

Abstract
Chronic neuroinflammation is evident in brain aging and neurodegenerative disorders and is often associated with excessive nitric oxide (NO) production within the central nervous system (CNS). Under such conditions, increased NO levels are observed at the choroid plexus (CP), an epithelial layer that forms the blood–cerebrospinal fluid barrier (BCSFB) and serves as a selective gateway for leukocyte entry to the CNS in homeostasis and following injury. Here, we hypothesized that elevated cerebral NO levels interfere with CP gateway activity. We found that induction of leukocyte trafficking determinants by the CP and sequential leukocyte entry to the CSF are dependent on the CP epithelial NFκB/p65 signaling pathway, which was inhibited upon exposure to NO. Examining the CP in 5XFAD transgenic mouse model of Alzheimer's disease (AD-Tg) revealed impaired ability to mount an NFκB/p65-dependent response. Systemic administration of an NO scavenger in AD-Tg mice alleviated NFκB/p65 suppression at the CP and augmented its gateway activity. Together, our findings identify cerebral NO as a negative regulator of CP gateway activity for immune cell trafficking to the CNS.
Keywords: BCSFB, choroid plexus, NFκB, nitric oxide
Introduction
Chronic neuroinflammation is a common feature in brain aging and in numerous neurological disorders. One of the hallmarks of the neuroinflammatory response is the elevation of toxic molecules within the territory of the central nervous system (CNS). As such, excessive production of nitric oxide (NO) has been implicated in the pathophysiology of a number of neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's diseases (PD), and amyotrophic lateral sclerosis (ALS) (Smith et al, 1997; Aliyev et al, 2004; Duncan & Heales, 2005; Brown, 2007). In their activated state, glial cells, such as microglia and astrocytes, express inducible forms of nitric oxide synthase and are a major source of NO in the brain (Bal-Price et al, 2002); thus, under neurodegenerative conditions, chronic glial stimulation is associated with overwhelming levels of NO species in the CNS parenchyma and the cerebrospinal fluid (CSF) (Qureshi et al, 1995; Tohgi et al, 1999; Taskiran et al, 2000; Moss & Bates, 2001; Liu et al, 2002; Block et al, 2007).
Circulating immune cells play a pivotal role in controlling the local inflammatory response within the CNS in both acute and chronic CNS pathologies, though their recruitment to the inflamed brain parenchyma is often delayed or suboptimal (Butovsky et al, 2006, 2007; Simard et al, 2006; Trivedi et al, 2006; Beers et al, 2008; Town et al, 2008; Shechter et al, 2009; Mildner et al, 2011; Vaknin et al, 2011; Falcao et al, 2012; Raposo et al, 2014). Using an experimental model of acute spinal cord injury, our group recently showed that recruitment of inflammation-resolving monocyte-derived macrophages (mo-MΦ) to the CNS is orchestrated through a remote gateway (Shechter et al, 2013)—the brain's choroid plexus (CP), which forms the blood–CSF barrier (BCSFB); we further suggested that suboptimal trafficking of immune cells via the CP might be an underlying mechanism shared in the pathophysiology of neurodegenerative conditions (Schwartz & Baruch, 2014b).
The CP epithelium is exposed to brain-derived signals from its apical side, via the CSF, and to peripheral signals from its basal side, via the circulation (Johanson et al, 2011; Ransohoff & Engelhardt, 2012; Baruch et al, 2014). Specifically, the activity of the CP in supporting immune cell trafficking through this gate was found to be dependent on a local synergistic effect between tumor necrosis factor (TNF)α and immune cell-derived interferon (IFN)γ, whereby TNFα was suggested to act as a ‘danger’ signal emerging from the brain parenchyma under neuroinflammatory conditions (Kunis et al, 2013). Here, we proposed that this reaction is mediated by an epithelial NFκB response and that under neurodegenerative conditions, this signaling pathway might be inhibited. We further hypothesized that one such inhibitor is NO (Matthews et al, 1996; Marshall & Stamler, 2001), the levels of which were reported to be elevated at the CP of AD transgenic mice (AD-Tg) and in human patients (Vargas et al, 2010).
In the present study, we demonstrate that CP gateway activity for leukocyte trafficking to the CSF involves epithelial NFκB/p65 nuclear translocation, which was experimentally suppressed upon exposure to NO. We further show that in AD-Tg mice, elevated NO levels at the CP were associated with epithelial cytoplasmic retention of the NFκB/p65 subunit. Systemic administration of an NO scavenger enabled CP epithelial NFκB/p65 nuclear translocation, induced expression of CP leukocyte trafficking determinants, and was associated with enhanced recruitment of mo-MΦ to the brain parenchyma.
Results
Intracerebroventricular administration of TNFα induces leukocyte trafficking to the CSF via the CP
Intracerebroventricular (ICV) injections of TNFα were previously shown to elicit an inflammatory response throughout the brain, which was associated with T-cell accumulation at the CP (Xu et al, 2010). Here, we first tested whether TNFα, as a cerebroventricular signal, would suffice to induce leukocyte trafficking to the CSF. To this end, we examined by flow cytometry the CSF cellular composition in mice, 24 h following ICV administration of TNFα in escalating dosages, in comparison with two control groups, mice that were ICV-injected with PBS and untreated mice (Fig1A–D). ICV administration of 100 ng or 150 ng of TNFα resulted in significantly higher leukocyte numbers in the CSF (Fig1B), including CD4+ T cells (Fig1C) and CD11b+ monocytes/neutrophils (Fig1D), compared to either PBS-injected or untreated mice.
