging needs to be utilized to study pharmacodynamics of these compounds entering the brain and occupying D3R. Nevertheless, these pharmacokinetic parameters appeared to be promising for a translational candidate, justifying further exploration in both rat and nonhuman primate models of drug addiction.
Previous studies have demonstrated a trend in which D3R antagonists and partial agonists typically do not affect psychostimulant self-administration with low FR response requirements but attenuate drug taking in paradigms that increase the response requirement (e.g., high FR or PR models) or second-order schedules,29−31,126,155−157 suggesting that D3R antagonism does not affect the primary reinforcing effects of drugs but reduces the motivation to self-administer drugs.48,158 Consistent with that view is the success of D3R antagonists in rodent models of relapse, such as drug-, cue- and stress-induced reinstatement.28−31,157,159 Compound 1 significantly reduced PR responding for METH, as measured by break point. There was no clear difference in potency seen between the (R)- and (S)-enantiomers; however, this study did not test a sufficient range of doses to reliably determine whether one enantiomer was more potent than the other in reducing PR break point in rats. Racemic 1 significantly attenuated METH-primed reinstatement as well. Overall, (±)-1 (3.0–10 mg/kg, i.p.) was efficacious in two rat models of drug self-administration and drug seeking (i.e., reinstatement).
In the food–cocaine choice model, cynomolgus monkeys were allowed to choose either a food reward or an i.v. cocaine infusion, with the cocaine dose varied across trials. Overall, 1 had inconsistent effects; when all data were combined, the general effect that emerged was that 1 shifted the dose–response curve of cocaine to the left, i.e., it potentiated cocaine choice over food. In the food–METH choice model using rhesus monkeys, the result was similar: 1 tended to shift the METH dose–response to the left or have no effect. One subject (R-1691) appeared to develop tolerance to the effect of 1. The results of these studies may not appear to support further investigation. However, it must be noted that the food–drug choice model is not directed toward relapse, which the rodent studies with 1 and other D3R antagonists most support. Further evaluation of 1 and newer generation D3R antagonists and partial agonists are underway in nonhuman primate reinstatement to drug seeking models to determine if the rodent studies can be replicated with these agents in primates with a history of chronic drug taking. Chronic dosing with the treatment agent can also be evaluated in the nonhuman primates, which will also inform human studies.
In contrast to D3R antagonists such as (±)-1 and 7, D3R partial agonists pose several interesting questions. For example, it is known that chronic administration of receptor agonists commonly results in receptor downregulation, whereas chronic administration of receptor antagonists commonly results in receptor upregulation. With regard to partial agonists, a recent PET imaging study in cynomolgus monkeys found that chronic aripiprazole, a D2R partial agonist, produced different effects on D2R binding potential: it appeared to increase binding when the subject had, on average, low D2R availability and decreased binding potential in monkeys with higher average D2R availability.160 Thus, depending on the long-term consequences of cocaine or METH abuse on receptor availability, the effects of receptor partial agonists may be different than antagonists. There are behavioral nuances as well. Recently, it was reported that the effects of aripiprazole on cocaine–food choice varied depending on whether monkeys were given access to cocaine during aripiprazole treatment.112 Thus, it remains possible that in nonhuman primates and humans D3R partial agonists would be more effective in a setting in which cocaine or METH were not available (e.g., residential treatment facility), at least in the early stages of treatment. Finally, there are ongoing studies to evaluate the effects of antagonists and partial agonists on peripheral D3Rs, particularly in regard to potential synergistic effects in combination with cocaine- and METH-induced elevations in heart rate and blood pressure, a key treatment risk that must be thoroughly evaluated and thoughtfully considered when describing potential treatment compounds.
5. Overall Conclusions and Our Perspective
The D3R remains an enigmatic target for addiction pharmacotherapy. The translation between rodent and nonhuman primate addiction models and again between nonhuman primate addiction models and humans in a clinical setting is fraught with pitfalls. Predecessors of 1 and 7 have failed to successfully clear the hurdles that lead to successful clinical trials. The partial agonist 8 was efficacious in rodent and primate models of addiction, but it failed in clinical trials for cocaine abuse.118,161,162 Compound 4 looked promising in rodent models of drug taking and drug seeking, but poor bioavailability and short half-life in primates led GlaxoSmithKline to halt further development of the compound.118 The recent failure of buspirone, as described above, may also bode poorly for continuing to pursue this target.
Compound 1 will likely remain a research tool for basic and preclinical studies. Its inconsistent behavioral profile in nonhuman primates thus far may be directly related to a poor pharmacokinetic profile, lack of target engagement at the doses and time points tested, or simply lack of efficacy in these models. Future studies to parse out the contributing factors to its demise, before reaching the clinic, will be used for the next generation of drug design and choice of behavioral models. Nevertheless, newer generation agents in our laboratory and others are already making their way forward in development and will soon be evaluated in these and other models of drug abuse and neuropsychiatric disorders. Only continued drug design using structure–activity relationships and in vivo data will ultimately lead us to successfully identify D3R-selective compounds that have appropriate drug-like properties. So far, this goal remains to be achieved.
A key concern in the field of addiction medicine and indeed all neuropsychiatric drug discovery is the effectiveness of animal models to evaluate preclinical candidates.163,164 In one of the first studies using D3R partial agonists, it was reported that the effects of 8 were different in models of drug seeking compared to drug self-administration.70 The study of D3R compounds has provided a better understanding of the importance of models that incorporate long-term cocaine and METH self-administration and a clearer perception of the importance of behavioral phenotype. For example, it appears that the behavioral pharmacology of D3R partial agonists are different depending on whether the subjects are drug-naïve or have a drug history123 and, for D3R agonists, whether the drug is cocaine or METH.124 Moreover, individual differences in the efficacy of D3R antagonists to reduce drug self-administration have been found involving acute vs chronic treatment,165 drug history,165 and social rank.107 These findings are reflected in human imaging studies involving the D3R-preferential PET ligand [11C]PHNO166 in cocaine and METH abusers.42−44,167
There are important temporal differences in psychostimulant-induced D3R dysregulation that should be taken into account for the D3R’s therapeutic potential. For instance, studies designed to examine the time course of changes in D3R binding and sensitivity observe D3R upregulation after prolonged abstinence (30–45 days after the last cocaine administration) but not during self-administration or within 7 days of abstinence.133,168,169 Furthermore, the subjects in some of the recent human imaging studies demonstrating D3R upregulation following chronic cocaine or METH use were scanned on average 18.5 ± 20.542 and 50.1 ± 64.443 days, respectively, after their most recent drug use, which appear to reflect substantial periods of abstinence. Therefore, although it is clear that D3R expression is significantly altered in drug abusers, perhaps there is a specific window of time in the addiction cycle in which the therapeutic potential of D3R antagonists and partial agonists could be maximally beneficial.
The difficulties of treating drug addiction are compounded by the high incidence of comorbid neuropsychiatric disorders with substance use disorders, including depression, anxiety, ADHD, and schizophrenia.170−173 Further complicating the search for translational medications for addiction is the complexity of D3R receptor signaling and genetics. There is not yet a clear understanding of the role of D1R–D3R heteromers in the striatum or whether receptor heteromers are a viable target for drug development. We have recently speculated on the possible role of the D1R–D3R heteromer in drug memory reconsolidation, and future studies in this area are of great interest.174 Additionally, there are a number of studies linking various D3R gene polymorphisms with neuropsychiatric disorders. In particular, rs6280, which encodes the functional missense mutation Ser9Gly, may enhance reward-related DA release.175 This particular polymorphism has been associated with nicotine dependence,176 alcohol dependence,177 and early onset heroin dependence.178 Further study will be necessary to determine the significance of receptor heteromerization and genetic variation at D3R in the development of substance use disorders or in the response to an antiaddiction pharmacotherapeutic.
