

Abstract
The second messenger NAADP triggers Ca2+ release from endo-lysosomes. Although two-pore channels (TPCs) have been proposed to be regulated by NAADP, recent studies have challenged this. By generating the first mouse line with demonstrable absence of both Tpcn1 and Tpcn2 expression (Tpcn1/2−/−), we show that the loss of endogenous TPCs abolished NAADP-dependent Ca2+ responses as assessed by single-cell Ca2+ imaging or patch-clamp of single endo-lysosomes. In contrast, currents stimulated by PI(3,5)P2 were only partially dependent on TPCs. In Tpcn1/2−/− cells, NAADP sensitivity was restored by re-expressing wild-type TPCs, but not by mutant versions with impaired Ca2+-permeability, nor by TRPML1. Another mouse line formerly reported as TPC-null likely expresses truncated TPCs, but we now show that these truncated proteins still support NAADP-induced Ca2+ release. High-affinity [32P]NAADP binding still occurs in Tpcn1/2−/− tissue, suggesting that NAADP regulation is conferred by an accessory protein. Altogether, our data establish TPCs as Ca2+-permeable channels indispensable for NAADP signalling.
Keywords: Ca2+, electrophysiology, endo-lysosome, NAADP, TPC
See also: TJ Jentsch et al (July 2015)
Introduction
Ca2+ release from intracellular Ca2+ stores constitutes a universal cell signalling mechanism and is evoked by any of three principal Ca2+-mobilizing messengers: inositol 1,4,5-trisphosphate (IP3), cyclic ADP ribose (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP) (Berridge et al, 2003). Recruited by extracellular stimuli as diverse as cell–cell contact and GPCR activation, NAADP has been implicated in processes such as fertilization, exocytosis, autophagy, cardiac and neural function, and cell differentiation (Galione, 2014). NAADP differs from IP3 and cADPR, which regulate IP3 receptors and ryanodine receptors, respectively, in the ER, by primarily targeting a different Ca2+ store (acidic endo-lysosomal organelles) (Churchill et al, 2002) and a different Ca2+-permeable channel (Galione, 2011). However, the molecular identity of this NAADP-regulated channel has proven controversial, with several candidate channel families being proposed without a common consensus being reached (Morgan et al, 2011; Guse, 2012; Marchant & Patel, 2013).
Therefore, the proposal that the two-pore channel (TPC) family are Ca2+-permeable channels regulated by NAADP was a promising development (Brailoiu et al, 2009; Calcraft et al, 2009; Zong et al, 2009); TPCs are endo-lysosomal channels with homologies to TRP (one-domain) and CaV (four-domain) channels, with a predicted intermediate two-domain structure that probably assembles as dimers (Rietdorf et al, 2011; Churamani et al, 2012). Although a three-gene family, several species, including mice and humans, only have Tpcn1 and Tpcn2 genes.
TPCs are emerging as physiologically important channels mediating NAADP signalling in diverse contexts, for example cell differentiation, angiogenesis, immune cell signalling, smooth muscle contraction, autophagy, and cardiovascular and liver physiology (Aley et al, 2010; Tugba Durlu-Kandilci et al, 2010; Esposito et al, 2011; Davis et al, 2012; Lu et al, 2013; Zhang et al, 2013; Favia et al, 2014; Grimm et al, 2014). Moreover, TPCs are the only known Ca2+-release channels in plants, where they mediate long-range Ca2+ waves (Choi et al, 2014).
Several lines of evidence from different groups support TPCs as NAADP-regulated channels with many of the expected properties: manipulation of TPC expression (by overexpression, RNAi or gene disruption) paralleled NAADP-dependent responses in multiple systems (Morgan & Galione, 2014), and NAADP-dependent currents were observed with both over-expressed TPCs and affinity-purified TPCs in lipid bilayers (Pitt et al, 2010, 2014; Rybalchenko et al, 2012), with single-organelle planar patch-clamp (Schieder et al, 2010) or with cells in which TPCs were re-directed to the plasma membrane (Brailoiu et al, 2010; Yamaguchi et al, 2011; Jha et al, 2014). Furthermore, recent studies have suggested that TPCs may not bind NAADP directly but rather require an accessory protein (Lin-Moshier et al, 2012; Walseth et al, 2012a,b) that co-immunoprecipitates with TPCs (Ruas et al, 2010; Walseth et al, 2012a).
Against this compelling body of evidence, recent papers challenged the status of TPCs as NAADP-regulated Ca2+-permeable channels by proposing that TPCs are instead Na+-selective channels activated by the phosphoinositide lipid PI(3,5)P2 (phosphatidylinositol 3,5-bisphosphate) but not by NAADP (Wang et al, 2012; Cang et al, 2013). Their conclusions were drawn from the use of a mouse line designed to knockout both Tpcn1 and Tpcn2 expression in combination with conventional patch-clamp of endo-lysosomes and Ca2+ imaging. However, whether these mice are bona fide TPC-null is open to debate as they have the potential to express ≥ 91% of the full-length TPC sequences (Morgan & Galione, 2014; Ruas et al, 2014).
In view of these conflicting findings, and given the emerging importance of NAADP and TPCs in cell signalling, it is a matter of urgency to rigorously define the relationship between TPCs and NAADP-regulated Ca2+ release. Therefore, we have generated and fully characterized a new transgenic mouse line with a demonstrable absence of both Tpcn1 and Tpcn2 expression. This has allowed us to examine for the first time the effect of loss of endogenous TPC1 and TPC2 proteins on single-cell Ca2+ release or native currents from single endo-lysosomes and the effects of their re-expression. Our data reaffirm that TPCs are essential for NAADP-induced Ca2+ signalling and NAADP-stimulated endo-lysosomal Ca2+-permeable currents, but are not essential for PI(3,5)P2-mediated currents.
Results
Generation of Tpcn1/2−/− mice with demonstrable lack of Tpcn1 and Tpcn2 expression
We generated a mouse line carrying Tpcn1T159 (Ruas et al, 2014) and Tpcn2YHD437 (Calcraft et al, 2009) mutant alleles (Fig1A and B) and have prepared mouse embryonic fibroblasts (MEF) from Tpcn1T159/Tpcn2YHD437 animals. RT–qPCR analysis revealed that MEFs express both Tpcn1 and Tpcn2 (Fig1C); no detectable levels of Tpcn1 or Tpcn2 mRNAs were observed in MEFs from Tpcn1T159/Tpcn2YHD437 animals, including a newly identified Tpcn1B isoform arising from an alternative promoter (Ruas et al, 2014) (Fig1D–G). Expression from the Tpcn mutant alleles in Tpcn1T159/Tpcn2YHD437 animals is predicted to result in production of only small portions of the N-terminal tails of the respective TPC proteins (Fig1E), corresponding to only the first 102 (for TPC1) or 20 (for TPC2) amino acid residues (≤ 12% of the full-length sequence). This contrasts with a mutant mouse line (developed by D. Ren and referred to hereafter as Tpcn1/2Dren) used in recent studies in which ≥ 91% of the full-length TPC sequence could be still expressed, that is 748 (for TPC1; equivalent to TPC1B) or 682 (for TPC2) amino acid residues (Wang et al, 2012; Cang et al, 2013) (see below and Fig 7A).
Figure 1.
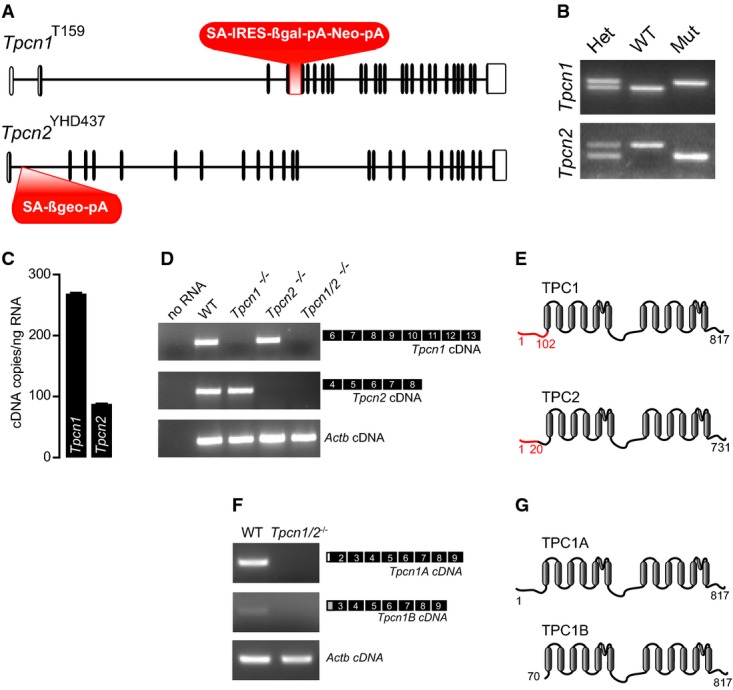
- Gene structure of Tpcn1T159 and Tpcn2YHD437 alleles in transgenic mice. Exons are represented as vertical segments (UTRs, unfilled boxes); knockout and gene trap cassettes are represented in red. Splice acceptor (SA), internal ribosomal entry site (IRES), β-galactosidase gene (βgal), neomycin resistance gene (Neo), β-galactosidase/neomycin resistance chimeric gene (βgeo), polyadenylation signal (pA).
- Genotyping results for homozygote wild-type (WT), homozygote mutant Tpcn1T159 or Tpcn2YHD437 (Mut), and heterozygote animals (Het).
- RT–qPCR analysis of absolute levels of Tpcn1 and Tpcn2 transcripts in WT MEFs. Tpcn1/Tpcn2 ratio of expression corresponds to 3.0; n = 6; mean ± SEM.
- RT–PCR analysis of Tpcn1 and Tpcn2 expression in MEFs from WT or homozygote transgenic embryos. Amplified cDNAs correspond to exons shown in black. Expression of Actb was used as a control.