Figure 1.

- A Gating strategy and representative flow cytometry plots of CD4+ and CD11b+ leukocytes (CD45+) in CSF aspirated from mice, 24 h following ICV injection of TNFα (100 ng/mouse), or from naïve mice (n = 4 per group; SSC, side scatter; FSC, forward scatter).
- B–D Quantitative analysis of leukocytes (total CD45+ leukocytes (B); CD4+ and CD11b+ (C, D) cells out of total CD45+ cells) in the CSF, 24 h following ICV injection of 50, 100, or 150 ng of TNFα (n = 4 per group; cells per μl; one-way ANOVA followed by Newman–Keuls post hoc test).
- E Representative flow cytometry plots and fraction analysis of the kinetics of CSF cellular composition during the first 24 h following ICV administration of TNFα (100 ng/mouse; n = 4 per group).
- F, G Representative microscopic images of the brain's third ventricle (3v), immunostained for the myeloid markers, Mac-2 or IBA-1, together with cytokeratin or E-cadherin, 24 h following ICV injection of TNFα (100 ng/mouse; n = 5 per group; scale bar, 50 μm; inserts show PBS-injected controls and higher magnification of the boxed area; arrows indicate Mac-2-positive myeloid cells).
Next, we examined the dynamics of leukocyte infiltration to the CSF following ICV administration of TNFα, by analyzing the CSF cellular composition at different time points. Starting from 4 h following the injection, we observed infiltration of neutrophils (CD11bhigh) to the CSF, which in the following hours (8 h, 12 h, 24 h) gradually shifted toward a preponderance of monocytes (CD11bint) and CD4+ T cells (Fig1E). Immunohistochemical analysis confirmed the accumulation of Mac-2+ and IBA-1+ macrophages at the CP and the adjacent ventricular spaces (Fig1F and G), indicating their trafficking through the CP-CSF migratory pathway (Shechter et al, 2013). These results demonstrated that as a cerebroventricular signal, TNFα is capable of inducing orchestrated and sequential entry of different leukocyte subsets to the CSF, and encouraged us to further use TNFα to experimentally stimulate CP activity.
Nitric oxide represses NFκB/p65 nuclear translocation in choroid plexus epithelial cells
NO acts as a non-canonical repressor of the NFκB signaling pathway (Matthews et al, 1996; Marshall & Stamler, 2001). Taken together with the fact that CP epithelial cells express TNFα receptor I (TNF-R1) (Kunis et al, 2013), which classically channels its signaling cascade through the NFκB pathway (Li & Lin, 2008), we hypothesized that exposure to increased levels of NO, as evident in the pathophysiology of AD (Fernandez-Vizarra et al, 2004; Nathan et al, 2005; Vargas et al, 2010; Kummer et al, 2011), might interfere with the ability of the CP to respond to TNFα. Testing this possibility in vitro, CP epithelial cells were cultured for 3 days to establish a confluent monolayer and then treated for 48 h with the NO donor, DETA/NONOate (henceforth, DETA), or left untreated (schematically depicted in Fig2A). Following 48 h of preconditioning with NO, the cultures were stimulated for 10 min with 20 ng/ml TNFα (a dose corresponding to the linear range of the dose-dependent response curve of the CP to this cytokine; Supplementary Fig S1) and were immediately dissociated into a single-cell suspension and fixed for intracellular immunostaining of NFκB/p65, cytokeratin, and Hoechst nuclear staining. Translocation of the p65 subunit was quantitatively examined using high-throughput single-cell flow cytometry image analysis (ImageStream) (Maguire et al, 2011). While DETA pre-treatment alone did not affect p65 subunit cellular localization, it completely inhibited its TNFα-induced nuclear translocation (Fig2B and C). Assessing p65 nuclear translocation in undissociated CP epithelium cultured monolayers by immunohistochemical analysis further confirmed this effect (Supplementary Fig S2).
Figure 2.
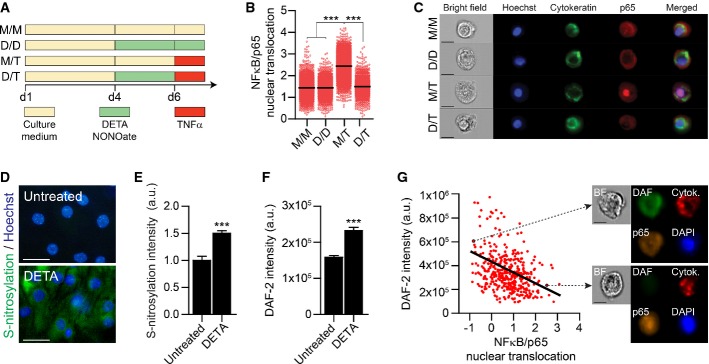
- A Schematic representation of the experimental groups. CP epithelial cells were cultured for 4 days (reaching confluence). On day 4, groups were either treated with the NO donor, DETA/NONOate (150 μM) or left untreated. On day 6, the groups were either stimulated with TNFα (20 ng/ml) or left untreated. Next, cell cultures were dissociated into a single-cell suspension and fixed for intracellular staining (M/M, medium only; D/D, DETA/NONOate only; M/T, medium followed by TNFα; D/T, DETA/NONOate followed by TNFα).
- B Similarity index between NFκB/p65 and nuclear (Hoechst) localization, which is a measure of NFκB/p65 nuclear translocation, examined by ImageStream (see Materials and Methods) (n > 2,000 cells per group; one-way ANOVA followed by Newman–Keuls post hoc test).