Given these considerations, it is important that the preclinical and clinical studies showing negative results with D3R antagonists and partial agonists thus far not be used to prematurely rule out the utilization of these pharmacotherapies for addiction. Instead, these studies should be used to guide future efforts in designing more appropriate behavioral models to evaluate D3R compounds and determining the subsets of individuals in which D3R pharmacotherapies can be most effective. To reiterate, assessing a medication that may have its greatest utility in the prevention of relapse, as may be the case for D3R pharmacotherapy, requires that clinical subjects abstain from drug taking for a period of time. On the basis of preclinical results thus far, D3R antagonists do not effectively reduce active drug taking, but they do reduce relapse-like behavior. Therefore, if D3R antagonists are best suited for a role as a relapse-prevention treatment, then they will likely only be successful in the framework of a larger treatment program in which subjects have already attained abstinence. Trials designed to test D3R antagonists in patients actively using drugs may not see any attenuation in drug taking and would not be well-suited to evaluate the abstinence-maintaining therapeutic potential of this compound class. Hence, lessons learned from the buspirone case described in Section 2.3 should be used in future psychostimulant medication development.
This Perspective highlights the importance of developing selective compounds to better understand the role of D3R in addiction. It is our opinion that repurposing drugs that are clinically available has utility in guiding future research. However, the use of drugs with D3R affinity, but also many other targets (e.g., buspirone), should not be used to exemplify the pharmacology, biology, and physiology related to this important target. Recently, a new D3R-preferential partial agonist, 23 (BP1.4979 in Chart 3, communicated by Jean-Charles Schwartz, Dopamine 2013 meeting), was introduced by Bioprojet, and a clinical trial on smoking cessation has begun (ClinicalTrials.gov identifier NCT01785147). Although data remain unpublished, presumably this compound has been vetted through all of the in vitro and in vivo assays that Pharma has at its disposal (as compared to academic drug discovery programs such as ours) to be considered for translation to human studies. The fact that 23 is a partial agonist is of further interest and will provide answers to some of the questions regarding D3R efficacy posed above. Results of these clinical investigations will be of great interest to our community of addiction researchers and will very likely determine if the D3R remains a target to pursue for smoking cessation as well as other substance use disorders. One final thought is that D3R antagonists and partial agonists may not prove to be efficacious or safe for treatment of cocaine or METH addiction but may still hold promise for addictions to other substances, such as nicotine, opiates (including prescription pain killers, e.g., oxycodone), alcohol, food and Δ9-tetrahydrocannabinol (THC), the active ingredient in marijuana. Hence, it is our perspective that the development of selective D3R ligands, with varying efficacies, as research tools that are active and metabolically stable in vivo, can help to elucidate underpinnings of addiction and other neuropsychiatric disorders. Hence, future research and development of these agents toward these therapeutic end points remains vitally important. Further, developing animal models that can translate preclinical research is challenging and yet key to finding pharmacotherapeutic treatments for this underserved patient population.
Acknowledgments
We acknowledge Jianjing Cao and Drs. Peter Grundt, Ashwini K. Banala, and Vivek Kumar for synthetic contributions to this project. We acknowledge Catherine Schweppe and Caitlin Burzynski for assistance in obtaining binding data. We acknowledge Drs. Barbara Slusher and Rana Rais for the pharmacokinetic analyses and Michael Coller and Michelle Bell for excellent assistance in collecting data from monkeys. This work was funded by the NIDA-IRP and grants DA012460 and DA010584. W.S.J. is supported by T32 AA-007565.
Glossary
Abbreviations Used
- D3R
dopamine D3 receptor
- DA
dopamine
- METH
methamphetamine
- SAR
structure–activity relationships
- DAT
dopamine transporter
- GPCR
G protein-coupled receptors
- cAMP
cyclic AMP
- PR
progressive ratio
- FR
fixed ratio
- Cmax
peak plasma concentration
- t1/2
plasma half-life
- F%
bioavailability fraction
- PK
pharmacokinetics
Biographies
Thomas M. Keck received his doctorate in Physiology and Pharmacology in 2009 from Oregon Health & Science University in Portland, Oregon. He did his postdoctoral studies at the National Institute on Drug Abuse (NIDA-IRP), NIH, in the Medicinal Chemistry Section, Molecular Targets and Medications Discovery Research Branch, under the mentorship of Dr. Newman. In 2014, he became an Assistant Professor at Rowan University in Glassboro, New Jersey, with joint appointments in the Department of Chemistry & Biochemistry and the Department of Biomedical & Translational Sciences. His research focuses on the evaluation of novel ligands in preclinical models of neuropsychiatric disorders.
William S. John received his B.S. in Biology at the University of North Carolina at Greensboro in 2010. He is currently a doctoral student in the Department of Physiology & Pharmacology at Wake Forest School of Medicine under the mentorship of Dr. Nader. His dissertation research focuses on the role of DA D3 receptors in mediating the behavioral effects of substance abuse in nonhuman primate models of cocaine and METH self-administration, with the goal of identifying novel treatment strategies.
Paul W. Czoty received his doctorate in Neuroscience from Emory University in Atlanta, Georgia, in 2000. After his postdoctoral work at Harvard Medical School’s McLean Hospital in Belmont, Massachusetts, he went to the Wake Forest School of Medicine, where he is an Associate Professor of Physiology & Pharmacology. He has written over 50 journal articles describing research using nonhuman primate models of substance abuse.
Michael A. Nader received his doctorate in Experimental Psychology from the University of Minnesota, Minneapolis, Minnesota, in 1985. After his postdoctoral work at Uniformed Services University of the Health Sciences in Bethesda, Maryland, he went to the University of Chicago, where he was a member of the Research Faculty. He has been at Wake Forest University School of Medicine since 1992, where he is a Professor of Physiology & Pharmacology and Radiology. He has over 140 journal articles describing preclinical models of drug abuse.
Amy Hauck Newman received her doctorate in Medicinal Chemistry from the Medical College of Virginia, Virginia Commonwealth University. After postdoctoral studies at the National Institute on Diabetes, Digestive and Kidney Diseases, National Institutes of Health (NIH), and a brief time at Walter Reed Army Institute of Research, she joined the Intramural Research Program of the National Institute on Drug Abuse (NIDA-IRP), NIH. She is currently a Senior Investigator, Deputy Scientific Director, and Chief of the Molecular Targets and Medications Discovery Research Branch and the Medicinal Chemistry Section. She has coauthored more than 230 original articles and reviews on the design, synthesis, and pharmacological evaluation of CNS active agents, with an emphasis on selective ligands for the dopaminergic system, as leads toward potential treatment medications for psychostimulant abuse.