- Two-domain organization of TPC1 and TPC2 proteins showing transmembrane helices (grey) and amino acid residues (numbers). Predicted residual expression of TPC proteins from transgenic animals is represented in red.
- RT–PCR analysis of Tpcn1A and Tpcn1B expression in MEFs from WT or Tpcn1/2−/− embryos. Amplified cDNAs correspond to the exons shown in black including isoform-specific 5′-UTRs (white box for Tpcn1A and grey box for Tpcn1B). Expression of Actb was used as a control.
- TPC1 protein variants expressed from Tpcn1A and Tpcn1B transcripts.
Figure 7.

- A Schematic representation of TPC2 protein with highlighted pore mutations.
- B Immunoblotting analysis of Tpcn1/2−/− (DKO) MEFs expressing mCherry-tagged mouse wild-type TPC2 (WT) and pore mutants N257A and E643A (mock, empty vector). Blot was probed for mCherry and for β-actin as a loading control.
- C, D Live-cell imaging of MEF cells expressing mCherry-tagged TPC2 mutant pore proteins (LTG, LysoTracker Green signal; mCh, mCherry signal). Scale bar, 100 μm (C; larger images are shown in Supplementary Fig S5) or 10 μm (D). Images in (C) were taken with the same acquisition settings as in Figs5C and 7C.
- E, F Representative fura-2 Ca2+ traces from DKO MEFs expressing mCherry-tagged TPC2 proteins (E) and maximum Ca2+ responses induced by 10 μM NAADP/AM (F); n = 311–413; ***P < 0.001, ns, P > 0.05 relative to DKO/DMSO using the ANOVA–Tukey test.
These results indicate unequivocally that the mice we have generated have knocked-out expression for both of the Tpcn genes, which we therefore refer to as Tpcn1/Tpcn2 double knockout (Tpcn1/2−/−).
NAADP induces Ca2+ release from acidic Ca2+ stores
MEFs were analysed for their ability to respond to NAADP. Cytosolic Ca2+ was monitored with fura-2, and NAADP was bath-applied as its cell-permeant ester form, NAADP/AM. In wild-type MEFs, NAADP/AM evoked robust Ca2+ signals which were inhibited by pre-treatment with bafilomycin A1, GPN, and nigericin, agents that deplete acidic Ca2+ stores, and by the NAADP antagonist trans-Ned-19 (Fig2A and B). This is consistent with NAADP releasing Ca2+ from endo-lysosomes.
Figure 2.
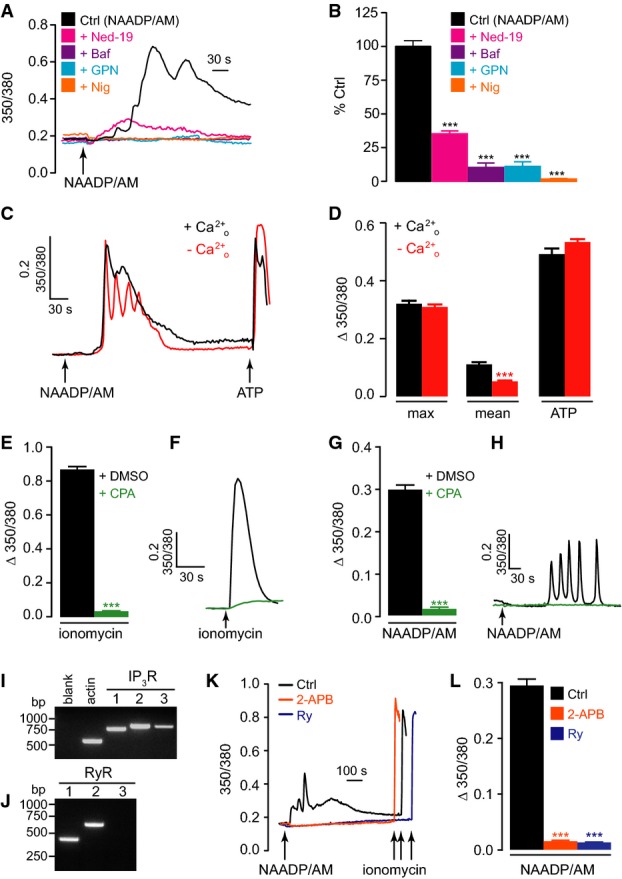
- A, B Representative single-cell Ca2+ traces showing 350/380 ratios of fura-2 fluorescence (A) and maximum Ca2+ changes (B) upon addition of 10 μM extracellular NAADP/AM in WT MEFs, which were blocked by pre-incubation with 10 μM trans-Ned-19 (Ned-19; 45 min), 1 μM bafilomycin A1 (Baf; 45 min), 5 μM nigericin (Nig; 30 min), or 200 μM GPN (5 min); control (Ctrl) was pre-incubated with DMSO (vehicle); n = 121–272; ***P < 0.001 relative to control using the ANOVA-Tukey test.
- C, D Ca2+ signals with 10 μM NAADP/AM in 1.8 mM extracellular Ca2+ (+ Ca2+o) or Ca2+-free medium containing 100 μM EGTA (− Ca2+o) in WT MEFs. (C) Representative single-cell fura-2 Ca2+ traces upon addition of 10 μM NAADP/AM and 100 μM ATP. (D) Maximum Ca2+ changes (max) and mean Ca2+ release over a period of 300 s post-addition of 10 μM NAADP/AM;n = 233–385 cells; ***P < 0.001 relative to + Ca2+o using an unpaired t-test.
- E–H Cells treated with 200 μM CPA or 0.1% DMSO in medium + Ca2+o for 50 min. Cells were then briefly washed and maintained in Ca2+-free medium (+100 µM EGTA) in which they were stimulated with 2 μM ionomycin (E, F) or 10 μM NAADP/AM (G, H). Maximum Ca2+ changes (E, G) and representative single-cell fura-2 Ca2+ traces (F, H); n = 49–148 cells; ***P < 0.001 relative to DMSO control, using the unpaired t-test.
- I, J MEFs express all three IP3 receptor subtypes (IP3R 1–3) and ryanodine receptor (RyR) types 1 and 2, detected by RT–PCR analysis. Blank refers to no RNA. Positive control for expression for RyR type 3 is shown in Supplementary Fig S1.
- K, L Cells treated with 2 μM 2-APB, 20 μM ryanodine, or 0.1% DMSO prior to application of 10 μM NAADP/AM. Representative single-cell fura-2 Ca2+ traces (K) and maximum Ca2+ changes (L); n = 142–374; ***P < 0.001 relative to control using the unpaired t-test.
To ascertain whether Ca2+ influx contributed to the NAADP response, we repeated experiments in Ca2+-free medium (Fig2C and D). The maximum amplitude of the NAADP-induced Ca2+ release was unaffected by removing external Ca2+ confirming that this early phase of the response is entirely due to intracellular Ca2+ release. That the mean Ca2+ response was, overall, somewhat reduced in Ca2+-free medium (Fig2C and D) suggested that Ca2+ influx played a role in sustaining the response but that it was not essential for NAADP action.
The long-standing “trigger hypothesis” describes NAADP as a provider of an initial “trigger” of Ca2+ that is subsequently amplified by Ca2+ release from the ER by virtue of the Ca2+ sensitivity of the IP3 receptor (IP3R) or ryanodine receptor (RyR), that is, Ca2+-induced Ca2+ release (CICR). We confirmed the co-involvement of the ER in several ways, first by depleting the ER with the Ca2+-ATPase inhibitor cyclopiazonic acid (CPA) (Fig2E and F), which abrogated NAADP/AM responses (Fig2G and H). Given that IP3R1–3 and RyR1–2 were all detected by RT–PCR in our WT MEFs (Fig2I and J and Supplementary Fig S1), we tested which ER channel families were functionally important; IP3R and RyR blockade with 2-APB (2-aminoxydiphenylborate) and ryanodine, respectively, abolished NAADP/AM-stimulated Ca2+ signals (Fig2K and L). NAADP-induced responses in the well-characterized pancreatic acinar cell exhibit a similar pharmacology (Cancela et al, 1999). Together with the fact that NAADP required acidic Ca2+ stores (Fig2A and B), these data are consistent with the trigger hypothesis whereby NAADP provides the trigger Ca2+ from acidic stores that is subsequently amplified by IP3Rs and/or RyRs on the ER (Churchill & Galione, 2000).
TPC knockout abrogates NAADP-induced Ca2+ signals
Using MEFs obtained from TPC knockout animals, we tested the requirement of TPCs for NAADP-induced Ca2+ signals. In WT MEFs, NAADP/AM evoked robust Ca2+ signals (Fig3A–E) that were approximately 40% of the amplitude of that evoked by the purinergic agonist ATP (Fig3D). In single-knockout MEFs lacking either TPC1 or TPC2, the NAADP responses were still present but significantly reduced in terms of the maximum amplitude or the mean Ca2+ signal (Fig3A–C); TPC2 knockout also affected NAADP responses in macrophages derived from adult mice (Supplementary Fig S2), a cell type in which it was recently argued that TPCs were NAADP insensitive (Wang et al, 2012). Critically, in Tpcn1/2−/− MEFs, NAADP responses were eliminated while ATP responses remained robust (Fig3A–E). Note that the effects of TPC ablation cannot be due to altered Ca2+ influx because the peak responses to NAADP are independent of Ca2+ entry (Fig2D).
Figure 3.

- A–C Representative single-cell fura-2 Ca2+ traces (A), maximum Ca2+ changes (B), and mean Ca2+ release over a period of 500 s (C), post-addition of 10 μM NAADP/AM to wild-type (WT), Tpcn1−/− (TPC1 KO), Tpcn2−/− (TPC2 KO), and Tpcn1/2−/− (DKO) MEFs. Control corresponds to WT cells treated with DMSO;n = 384–621; ***P < 0.001 relative to WT using the ANOVA–Tukey test.