- C Representative images of NFκB/p65 cellular localization in the different experimental groups (cytokeratin in green; p65 in red; Hoechst nuclear staining in blue; scale bar, 10 μm).
- D, E Representative visualization (D) and quantitative analysis (E) of S-nitrosylated proteins in CP epithelial cells cultures, which were either exposed to DETA/NONOate or left untreated (S-nitrosylation in green, Hoechst nuclear staining in blue) (scale bar, 25 μm; n = 10 per group; Student's t-test).
- F Quantitative analysis of intracellular NO, measured by flow cytometry of DAF-2 DA florescence intensity, in DETA/NONOate-treated and untreated cells (n > 2,000 cells per group; Student's t-test).
- G Negative correlation between NFκB/p65 nuclear translocation and intracellular NO, as assessed by DAF-2 DA florescence intensity, on a single-cell level (Pearson's r = −0.2968, P < 0.0001). Arrows indicate representative images of the cellular spatial localization of NFκB/p65 (in orange), DAF-2 DA (in green), cytokeratin (in red), and Hoechst nuclear staining (in blue), as well as a brightfield (BF) overview of the cell.
To test whether the effect of exogenous NO on CP epithelial cells is mediated via post-translational protein modifications, we performed a biotin-switch assay, enabling visualization of nitrosative modifications on proteins and found increased levels of S-nitrosylation following DETA exposure (Fig2D and E). Next, to evaluate the causal relationship between NO and nuclear translocation of NFκB/p65 in CP epithelial cells, we used DAF-2 DA, a detector of intracellular NO (Kojima et al, 1998). Using the same experimental paradigm described above, we found that DETA pre-treated cells exhibited accumulation of intracellular NO (Fig2F), which negatively correlated with nuclear translocation of the p65 subunit (Fig2G). Together, these data indicated that both on the single-cell level and in undissociated tissues, exposure of the CP to NO could repress NFκB/p65 signaling pathway.
Nitric oxide inhibits induction of leukocyte trafficking determinants by the CP
Transepithelial migration of immune cells across the CP requires the expression of integrin ligands and chemokines by the epithelium (Szmydynger-Chodobska et al, 2009, 2012; Kunis et al, 2013; Shechter et al, 2013). To examine whether elevated NO levels interfere with the induction of leukocyte trafficking molecule expression, CP epithelial cells were cultured under the same conditions as described above, followed by a longer stimulation with TNFα (24 h), for assessment of gene expression and protein synthesis. Quantitative real-time PCR (qPCR) analysis revealed that exposure to DETA alone significantly decreased the expression levels of ifngr2 (Fig3A), previously shown to be involved in the synergistic effect of TNFα and IFNγ on the expression of trafficking determinants by the CP (Kunis et al, 2013). Upon TNFα stimulation, ifngr2 expression was upregulated by the CP epithelial cells, an effect that was inhibited when the cells were pre-treated with DETA (Fig3A). We further examined the effect of DETA on CP epithelial cell expression levels of the chemokines, ccl2 and cxcl10, and the integrin ligand, icam1, involved in transepithelial migration of immune cells across the CP (Szmydynger-Chodobska et al, 2009, 2012; Kunis et al, 2013; Shechter et al, 2013), and found that DETA pre-treatment suppressed their induction by TNFα (Fig3B). Immunohistochemical analysis of CP primary epithelial cultures further confirmed that ICAM-1 elevation following treatment with TNFα was inhibited by DETA pre-treatment (Fig3C and D). Closer examination of the epithelial monolayer morphology showed that exposure to TNFα resulted in disturbance of ZO-1 tight junction protein localization, as previously observed (Ma et al, 2004; Aveleira et al, 2010), and that this effect was attenuated by the DETA pre-treatment (Fig3E).
Figure 3.
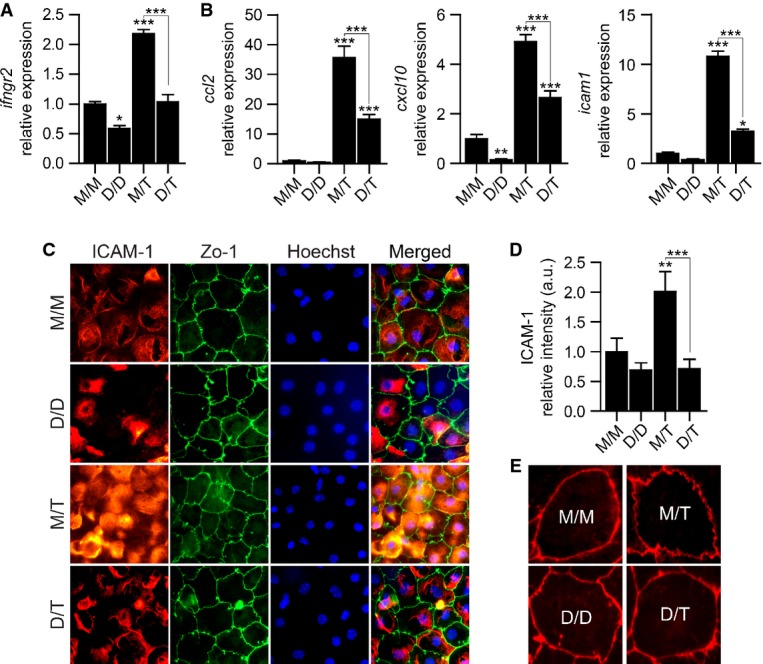
- A, B mRNA levels of the genes ifngr2, ccl2, cxcl10, and icam1, measured by qPCR, in CP epithelial cultures, treated as described in Fig2A. Expression levels are relative to untreated (M/M) cultures (n = 6 per group; one-way ANOVA followed by Newman–Keuls post hoc test; data are representative of at least three independently performed experiments in each case).