Supporting Information Available
Detailed methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Newman A. H.; Grundt P.; Nader M. A. Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J. Med. Chem. 2005, 48, 3663–3679. [DOI] [PubMed] [Google Scholar]
- Chien E. Y.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T. M.; Burzynski C.; Shi L.; Newman A. H. Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv. Pharmacol. 2014, 69, 267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z.; Hou T.; Li Y. Structure-based drug design for dopamine D3 receptor. Comb. Chem. High Throughput Screening 2012, 15, 775–791. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Grundt P.; Cyriac G.; Deschamps J. R.; Taylor M.; Kumar R.; Ho D.; Luedtke R. R. N-(4-(4-(2,3-Dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem. 2009, 52, 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto S.; Wise R. A. Mapping of chemical trigger zones for reward. Neuropharmacology 2004, 47, 190–201. [DOI] [PubMed] [Google Scholar]
- Koob G. F.; Volkow N. D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo S. J.; Nestler E. J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 2013, 14, 609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow N. D.; Wang G. J.; Fowler J. S.; Tomasi D.; Telang F. Addiction: beyond dopamine reward circuitry. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 15037–15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt K. C.; Reith M. E. Regulation of the dopamine transporter: aspects relevant to psychostimulant drugs of abuse. Ann. N.Y. Acad. Sci. 2010, 1187, 316–340. [DOI] [PubMed] [Google Scholar]
- Volkow N. D.; Wang G. J.; Fowler J. S.; Gatley S. J.; Logan J.; Ding Y. S.; Dewey S. L.; Hitzemann R.; Gifford A. N.; Pappas N. R. Blockade of striatal dopamine transporters by intravenous methylphenidate is not sufficient to induce self-reports of “high”. J. Pharmacol. Exp. Ther. 1999, 288, 14–20. [PubMed] [Google Scholar]
- Volkow N. D.; Ding Y. S.; Fowler J. S.; Wang G. J.; Logan J.; Gatley J. S.; Dewey S.; Ashby C.; Liebermann J.; Hitzemann R.; Wolf A. P. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch. Gen. Psychiatry 1995, 52, 456–463. [DOI] [PubMed] [Google Scholar]
- Ahlgren-Beckendorf J. A.; Levant B. Signaling mechanisms of the D3 dopamine receptor. J. Recept. Signal Transduction Res. 2004, 24, 117–130. [DOI] [PubMed] [Google Scholar]
- Sidhu A.; Niznik H. B. Coupling of dopamine receptor subtypes to multiple and diverse G proteins. Int. J. Dev. Neurosci. 2000, 18, 669–677. [DOI] [PubMed] [Google Scholar]
- Marcellino D.; Ferre S.; Casado V.; Cortes A.; Le Foll B.; Mazzola C.; Drago F.; Saur O.; Stark H.; Soriano A.; Barnes C.; Goldberg S. R.; Lluis C.; Fuxe K.; Franco R. Identification of dopamine D1–D3 receptor heteromers. Indications for a role of synergistic D1–D3 receptor interactions in the striatum. J. Biol. Chem. 2008, 283, 26016–26025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart X.; Navarro G.; Moreno E.; Yano H.; Cai N. S.; Sanchez-Soto M.; Kumar-Barodia S.; Naidu Y. T.; Mallol J.; Cortes A.; Lluis C.; Canela E. I.; Casado V.; McCormick P. J.; Ferre S. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: the dopamine D1–D3 receptor heterotetramer. Mol. Pharmacol. 2014, 86, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini C.; Savoia P.; Bono F.; Tallarico P.; Missale C. The D3 dopamine receptor: From structural interactions to function. Eur. Neuropsychopharmacol. 2014, 10.1016/j.euroneuro.2014.1011.1021. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Blaylock B. L.; Nader M. A.; Bergman J.; Sibley D. R.; Skolnick P. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem. Pharmacol. 2012, 84, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidbreder C. A.; Newman A. H. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N.Y. Acad. Sci. 2010, 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F. Recent advances in the development of dopamine D3 receptor antagonists: a medicinal chemistry perspective. ChemMedChem 2011, 6, 1152–1162. [DOI] [PubMed] [Google Scholar]
- Micheli F.; Heidbreder C. Dopamine D3 receptor antagonists: a patent review (2007–2012). Expert Opin. Ther. Pat. 2013, 23, 363–381. [DOI] [PubMed] [Google Scholar]
- Ye N.; Neumeyer J. L.; Baldessarini R. J.; Zhen X.; Zhang A. Update 1 of: Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders. Chem. Rev. 2013, 113, PR123–178. [DOI] [PubMed] [Google Scholar]
- Michino M.; Donthamsetti P.; Beuming T.; Banala A.; Duan L.; Roux T.; Han Y.; Trinquet E.; Newman A. H.; Javitch J. A.; Shi L. A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol. Pharmacol. 2013, 84, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthan S.; Saini S. K.; Zhou G.; Hobrath J. V.; Padmalayam I.; Zhai L.; Bostwick J. R.; Antonio T.; Reith M. E.; McDowell S.; Cho E.; McAleer L.; Taylor M.; Luedtke R. R. Design, synthesis, and structure–activity relationship studies of a series of [4-(4-carboxamidobutyl)]-1-arylpiperazines: insights into structural features contributing to dopamine D3 versus D2 receptor subtype selectivity. J. Med. Chem. 2014, 57, 7042–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundt P.; Carlson E. E.; Cao J.; Bennett C. J.; McElveen E.; Taylor M.; Luedtke R. R.; Newman A. H. Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J. Med. Chem. 2005, 48, 839–848. [DOI] [PubMed] [Google Scholar]
- Banala A. K.; Levy B. A.; Khatri S. S.; Furman C. A.; Roof R. A.; Mishra Y.; Griffin S. A.; Sibley D. R.; Luedtke R. R.; Newman A. H. N-(3-Fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J. Med. Chem. 2011, 54, 3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A. H.; Beuming T.; Banala A. K.; Donthamsetti P.; Pongetti K.; LaBounty A.; Levy B.; Cao J.; Michino M.; Luedtke R. R.; Javitch J. A.; Shi L. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012, 55, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J. G.; Newman A. H.; Gardner E. L.; Ashby C. R. Jr.; Heidbreder C. A.; Pak A. C.; Peng X. Q.; Xi Z. X. Acute administration of SB-277011A, NGB 2904, or BP 897 inhibits cocaine cue-induced reinstatement of drug-seeking behavior in rats: role of dopamine D3 receptors. Synapse 2005, 57, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z. X.; Newman A. H.; Gilbert J. G.; Pak A. C.; Peng X. Q.; Ashby C. R. Jr.; Gitajn L.; Gardner E. L. The novel dopamine D3 receptor antagonist NGB 2904 inhibits cocaine’s rewarding effects and cocaine-induced reinstatement of drug-seeking behavior in rats. Neuropsychopharmacology 2006, 31, 1393–1405. [DOI] [PubMed] [Google Scholar]
- Higley A. E.; Spiller K.; Grundt P.; Newman A. H.; Kiefer S. W.; Xi Z. X.; Gardner E. L. PG01037, a novel dopamine D3 receptor antagonist, inhibits the effects of methamphetamine in rats. J. Psychopharmacol. 2011, 25, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley A. E.; Kiefer S. W.; Li X.; Gaal J.; Xi Z. X.; Gardner E. L. Dopamine D(3) receptor antagonist SB-277011A inhibits methamphetamine self-administration and methamphetamine-induced reinstatement of drug-seeking in rats. Eur. J. Pharmacol. 2011, 659, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor P. G.; Sokol R. J.; D’Onofrio G. Addiction medicine: the birth of a new discipline. JAMA Int. Med. 2014, 174, 1717–1718. [DOI] [PubMed] [Google Scholar]
- Le Houezec J.; Aubin H. J. Pharmacotherapies and harm-reduction options for the treatment of tobacco dependence. Expert Opin. Pharmacother. 2013, 14, 1959–1967. [DOI] [PubMed] [Google Scholar]