- D, E Maximum amplitude (D) and mean Ca2+ (E) of the responses to different NAADP/AM concentrations in WT and DKO cells. The subsequent maximum response to 100 μM ATP (cf. (A)) after each NAADP/AM concentration is also plotted (D). 1 μM NAADP/AM induced a maximal Ca2+ peak corresponding to 39 ± 3% of the 100 μM ATP response; n = 41–105.
- F Maximum Ca2+ responses to 200 μM GPN or 10 μM nigericin; n = 111–285; P > 0.05 (ns) relative to WT using the ANOVA–Tukey test.
- G Endo-lysosomal luminal pH (pHL) by endocytosed fluorescently labelled dextrans in primary MEFs determined by single-cell measurements; n = 105 for WT or DKO.
Next, we checked whether TPC disruption simply shifted the NAADP concentration–response curve; in WT cells, addition of NAADP/AM over a wide range of concentrations produced the bell-shaped curve (Fig3D and E), that is a characteristic of mammalian NAADP-regulated Ca2+ signalling (Galione, 2011), and although Tpcn1/2−/− cells responded well to ATP, there was no response to NAADP at any concentration tested (Fig3D and E).
Finally, we checked Ca2+ storage and luminal pH (pHL) within the endo-lysosomal system, either of which could potentially affect NAADP-induced Ca2+ release (Pitt et al, 2010, 2014; Schieder et al, 2010; Rybalchenko et al, 2012; Wang et al, 2012). The lack of NAADP-induced Ca2+ release in Tpcn1/2−/− cells was not due to an absence of releasable Ca2+ because lysosomotropic agents evoked similar Ca2+ signals when compared to WT cells (Fig3F). Similarly, the pHL measured across the entire endo-lysosomal system was unaffected as determined by ratiometric pHL recordings (Fig3G and Supplementary Fig S3).
Taken together, these data indicate that TPC1 and TPC2 contribute to NAADP-evoked Ca2+ signalling and that removing both TPCs eradicates the ability of cells to respond to NAADP by directly affecting Ca2+ release, not endo-lysosomal Ca2+ storage or pHL.
TPCs are required for NAADP-evoked endo-lysosomal currents
Although the above data suggest that TPCs are essential for NAADP-induced Ca2+ signals, they do not explicitly demonstrate the activation of Ca2+-permeable channels on endo-lysosomes by NAADP. Therefore, we monitored native currents by planar patch-clamp of single whole endo-lysosomes swollen with vacuolin-1 and purified from WT or TPC knockout MEFs; importantly, such swelling does not affect NAADP-induced Ca2+ signalling (Supplementary Fig S4). In the presence of K+ and Ca2+ (but in the absence of Na+), cytosolic nanomolar concentrations of NAADP stimulated an inward current (lumen to cytoplasm) (Fig4A and B) with a reversal potential of +75 ± 7 mV, in WT MEFs. This is consistent with Ca2+ being the major permeant ion under these conditions (equilibrium potentials, EK = −16 mV, ECa = +73 mV). Importantly, NAADP-induced currents were undetectable in similar preparations from Tpcn1/2−/− and Tpcn2−/− cells, while they were still present (reversal potential of +75 ± 4 mV) in preparations from Tpcn1−/− cells (Fig4A and B). This implicates TPCs as the predominant Ca2+-permeant channels in endo-lysosomes regulated by NAADP, but largely carried by TPC2 in MEFs under our conditions.
Figure 4.
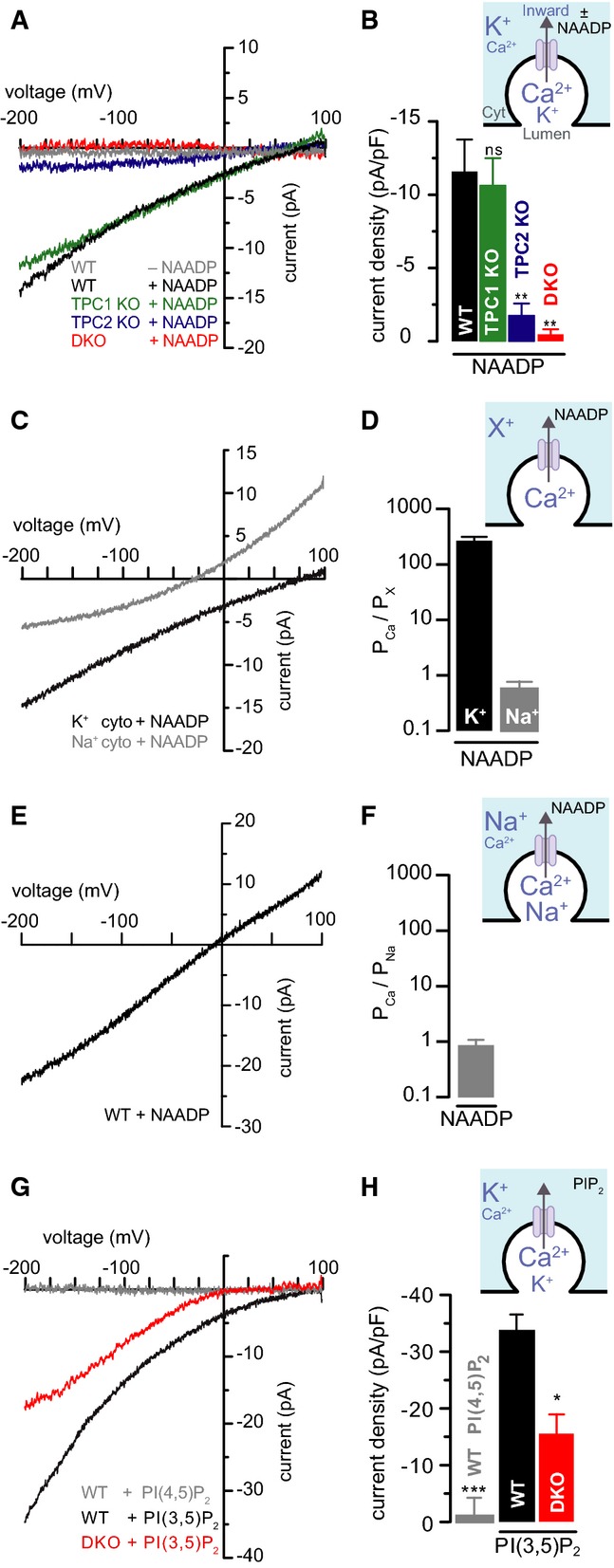
- A, B Single-lysosome currents from wild-type (WT), Tpcn1−/− (TPC1 KO), Tpcn2−/− (TPC2 KO), and Tpcn1/2−/− (DKO) MEFs in the presence or absence of cytosolic NAADP (50 nM); solutions contained Ca2+ (cytosol: 0.2 mM; lumen: 60 mM) plus K+ (cytosol: 130 mM; lumen: 70 mM). Inward currents are defined as lumen-to-cytosol. (A) Representative current–voltage (I–V) curves from single isolated lysosomes. (B) Population data were measured at −200 mV from (A); n = 5–6; ns, P > 0.05, **P < 0.01 relative to WT using the ANOVA-Tukey test.
- C, D NAADP (50 nM)-evoked single-lysosome currents from WT MEFs under bi-ionic conditions: 160 mM monovalent “X+” (either K+ or Na+) in the cytosol and 107 mM Ca2+ in the lumen. (C) Representative I–V curves from isolated lysosomes. (D) Population data of the relative Ca2+/monovalent permeability ratios; n = 9–11.
- E, F Single-lysosome currents from WT MEFs in the presence of cytosolic NAADP (50 nM); solutions contained Ca2+ (cytosol: 0.2 mM; lumen: 61 mM) and Na+ (cytosol: 160 mM; lumen: 70 mM). (E) Representative I–V curve from single lysosomes derived from WT MEFs. (F) Population data of the relative Ca2+/Na+ permeability ratios; n = 6.
- G, H Single-lysosome currents from WT or DKO MEFs in the presence of cytosolic PI(3,5)P2 (10 μM) or PI(4,5)P2 (10 μM); solutions contained Ca2+ (cytosol: 0.2 mM; lumen: 60 mM) plus K+ (cytosol: 130 mM; lumen: 70 mM). (G) Representative I–V curves from single lysosomes derived from WT or DKO MEFs. (H) Population data were measured at −200 mV from (G); n = 3–4; ***P < 0.001, *P < 0.05 relative to WT/PI(3,5)P2 using Student's t-test.
In view of recent proposals that TPCs also conduct Na+ (Wang et al, 2012; Cang et al, 2013, 2014; Boccaccio et al, 2014; Jha et al, 2014; Pitt et al, 2014), we quantified the ion selectivity of TPCs in our preparation, by performing experiments under bi-ionic conditions (luminal Ca2+, cytosolic monovalent). With cytosolic K+, the reversal potential was +76 ± 2 mV which equates to a PCa/PK permeability ratio of 268 ± 47 (Fig4C and D). By contrast, with Na+ as the monovalent ion, the reversal potential was −22 ± 5 mV which equates to a PCa/PNa permeability ratio of 0.57 ± 0.19 (Fig4C and D).
Additionally, we measured the relative Ca2+ permeability in the presence of luminal Na+. Because seal formation requires luminal Ca2+, currents were necessarily recorded with both Ca2+ and Na+ in the lumen. Under these conditions, NAADP stimulated an inward current with a reversal potential of −3.8 ± 2.9 mV (equilibrium potentials, ENa = −21 mV, ECa = +73 mV), which equates to a permeability ratio PCa/PNa of 0.86 ± 0.22 (Fig4E and F). Therefore, the permeability ratio was the same irrespective of whether Na+ was just cytosolic or on both sides of the membrane.
These results demonstrate that the permeability of TPCs to Na+ and Ca2+ is of the same order of magnitude, thus differing from the proposal that TPCs are highly Na+-selective channels (Wang et al, 2012; Cang et al, 2013, 2014). In other words, the NAADP-stimulated current displays a rank order of selectivity of Na+ ≥ Ca2+ ≫ K+. Furthermore, these results suggest that NAADP-induced Ca2+ currents are mediated by endogenous TPCs and not by other proposed NAADP-activated endo-lysosomal channels such as TRPML1 (Zhang et al, 2009) or TRPM2, the latter being activated by NAADP at much higher concentrations [EC50 100–730 μM (Lange et al, 2008)].
