

Abstract
More potent, but less known than IP3 that liberates Ca2+ from the ER, NAADP releases Ca2+ from acidic stores. The notion that TPC channels mediate this Ca2+ release was questioned recently by studies suggesting that TPCs are rather PI(3,5)P2-activated Na+ channels. Ruas et al (2015) now partially reconcile these views by showing that TPCs significantly conduct both cations and confirm their activation by both NAADP and PI(3,5)P2. They attribute the failure of others to observe TPC-dependent NAADP-induced Ca2+ release in vivo to inadequate mouse models that retain partial TPC function.
See also: M Ruas et al (July 2015)
Ca2+ is a powerful cellular second messenger, with low cytosolic levels. Whereas classical Ca2+ mobilizing factors, such as inositol 1,4,5-trisphosphate (IP3), cyclic ADP-ribose (cADPR) or Ca2+ itself, open Ca2+ channels of the endoplasmic reticulum (ER), nicotinic acid adenine dinucleotide phosphate (NAADP), the most recently discovered and most potent Ca2+-releasing factor, triggers Ca2+ release from acidic compartments (lysosomes in mammalian cells) at low nM concentrations (Morgan & Galione, 2014). Ca2+ release from acidic stores can, in turn, trigger a more “global” Ca2+ release from the ER. The physiological role of NAADP is incompletely understood, and the molecular NAADP target has remained controversial. The mystery surrounding NAADP-induced Ca2+ release seemed to be lifted in 2009 when three groups described “two-pore channels” 1 and 2 (TPC1/2) as mediating NAADP-induced lysosomal Ca2+ release (“two-pore channel” is a misnomer; TPCs resemble concatemers of two Kv-like channels and most likely form a single pore as a dimer). Their results were based on the following: (i) TPC1 and TPC2 localize to endolysosomal compartments, (ii) NAADP triggered large Ca2+ transients in TPC overexpressing, but not in wild-type (WT) cells (Brailoiu et al, 2009; Calcraft et al, 2009; Zong et al, 2009), (iii) NAADP activated a Ca2+-dependent plasma membrane conductance in pancreatic β cells from WT, but not from Tpcn2−/− mice (Calcraft et al, 2009), and (iv) NAADP triggered inward (lumen to cytosol) Ca2+ currents by planar patch clamp recordings of isolated, artificially enlarged endolysosomes from TPC2-overexpressing cells (Schieder et al, 2010).
These seemingly convincing results were questioned by Wang et al (2012) who could not observe NAADP-induced currents using conventional patch clamp of enlarged endolysosomes from TPC1- or TPC2-overexpressing cells. Instead, they found that phosphoinositol 3,5-bisphosphate (PI(3,5)P2), a PIP2 of endosomal and lysosomal membranes, activates endolysosomal currents in native and TPC-overexpressing cells. These were independent of TRPML1, a lysosomal channel known to be activated by PI(3,5)P2, and transported Na+ much better than Ca2+. In contrast to Steinberg et al (2010) who obtained a lysosomal Na+ concentration of ∼20 mM in ionophore-treated cells, Wang et al reported that lysosomes contained ∼140–150 mM Na+. However, this value is unlikely to reflect normal Na+ levels because their lysosomal preparation was obtained by several long centrifugation steps in non-physiological solutions. Based on their PCa/PNa value for TPC2 (∼0.1) and the assumed high luminal Na+ concentration, Wang et al (2012) suggested that TPCs physiologically function as Na+ channels. Contrasting with Calcraft et al (2009), NAADP-induced Ca2+ transients in pancreatic islets isolated from bona fide TPC1/2 double-knockout (Tpcn1/2−/−) mice were normal. However, the gene-trap strategy used to generate these mice hypothetically allowed for the expression of TPC1 and TPC2 proteins carrying deletions in the cytoplasmic N-termini. Since these truncated proteins failed to yield PI(3,5)P2-stimulated currents in heterologous expression, Wang et al (2012) concluded that their Tpcn1/2−/− mice lacked functional TPC proteins.