- C, D Representative microscopic images (C) and quantitative analysis (D) of CP cell cultures treated as described in Fig2A, immunostained for ICAM-1 (in red), ZO-1 (in green) and Hoechst nuclear staining (in blue) (n = 6 per group; one-way ANOVA followed by Newman–Keuls post hoc test).
- E Representative microscopic images of ZO-1 epithelial tight junction's disturbance in CP cell cultures, treated as described in Fig2A.
Elevated NO levels in the CP of AD transgenic mice are involved in repression of NFκB signaling pathway
The finding that CP gateway activity is NFκB dependent and could be experimentally suppressed by NO, prompted us to test in vivo our working hypothesis that elevated cerebral NO levels interfere with CP function in enabling leukocyte trafficking, and to determine whether this effect is relevant in the context of neurodegenerative diseases. We therefore evaluated the ability of the CP to mount an NFκB/p65 response in the 5XFAD transgenic mouse model of AD, co-expressing five mutations associated with familial AD (AD-Tg) (Oakley et al, 2006). To this end, CP tissues of AD-Tg mice, at the ages of 4–18 months (corresponding to the early and late progressive stages of disease pathology in this transgenic mouse model), and age-matched wild-type (WT) controls were freshly excised following intracardial perfusion, briefly stimulated ex vivo with TNFα, dissociated into a single-cell suspension, and analyzed for p65 nuclear translocation using ImageStream. To specifically detect the epithelial cells of the CP, cells were pre-gated by cytokeratin labeling. We found that CP epithelial cells of AD-Tg mice, at all examined ages, exhibited reduced responsiveness to TNFα stimulation, as assessed by nuclear translocation of the p65 subunit, relative to WT mice (Fig4A and B).
Figure 4.

- A Flow cytometry gating strategy to identify cytokeratin-positive CP epithelial cells, and measurement of the similarity index between NFκB/p65 and Hoechst nuclear staining, which is a measure for NFκB/p65 nuclear translocation, as assessed by ImageStream analysis, in AD-Tg and WT mice at various age groups following ex vivo stimulation with 20 ng/ml TNFα (n > 5,000 cells per group; one-way ANOVA followed by Newman–Keuls post hoc test).
- B Representative images (ImageStream) of NFκB/p65 cellular localization in CP epithelial cells from 9-month-old AD-Tg or WT mice, following ex vivo stimulation with 20 ng/ml TNFα (cytokeratin in green; p65 in red; Hoechst nuclear staining in blue; scale bar, 10 μm).
- C–E Representative microscopic images (C) of the brain of 10-month-old AD-Tg mice treated with either rutin or vehicle. Brain slices (6 μm) were immunostained for amyloid beta (Aβ) plaques (in red), GFAP (in green), and Hoechst nuclear staining (in blue). Mean Aβ plaque area (D) in the hippocampal dentate gyrus (HC) and the cortex (5th layer) were quantified. Astrogliosis was assessed in the cortex (5th layer) by GFAP immunoreactivity (E) (n = 8 per group; Student's t-test; scale bar, 250 μm).
- F Quantitative analysis of intracellular NO, measured by flow cytometry of DAF-2 DA mean florescence intensity (MFI), in CP of 4-month-old AD-Tg mice and age-matched WT controls, which were treated with either rutin or vehicle (drinking water) (n = 3–6 per group; one-way ANOVA followed by Newman–Keuls post hoc test).
- G Similarity index analysis, examined by ImageStream, of NFκB/p65 nuclear translocation, in CP of 4-month-old AD-Tg mice, which were treated with either rutin or vehicle (drinking water), and untreated WT controls (n = 3–4 per group; one-way ANOVA followed by Newman–Keuls post hoc test).
- H mRNA expression levels of the genes icam1, ccl2, and cxcl10, measured by qPCR, in CP of 4-month-old AD-Tg mice, which were treated with either rutin or vehicle (drinking water) (n = 5–6 per group; Student's t-test).
- I Representative flow cytometry plots and quantitative analysis of cells isolated from the brains of 10-month-old AD-Tg mice, treated with either rutin or vehicle (drinking water). CD11bhigh/CD45high mo-MΦ were gated and quantified (n = 5–7 per group; Student's t-test).
Prompted by these observations, we considered that the reduced responsiveness of the CP in AD-Tg mice could be an outcome of increased cerebral levels of NO, and thus, reducing local NO levels at the CP of AD-Tg mice might increase gateway activity for leukocyte trafficking. For this purpose, we chose to use the NO scavenger, rutin, which was previously reported to attenuate disease pathology in other AD-Tg murine models (Javed et al, 2012; Xu et al, 2014). First, to confirm that rutin administration could exert a similar effect in a 5XFAD AD-Tg model, we orally treated the mice with either rutin or vehicle, on a daily basis for a period of 4 weeks, and evaluated the effect on disease pathology. In line with previous observations (Javed et al, 2012; Xu et al, 2014), we found that rutin-treated AD-Tg mice had significantly lower cerebral Aβ plaque burden (Fig4C and D), as quantified in the hippocampal dentate gyrus, and the cerebral cortex (5th layer), two brain regions exhibiting robust Aβ plaque pathology in 5XFAD AD-Tg mice (Oakley et al, 2006). In addition, we observed a marked decrease in cerebral astrogliosis, as assessed by GFAP immunoreactivity (Fig4E), in the brains of rutin-treated AD-Tg mice.