The endo-lysosome-specific lipid, PI(3,5)P2, has been reported to regulate both TRPML1 (Dong et al, 2010) and TPC channels (Wang et al, 2012; Cang et al, 2013, 2014; Boccaccio et al, 2014; Grimm et al, 2014; Jha et al, 2014; Pitt et al, 2014). In WT endo-lysosomes, robust Ca2+ currents (reversal potential +70 ± 10 mV) were stimulated by PI(3,5)P2, whereas PI(4,5)P2 was without effect (Fig4G and H). Interestingly, PI(3,5)P2-stimulated currents were still seen in Tpcn1/2−/− endo-lysosomes, but were reduced (Fig4G and H), which suggests that both TPC-dependent and TPC-independent currents are modulated by the lipid; indeed, the residual TPC-independent currents unmasked in Tpcn1/2−/− endo-lysosomes were markedly inwardly rectifying with a reversal potential of −6 ± 13 mV and therefore consistent with TRPML1-mediated K+ currents (EK = −16 mV) (Dong et al, 2010).
Together, these data indicate that while NAADP-induced endo-lysosomal currents are wholly dependent on TPCs, PI(3,5)P2-induced currents can also be mediated by other endo-lysosomal channels as may be predicted for a permissive lipid endo-lysosomal channel modulator (Cang et al, 2014).
TPC expression rescues NAADP-induced Ca2+ release in Tpcn1/2−/− MEFs
To confirm that the loss of NAADP responsiveness in Tpcn1/2−/− MEFs was due to the specific lack of TPCs, we restored expression of TPCs and assessed NAADP-induced Ca2+ responses. Thus, Tpcn1/2−/− MEFs were transduced with lentiviruses for expression of either mouse TPC1 or TPC2 tagged with a C-terminal mCherry. Immunoblot analysis confirmed that transduction resulted in expression of TPC1 and TPC2 (Fig5A) and live-cell fluorescence verified that they were expressed in all cells (Fig5C and Supplementary Fig S5) with the expected pattern of localization; while TPC1 shows a more modest co-localization with LysoTracker Green and consistent with recycling endosomes (Calcraft et al, 2009; Ruas et al, 2014), TPC2 shows a strong co-localization with LysoTracker Green, indicative of late endosomal/lysosomal localization, as confirmed by other endo-lysosomal markers (Fig5D and Supplementary Fig S6).
Figure 5.
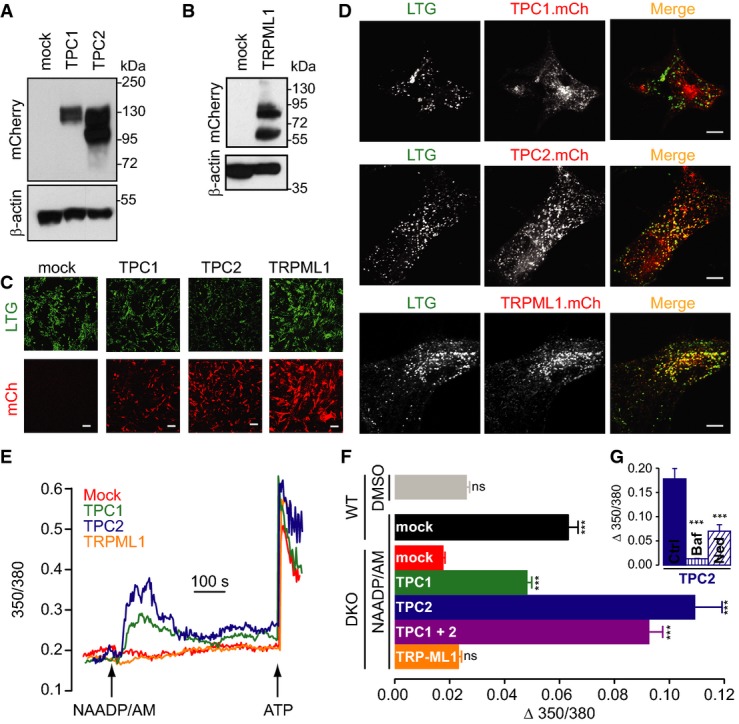
- A, B Immunoblotting analysis of Tpcn1/2−/− (DKO) MEFs expressing mCherry-tagged mouse TPC1 and TPC2 (mock, empty vector) (A) or mouse TRPML1 (B). The top half of the blot was probed for mCherry and the bottom half for β-actin as a loading control.
- C, D Live-cell imaging of MEF cells expressing mCherry-tagged proteins (LTG, LysoTracker Green signal; mCh, mCherry signal). Scale bar, 100 μm (C; larger images are shown in Supplementary Fig S5) or 10 μm (D). Images in (C) were taken with the same acquisition settings as in Figs6C and 7C.
- E, F Representative fura-2 Ca2+ traces from DKO MEFs expressing mCherry-tagged proteins (E) and maximum Ca2+ responses induced by 10 μM NAADP/AM (F); DMSO represents control for NAADP/AM addition; n = 137–468; ***P < 0.001, ns, P > 0.05 relative to DKO/mock using the ANOVA–Tukey test.
- G Maximum Ca2+ responses induced by 10 μM NAADP/AM in TPC2-transduced DKO MEFs are inhibited by pre-incubation with 1 μM bafilomycin A1 (Baf; 45 min) or 10 μM trans-Ned-19 (Ned-19; 45 min); n = 63–113; ***P < 0.001 relative to control (0.1% DMSO) using the ANOVA–Tukey test.
We then examined NAADP-induced Ca2+ signals in Tpcn1/2−/− MEFs after re-expression of TPC proteins and compared them to responses in mock-transduced cells. We observed no NAADP-induced Ca2+ signals in mock-transduced Tpcn1/2−/− cells, comparable to DMSO alone in WT cells (the vehicle control for NAADP/AM; Fig5E and F). Strikingly, re-expression of either TPC1 or TPC2 in Tpcn1/2−/− MEFs restored NAADP responsiveness, with TPC2 being the more efficient (Fig5E and F) and restoring Ca2+ responses beyond those observed in mock-transduced WT cells (Fig5F). Co-expression of both TPCs had no greater effect than TPC2 alone (Fig5F). Importantly, the Ca2+ responses observed in TPC2-rescued cells exhibit the expected pharmacology: they were inhibited by bafilomycin A1 and trans-Ned-19 (Fig5G). Additionally, the rescue was specific to TPCs because expression of the Ca2+-permeable endo-lysosomal TRPML1 in Tpcn1/2−/− MEFs (Fig5B–F) failed to have any effect, further arguing against its being an NAADP-regulated channel (Pryor et al, 2006; Yamaguchi et al, 2011).
Pore-mutant TPCs fail to rescue NAADP-induced Ca2+ release
To ascertain whether TPCs rescue NAADP responses in Tpcn1/2−/− MEFs by acting as Ca2+-permeable channels, we generated lentiviruses for expression of TPC2-containing point mutations that affect permeability of the channel to Ca2+: N257A acts as a pore-dead mutant, whereas E643A has a reduced Ca2+ selectivity (Schieder et al, 2010) (Fig6A). Both mutants of TPC2 were expressed at similar levels and in the same endo-lysosomal compartments as wild-type TPC2 (Fig6B–D). While expression of wild-type TPC2 completely restored NAADP responses in Tpcn1/2−/− MEFs, neither of the TPC2 mutants was able to rescue the response (Fig6E and F). This suggests that TPC2 must not only be a functional channel to restore NAADP action but one with a sufficient permeability to Ca2+.
Figure 6.

- A Schematic representation of TPC1 and TPC2 proteins corresponding to full-length (FL) and N-terminal truncations (ΔN) predicted to be expressed in the mutant Tpcn1/2Dren mice used in Cang et al (2014, 2013) and Wang et al (2012). Transmembrane helices are represented by vertical blocks, and numbers represent amino acid residues.
- B Immunoblotting analysis of Tpcn1/2−/− (DKO) MEFs expressing mCherry-tagged mouse TPC1 and TPC2 and full-length (FL) and N-terminal truncations (ΔN). Blot was probed for mCherry and for β-actin as a loading control. Further immunoblots from PNGase F-treated samples are shown in Supplementary Fig S7.
- C, D Live-cell imaging of MEF cells expressing mCherry-tagged proteins (LTG, LysoTracker Green signal; mCh, mCherry signal). Scale bar, 100 μm (C; larger images are shown in Supplementary Fig S5) or 10 μm (D). Images in (C) were taken under the same acquisition parameters as in Figs5C and 6C.
- E, F Representative fura-2 Ca2+ traces from DKO MEFs expressing mCherry-tagged proteins (mock, empty vector) (E) and maximum Ca2+ responses induced by 10 μM NAADP/AM (F); n = 171–224; ***P < 0.001 relative to mock whereas †††P < 0.001 comparing FL to ΔN using the ANOVA–Tukey test.
- G Comparison of number of responding cells to NAADP/AM treatment for each set of transduced DKO MEF cells. Only a cell showing a maximum NAADP/AM-induced Ca2+ response greater than the standard deviation of the basal 350/380 ratio for its set was considered as a responder; ***P < 0.001 relative to mock whereas †††P < 0.001 comparing FL to ΔN using contingency tables.