Ruas et al (2015) now re-examine this issue using mouse embryonic fibroblasts (MEFs) derived from WT and different Tpcn1/2−/− mice with confirmed absence of TPCs. In WT MEFs, NAADP induced Ca2+ signals that depended on acidic Ca2+ stores and were amplified by secondary Ca2+ release from the ER. Loss of either TPC1 or TPC2 diminished, and loss of both proteins abolished, the Ca2+ response. Endogenous TPC currents of vacuolin-enlarged endolysosomes from MEFs were investigated with the planar patch clamp technique. In the presence of Ca2+ and K+, nM concentrations of NAADP induced inward Ca2+ currents that were strongly reduced in Tpcn2−/− MEFs and virtually absent in Tpcn1/2−/− MEFs. (Because vacuolin indiscriminately enlarges endolysosomal vesicles, both endosomal TPC1 and lysosomal TPC2 must be disrupted to establish a role of TPCs with this technique.) PI(3,5)P2 also evoked inward currents which were reduced, but not abolished in Tpcn1/2−/− MEFs. This confirmed that TPCs are activated by both NAADP and PI(3,5)P2 (Jha et al, 2014) and that PI(3,5)P2 also activates other lysosomal channels such as TRPML1. Ruas et al (2015) determined the permeability ratios PCa/PK and PCa/PNa and confirmed the findings of Wang et al (2012) that TPCs, while being almost impermeable for K+, are indeed permeable for Na+. However, whereas Wang et al (2012) determined PCa/PNa of overexpressed TPC2 to be ∼0.1, Ruas et al (2015) found the permeabilities for Ca2+ and Na+ of endogeneous TPCs to be almost equal. NAADP-induced Ca2+ release in Tpcn1/2−/− MEFs was rescued by expression of WT TPCs, but neither by a transport-deficient TPC2 mutant nor by a mutant with strongly reduced Ca2+ permeability. The N-terminally truncated TPC1 and TPC2 proteins that may be expressed in the “Tpcn1/2−/−” mice of Wang et al (2012) restored NAADP-induced Ca2+ signals, likely explaining the unchanged NAADP-induced Ca2+ signals in those mice. The complete loss of TPCs did not affect binding of NAADP to liver homogenates (Ruas et al, 2015), agreeing with previous findings that NAADP does not bind TPCs directly but through an unknown auxiliary protein.
The question remains why Wang et al (2012) did not observe NAADP-induced currents in enlarged endolysosomes from TPC-overexpressing cells. Possibly, their tagged TPC constructs interfered with TPC NAADP sensitivity, as known from N-terminally tagged TPC1 (Morgan & Galione, 2014). Recent results show that Mg2+ strongly inhibits NAADP- and PI(3,5)P2-induced TPC2 currents in a pH-dependent manner (Jha et al, 2014). However, differences in Mg2+ concentrations are unlikely to explain the divergent results. The hypothesis that Wang et al (2012), but not Ruas et al (2015), lost a putative NAADP-binding protein in their patch clamp preparations seems implausible as the former studies were done under more physiological conditions.
The biological functions of TPCs are poorly understood. Lysosomal degradation of endocytosed proteins is impaired in Tpcn2−/− cells, and Tpcn2−/− mice are susceptible to fatty liver disease (Grimm et al, 2014). These phenotypes hint at a role of TPC2 in the fusion of late endosomes and lysosomes. Similarly, cells from Tpcn1−/− or Tpcn2−/− mice are resistant to Ebola virus infection due to perturbed fusion between viral and endosomal membranes (Sakurai et al, 2015). Interestingly, both TPC (Cang et al, 2013) and TRPML1 (Medina et al, 2015) channels were linked to mTOR-dependent cellular nutrient sensing.
By studying TPC channels in a native context and using mouse models with confirmed absence of both TPC1 and TPC2, Ruas et al (2015) eliminate remaining doubts on their role in NAADP-induced lysosomal Ca2+ release. Whereas PI(3,5)P2 in endolysosomal membranes may be a permissive factor for channels such as TPCs and TRPML1, NAADP likely plays a role in signal transduction (Fig 1). TPCs may transport both Ca2+ and Na+ from acidic vesicles into the cytosol. The large driving force for Ca2+ exit, the low cytosolic Ca2+ concentration that is raised drastically even with a rather small Ca2+ flux, together with the known messenger role of Ca2+ suggests that acute opening of TPCs exerts its effects mainly through Ca2+. Localized Ca2+ transients play a crucial role in vesicle fusion and also open Ca2+ channels in the ER membrane. Lysosomal Na+ efflux may additionally depolarize the lysosomal membrane and contribute to membrane fusion.
Figure 1.