We next examined whether local NO levels at the CP were affected by the systemic administration of rutin. To this end, intracellular levels of NO were quantified in AD-Tg and WT mice, using the fluorescent detector DAF-2 DA, as performed in vitro (Fig2D and E). Flow cytometry analysis confirmed higher intracellular NO levels in the CP of AD-Tg mice, as compared to age-matched WT controls, and that the levels of NO were decreased following rutin treatment (Fig4F). Evaluating the responsiveness of the CP epithelium of WT and AD-Tg mice to TNFα re-stimulation following rutin treatment revealed that the reduction in NO levels at the CP resulted in a restored potency for NFκB/p65 nuclear translocation (Fig4G). Furthermore, examining CP gateway activity, we found upregulation in the expression levels of the NFκB-dependent leukocyte trafficking determinants, ccl2 and cxcl10, but not of icam1 (Fig4H). Finally, to examine whether this effect was accompanied with immune cell recruitment to the CNS, we analyzed the brains of rutin-treated AD-Tg mice using flow cytometry and found increased number of CD45highCD11bhigh cells, corresponding to a population of infiltrating mo-MΦ (Shechter et al, 2013) (Fig4I). Collectively, these findings demonstrated that the neuroprotective effect exerted by the administration of an NO scavenger to AD-Tg mice involved, at least in part, augmentation of CP gateway activity for leukocyte trafficking.
Discussion
Owing to its unique location at the brain ventricles, the CP epithelium is exposed to brain-derived signals from its apical side, via the CSF, and to peripheral signals from its basal side, via the circulation, both of which affect its activity (Baruch et al, 2014). In aging and neurodegenerative conditions, accumulation of toxic molecules in the CSF was suggested to affect CP function (Lin et al, 1996; Johanson et al, 2004; Emerich et al, 2005; Marques et al, 2013); yet, their interactions with the CP or potential involvement in pathophysiology are poorly understood. Here, we focused on the NFκB signaling pathway at the CP and explored how two different cerebroventricular signals can modulate its function.
Orchestrated leukocyte trafficking to the CSF via the choroid plexus
Inflammation and immune cell recruitment are fundamental responses characterizing any wound healing process (Singer & Clark, 1999). These responses involve sequential phases, in which early neutrophil recruitment is followed by myeloid infiltration and delayed lymphocyte entry (Eming et al, 2007). However, whether similar events are involved in CNS repair has been a subject of debate, due to structural and spatial characteristics of the CNS as an immune privileged site (Shechter & Schwartz, 2013). Here, we found that similar to the situation in peripheral tissue-specific inflammatory immune cascade, entry of leukocytes via the CP to the CSF follows a synchronized, sequential pattern. In the hours following ICV administration of TNFα, the cellular composition of the CSF changed rapidly, being initially dominated by neutrophils (4–8 h), later shifting toward myeloid and T-cell entry (12–24 h). These results are consistent with the kinetics of both neutrophil (Szmydynger-Chodobska et al, 2009) and myeloid cell (Szmydynger-Chodobska et al, 2012; Shechter et al, 2013) recruitment to the damaged CNS, whereby immune cells were found to appear at the CP within hours, and in the CNS parenchyma within days, after the insult. These data indicate that following acute CNS damage, TNFα, which was shown to be elevated in the CSF of patients following spinal cord injury (Wang et al, 1996) and stroke (Zaremba & Losy, 2001), may serve as a CSF-borne danger signal, which is sufficient to induce activation of the CP to support immune cell recruitment to the CNS.
Choroid plexus epithelial NFκB signaling pathway inhibition by nitric oxide
TNFα stimulation classically involves the activation of an NFκB signaling pathway (Li & Lin, 2008; Lawrence, 2009), which we show here to mediate the induction of leukocyte homing and trafficking determinants expression by the CP epithelium. We found this process to be suppressed, both in vitro and in vivo, when local NO levels at the CP were elevated.
Although generally considered a pro-inflammatory mediator, NO acts through a non-canonical S-nitrosylation mechanism on residues of NFκB, suppressing its nuclear translocation and inhibiting inflammation-mediated gene transcription (Peng et al, 1995; Katsuyama et al, 1998; Marshall & Stamler, 2001; Pineda-Molina et al, 2001). Importantly, both neuronal NO and epithelium-derived NO were suggested to affect CP function (Lin et al, 1996). Our present in vitro findings revealed that treatment of primary cultures of CP epithelial cells with NO resulted in NFκB/p65 subunit cytoplasmic retention and led to reduced expression of IFNγ-R by the epithelial cells, thereby potentially affecting CP responsiveness to IFNγ, a key regulatory signal for leukocyte trafficking via the CP (Kunis et al, 2013). The induction of trafficking determinants by the CP following TNFα stimulation was suppressed when cell cultures were pre-exposed to NO. Examining this process at the single-cell level, using high-throughput flow cytometry image analysis, we found that NFκB/p65 nuclear translocation was negatively correlated with intracellular NO levels in cytokeratin-positive CP epithelial cells. Furthermore, we observed that the NFκB-mediated process of tight junction disruption, associated with trans-endothelial leukocyte trafficking (Aveleira et al, 2010), was moderated in CP epithelial cultures following NO pre-treatment.