N-terminally truncated TPCs rescue NAADP-induced Ca2+ release
In the recent studies challenging TPCs as NAADP-regulated Ca2+-permeable channels, the Tpcn1 and Tpcn2 gene disruptions present in the Tpcn1/2Dren line were proposed to potentially result in expression of truncated, dysfunctional versions of TPC1 and TPC2 (Wang et al, 2012; Cang et al, 2013). However, that they were indeed dysfunctional was not confirmed at the level of cytosolic Ca2+ signals, and so we generated and tested the self-same N-terminal truncated forms of mouse TPC1 or TPC2 in which only the first 69 or 49 respective amino acid residues are missing (Fig7A); it is important to note that ΔN69-TPC1 is equivalent to TPC1B, a protein predicted to be translated from a naturally occurring Tpcn1B isoform (Ruas et al, 2014) (Fig1F and G). The maximum expression level attained with either truncated form was lower than their full-length counterparts (Fig7B and C and Supplementary Fig S7), but nonetheless they were endo-lysosomal, showing a strong co-localization with LysoTracker Green (Fig7D). In spite of the lower expression, each truncated TPC remained able to rescue NAADP responsiveness, both in amplitude of Ca2+ signals (50–65% of that seen with their full-length equivalents) and in the number of responding cells (70–100% of transduced cells) (Fig7E–G).
These data raise doubts about whether the Tpcn1/2Dren mice used in the previous studies (Wang et al, 2012; Cang et al, 2013, 2014) were TPC-null animals, and this may explain why preparations from pancreatic islets from these animals still retained NAADP-induced Ca2+ signals (Wang et al, 2012).
Tpcn1/2−/− mouse liver retains high-affinity NAADP-binding proteins
Recent studies using a radiolabelled NAADP photoaffinity probe identified putative NAADP-binding proteins in several cell preparations (Lin-Moshier et al, 2012; Walseth et al, 2012a,b) that interact with TPCs and show high-affinity specific binding to NAADP (Ruas et al, 2010; Walseth et al, 2012a). Based on their apparent molecular weights, which are lower than those predicted for TPCs and on results from transgenic mouse lines with gene trap insertions in either Tpcn1 or Tpcn2 genes, it was suggested that these proteins were distinct from TPCs and that an accessory NAADP-binding protein confers regulation by NAADP (Lin-Moshier et al, 2012; Walseth et al, 2012a). However, the conclusive proof that NAADP binding does not require TPC proteins demands the analysis of tissue with complete absence of both TPC1 and TPC2 proteins.
We therefore compared NAADP binding in mouse liver from WT or Tpcn1/2−/− mice, using a [32P]NAADP-binding assay. Liver was chosen, as we have previously shown that this tissue shows high levels of NAADP binding (Calcraft et al, 2009). Quantitative RT–PCR revealed that in liver from WT mice both Tpcn1 and Tpcn2 are expressed, albeit at different levels, with Tpcn1 mRNA being approximately 40-fold more abundant than Tpcn2 mRNA (Fig8A). As expected, Tpcn1 and Tpcn2 mRNAs were not detected in liver preparations from Tpcn1/2−/− animals (Fig8B).
Figure 8.
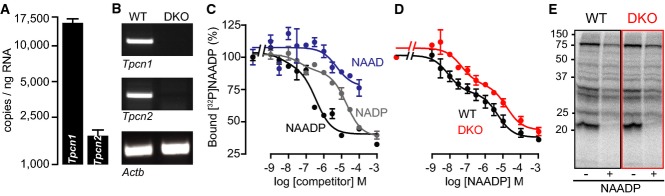
- A RT–qPCR analysis of absolute levels of Tpcn1 and Tpcn2 transcripts in liver from WT animals. Tpcn1/Tpcn2 ratio of expression corresponds to 43.9; n = 6.
- B RT–PCR analysis of Tpcn1 and Tpcn2 expression in wild-type (WT) or Tpcn1/2−/− (DKO) liver preparations. Expression of Actb was used as a control. Amplified cDNA regions correspond to the same exons as in Fig1D.
- C, D [32P]NAADP-binding assay with competition by NAADP and NAADP-related dinucleotides performed with liver homogenates from WT (C, D) or DKO (D) animals. Data are expressed as values relative to total binding performed in the absence of unlabelled dinucleotide. (D) The IC50 values for the high-affinity binding site were WT: 13.7 ± 7.5 nM and DKO: 146.3 ± 60.5 nM (P > 0.09) and for the low-affinity site WT: 6.9 ± 4.1 μM and DKO: 27.4 ± 8.3 μM (P > 0.08); N (number of animals) = 5–7; n (number of binding reactions) = 10–14.
- E Photoaffinity labelling of liver homogenates from WT or DKO animals performed with [32P]5N3-NAADP in the presence or absence of unlabelled NAADP (1 μM).
[32P]NAADP binding with unlabelled NAADP competition performed in liver homogenates from WT animals shows the characteristic binding curve revealing two populations of binding sites (Calcraft et al, 2009) (Fig8C and D) with higher affinity for NAADP when compared to related pyridine dinucleotides such as NADP or NAAD (Fig8C). Importantly, [32P]NAADP binding was retained in similar preparations from Tpcn1/2−/− animals and showed similar IC50 values for both the high-affinity and low-affinity binding sites (Fig8D). Furthermore, photoaffinity labelling of NAADP-binding proteins in liver homogenates carried out using [32P]5N3-NAADP revealed no differences in the pattern of specifically labelled proteins, as assessed by competition with unlabelled NAADP (Fig8E).
Together, the data indicate that high-affinity NAADP binding does not require TPCs and support the hypothesis that an auxiliary NAADP-binding protein confers NAADP regulation.
Discussion
In spite of compelling evidence from different groups (Morgan & Galione, 2014), recent studies have challenged the fundamental premise that TPCs are essential components of the NAADP-regulated channel, either by putting forward other target channels (Zhang et al, 2009; Guse, 2012) or, more recently, by suggesting that TPCs are lipid-activated Na+-selective channels entirely dispensable for NAADP action (Wang et al, 2012).
In view of such contentions, we have investigated the role of TPCs in NAADP-dependent signalling in embryonic fibroblasts from Tpcn1/2−/− mice that we have developed (the first demonstrable TPC1/2-null system). This has allowed us to express various channels on a null background, record endogenous endo-lysosomal TPC currents, and image Ca2+ signals in the same cell type, permitting a direct comparison of results.
TPCs are essential effectors of NAADP action
Our data overwhelmingly suggest that TPCs are essential for NAADP-induced Ca2+ signalling. We conclude this because: (i) NAADP-dependent Ca2+ responses were eliminated in Tpcn1/2−/− cells whereas Ca2+ storage, pHL, and PI(3,5)P2 responsiveness were preserved; (ii) NAADP responses were selectively rescued by TPCs and not by another Ca2+-permeant endo-lysosomal channel, TRPML1 (Zong et al, 2009; Dong et al, 2010; Yamaguchi et al, 2011); and (iii) eradication of NAADP-regulated Ca2+ signalling in Tpcn1/2−/− cells cannot be explained by incidental loss of NAADP-binding proteins since they are still present in Tpcn1/2−/− preparations. Our data thus reinforce conclusions reached in our previous studies where NAADP responses were abrogated in cells from Tpcn2−/− mice (Calcraft et al, 2009; Tugba Durlu-Kandilci et al, 2010).
Although clearly essential, are TPCs actually activated by NAADP? Our electrophysiological recordings suggest that they are. NAADP-evoked currents were robust in planar patch-clamp recordings of single endo-lysosomes from WT but undetectable in Tpcn1/2−/− or Tpcn2−/− preparations. Hence, TPC activation is relatively direct and not secondary to NAADP-induced changes in membrane potential since recordings were carried out under voltage-clamp. Under these conditions, TPC2 appears to be the predominant NAADP-activated channel; we do not currently understand why endogenous TPC1 does not contribute currents in this system (as evidenced from Tpcn1−/− and Tpcn2−/− preparations), even though TPC1 supports NAADP-induced Ca2+ release as we have shown in the rescue experiments; it is possible that TPC1-decorated endosomes are simply absent from the organelle preparation or its coupling to NAADP is less robust and lost upon purification.
Our recordings differ from those of the recent papers in several key ways: first, we successfully observed NAADP-stimulated currents in endo-lysosomal preparations, which mirrors previous work (Zhang et al, 2009; Pitt et al, 2010, 2014; Schieder et al, 2010; Rybalchenko et al, 2012; Grimm et al, 2014; Jha et al, 2014), whereas others, surprisingly, could not detect NAADP-dependent currents (irrespective of TPC expression) (Wang et al, 2012; Cang et al, 2013). Second, the scale of endo-lysosomal currents is different: our endogenous NAADP-dependent currents are in the pA range, whereas lipid-stimulated currents were in the nA range in other studies (Wang et al, 2012; Cang et al, 2013; Jha et al, 2014).
It is unlikely that the ability to observe NAADP-induced currents is a function of the patch-clamp technique used; others using a conventional patch-clamp technique have also been able to record NAADP-stimulated currents in endo-lysosomal preparations (Jha et al, 2014). However, it is possible that under some experimental conditions, necessary components of the NAADP-regulatory pathway are lost and/or inhibitory factors such as Mg2+ or TPC phosphorylation state (Jha et al, 2014) are more prevalent.
Validity of Tpcn knockout mouse models
The recent conclusion that TPCs are not activated by NAADP (Wang et al, 2012) arose from the assumption that the Tpcn1/2Dren mice were TPC-null, but we raise doubts as to whether their mice were true knockouts. First, no mRNA or protein expression data were presented. Second, these mice may still express functional, shorter TPC variants as we shall now discuss.
The authors' Cre-Lox strategy excised exons 1 and 2 of Tpcn1 and exon 1 of Tpcn2 (Wang et al, 2012; Cang et al, 2013, 2014), thereby removing the initiating ATG codon. Consequently, as the authors conceded, N-terminally truncated proteins (≥ 91% of the full-length sequence) could still be produced via initiation of translation at a downstream ATG codon (positions 70 and 50 for TPC1 and TPC2, respectively). Although these variants were dismissed as inactive channels on the basis of their PI(3,5)P2 insensitivity (Wang et al, 2012), we clearly show that these ΔN69-TPC1 or ΔN49-TPC2 proteins are functional in response to NAADP; these proteins correctly localized to endo-lysosomes (see also Ruas et al, 2014) and supported NAADP-induced Ca2+ signals in our Tpcn1/2−/− MEFs.