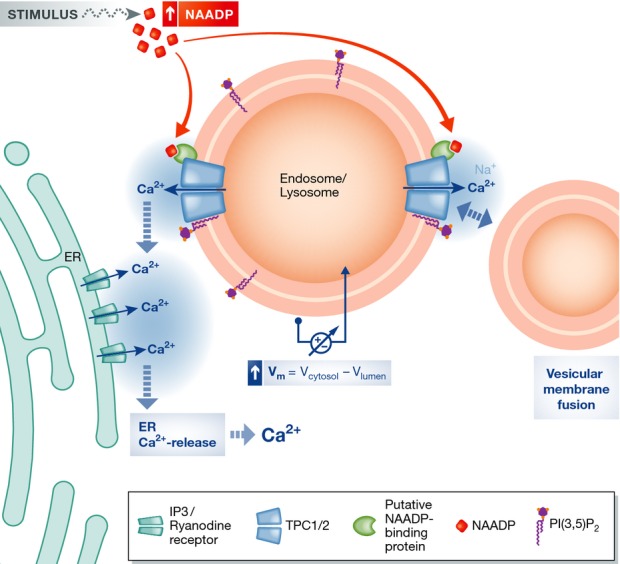
Proposed model for TPC function in NAADP-induced Ca2+ release from endolysosomal compartments
Cytosolic NAADP levels increase in response to certain stimuli in a poorly understood way. NAADP binds to a yet unidentified protein, which associates with TPCs leading to their opening. Opening of TPCs also requires PI(3,5)P2, a lipid component of endolysosomal membranes. Open TPCs release Ca2+ and possibly Na+ into the cytosol, leading to a local increase in Ca2+ and potentially a change in the vesicular membrane potential. This localized Ca2+ transient may have two effects: It can lead to opening of Ca2+ channels (IP3 and ryanodine receptors) in the ER membrane by Ca2+-induced Ca2+ release, which in turn causes a global Ca2+ increase. In addition, it may stimulate the fusion of vesicular membranes and thereby affect trafficking in the endolysosomal system. Whether vesicular Na+ release also stimulates vesicle fusion (Wang et al, 2012), either by increasing local Na+ concentrations (with relative concentration changes much less than with Ca2+) or by changing the vesicular voltage, is under debate.
The identification of TPCs as channels targeted by NAADP allows rigorous investigation into the role of this interesting messenger molecule in spatially and temporally complex Ca2+ signaling. The conceptual integration of vesicular ion gradients, channels, transporters and their metabolic regulation, into a functional module that interacts with the rest of the cell, is a challenge which is being tackled by an increasing number of groups.
References
- Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, Patel S. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186:201–209. doi: 10.1083/jcb.200904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang C, Zhou Y, Navarro B, Seo YJ, Aranda K, Shi L, Battaglia-Hsu S, Nissim I, Clapham DE, Ren D. mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell. 2013;152:778–790. doi: 10.1016/j.cell.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C, Holdt LM, Chen CC, Hassan S, Muller C, Jors S, Cuny H, Kissing S, Schroder B, Butz E, Northoff B, Castonguay J, Luber CA, Moser M, Spahn S, Lullmann-Rauch R, Fendel C, Klugbauer N, Griesbeck O, Haas A, et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat Commun. 2014;5:4699. doi: 10.1038/ncomms5699. [DOI] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S. Convergent regulation of the lysosomal two-pore channel-2 by Mg2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO J. 2014;33:501–511. doi: 10.1002/embj.201387035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AJ, Galione A. Two-pore channels (TPCs): current controversies. BioEssays. 2014;36:173–183. doi: 10.1002/bies.201300118. [DOI] [PubMed] [Google Scholar]
- Ruas M, Davis LC, Chen CC, Morgan AJ, Chuang KT, Walseth TF, Grimm C, Garnham C, Powell T, Platt N, Platt FM, Biel M, Wahl-Schott C, Parrington J, Galione A. Expression of Ca2+-permeable two-pore channels rescues NAADP signalling in TPC-deficient cells. EMBO J. 2015;34:1743–1758. doi: 10.15252/embj.201490009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai Y, Kolokoltsov AA, Chen CC, Tidwell MW, Bauta WE, Klugbauer N, Grimm C, Wahl-Schott C, Biel M, Davey RA. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science. 2015;347:995–998. doi: 10.1126/science.1258758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieder M, Rotzer K, Bruggemann A, Biel M, Wahl-Schott CA. Characterization of two-pore channel 2 (TPCN2)-mediated Ca2+ currents in isolated lysosomes. J Biol Chem. 2010;285:21219–21222. doi: 10.1074/jbc.C110.143123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg BE, Huynh KK, Brodovitch A, Jabs S, Stauber T, Jentsch TJ, Grinstein S. A cation counterflux supports lysosomal acidification. J Cell Biol. 2010;189:1171–1186. doi: 10.1083/jcb.200911083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong XP, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D, Xu H. TPC proteins are phosphoinositide-activated sodium-selective ion channels in endosomes and lysosomes. Cell. 2012;151:372–383. doi: 10.1016/j.cell.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rotzer K, Griesbeck O, Harz H, Biel M, Wahl-Schott C. The two-pore channel TPCN2 mediates NAADP-dependent Ca2+-release from lysosomal stores. Pflugers Arch. 2009;458:891–899. doi: 10.1007/s00424-009-0690-y. [DOI] [PMC free article] [PubMed] [Google Scholar]