Choroid plexus epithelial NFκB/p65 repression in AD transgenic mice
In general, resolution of inflammation is an active process, which depends on well-orchestrated innate and adaptive immune responses. In chronic neurodegenerative diseases, CNS-infiltrating leukocytes were suggested to have a beneficial role in mitigation of the neuroinflammatory response, though their spontaneous entry to the CNS is often insufficient or suboptimal (Simard et al, 2006; Butovsky et al, 2007; Beers et al, 2008; Town et al, 2008; Finkelstein et al, 2011; Mildner et al, 2011; Vaknin et al, 2011; Kunis et al, 2015); under those conditions, augmenting their recruitment to the CNS has been considered as a therapeutic approach (Britschgi & Wyss-Coray, 2007; Popovich & Longbrake, 2008; Prinz et al, 2011; Derecki et al, 2012; Prinz & Priller, 2014; Schwartz & Baruch, 2014a). Here, we found that the NFκB signaling pathway involvement in CP activation for leukocyte trafficking is impaired in AD-Tg mice. Thus, comparison of AD-Tg and WT mice revealed that upon ex vivo TNFα stimulation, the CP of AD-Tg mice exhibit an impaired ability to mount NFκB response. Notably, this effect was observed along both the early and late progressive stages of the disease in this AD-Tg line. These findings support our contention that CP dysfunction as a gateway for leukocyte entry to the CNS might be an underlying mechanism in the pathophysiology of neurodegenerative diseases (Schwartz & Baruch, 2014b).
Recent studies have suggested NO as a negative player in the progressive pathological nature of AD (Fernandez et al, 2010), and genetic ablation of inducible nitric oxide synthase (iNOS) was shown to be neuroprotective in this pathology (Nathan et al, 2005). In line with these reports, the NO scavenger, rutin, was shown to have a therapeutic effect in attenuating cognitive impairments in murine models of AD by reducing NO stress (Javed et al, 2012; Xu et al, 2014). Here, adopting a systemic rutin administration protocol in 5XFAD AD-Tg mice confirmed its effect on disease pathology. Importantly, the CP of treated AD-Tg mice showed reduced NO levels, and this reduction was accompanied by improved capacity of the CP to be stimulated by TNFα, resulting in NFκB/p65 nuclear translocation, and induction of epithelial leukocyte homing and trafficking determinants. Notably, not all tested trafficking molecules that were upregulated in vitro by TNFα were found to be affected; specifically, icam1 was not upregulated following rutin treatment, suggesting that additional mechanisms are involved in this response. Nevertheless, the activation of the CP for leukocyte trafficking following rutin treatment was associated with enhanced accumulation of mo-MΦ in the CNS. These findings indicate that in AD-Tg mice, elevated cerebroventricular levels of NO are involved in decreased CP gateway activity and that systemic NO scavenging can ameliorate this effect.
Taken together, our findings attribute a novel negative role for NO in CNS pathophysiology and propose a model according to which CSF-borne signals regulate CP gateway activity for leukocyte entry to the CNS. Accordingly, while CP gateway activity is sensitive to CSF-borne danger signals, such as TNFα, a NO-mediated local repression of the epithelial NFκB signaling pathway dampens CP responsiveness in chronic neurodegenerative diseases (Fig5). It is therefore possible that alleviating the repression of NFκB signaling pathway at the CP to support the recruitment of inflammation-resolving leukocytes to the CNS can serve as a therapeutic approach for chronic neurodegenerative conditions.
Figure 5.

Proposed model for CP gateway dysfunction for leukocyte trafficking following cerebroventricular nitric oxide elevation
Schematic illustration of the intracellular cascade of events at the CP compartment, following stimulation with TNFα, in physiology, and under pathological conditions of chronic exposure to nitric oxide. (1) In the steady state, the CP senses CSF-borne danger/pro-inflammatory signals, derived from the brain parenchyma. TNFα, as a particular example of those signals, is sensed by the CP via the TNFα receptor (TNF-R). (2) Upon TNFα stimulation, TNF-R signaling cascade is funneled, through the NFκB pathway, into translocation of the p65 subunit to the nucleus, which initiates a cellular response involving the upregulation of IFNγ-R on the CP epithelium, leukocyte trafficking determinant expression, and disruption of the epithelial tight junctions. (3) IFNγ, secreted by CP stromal Th1 cells, acts in synergy with the NFκB signaling pathway, in inducing the expression of specific leukocyte trafficking molecules such as ICAM-1, CCL2, and CXCL10. Together, these events support CP-mediated leukocyte entry to the CNS. (4) Under neuroinflammatory conditions, which involve cerebroventricular elevation of nitric oxide levels and its accumulation at the CP, nitrosative modifications of the NFκB complex prevent its nuclear translocation following TNFα stimulation.
Materials and Methods
Animals
5XFAD transgenic mice (Tg6799) that co-overexpress familial AD mutant forms of human APP (the Swedish mutation, K670N/M671L; the Florida mutation, I716V; and the London mutation, V717I) and PS1 (M146L/L286V) transgenes under transcriptional control of the neuron-specific mouse Thy-1 promoter (Oakley et al, 2006), and AD double transgenic B6.Cg-Tg (APPswe, PSEN1dE9) 85Dbo/J mice (Borchelt et al, 1997) were purchased from The Jackson Laboratory. Genotyping was performed by PCR analysis of tail DNA, as previously described (Oakley et al, 2006). Adult male and female wild-type (WT) C57Bl/6J mice were supplied by Harlan Biotech (Jerusalem, Israel). All experiments were in compliance with the regulations formulated by the Institutional Animal Care and Use Committee of the Weizmann Institute of Science.