Moreover, the expression of truncated TPCs can indeed occur physiologically; at least for Tpcn1, there is an alternative promoter downstream of exon 2 (Ruas et al, 2014), and mRNA for this novel shorter variant Tpcn1B (which gives rise to TPC1B, equivalent to ΔN69-TPC1) is present in MEFs from WT mice (but not in MEFs from our Tpcn1/2−/− mice).
The presence of either (or both) of these shorter functional TPC proteins in the Tpcn1/2Dren mice (Wang et al, 2012; Cang et al, 2013, 2014) would mean that they are not bona fide Tpcn1/2 double knockouts; these studies could potentially be misleading in their claims that TPCs are not essential for NAADP-evoked Ca2+ signals.
TPCs as Ca2+-permeable channels
Another recent controversy has been whether TPCs are Ca2+-permeable channels (Wang et al, 2012; Cang et al, 2013), despite different groups describing TPCs as permeant to Ca2+, or to Ca2+ surrogates, in lipid bilayers (Pitt et al, 2010, 2014; Rybalchenko et al, 2012), single-organelle planar patch-clamp (Schieder et al, 2010), or TPCs targeted to the plasma membrane (Brailoiu et al, 2010; Yamaguchi et al, 2011; Jha et al, 2014). By necessity, such experiments relied on TPC over-expression, but it is unclear whether heterologous expression truly replicates the properties of endogenous TPCs, a known complication in the TRP or Orai channel fields where different expression levels influence channel regulation, oligomerization states, and, crucially, ion selectivity (Putney, 2004; Thompson & Shuttleworth, 2013).
We conclude that TPCs are indeed Ca2+ permeant from multiple lines of evidence. First, Ca2+ fluxes through TPCs are critical for supporting NAADP-induced Ca2+ release because mutant TPC2 channels with a reduced or negligible Ca2+ permeability (Schieder et al, 2010) fail to rescue NAADP responses in Tpcn1/2−/− cells. Importantly, the E643A mutant is a proven active cation channel—albeit with an altered selectivity filter (Schieder et al, 2010)—providing evidence that cation fluxes per se are not enough to support NAADP responses and that a sufficient Ca2+ flux is required. This is further underscored by the lack of rescue by another cation channel, TRPML1.
More direct evidence for Ca2+ permeability came from endo-lysosomal patch-clamp studies. Fortuitously, the endogenous endo-lysosomal NAADP-stimulated currents in MEFs are larger than those of endogenous currents in non-transfected HEK293 cells used previously (Schieder et al, 2010), allowing us to directly address whether endogenous TPCs are permeant to Ca2+. We recorded whole-lysosome native currents with Ca2+ and K+ in the lumen: in a mixed solution protocol, NAADP-stimulated currents exhibited a high permeability of Ca2+ over K+ with a reversal potential of +75 mV that was in excellent agreement with the equilibrium potential calculated for Ca2+ (ECa +73 mV); under bi-ionic conditions, the PCa/PK was quantified as ∼270. These indicate that, under these conditions, endogenous mouse TPCs are highly selective for Ca2+ over K+, in agreement with our previous results with mouse channels (Schieder et al, 2010; Grimm et al, 2014).
By contrast, the overall NAADP-stimulated TPC current is less discriminatory between Na+ and Ca2+ with a PCa/PNa of 0.6–0.8. Therefore, we agree that TPCs are permeable to Na+ (Wang et al, 2012; Cang et al, 2013, 2014; Boccaccio et al, 2014; Grimm et al, 2014; Jha et al, 2014; Pitt et al, 2014), but under our experimental conditions, we still observe a comparable Ca2+ flux. The simplest explanation of our data is that TPCs are Ca2+-permeable cation channels (and not highly Na+-selective), which broadly agrees with other studies showing permeability of mammalian TPCs to various cations such as K+, Cs+, Ba2+, Ca2+, Na+ and H+ (Brailoiu et al, 2010; Pitt et al, 2010, 2014; Schieder et al, 2010; Yamaguchi et al, 2011; Rybalchenko et al, 2012; Boccaccio et al, 2014; Grimm et al, 2014; Jha et al, 2014).
The alternative model for NAADP-induced Ca2+ release states that any stimulation of NAADP-induced Ca2+ release by TPCs could be an indirect consequence of TPC-mediated Na+ fluxes (Wang et al, 2012; Cang et al, 2013). However, such Na+ currents would inhibit Ca2+ release by depolarizing endo-lysosomes and reducing the electrochemical gradient for Ca2+ (Morgan & Galione, 2014). To accommodate TPCs as Na+-selective channels in NAADP-induced Ca2+ release would require a more complex circuit, for example involving voltage-gated or Na+-stimulated Ca2+-permeable channels (Morgan & Galione, 2014) for which there is currently no electrophysiological evidence. Moreover, NAADP signalling does not appear to require Na+ because it evokes a robust Ca2+ release from sea urchin egg homogenates in Na+-free media (Genazzani et al, 1997).
Taken together, we conclude that endogenous TPCs act as Ca2+-permeable channels stimulated by NAADP, consistent with the original model (Brailoiu et al, 2009; Calcraft et al, 2009; Zong et al, 2009) and that they are not Na+-selective counter-ion current facilitators.
Modulation by PI(3,5)P2
Recent reports demonstrated that TPCs, like TRPML1, are regulated by the endo-lysosome-specific lipid, PI(3,5)P2 (Dong et al, 2010; Wang et al, 2012; Cang et al, 2013; Boccaccio et al, 2014; Jha et al, 2014; Pitt et al, 2014), and our data agree with this conclusion: PI(3,5)P2 stimulated robust Ca2+-permeable endo-lysosomal currents, and the lipid-stimulated currents were reduced in Tpcn1/2−/− MEFs, consistent with a TPC-dependent component of the PI(3,5)P2 response. The residual PI(3,5)P2-stimulated current is attributable to other endogenous channels, a likely candidate being TRPML1 given the characteristic inward rectifying curve (Dong et al, 2010).
Therefore, PI(3,5)P2 activates multiple channel families such as TPCs, TRPML1 and RyR (Dong et al, 2010; Touchberry et al, 2010; Wang et al, 2012; Feng et al, 2014) consistent with its being a permissive lipid factor [analogous to PI(4,5)P2 in the plasma membrane (Suh & Hille, 2008)], whereas NAADP effects on endo-lysosomes appear to be uniquely dependent upon one channel family, the TPCs.
TPCs and NAADP binding
Recent studies suggest that NAADP may not bind to TPCs directly but via a smaller molecular weight NAADP-binding protein(s) (Lin-Moshier et al, 2012; Walseth et al, 2012a,b) that co-immunoprecipitates with TPCs as part of a channel complex (Ruas et al, 2010; Walseth et al, 2012a). However, it is difficult to rule out whether NAADP binds TPCs directly, because in a previous study, single Tpcn1 or Tpcn2 knockout mice were used and the gene disruption strategy used to generate them meant that large portions of TPC proteins could potentially still be produced (Lin-Moshier et al, 2012). Therefore, the use of our bona fide double TPC1/2-null system has allowed us to conclude that TPCs are not required for high-affinity NAADP binding, as judged by crude homogenate binding studies or photoaffinity radiolabelling of mouse liver proteins, with the caveat that low-abundance TPCs may not be detected via photoaffinity labelling and/or if other more abundant NAADP-binding proteins (not related to its Ca2+-release properties) mask any TPC contribution.
In conclusion, the use of the first demonstrable TPC double-knockout mice affirms TPCs as Ca2+-permeable channels that are absolutely required for NAADP-stimulated Ca2+ signalling and supports PI(3,5)P2 as a non-selective modulator of endo-lysosomal channels. Expression of various channels in this TPC-null background reinforces this conclusion in demonstrating that only Ca2+-permeable TPCs can rescue NAADP signals. Our data contradict recent assertions that TPCs are NAADP-insensitive Na+-selective channels and establish TPCs as NAADP-regulated Ca2+-permeable channels.
Materials and Methods
Generation of Tpcn1/2−/− mice
Homozygote Tpcn1T159 (mutant allele nomenclature: Tpcn1tm1Dgen) mice (Ruas et al, 2014) carrying a targeted disruption of exons 4 to 5 were obtained from the European Mouse Mutagenesis Archive (EMMA) and were used with homozygote Tpcn2YHD437 (mutant allele nomenclature: Tpcn2Gt(YHD437)Byg) mice (Calcraft et al, 2009) for dihybrid crosses to generate mice carrying knocked-out expression for both Tpcn1 and Tpcn2 genes. The genotyping of animals was performed on DNA extracted from ear biopsies using the following primers: Tpcn1 (Intron 4F: CTGGCATCTTGAGGTTTGGT; Intron 5 R: GGGCTACACTCCCAAGCATA; KO cassette F: CCAGCTCATTCCTCCCACTC; WT product size: 376 bp; Mut product size: 459); Tpcn2 (Intron 1F: CTTCGGAGCCTTCTTTCCTT; Intron 1 R: CTGTCCCTGACGAGTGGTTT; Gene trap cassette F: GTCGGGGCTGGCTTAACTATG; WT product size: 493 bp; Mut product size: 336). Reaction products were analysed by agarose gel electrophoresis. Mice with genotype corresponding to Tpcn1T159/Tpcn2YHD437 were born at the expected Mendelian proportion (8/126; 6.35%).
Gene expression analysis
For analysis of gene expression, RNA was extracted following an RNeasy QiaRNA extraction procedure (Qiagen) with an in-column DNase I treatment. One-step RT–PCR was performed in a reaction containing extracted total RNA, SuperScript III RT/Platinum Taq High Fidelity Enzyme Mix (Invitrogen), and gene-specific primers: Tpcn1 (F: ATTTTCCTGGTGGACTGTCG; R: CAGAGCAGCGACTTCGTAAA; product size: 606 bp); Tpcn2 (F: GGGCTTCATCATTTTCCTGA; R: TTGTTGGAAGTCGTCAGCAG; product size: 564 bp); Actb (F: TGTTACCAACTGGGACGACA; R: AAGGAAGGCTGGAAAAGAGC; product size: 573 bp). Reaction products were analysed by agarose gel electrophoresis.