- Jonas D. E.; Amick H. R.; Feltner C.; Bobashev G.; Thomas K.; Wines R.; Kim M. M.; Shanahan E.; Gass C. E.; Rowe C. J.; Garbutt J. C. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 2014, 311, 1889–1900. [DOI] [PubMed] [Google Scholar]
- Raisch D. W.; Fye C. L.; Boardman K. D.; Sather M. R. Opioid dependence treatment, including buprenorphine/naloxone. Ann. Pharmacother. 2002, 36, 312–321. [DOI] [PubMed] [Google Scholar]
- Fudala P. J.; Bridge T. P.; Herbert S.; Williford W. O.; Chiang C. N.; Jones K.; Collins J.; Raisch D.; Casadonte P.; Goldsmith R. J.; Ling W.; Malkerneker U.; McNicholas L.; Renner J.; Stine S.; Tusel D.; Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N. Engl. J. Med. 2003, 349, 949–958. [DOI] [PubMed] [Google Scholar]
- Koob G. F. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction 2006, 101, 23–30. [DOI] [PubMed] [Google Scholar]
- Staley J. K.; Mash D. C. Adaptive increase in D3 dopamine receptors in the brain reward circuits of human cocaine fatalities. J. Neurosci. 1996, 16, 6100–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal D. M.; Moraes C. T.; Mash D. C. Up-regulation of D3 dopamine receptor mRNA in the nucleus accumbens of human cocaine fatalities. Brain Res. Mol. Brain Res. 1997, 45, 335–339. [DOI] [PubMed] [Google Scholar]
- Mash D. C.; Staley J. K. D3 dopamine and kappa opioid receptor alterations in human brain of cocaine-overdose victims. Ann. N.Y. Acad. Sci. 1999, 877, 507–522. [DOI] [PubMed] [Google Scholar]
- Le Foll B.; Collo G.; Rabiner E. A.; Boileau I.; Merlo Pich E.; Sokoloff P. Dopamine D3 receptor ligands for drug addiction treatment: update on recent findings. Prog. Brain Res. 2014, 211, 255–275. [DOI] [PubMed] [Google Scholar]
- Boileau I.; Payer D.; Houle S.; Behzadi A.; Rusjan P. M.; Tong J.; Wilkins D.; Selby P.; George T. P.; Zack M.; Furukawa Y.; McCluskey T.; Wilson A. A.; Kish S. J. Higher binding of the dopamine D3 receptor-preferring ligand [11C]-(+)-propyl-hexahydro-naphtho-oxazin in methamphetamine polydrug users: a positron emission tomography study. J. Neurosci. 2012, 32, 1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer D. E.; Behzadi A.; Kish S. J.; Houle S.; Wilson A. A.; Rusjan P. M.; Tong J.; Selby P.; George T. P.; McCluskey T.; Boileau I. Heightened D3 dopamine receptor levels in cocaine dependence and contributions to the addiction behavioral phenotype: a positron emission tomography study with [11C]-+-PHNO. Neuropsychopharmacology 2014, 39, 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer D.; Balasubramaniam G.; Boileau I. What is the role of the D3 receptor in addiction? A mini review of PET studies with [11C]-(+)-PHNO. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 52, 4–8. [DOI] [PubMed] [Google Scholar]
- Erritzoe D.; Tziortzi A.; Bargiela D.; Colasanti A.; Searle G. E.; Gunn R. N.; Beaver J. D.; Waldman A.; Nutt D. J.; Bani M.; Merlo-Pich E.; Rabiner E. A.; Lingford-Hughes A. In vivo imaging of cerebral dopamine D3 receptors in alcoholism. Neuropsychopharmacology 2014, 39, 1703–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B.; Wilson A. A.; Graff A.; Boileau I.; Di Ciano P. Recent methods for measuring dopamine D3 receptor occupancy in vivo: importance for drug development. Front. Pharmacol. 2014, 5, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidbreder C. Rationale in support of the use of selective dopamine D(3) receptor antagonists for the pharmacotherapeutic management of substance use disorders. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2013, 386, 167–176. [DOI] [PubMed] [Google Scholar]
- Heidbreder C. Selective antagonism at dopamine D3 receptors as a target for drug addiction pharmacotherapy: a review of preclinical evidence. CNS Neurol. Disord.: Drug Targets 2008, 7, 410–421. [DOI] [PubMed] [Google Scholar]
- Heidbreder C. A.; Andreoli M.; Marcon C.; Thanos P. K.; Ashby C. R. Jr.; Gardner E. L. Role of dopamine D3 receptors in the addictive properties of ethanol. Drugs Today 2004, 40, 355–365. [DOI] [PubMed] [Google Scholar]
- Le Foll B.; Goldberg S. R.; Sokoloff P. The dopamine D3 receptor and drug dependence: effects on reward or beyond?. Neuropharmacology 2005, 49, 525–541. [DOI] [PubMed] [Google Scholar]
- Sokoloff P.; Giros B.; Martres M. P.; Bouthenet M. L.; Schwartz J. C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [DOI] [PubMed] [Google Scholar]
- Gurevich E. V.; Joyce J. N. Distribution of dopamine D3 receptor expressing neurons in the human forebrain: comparison with D2 receptor expressing neurons. Neuropsychopharmacology 1999, 20, 60–80. [DOI] [PubMed] [Google Scholar]
- Searle G.; Beaver J. D.; Comley R. A.; Bani M.; Tziortzi A.; Slifstein M.; Mugnaini M.; Griffante C.; Wilson A. A.; Merlo-Pich E.; Houle S.; Gunn R.; Rabiner E. A.; Laruelle M. Imaging dopamine D3 receptors in the human brain with positron emission tomography, [11C]PHNO, and a selective D3 receptor antagonist. Biol. Psychiatry 2010, 68, 392–399. [DOI] [PubMed] [Google Scholar]
- Sun J.; Xu J.; Cairns N. J.; Perlmutter J. S.; Mach R. H. Dopamine D1, D2, D3 receptors, vesicular monoamine transporter type-2 (VMAT2) and dopamine transporter (DAT) densities in aged human brain. PLoS One 2012, 7, e49483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Barajas C.; Malik M.; Taylor M.; Neve K. A.; Mach R. H.; Luedtke R. R. Characterization of [3H]LS-3-134, a novel arylamide phenylpiperazine D3 dopamine receptor selective radioligand. J. Neurochem. 2014, 131, 418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan M. J.; Peglion J. L.; Vian J.; Rivet J. M.; Brocco M.; Gobert A.; Newman-Tancredi A.; Dacquet C.; Bervoets K.; Girardon S.; Jacques V.; Chaput C.; Audinot V. Functional correlates of dopamine D3 receptor activation in the rat in vivo and their modulation by the selective antagonist, (+)-S 14297:1. Activation of postsynaptic D3 receptors mediates hypothermia, whereas blockade of D2 receptors elicits prolactin secretion and catalepsy. J. Pharmacol. Exp. Ther. 1995, 275, 885–898. [PubMed] [Google Scholar]
- Cho D. I.; Zheng M.; Kim K. M. Current perspectives on the selective regulation of dopamine D(2) and D(3) receptors. Arch. Pharm. Res. 2010, 33, 1521–1538. [DOI] [PubMed] [Google Scholar]
- Kishi T.; Matsuda Y.; Iwata N.; Correll C. U. Antipsychotics for cocaine or psychostimulant dependence: systematic review and meta-analysis of randomized, placebo-controlled trials. J. Clin. Psychiatry 2013, 74, e1169–1180. [DOI] [PubMed] [Google Scholar]
- Veselinovic T.; Paulzen M.; Grunder G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev. Neurother. 2013, 13, 1141–1159. [DOI] [PubMed] [Google Scholar]
- Roman V.; Gyertyan I.; Saghy K.; Kiss B.; Szombathelyi Z. Cariprazine (RGH-188), a D(3)-preferring dopamine D(3)/D(2) receptor partial agonist antipsychotic candidate demonstrates anti-abuse potential in rats. Psychopharmacology 2013, 226, 285–293. [DOI] [PubMed] [Google Scholar]
- Gould R. W.; Duke A. N.; Nader M. A. PET studies in nonhuman primate models of cocaine abuse: translational research related to vulnerability and neuroadaptations. Neuropharmacology 2014, 84, 138–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszy J.; Laszlovszky I.; Gyertyan I. Dopamine D3 receptor antagonists improve the learning performance in memory-impaired rats. Psychopharmacology 2005, 179, 567–575. [DOI] [PubMed] [Google Scholar]