Rutin administration
The nitric oxide scavenger, rutin (Sigma-Aldrich), was dissolved in drinking water and orally administered at a daily dose of 100 mg/kg, for 4 weeks, as previously described (Xu et al, 2014).
Intracerebroventricular (ICV) injections
TNFα (50, 100, or 150 ng) dissolved in PBS to a final volume of 15 μl was injected ICV (0.4 mm posterior to the bregma, 1.0 mm lateral to the midline, and 2.0 mm in depth from the brain surface), as described (Baruch et al, 2014).
Primary culture of choroid plexus cells
Mice were deeply anesthetized and intracardially perfused with PBS; the CPs were then removed under a dissecting microscope (Stemi DV4; Zeiss) in PBS into tubes containing 0.25% trypsin and kept on ice. When all CPs were collected, the tubes were shaken for 20 min at 37°C, and cells were then manually dissociated. The cell suspension was washed in culture medium for epithelial cells (DMEM/HAM's F12 (Invitrogen Corp) supplemented with 10% FCS (Sigma-Aldrich), 1 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 mg/ml streptomycin, 5 μg/ml insulin, 20 μM Ara-C, 5 ng/ml sodium selenite, and 10 ng/ml EGF) and cultured (2.5 × 105 cells/well) at 37°C, 5% CO2 in 24-well plates (Nunc). The medium was refreshed after 24 h, and after 72 h, the cells were either left untreated or treated with the nitric oxide donor DETA/NONOate (150 μM; Cayman Chemical) for 48 h, followed by brief (10 min) or long (24 h) stimulation with TNFα (PeproTech). Cell viability was quantified by Trypan blue staining after detachment of the cells with 0.25% trypsin for 10 min at 37°C.
RNA purification, cDNA synthesis, and real-time quantitative PCR
Total RNA of the choroid plexus tissues, or from cell cultures, was extracted using the ZR RNA MicroPrep kit (Zymo Research). mRNA (1 μg) was converted to cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The expression of specific mRNAs was assayed using fluorescence-based real-time quantitative PCR (qPCR). qPCRs were performed using Power SYBR Green PCR Master Mix (Applied Biosystems). Reactions were performed in triplicate for each sample using the standard curve method. Peptidylprolyl isomerase A (ppia) was chosen as a reference gene according to its stability in the target tissue. The amplification cycles were 95°C for 5 s, 60°C for 20 s, and 72°C for 15 s. At the end of the assay, a melting curve was constructed to evaluate the specificity of the reaction. All quantitative real-time PCRs were performed and analyzed using the StepOnePlus PCR System (Applied Biosystems). The following primers were used:
ppia forward 5′-AGCATACAGGTCCTGGCATCTTGT-3′ and reverse 5′-CAAAGACCACATGCTTGCCATCCA-3′;
ifngr2 forward 5′-TCCCACACCCATTCACAG-3′ and reverse 5′-AGGTCCAACAGTAACATTCTC-3′;
icam1 forward 5′-AGATCACATTCACGGTGCTGGCTA-3′ and reverse 5′-AGCTTTGGGATGGTAGCTGGAAGA-3′;
ccl2 forward 5′-CATCCACGTGTTGGCTCA-3′ and reverse 5′-GATCATCTTGCTGGTGAATGAGT-3′; and
cxcl10 forward 5′-AACTGCATCCATATCGATGAC-3′ and reverse 5′-GTGGCAATGATCTCAACAC-3′.
Immunohistochemistry and immunocytochemistry
For immunoflouorescent staining, paraffin-embedded sections (6 μm thick) of mouse brains underwent deparaffinization and were blocked with M.O.M. immunodetection kit reagent (Vector Laboratories) containing 0.1% Triton X-100 (Sigma-Aldrich) and stained with different combinations of the following primary antibodies: mouse anti-E-cadherin (1:100, Invitrogen), mouse anti-cytokeratin (1:100, AbCam), rabbit anti-Mac-2 (1:200, Cedarlane), rabbit anti-IBA1 (1:200, Wako), mouse anti-Aβ (1:300, Covance), and rabbit anti-GFAP (1:100, Dako). Secondary antibodies included Cy2/Cy3 anti-rabbit/mouse antibodies (1:200; all from Jackson ImmunoResearch). The slides were exposed to Hoechst for nuclear staining (1:2,000; Invitrogen Probes) for 30 s. For immunocytochemistry, CP cells were isolated and grown on cover slips to confluence, as described before (Kunis et al, 2013). Cytokines were added for the last 24 h of culture, the wells were then washed with PBS, and the cells were fixed with methanol–acetone (1:1) for 10 min at −20°C, followed by two washing steps with PBS. The cover slips of the cultured CP cells were blocked with M.O.M. immunodetection kit reagent (Vector Laboratories) containing 0.1% Triton X-100 (Sigma-Aldrich) and stained with the following antibodies: rat anti-ICAM-1 (1:100; Abcam), mouse anti-occludin (1:100, Invitrogen), rabbit anti-NFκB/p65 (1:200; Santa Cruz), and mouse anti-ZO-1 (1:100; Invitrogen). Secondary antibodies included Cy2/Cy3-conjugated donkey anti-rat and rabbit or mouse antibody (1:200; Jackson ImmunoResearch, West Grove, PA). The cover slips were exposed to Hoechst stain (1:2,000; Invitrogen) for 1 min and mounted onto slides. A fluorescence microscope (Nikon Eclipse 80i) was used for microscopic analysis. The fluorescence microscope was equipped with a digital camera (DXM 1200F; Nikon) and with 20× NA 0.50 and 40× NA 0.75 objective lenses (Plan Fluor; Nikon). Recordings were made using acquisition software (NIS-Elements, F3). Images were cropped, merged, and optimized using Photoshop CS6 13.0 (Adobe) and were arranged using Illustrator CS5 15.1 (Adobe). For quantification of staining intensity, cell borders (20–25 cells per picture) were marked, and the corrected total cell fluorescence (CTCF) was quantified using Image-Pro Plus software (Media Cybernetics, Inc.), as previously described (Burgess et al, 2010).