For RT–qPCR, cDNA was synthesized from RNA using high-capacity cDNA Reverse Transcription kit (Applied Biosystems). cDNA was subjected to qPCR using gene-specific, intron-flanking primers for Tpcn1 (F: CTGTCCTCTGGATGGAACCT; R: TCCATGTTGAGCGTCAGTG) and Tpcn2 (F: CCCTGGCTGTATACCGATTG; R: GTCCCAGAGCGACAGTGG) with Universal Probes (#95 for Tpcn1 and #106 for Tpcn2) in a Light Cycler 480 System (Roche). cDNA copy numbers were determined against a standard curve using a custom-made double-stranded DNA fragment containing the amplicon sequences for Tpcn1 and Tpcn2 (GeneArt Strings, Life Technologies).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde in PBS and permeabilized/blocked with 0.1% saponin/5% goat serum in PBS (a methanol permeabilization step was included for anti-PDI labelling). Antibody incubations were performed in PBS/0.01% saponin/5% goat serum. The primary antibodies used were anti-RFP (rat monoclonal 5F8; antibodies-online.com), anti-mCherry (mouse monoclonal 1C51; Novus Biologicals), anti-Lamp1 (rat monoclonal 1D4B; DSHB), anti-TfR (mouse monoclonal H68.4; Invitrogen), anti-EEA1 (rabbit monoclonal, C45B10; Cell Signalling Technology), and anti-PDI (rabbit monoclonal, C81H6; Cell Signalling Technology). The secondary antibodies used were derived from goat serum, cross-absorbed, and conjugates of Alexa 488 (for organelle markers) or Alexa 546 (for mCherry) (Invitrogen). Cells were viewed on a Zeiss 510 META confocal microscope, in multitrack mode, using the following excitation/emission parameters (nm): Alexa 488 (488/505–530) and Alexa 546 (543/>560).
Intracellular Ca2+ measurements
MEFs were loaded with the ratiometric Ca2+ indicator Fura 2-AM and where indicated pre-treated with pharmacological agents before addition of NAADP/AM, followed by ATP. The maximum amplitude and the mean [Ca2+] were calculated on a single-cell basis. Further details are given in Supplementary Materials and Methods.
Lysosomal currents
Whole-lysosome planar patch-clamp recordings were performed in vacuolin-enlarged lysosomes from MEFLTAs isolated using differential centrifugation (Schieder et al, 2010). The planar patch-clamp technology combined with a pressure control system (Port-a-Patch, Nanion Technologies) was applied as previously described (Schieder et al, 2010). Currents were recorded at room temperature (21–23°C) using an EPC-10 patch-clamp amplifier and PatchMaster acquisition software (HEKA). Data were digitized at 40 kHz and filtered at 2.8 kHz. Seal resistance was 1–3 GΩ, and the mean endo-lysosomal capacitance was 0.82 ± 0.06 pF (n = 27). Inward currents are defined as ion movement from the endo-lysosomal lumen to cytoplasm (Bertl et al, 1992).
For experiments using mixed Ca2+/K+ solutions, the cytoplasmic solution contained 60 mM KF, 70 mM K-MSA (methanesulfonate), 0.2 mM Ca-MSA, and 10 mM HEPES (pH adjusted with KOH to 7.2); luminal solution was 70 mM K-MSA, 60 mM Ca-MSA, 1 mM MgCl2, and 10 mM HEPES (pH adjusted with MSA to 4.6). Mannitol was used to adjust osmolarity.
For experiments using mixed Ca2+/Na+ solutions, the cytoplasmic solution contained 60 mM NaF, 100 mM Na-MSA, 0.2 mM Ca-MSA, 5 mM Hepes, and 5 mM MES (pH adjusted with NaOH to 7.2). Luminal solution was 70 mM Na-MSA, 60 mM Ca-MSA, 1 mM CaCl2, 5 mM Hepes and 5 mM MES (pH 4.6).
For the bi-ionic experiments, the cytoplasmic solution contained 60 mM KF, 100 mM K-MSA, 5 mM Hepes, and 5 mM MES (pH 7.2 with KOH), whereas the luminal solution was 105 mM Ca-MSA, 2 mM CaCl2, 5 mM Hepes, and 5 mM MES (pH 4.6). For Na+ experiments, all K+ salts were replaced by their equimolar Na+ version.
Currents in the absence of NAADP (or phosphoinositides) were subtracted from the currents in the presence of these stimulators as previously described (Schieder et al, 2010). Water-soluble diC8-PIP2, PI(3,5)P2, and PI(4,5)P2 were from A.G. Scientific. NAADP was from Tocris Bioscience.
Radioligand binding assays
[32P]NAADP was incubated with liver homogenate samples adsorbed to nitrocellulose filters and bound radionucleotide detected and quantified by phosphor imaging. Further details are given in Supplementary Materials and Methods.
Photoaffinity labelling
Liver homogenate samples were photo-labelled with [32P-5N3]NAADP and proteins separated by SDS–PAGE. Signal from dried gels was detected by phosphor imaging. Further details are given in Supplementary Materials and Methods.
Statistical analysis
Data are presented as mean ± SEM and analysed by Student's t-test or a one-way ANOVA (with Tukey–Kramer, Dunnett's, or Kruskal–Wallis post-tests) where appropriate and significance determined as P < 0.05. Graphs were usually annotated using the following conventions: P > 0.05 (ns), P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***). The number of responding cells (Fig7) was assessed by multiple 2 × 2 contingency tables (Fisher's exact test) with the significance threshold (α) corrected to α′ using α′ = α/[2(c−1)] where c = number of columns and significance therefore only accepted when P < 0.00625.
Acknowledgments
This work was supported by grants from The Wellcome Trust (084101/Z/07/Z), MRC (G0901521), NIH (R15 GM100444), Bavarian Research Foundation (DKO-125-10), and Deutsche Forschungsgemeinschaft (SFB TRR 152 TP04, TP06, and TP12). AG is a Wellcome Trust Senior Investigator (Ref: 102828/Z/13/Z). FMP is a Royal Society Wolfson Research Merit Award holder. We thank Dr. Grant Churchill for advice and help on the NAADP/AM synthesis. We thank the Oxford Biomedical Science Building staff for animal housing and care.
Author contributions
MR performed Tpcn expression, MEF preparation, lentiviral expression and localization studies, immunoblotting, and radioligand binding assays; LCD performed RyR and IP3R expression, macrophage preparation, intracellular Ca2+ measurements, and luminal pH determinations; CCC, CGr, CWS, and MB performed lysosomal-current experiments; and CGa performed NAADP/AM and [32P]NAADP synthesis. MR, LCD, AJM, and TP designed experiments and analysed data; KTC performed RT–PCR and RT–qPCR experiments; TFW performed photoaffinity labelling; NP and FMP supervised macrophage work; and JP and AG were responsible for overall supervision. MR, LCD, AJM, JP, and AG wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Materials and Methods
Source Data for Supplementary Figure S1
Source Data for Supplementary Figure S2
Source Data for Supplementary Figure S7
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
References
- Aley PK, Mikolajczyk AM, Munz B, Churchill GC, Galione A, Berger F. Nicotinic acid adenine dinucleotide phosphate regulates skeletal muscle differentiation via action at two-pore channels. Proc Natl Acad Sci USA. 2010;107:19927–19932. doi: 10.1073/pnas.1007381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Bertl A, Blumwald E, Coronado R, Eisenberg R, Findlay G, Gradmann D, Hille B, Köhler K, Kolb HA, MacRobbie E. Electrical measurements on endomembranes. Science. 1992;258:873–874. doi: 10.1126/science.1439795. [DOI] [PubMed] [Google Scholar]
- Boccaccio A, Scholz-Starke J, Hamamoto S, Larisch N, Festa M, Gutla PVK, Costa A, Dietrich P, Uozumi N, Carpaneto A. The phosphoinositide PI(3,5)P2 mediates activation of mammalian but not plant TPC proteins: functional expression of endolysosomal channels in yeast and plant cells. Cell Mol Life Sci. 2014;71:4275–4283. doi: 10.1007/s00018-014-1623-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, Patel S. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186:201–209. doi: 10.1083/jcb.200904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu E, Rahman T, Churamani D, Prole DL, Brailoiu GC, Hooper R, Taylor CW, Patel S. An NAADP-gated two-pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J Biol Chem. 2010;285:38511–38516. doi: 10.1074/jbc.M110.162073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang K-T, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancela JM, Churchill GC, Galione A. Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature. 1999;398:74–76. doi: 10.1038/18032. [DOI] [PubMed] [Google Scholar]
- Cang C, Zhou Y, Navarro B, Seo Y-J, Aranda K, Shi L, Battaglia-Hsu S, Nissim I, Clapham DE, Ren D. mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell. 2013;152:778–790. doi: 10.1016/j.cell.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang C, Bekele B, Ren D. The voltage-gated sodium channel TPC1 confers endolysosomal excitability. Nat Chem Biol. 2014;10:463–469. doi: 10.1038/nchembio.1522. [DOI] [PubMed] [Google Scholar]
- Choi W-G, Toyota M, Kim S-H, Hilleary R, Gilroy S. Salt stress-induced Ca2+ waves are associated with rapid, long-distance root-to-shoot signaling in plants. Proc Natl Acad Sci USA. 2014;111:6497–6502. doi: 10.1073/pnas.1319955111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churamani D, Hooper R, Brailoiu E, Patel S. Domain assembly of NAADP-gated two-pore channels. Biochem J. 2012;441:317–323. doi: 10.1042/BJ20111617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GC, Galione A. Spatial control of Ca2+ signaling by nicotinic acid adenine dinucleotide phosphate diffusion and gradients. J Biol Chem. 2000;275:38687–38692. doi: 10.1074/jbc.M005827200. [DOI] [PubMed] [Google Scholar]
- Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A. NAADP mobilizes Ca2+ from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell. 2002;111:703–708. doi: 10.1016/s0092-8674(02)01082-6. [DOI] [PubMed] [Google Scholar]