- Mugnaini M.; Iavarone L.; Cavallini P.; Griffante C.; Oliosi B.; Savoia C.; Beaver J.; Rabiner E. A.; Micheli F.; Heidbreder C.; Andorn A.; Merlo Pich E.; Bani M. Occupancy of brain dopamine D3 receptors and drug craving: a translational approach. Neuropsychopharmacology 2013, 38, 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S.; Gerretsen P.; Takeuchi H.; Caravaggio F.; Chow T.; Le Foll B.; Mulsant B.; Pollock B.; Graff-Guerrero A. The potential role of dopamine D(3) receptor neurotransmission in cognition. Eur. Neuropsychopharmacol. 2013, 23, 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix L. P.; Hows M. E.; Shah A. J.; Hagan J. J.; Heidbreder C. A. Selective antagonism at dopamine D3 receptors enhances monoaminergic and cholinergic neurotransmission in the rat anterior cingulate cortex. Neuropsychopharmacology 2003, 28, 839–849. [DOI] [PubMed] [Google Scholar]
- Millan M. J.; Di Cara B.; Dekeyne A.; Panayi F.; De Groote L.; Sicard D.; Cistarelli L.; Billiras R.; Gobert A. Selective blockade of dopamine D(3) versus D(2) receptors enhances frontocortical cholinergic transmission and social memory in rats: a parallel neurochemical and behavioural analysis. J. Neurochem. 2007, 100, 1047–1061. [DOI] [PubMed] [Google Scholar]
- Loiseau F.; Millan M. J. Blockade of dopamine D(3) receptors in frontal cortex, but not in sub-cortical structures, enhances social recognition in rats: similar actions of D(1) receptor agonists, but not of D(2) antagonists. Eur. Neuropsychopharmacol. 2009, 19, 23–33. [DOI] [PubMed] [Google Scholar]
- Xing B.; Meng X.; Wei S.; Li S. Influence of dopamine D3 receptor knockout on age-related decline of spatial memory. Neurosci. Lett. 2010, 481, 149–153. [DOI] [PubMed] [Google Scholar]
- Sokoloff P.; Leriche L.; Diaz J.; Louvel J.; Pumain R. Direct and indirect interactions of the dopamine D(3) receptor with glutamate pathways: implications for the treatment of schizophrenia. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2013, 386, 107–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla M.; Perachon S.; Sautel F.; Garrido F.; Mann A.; Wermuth C. G.; Schwartz J. C.; Everitt B. J.; Sokoloff P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 1999, 400, 371–375. [DOI] [PubMed] [Google Scholar]
- Hackling A. E.; Stark H. Dopamine D3 receptor ligands with antagonist properties. ChemBioChem 2002, 3, 946–961. [DOI] [PubMed] [Google Scholar]
- Boeckler F.; Gmeiner P. Dopamine D3 receptor ligands: recent advances in the control of subtype selectivity and intrinsic activity. Biochim. Biophys. Acta 2007, 1768, 871–887. [DOI] [PubMed] [Google Scholar]
- Micheli F.; Heidbreder C. Selective dopamine D3 receptor antagonists: a review 2001–2005. Recent Pat. CNS Drug Discovery 2006, 1, 271–288. [DOI] [PubMed] [Google Scholar]
- Boeckler F.; Gmeiner P. The structural evolution of dopamine D3 receptor ligands: structure–activity relationships and selected neuropharmacological aspects. Pharmacol. Ther. 2006, 112, 281–333. [DOI] [PubMed] [Google Scholar]
- Grundt P.; Prevatt K. M.; Cao J.; Taylor M.; Floresca C. Z.; Choi J. K.; Jenkins B. G.; Luedtke R. R.; Newman A. H. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J. Med. Chem. 2007, 50, 4135–4146. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Banala A. K.; Garcia E. G.; Cao J.; Keck T. M.; Bonifazi A.; Deschamps J. R.; Newman A. H. Chiral resolution and serendipitous fluorination reaction for the selective dopamine D3 receptor antagonist BAK2-66. ACS Med. Chem. Lett. 2014, 5, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z.; Li S.; Cui J.; Xu J.; Taylor M.; Ho D.; Luedtke R. R.; Mach R. H. Synthesis and pharmacological evaluation of fluorine-containing D(3) dopamine receptor ligands. J. Med. Chem. 2011, 54, 1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Levant B.; Jiang C.; Keck T. M.; Newman A. H.; Wang S. Tranylcypromine substituted cis-hydroxycyclobutylnaphthamides as potent and selective dopamine D(3) receptor antagonists. J. Med. Chem. 2014, 57, 4962–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F.; Arista L.; Bonanomi G.; Blaney F. E.; Braggio S.; Capelli A. M.; Checchia A.; Damiani F.; Di-Fabio R.; Fontana S.; Gentile G.; Griffante C.; Hamprecht D.; Marchioro C.; Mugnaini M.; Piner J.; Ratti E.; Tedesco G.; Tarsi L.; Terreni S.; Worby A.; Ashby C. R. Jr.; Heidbreder C. 1,2,4-Triazolyl azabicyclo[3.1.0]hexanes: a new series of potent and selective dopamine D(3) receptor antagonists. J. Med. Chem. 2010, 53, 374–391. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Zhang S.; Fernandez F.; Ghosh B.; Zhen J.; Kuzhikandathil E.; Reith M. E.; Dutta A. K. Further structure--activity relationships study of hybrid 7-{[2-(4-phenylpiperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2-ol analogues: identification of a high-affinity D3-preferring agonist with potent in vivo activity with long duration of action. J. Med. Chem. 2008, 51, 101–117. [DOI] [PubMed] [Google Scholar]
- Chen J.; Collins G. T.; Zhang J.; Yang C. Y.; Levant B.; Woods J.; Wang S. Design, synthesis, and evaluation of potent and selective ligands for the dopamine 3 (D3) receptor with a novel in vivo behavioral profile. J. Med. Chem. 2008, 51, 5905–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.; Antonio T.; Reith M. E.; Dutta A. K. Structure–activity relationship study of N6-(2-(4-(1H-indol-5-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydrobenzo[d]thiazole-2,6-diamine analogues: development of highly selective D3 dopamine receptor agonists along with a highly potent D2/D3 agonist and their pharmacological characterization. J. Med. Chem. 2012, 55, 5826–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschammer N.; Elsner J.; Goetz A.; Ehrlich K.; Schuster S.; Ruberg M.; Kuhhorn J.; Thompson D.; Whistler J.; Hubner H.; Gmeiner P. Highly potent 5-aminotetrahydropyrazolopyridines: enantioselective dopamine D3 receptor binding, functional selectivity, and analysis of receptor–ligand interactions. J. Med. Chem. 2011, 54, 2477–2491. [DOI] [PubMed] [Google Scholar]
- Correll C. C.; McKittrick B. A. Biased ligand modulation of seven transmembrane receptors (7TMRs): functional implications for drug discovery. J. Med. Chem. 2014, 57, 6887–6896. [DOI] [PubMed] [Google Scholar]
- Shonberg J.; Lopez L.; Scammells P. J.; Christopoulos A.; Capuano B.; Lane J. R. Biased agonism at G protein-coupled receptors: the promise and the challenges—a medicinal chemistry perspective. Med. Res. Rev. 2014, 34, 1286–1330. [DOI] [PubMed] [Google Scholar]
- Hiller C.; Kling R. C.; Heinemann F. W.; Meyer K.; Hubner H.; Gmeiner P. Functionally selective dopamine D2/D3 receptor agonists comprising an enyne moiety. J. Med. Chem. 2013, 56, 5130–5141. [DOI] [PubMed] [Google Scholar]
- Möller D.; Kling R. C.; Skultety M.; Leuner K.; Hubner H.; Gmeiner P. Functionally selective dopamine D(2), D(3) receptor partial agonists. J. Med. Chem. 2014, 57, 4861–4875. [DOI] [PubMed] [Google Scholar]
- te Beek E. T.; Zoethout R. W.; Bani M. S.; Andorn A.; Iavarone L.; Klaassen E. S.; Fina P.; van Gerven J. M. Pharmacokinetics and central nervous system effects of the novel dopamine D3 receptor antagonist GSK598809 and intravenous alcohol infusion at pseudo-steady state. J. Psychopharmacol. 2012, 26, 303–314. [DOI] [PubMed] [Google Scholar]
- Mogg K.; Bradley B. P.; O’Neill B.; Bani M.; Merlo-Pich E.; Koch A.; Bullmore E. T.; Nathan P. J. Effect of dopamine D(3) receptor antagonism on approach responses to food cues in overweight and obese individuals. Behav. Pharmacol. 2012, 23, 603–608. [DOI] [PubMed] [Google Scholar]
- Nathan P. J.; O’Neill B. V.; Mogg K.; Bradley B. P.; Beaver J.; Bani M.; Merlo-Pich E.; Fletcher P. C.; Swirski B.; Koch A.; Dodds C. M.; Bullmore E. T. The effects of the dopamine D(3) receptor antagonist GSK598809 on attentional bias to palatable food cues in overweight and obese subjects. Int. J. Neuropsychopharmacol. 2012, 15, 149–161. [DOI] [PubMed] [Google Scholar]