- Davis LC, Morgan AJ, Chen J-L, Snead CM, Bloor-Young D, Shenderov E, Stanton-Humphreys MN, Conway SJ, Churchill GC, Parrington J, Cerundolo V, Galione A. NAADP activates two-pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr Biol. 2012;22:2331–2337. doi: 10.1016/j.cub.2012.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito B, Gambara G, Lewis AM, Palombi F, D'Alessio A, Taylor LX, Genazzani AA, Ziparo E, Galione A, Churchill GC, Filippini A. NAADP links histamine H1 receptors to secretion of von Willebrand factor in human endothelial cells. Blood. 2011;117:4968–4977. doi: 10.1182/blood-2010-02-266338. [DOI] [PubMed] [Google Scholar]
- Favia A, Desideri M, Gambara G, D'Alessio A, Ruas M, Esposito B, Bufalo DD, Parrington J, Ziparo E, Palombi F, Galione A, Filippini A. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2–dependent Ca2+ signaling. Proc Natl Acad Sci USA. 2014;111:E4706–E4715. doi: 10.1073/pnas.1406029111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Huang Y, Lu Y, Xiong J, Wong C-O, Yang P, Xia J, Chen D, Du G, Venkatachalam K, Xia X, Zhu MX. Drosophila TRPML Forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane. J Biol Chem. 2014;289:4262–4272. doi: 10.1074/jbc.M113.506501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galione A. NAADP receptors. Cold Spring Harb Perspect Biol. 2011;3:a004036. doi: 10.1101/cshperspect.a004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galione A. A primer of NAADP-mediated Ca2+ signalling: from sea urchin eggs to mammalian cells. Cell Calcium. 2014;58:27–47. doi: 10.1016/j.ceca.2014.09.010. [DOI] [PubMed] [Google Scholar]
- Genazzani AA, Mezna M, Dickey DM, Michelangeli F, Walseth TF, Galione A. Pharmacological properties of the Ca2+-release mechanism sensitive to NAADP in the sea urchin egg. Br J Pharmacol. 1997;121:1489–1495. doi: 10.1038/sj.bjp.0701295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C, Holdt LM, Chen C-C, Hassan S, Müller C, Jörs S, Cuny H, Kissing S, Schröder B, Butz E, Northoff B, Castonguay J, Luber CA, Moser M, Spahn S, Lüllmann-Rauch R, Fendel C, Klugbauer N, Griesbeck O, Haas A, et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat Commun. 2014;5 doi: 10.1038/ncomms5699. : Article number: 4699. [DOI] [PubMed] [Google Scholar]
- Guse AH. Linking NAADP to ion channel activity: a unifying hypothesis. Sci Signal. 2012;5:pe18. doi: 10.1126/scisignal.2002890. [DOI] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S. Convergent regulation of the lysosomal two-pore channel-2 by Mg2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO J. 2014;33:501–511. doi: 10.1002/embj.201387035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange I, Penner R, Fleig A, Beck A. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium. 2008;44:604–615. doi: 10.1016/j.ceca.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, Brailoiu E, Patel S, Marchant JS. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem. 2012;287:2296–2307. doi: 10.1074/jbc.M111.305813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Hao B-X, Graeff R, Wong CWM, Wu W-T, Yue J. Two-pore channel 2 (TPC2) inhibits autophagosomal-lysosomal fusion by alkalinizing lysosomal pH. J Biol Chem. 2013;288:24247–24263. doi: 10.1074/jbc.M113.484253. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Marchant JS, Patel S. Questioning regulation of two-pore channels by NAADP. Messenger. 2013;2:113–119. doi: 10.1166/msr.2013.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AJ, Platt FM, Lloyd-Evans E, Galione A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J. 2011;439:349–374. doi: 10.1042/BJ20110949. [DOI] [PubMed] [Google Scholar]
- Morgan AJ, Galione A. Two-pore channels (TPCs): current controversies. BioEssays. 2014;36:173–183. doi: 10.1002/bies.201300118. [DOI] [PubMed] [Google Scholar]
- Pitt SJ, Funnell TM, Sitsapesan M, Venturi E, Rietdorf K, Ruas M, Ganesan A, Gosain R, Churchill GC, Zhu MX, Parrington J, Galione A, Sitsapesan R. TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+ J Biol Chem. 2010;285:35039–35046. doi: 10.1074/jbc.M110.156927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt SJ, Lam AKM, Rietdorf K, Galione A, Sitsapesan R. Reconstituted human TPC1 is a proton-permeable ion channel and is activated by NAADP or Ca2+ Sci Signal. 2014;7:ra46. doi: 10.1126/scisignal.2004854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor PR, Reimann F, Gribble FM, Luzio JP. Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic. 2006;7:1388–1398. doi: 10.1111/j.1600-0854.2006.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW., Jr The enigmatic TRPCs: multifunctional cation channels. Trends Cell Biol. 2004;14:282–286. doi: 10.1016/j.tcb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Rietdorf K, Funnell TM, Ruas M, Heinemann J, Parrington J, Galione A. Two-pore channels form homo- and heterodimers. J Biol Chem. 2011;286:37058–37062. doi: 10.1074/jbc.C111.289835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruas M, Rietdorf K, Arredouani A, Davis LC, Lloyd-Evans E, Koegel H, Funnell TM, Morgan AJ, Ward JA, Watanabe K, Cheng X, Churchill GC, Zhu MX, Platt FM, Wessel GM, Parrington J, Galione A. Purified TPC isoforms form NAADP receptors with distinct roles for Ca2+ signaling and endolysosomal trafficking. Curr Biol. 2010;20:703–709. doi: 10.1016/j.cub.2010.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruas M, Chuang K-T, Davis LC, Al-Douri A, Tynan PW, Tunn R, Teboul L, Galione A, Parrington J. TPC1 has two variant isoforms and their removal has different effects on endo-lysosomal functions compared to loss of TPC2. Mol Cell Biol. 2014;34:3981–3992. doi: 10.1128/MCB.00113-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalchenko V, Ahuja M, Coblentz J, Churamani D, Patel S, Kiselyov K, Muallem S. Membrane potential regulates nicotinic acid adenine dinucleotide phosphate (NAADP) dependence of the pH- and Ca2+-sensitive organellar two-pore channel TPC1. J Biol Chem. 2012;287:20407–20416. doi: 10.1074/jbc.M112.359612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieder M, Rötzer K, Brüggemann A, Biel M, Wahl-Schott CA. Characterization of two-pore channel 2 (TPCN2)-mediated Ca2+ currents in isolated lysosomes. J Biol Chem. 2010;285:21219–21222. doi: 10.1074/jbc.C110.143123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B-C, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL, Shuttleworth TJ. How many Orai's does it take to make a CRAC channel? Sci Rep. 2013;3:1961. doi: 10.1038/srep01961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchberry CD, Bales IK, Stone JK, Rohrberg TJ, Parelkar NK, Nguyen T, Fuentes O, Liu X, Qu C-K, Andresen JJ, Valdivia HH, Brotto M, Wacker MJ. Phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) potentiates cardiac contractility via activation of the ryanodine receptor. J Biol Chem. 2010;285:40312–40321. doi: 10.1074/jbc.M110.179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugba Durlu-Kandilci N, Ruas M, Chuang K-T, Brading A, Parrington J, Galione A. TPC2 proteins mediate nicotinic acid adenine dinucleotide phosphate (NAADP)- and agonist-evoked contractions of smooth muscle. J Biol Chem. 2010;285:24925–24932. doi: 10.1074/jbc.M110.129833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walseth TF, Lin-Moshier Y, Jain P, Ruas M, Parrington J, Galione A, Marchant JS, Slama JT. Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide phosphate (NAADP)-binding proteins in sea urchin egg. J Biol Chem. 2012a;287:2308–2315. doi: 10.1074/jbc.M111.306563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walseth TF, Lin-Moshier Y, Weber K, Marchant JS, Slama JT, Guse AH. Nicotinic acid adenine dinucleotide 2′-phosphate (NAADP) binding proteins in T-lymphocytes. Messenger. 2012b;1:86–94. doi: 10.1166/msr.2012.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong X, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D, Xu H. TPC proteins are phosphoinositide-activated sodium-selective ion channels in endosomes and lysosomes. Cell. 2012;151:372–383. doi: 10.1016/j.cell.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Jha A, Li Q, Soyombo AA, Dickinson GD, Churamani D, Brailoiu E, Patel S, Muallem S. Transient receptor potential mucolipin 1 (TRPML1) and two-pore channels are functionally independent organellar ion channels. J Biol Chem. 2011;286:22934–22942. doi: 10.1074/jbc.M110.210930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Jin S, Yi F, Li P-L. TRP-ML1 functions as a lysosomal NAADP-sensitive Ca2+ release channel in coronary arterial myocytes. J Cell Mol Med. 2009;13:3174–3185. doi: 10.1111/j.1582-4934.2008.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z-H, Lu Y-Y, Yue J. Two-pore channel 2 differentially modulates neural differentiation of mouse embryonic stem cells. PLoS ONE. 2013;8:e66077. doi: 10.1371/journal.pone.0066077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rötzer K, Griesbeck O, Harz H, Biel M, Wahl-Schott C. The two-pore channel TPCN2 mediates NAADP-dependent Ca2+-release from lysosomal stores. Pflugers Arch. 2009;458:891–899. doi: 10.1007/s00424-009-0690-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Materials and Methods
Source Data for Supplementary Figure S1
Source Data for Supplementary Figure S2
Source Data for Supplementary Figure S7
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8