- Dodds C. M.; O’Neill B.; Beaver J.; Makwana A.; Bani M.; Merlo-Pich E.; Fletcher P. C.; Koch A.; Bullmore E. T.; Nathan P. J. Effect of the dopamine D3 receptor antagonist GSK598809 on brain responses to rewarding food images in overweight and obese binge eaters. Appetite 2012, 59, 27–33. [DOI] [PubMed] [Google Scholar]
- O’Connell D. P.; Vaughan C. J.; Aherne A. M.; Botkin S. J.; Wang Z. Q.; Felder R. A.; Carey R. M. Expression of the dopamine D3 receptor protein in the rat kidney. Hypertension 1998, 32, 886–895. [DOI] [PubMed] [Google Scholar]
- Hussain T.; Lokhandwala M. F. Renal dopamine receptors and hypertension. Exp. Biol. Med. 2003, 228, 134–142. [DOI] [PubMed] [Google Scholar]
- Zeng C.; Jose P. A. Dopamine receptors: important antihypertensive counterbalance against hypertensive factors. Hypertension 2011, 57, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armando I.; Villar V. A.; Jose P. A. Dopamine and renal function and blood pressure regulation. Compr. Physiol. 2011, 1, 1075–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C. W. Cocaine and cardiovascular toxicity. Addict. Biol. 1996, 1, 31–47. [DOI] [PubMed] [Google Scholar]
- Brecklin C. S.; Bauman J. L. Cardiovascular effects of cocaine: focus on hypertension. J. Clin. Hypertens. 1999, 1, 212–217. [PubMed] [Google Scholar]
- Haney M.; Rubin E.; Foltin R. W. Aripiprazole maintenance increases smoked cocaine self-administration in humans. Psychopharmacology 2011, 216, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lile J. A.; Stoops W. W.; Glaser P. E.; Hays L. R.; Rush C. R. Discriminative stimulus, subject-rated and cardiovascular effects of cocaine alone and in combination with aripiprazole in humans. J. Psychopharmacol. 2011, 25, 1469–1479. [DOI] [PubMed] [Google Scholar]
- Bergman J.; Roof R. A.; Furman C. A.; Conroy J. L.; Mello N. K.; Sibley D. R.; Skolnick P. Modification of cocaine self-administration by buspirone (buspar®): potential involvement of D3 and D4 dopamine receptors. Int. J. Neuropsychopharmacol. 2013, 16, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello N. K.; Fivel P. A.; Kohut S. J.; Bergman J. Effects of chronic buspirone treatment on cocaine self-administration. Neuropsychopharmacology 2013, 38, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winhusen T. M.; Kropp F.; Lindblad R.; Douaihy A.; Haynes L.; Hodgkins C.; Chartier K.; Kampman K. M.; Sharma G.; Lewis D. F.; VanVeldhuisen P.; Theobald J.; May J.; Brigham G. S. Multisite, randomized, double-blind, placebo-controlled pilot clinical trial to evaluate the efficacy of buspirone as a relapse-prevention treatment for cocaine dependence. J. Clin. Psychiatry 2014, 75, 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. W.; Fowler J. S.; Skolnick P.; Muench L.; Kang Y.; Shea C.; Logan J.; Kim D.; Carter P.; King P.; Alexoff D.; Volkow N. D. Therapeutic doses of buspirone block D3 receptors in the living primate brain. Int. J. Neuropsychopharmacol. 2014, 17, 1257–1267. [DOI] [PubMed] [Google Scholar]
- Gobert A.; Rivet J. M.; Cistarelli L.; Melon C.; Millan M. J. Buspirone modulates basal and fluoxetine-stimulated dialysate levels of dopamine, noradrenaline and serotonin in the frontal cortex of freely moving rats: activation of serotonin1A receptors and blockade of alpha2-adrenergic receptors underlie its actions. Neuroscience 1999, 93, 1251–1262. [DOI] [PubMed] [Google Scholar]
- Algeri S.; De Luigi A.; De Simoni M. G.; Imeri L.; Marconi M.; Nava S.; Perego C.; Sacchetti G. Multiple and complex effects of buspirone on central dopaminergic system. Pharmacol., Biochem. Behav. 1988, 29, 823–826. [DOI] [PubMed] [Google Scholar]
- John W. S.; Banala A. K.; Newman A. H.; Nader M. A. Effects of buspirone and the dopamine D3 receptor compound PG619 on cocaine and methamphetamine self-administration in rhesus monkeys using a food-drug choice paradigm. Psychopharmacology 2014, 232, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czoty P. W.; Nader M. A. Effects of oral and intravenous administration of buspirone on food-cocaine choice in socially housed male cynomolgus monkeys. Neuropsychopharmacology 2014, 40, 1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A. H.; Cao J.; Bennett C. J.; Robarge M. J.; Freeman R. A.; Luedtke R. R. N-(4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butyl, butenyl and butynyl)arylcarboxamides as novel dopamine D(3) receptor antagonists. Bioorg. Med. Chem. Lett. 2003, 13, 2179–2183. [DOI] [PubMed] [Google Scholar]
- Sorensen G.; Sager T. N.; Petersen J. H.; Brennum L. T.; Thogersen P.; Hee Bengtsen C.; Thomsen M.; Wortwein G.; Fink-Jensen A.; Woldbye D. P. Aripiprazole blocks acute self-administration of cocaine and is not self-administered in mice. Psychopharmacology 2008, 199, 37–46. [DOI] [PubMed] [Google Scholar]
- Thomsen M.; Fink-Jensen A.; Woldbye D. P.; Wortwein G.; Sager T. N.; Holm R.; Pepe L. M.; Caine S. B. Effects of acute and chronic aripiprazole treatment on choice between cocaine self-administration and food under a concurrent schedule of reinforcement in rats. Psychopharmacology 2008, 201, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltenstein M. W.; Do P. H.; See R. E. Repeated aripiprazole administration attenuates cocaine seeking in a rat model of relapse. Psychopharmacology 2009, 207, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czoty P. W.; Nader M. A. Effects of dopamine D2/D3 receptor ligands on food-cocaine choice in socially housed male cynomolgus monkeys. J. Pharmacol. Exp. Ther. 2013, 344, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoops W. W.; Lile J. A.; Lofwall M. R.; Rush C. R. The safety, tolerability, and subject-rated effects of acute intranasal cocaine administration during aripiprazole maintenance. Am. J. Drug Alcohol Abuse 2007, 33, 769–776. [DOI] [PubMed] [Google Scholar]
- Meini M.; Moncini M.; Cecconi D.; Cellesi V.; Biasci L.; Simoni G.; Ameglio M.; Pellegrini M.; Forgione R. N.; Rucci P. Aripiprazole and ropinirole treatment for cocaine dependence: evidence from a pilot study. Curr. Pharm. Des. 2011, 17, 1376–1383. [DOI] [PubMed] [Google Scholar]
- Song R.; Yang R. F.; Wu N.; Su R. B.; Li J.; Peng X. Q.; Li X.; Gaal J.; Xi Z. X.; Gardner E. L. YQA14: a novel dopamine D3 receptor antagonist that inhibits cocaine self-administration in rats and mice, but not in D3 receptor-knockout mice. Addict. Biol. 2012, 17, 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J.; Chen X.; Brodbeck R.; Primus R.; Braun J.; Wasley J. W.; Thurkauf A. NGB 2904 and NGB 2849: two highly selective dopamine D3 receptor antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 2715–2718. [DOI] [PubMed] [Google Scholar]
- Stafford D.; LeSage M. G.; Glowa J. R. Progressive-ratio schedules of drug delivery in the analysis of drug self-administration: a review. Psychopharmacology 1998, 139, 169–184. [DOI] [PubMed] [Google Scholar]
- Xi Z. X.; Gardner E. L. Pharmacological actions of NGB 2904, a selective dopamine D3 receptor antagonist, in animal models of drug addiction. CNS Drug Rev. 2007, 13, 240–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller K.; Xi Z. X.; Peng X. Q.; Newman A. H.; Ashby C. R. Jr.; Heidbreder C.; Gaal J.; Gardner E. L. The selective dopamine D3 receptor antagonists SB-277011A and NGB 2904 and the putative partial D3 receptor agonist BP-897 attenuate methamphetamine-enhanced brain stimulation reward in rats. Psychopharmacology 2008, 196, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robarge M. J.; Husbands S. M.; Kieltyka A.; Brodbeck R.; Thurkauf A.; Newman A. H. Design and synthesis of [(2,3-dichlorophenyl)piperazin-1-yl]alkylfluorenylcarboxamides as novel ligands selective for the dopamine D3 receptor subtype. J. Med. Chem. 2001, 44, 3175–3186. [DOI] [PubMed] [Google Scholar]
- Martelle J. L.; Claytor R.; Ross J. T.; Reboussin B. A.; Newman A. H.; Nader M. A. Effects of two novel D3-selective compounds, NGB 2904 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxamide] and CJB 090 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-4-(pyridin-2-yl)benzamide], on the reinforcing and discriminative stimulus effects of cocaine in rhesus monkeys. J. Pharmacol. Exp. Ther. 2007, 321, 573–582. [DOI] [PubMed] [Google Scholar]
- Achat-Mendes C.; Platt D. M.; Newman A. H.; Spealman R. D. The dopamine D3 receptor partial agonist CJB 090 inhibits the discriminative stimulus but not the reinforcing or priming effects of cocaine in squirrel monkeys. Psychopharmacology 2009, 206, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaylock B. L.; Gould R. W.; Banala A.; Grundt P.; Luedtke R. R.; Newman A. H.; Nader M. A. Influence of cocaine history on the behavioral effects of dopamine D(3) receptor-selective compounds in monkeys. Neuropsychopharmacology 2011, 36, 1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelle S. E.; Nader S. H.; Czoty P. W.; John W. S.; Duke A. N.; Garg P. K.; Garg S.; Newman A. H.; Nader M. A. Further characterization of quinpirole-elicited yawning as a model of dopamine D3 receptor activation in male and female monkeys. J. Pharmacol. Exp. Ther. 2014, 350, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton L. R.; Czoty P. W.; Gage H. D.; Nader M. A. Characterization of the dopamine receptor system in adult rhesus monkeys exposed to cocaine throughout gestation. Psychopharmacology 2010, 210, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio L.; Wee S.; Newman A. H.; Pulvirenti L.; Koob G. F. The dopamine D3 receptor partial agonist CJB090 and antagonist PG01037 decrease progressive ratio responding for methamphetamine in rats with extended-access. Addict. Biol. 2010, 15, 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason C. W.; Hassan H. E.; Kim K. P.; Cao J.; Eddington N. D.; Newman A. H.; Voulalas P. J. Characterization of the transport, metabolism, and pharmacokinetics of the dopamine D3 receptor-selective fluorenyl- and 2-pyridylphenyl amides developed for treatment of psychostimulant abuse. J. Pharmacol. Exp. Ther. 2010, 333, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung T. H.; Nolan B. C.; Hammerslag L. R.; Weber S. M.; Durbin J. P.; Peartree N. A.; Mach R. H.; Luedtke R. R.; Neisewander J. L. Phenylpiperazine derivatives with selectivity for dopamine D3 receptors modulate cocaine self-administration in rats. Neuropharmacology 2012, 63, 1346–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung T. H.; Loriaux A. L.; Weber S. M.; Chandler K. N.; Lenz J. D.; Schaan R. F.; Mach R. H.; Luedtke R. R.; Neisewander J. L. Reduction of cocaine self-administration and D3 receptor-mediated behavior by two novel dopamine D3 receptor-selective partial agonists, OS-3-106 and WW-III-55. J. Pharmacol. Exp. Ther. 2013, 347, 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M.; Chang W. L.; Durbin J. P.; Park P. E.; Luedtke R. R.; Mach R. H.; Swerdlow N. R. Using prepulse inhibition to detect functional D3 receptor antagonism: effects of WC10 and WC44. Pharmacol., Biochem. Behav. 2009, 93, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins G. T.; Witkin J. M.; Newman A. H.; Svensson K. A.; Grundt P.; Cao J.; Woods J. H. Dopamine agonist-induced yawning in rats: a dopamine D3 receptor-mediated behavior. J. Pharmacol. Exp. Ther. 2005, 314, 310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins G. T.; Newman A. H.; Grundt P.; Rice K. C.; Husbands S. M.; Chauvignac C.; Chen J.; Wang S.; Woods J. H. Yawning and hypothermia in rats: effects of dopamine D3 and D2 agonists and antagonists. Psychopharmacology 2007, 193, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisewander J. L.; Fuchs R. A.; Tran-Nguyen L. T.; Weber S. M.; Coffey G. P.; Joyce J. N. Increases in dopamine D3 receptor binding in rats receiving a cocaine challenge at various time points after cocaine self-administration: implications for cocaine-seeking behavior. Neuropsychopharmacology 2004, 29, 1479–1487. [DOI] [PubMed] [Google Scholar]
- Huang X. P.; Mangano T.; Hufeisen S.; Setola V.; Roth B. L. Identification of human Ether-a-go-go related gene modulators by three screening platforms in an academic drug-discovery setting. Assay Drug Dev. Technol. 2010, 8, 727–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T. M.; Yang H. J.; Bi G. H.; Huang Y.; Zhang H. Y.; Srivastava R.; Gardner E. L.; Newman A. H.; Xi Z. X. Fenobam sulfate inhibits cocaine-taking and cocaine-seeking behavior in rats: implications for addiction treatment in humans. Psychopharmacology 2013, 229, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T. M.; Zou M. F.; Bi G. H.; Zhang H. Y.; Wang X. F.; Yang H. J.; Srivastava R.; Gardner E. L.; Xi Z. X.; Newman A. H. A novel mGluR5 antagonist, MFZ 10-7, inhibits cocaine-taking and cocaine-seeking behavior in rats. Addict. Biol. 2014, 19, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson N. R.; Roberts D. C. Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J. Neurosci. Methods 1996, 66, 1–11. [DOI] [PubMed] [Google Scholar]
- Arnold J. M.; Roberts D. C. A critique of fixed and progressive ratio schedules used to examine the neural substrates of drug reinforcement. Pharmacol., Biochem. Behav. 1997, 57, 441–447. [DOI] [PubMed] [Google Scholar]
- Roberts D. C.; Morgan D.; Liu Y. How to make a rat addicted to cocaine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 1614–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roane H. S. On the applied use of progressive-ratio schedules of reinforcement. J. Appl. Behav. Anal. 2008, 41, 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer S. D.; Sullivan M. A.; Whittington R. A.; Vosburg S. K.; Kowalczyk W. J. Abuse liability of prescription opioids compared to heroin in morphine-maintained heroin abusers. Neuropsychopharmacology 2008, 33, 1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. D.; Comer S. D. A review of human drug self-administration procedures. Behav. Pharmacol. 2013, 24, 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoops W. W.; Lile J. A.; Glaser P. E.; Hays L. R.; Rush C. R. Intranasal cocaine functions as reinforcer on a progressive ratio schedule in humans. Eur. J. Pharmacol. 2010, 644, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein D. H.; Preston K. L.; Stewart J.; Shaham Y. Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology 2006, 189, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y.; Shalev U.; Lu L.; De Wit H.; Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology 2003, 168, 3–20. [DOI] [PubMed] [Google Scholar]
- Bossert J. M.; Marchant N. J.; Calu D. J.; Shaham Y. The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology 2013, 229, 453–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz J. L.; Higgins S. T. The validity of the reinstatement model of craving and relapse to drug use. Psychopharmacology 2003, 168, 21–30. [DOI] [PubMed] [Google Scholar]
- Koob G. F.; Kenneth Lloyd G.; Mason B. J. Development of pharmacotherapies for drug addiction: a Rosetta stone approach. Nat. Rev. Drug Discovery 2009, 8, 500–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolverton W. L.; Nader M. A.. Experimental evaluation of the reinforcing effects of drugs. In Testing and Evaluation of Drugs of Abuse; Adler M. W., Cowan A., Eds.; John Wiley & Sons: Oxford, England, 1990; pp 165–192. [Google Scholar]
- Lile J. A.; Morgan D.; Birmingham A. M.; Wang Z.; Woolverton W. L.; Davies H. M.; Nader M. A. The reinforcing efficacy of the dopamine reuptake inhibitor 2beta-propanoyl-3beta-(4-tolyl)-tropane (PTT) as measured by a progressive-ratio schedule and a choice procedure in rhesus monkeys. J. Pharmacol. Exp. Ther. 2002, 303, 640–648. [DOI] [PubMed] [Google Scholar]
- Woolverton W. L.; Balster R. L. Effects of antipsychotic compounds in rhesus monkeys given a choice between cocaine and food. Drug Alcohol Depend. 1981, 8, 69–78. [DOI] [PubMed] [Google Scholar]
- Negus S. S. Rapid assessment of choice between cocaine and food in rhesus monkeys: effects of environmental manipulations and treatment with d-amphetamine and flupenthixol. Neuropsychopharmacology 2003, 28, 919–931. [DOI] [PubMed] [Google Scholar]
- Levant B. Differential distribution of D3 dopamine receptors in the brains of several mammalian species. Brain Res. 1998, 800, 269–274. [DOI] [PubMed] [Google Scholar]
- Mach R. H.; Tu Z.; Xu J.; Li S.; Jones L. A.; Taylor M.; Luedtke R. R.; Derdeyn C. P.; Perlmutter J. S.; Mintun M. A. Endogenous dopamine (DA) competes with the binding of a radiolabeled D(3) receptor partial agonist in vivo: a positron emission tomography study. Synapse 2011, 65, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Ciano P.; Underwood R. J.; Hagan J. J.; Everitt B. J. Attenuation of cue-controlled cocaine-seeking by a selective D3 dopamine receptor antagonist SB-277011-A. Neuropsychopharmacology 2003, 28, 329–338. [DOI] [PubMed] [Google Scholar]
- Xi Z. X.; Gilbert J. G.; Pak A. C.; Ashby C. R. Jr.; Heidbreder C. A.; Gardner E. L. Selective dopamine D3 receptor antagonism by SB-277011A attenuates cocaine reinforcement as assessed by progressive-ratio and variable-cost-variable-payoff fixed-ratio cocaine self-administration in rats. Eur. J. Neurosci. 2005, 21, 3427–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyertyan I.; Kiss B.; Gal K.; Laszlovszky I.; Horvath A.; Gemesi L. I.; Saghy K.; Pasztor G.; Zajer M.; Kapas M.; Csongor E. A.; Domany G.; Tihanyi K.; Szombathelyi Z. Effects of RGH-237 [N-{4-[4-(3-aminocarbonyl-phenyl)-piperazin-1-yl]-butyl}-4-bromo-benzamide], an orally active, selective dopamine D(3) receptor partial agonist in animal models of cocaine abuse. J. Pharmacol. Exp. Ther. 2007, 320, 1268–1278. [DOI] [PubMed] [Google Scholar]
- Di Ciano P. Drug seeking under a second-order schedule of reinforcement depends on dopamine D3 receptors in the basolateral amygdala. Behav. Neurosci. 2008, 122, 129–139. [DOI] [PubMed] [Google Scholar]
- Vorel S. R.; Ashby C. R. Jr.; Paul M.; Liu X.; Hayes R.; Hagan J. J.; Middlemiss D. N.; Stemp G.; Gardner E. L. Dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-enhanced brain reward in rats. J. Neurosci. 2002, 22, 9595–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czoty P. W.; Gage H. D.; Garg P. K.; Garg S.; Nader M. A. Effects of repeated treatment with the dopamine D2/D3 receptor partial agonist aripiprazole on striatal D2/D3 receptor availability in monkeys. Psychopharmacology 2014, 231, 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ladona F. J.; Cox B. F. BP 897, a selective dopamine D3 receptor ligand with therapeutic potential for the treatment of cocaine-addiction. CNS Drug Rev. 2003, 9, 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preti A. New developments in the pharmacotherapy of cocaine abuse. Addict. Biol. 2007, 12, 133–151. [DOI] [PubMed] [Google Scholar]
- Pierce R. C.; O’Brien C. P.; Kenny P. J.; Vanderschuren L. J. Rational development of addiction pharmacotherapies: successes, failures, and prospects. Cold Spring Harbor Perspect. Med. 2012, 2, a012880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S. H. Validation crisis in animal models of drug addiction: beyond non-disordered drug use toward drug addiction. Neurosci. Biobehav. Rev. 2010, 35, 172–184. [DOI] [PubMed] [Google Scholar]
- John W. S.; Newman A. H.; Nader M. A. Differential effects of the dopamine D3 receptor antagonist PG01037 on cocaine and methamphetamine self-administration in rhesus monkeys. Neuropharmacology 2015, 92, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle G. E.; Beaver J. D.; Tziortzi A.; Comley R. A.; Bani M.; Ghibellini G.; Merlo-Pich E.; Rabiner E. A.; Laruelle M.; Gunn R. N. Mathematical modelling of [11C]-(+)-PHNO human competition studies. NeuroImage 2013, 68, 119–132. [DOI] [PubMed] [Google Scholar]
- Matuskey D.; Gallezot J. D.; Pittman B.; Williams W.; Wanyiri J.; Gaiser E.; Lee D. E.; Hannestad J.; Lim K.; Zheng M. Q.; Lin S. F.; Labaree D.; Potenza M. N.; Carson R. E.; Malison R. T.; Ding Y. S. Dopamine D(3) receptor alterations in cocaine-dependent humans imaged with [11C](+)PHNO. Drug Alcohol Depend. 2014, 139, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins G. T.; Truong Y. N.; Levant B.; Chen J.; Wang S.; Woods J. H. Behavioral sensitization to cocaine in rats: evidence for temporal differences in dopamine D3 and D2 receptor sensitivity. Psychopharmacology 2011, 215, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad K. L.; Ford K.; Marinelli M.; Wolf M. E. Dopamine receptor expression and distribution dynamically change in the rat nucleus accumbens after withdrawal from cocaine self-administration. Neuroscience 2010, 169, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady K. T.; Verduin M. L.; Tolliver B. K. Treatment of patients comorbid for addiction and other psychiatric disorders. Curr. Psychiatry Rep. 2007, 9, 374–380. [DOI] [PubMed] [Google Scholar]
- Schuckit M. A. Comorbidity between substance use disorders and psychiatric conditions. Addiction 2006, 101, 76–88. [DOI] [PubMed] [Google Scholar]
- Urbanoski K.; Kenaszchuk C.; Veldhuizen S.; Rush B. The clustering of psychopathology among adults seeking treatment for alcohol and drug addiction. J. Subst. Abuse Treat. 2015, 49, 21–26. [DOI] [PubMed] [Google Scholar]
- Compton M. T.; Weiss P. S.; West J. C.; Kaslow N. J. The associations between substance use disorders, schizophrenia-spectrum disorders, and Axis IV psychosocial problems. Soc. Psychiatry Psychiatr. Epidemiol. 2005, 40, 939–946. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Newman A. H.; Xu M. Dopamine D1 and D3 receptors mediate reconsolidation of cocaine memories in mouse models of drug self-administration. Neuroscience 2014, 278, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitz J.; Hodgkinson C. A.; Martin-Soelch C.; Shen P. H.; Szczepanik J.; Nugent A.; Herscovitch P.; Grace A. A.; Goldman D.; Drevets W. C. The functional DRD3 Ser9Gly polymorphism (rs6280) is pleiotropic, affecting reward as well as movement. PLoS One 2013, 8, e54108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Payne T. J.; Ma J. Z.; Li M. D. A functional polymorphism, rs6280, in DRD3 is significantly associated with nicotine dependence in European-American smokers. Am. J. Med. Genet., Part B 2008, 147, 1109–1115. [DOI] [PubMed] [Google Scholar]
- Kang S. G.; Lee B. H.; Lee J. S.; Chai Y. G.; Ko K. P.; Lee H. J.; Han D. M.; Ji H.; Jang G. H.; Shin H. E. DRD3 Gene rs6280 polymorphism may be associated with alcohol dependence overall and with Lesch type I alcohol dependence in Koreans. Neuropsychobiology 2014, 69, 140–146. [DOI] [PubMed] [Google Scholar]
- Kuo S. C.; Yeh Y. W.; Chen C. Y.; Huang C. C.; Chang H. A.; Yen C. H.; Ho P. S.; Liang C. S.; Chou H. W.; Lu R. B.; Huang S. Y. DRD3 variation associates with early-onset heroin dependence, but not specific personality traits. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 51, 1–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.