Cerebral amyloid beta plaque load quantitation
From each brain, 6-μm coronal slices were collected, and eight sections per mouse, from four different predetermined depths throughout the region of interest (dentate gyrus or cerebral cortex), were immunostained. Histogram-based segmentation of positively stained pixels was performed using the Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA). The segmentation algorithm was manually applied to each image, in the dentate gyrus area or in the cortical layer V, and the percentage of the area occupied by total Aβ immunostaining was determined. Prior to quantification, slices were coded to mask the identity of the experimental groups, and plaque burden was quantified by an observer blinded to the identity of the groups.
CSF collection
CSF was collected by the cisterna magna puncture technique, as previously performed (Baruch et al, 2014). In brief, mice were anesthetized and placed on a stereotactic instrument so that the head formed a 135° angle with the body. A sagittal incision of the skin was made inferior to the occiput and the subcutaneous tissue and muscle were separated. A capillary was then inserted into the cisterna magna, through the dura matter lateral to the arteria dorsalis spinalis. Approximately 10 μl CSF could be aspirated from each mouse. The collected CSF was taken for analysis by flow cytometry.
Flow cytometry sample preparation and analysis
Prior to tissue collection, mice were intracardially perfused with PBS. Choroid plexus tissues were isolated from the lateral, third and fourth ventricles of the brain, incubated at 37°C for 45 min in PBS (with Ca2+/Mg2+) containing 400 units/ml collagenase type IV (Worthington Biochemical Corporation), and then manually homogenized by pipettation. Brains were dissected, dissociated using the gentleMACS™ dissociator (Miltenyi Biotec), and loaded on Percoll gradient (GE Healthcare) to isolate leukocytes. The following fluorochrome-labeled mAbs were used according to the manufacturers' protocols: PE-conjugated anti-CD11b, FITC-conjugated anti-CD45 (both from BioLegend), v450-conjugated anti-CD4 (BD Bioscience), and DAF-2 DA (Sigma-Aldrich). Flow cytometry analysis was performed on each sample using a BD Biosciences LSRII flow cytometer, and the acquired data were analyzed using FlowJo software (Tree Star).
High-throughput single-cell flow cytometry image analysis
CP tissues freshly extracted following intracardial perfusion were ex vivo stimulated in a Petri dish with TNFα (20 ng/ml) for 10 min. Tissues were transferred into tubes containing 0.25% trypsin, incubated with agitation for 20 min at 37°C, manually dissociated into a single-cell suspension, and fixed using methanol–acetone (1:1) for 10 min at −20°C. Following fixation, cells were permeabilized with 0.2% Triton X-100 diluted in FACS buffer (10% FCS, 2 mM EDTA, PBS) and intracellularly immunostained with rabbit anti-NFκB/p65 (1:200; Santa Cruz) and mouse anti-cytokeratin (1:100; Covance). For nuclear visualization, cells were exposed to Hoechst stain (1:2,000; Invitrogen) for 1 min prior to imaging flow cytometry examination. For in vitro analysis, primary CP epithelial cells were cultured as described above. Corresponding groups were stimulated with 20 ng/ml TNFα for 10 min, washed, and re-suspended using 0.25% trypsin at 37°C for 10 min. The cells were then washed with FACS buffer and immediately fixed using methanol–acetone (1:1) for 10 min at −20°C. Intracellular labeling was done as with the ex vivo protocol. The cells (minimum 2,000 for each sample) were examined by imaging flow cytometry using the ImageStreamX (Amnis—part of EMD Millipore, Seattle, WA). Images were analyzed using IDEAS 6.0 software (Amnis). Cells were gated for single cells using the area and aspect ratio features and for focused cells using the Gradient RMS feature (George et al, 2006). Cells were gated according to the area and intensity of the Hoechst staining, and only cytokeratin-positive cells (based on their staining intensity) were analyzed. The nuclear translocation of NFκB/p65 was calculated using the ‘Similarity’ feature, which is the log-transformed Pearson's correlation coefficient between the NFκB/p65 and the nuclear staining images. The similarity index provides a measure of the degree to which two images (NFκB/p65 and Hoechst in this case) are linearly correlated within the masked region. Higher similarity means higher correlation and thus reflects co-localization of the two stains—that is, a higher level of NFκB/p65 nuclear localization.
S-Nitrosylated protein detection assay
Visualization of protein nitrosylation was performed using a ‘biotin-switch’ method S-nitrosylation assay kit (Cayman, #10006518). The assay was used on CP epithelial cell cultures, according to the manufacturer's protocol. Florescence intensity was quantified by an observer blinded to the identity of the groups.
Intracellular nitric oxide staining
To quantify intracellular nitric oxide in vitro, cells were incubated with 10 μM DAF-2 DA (Santa Cruz), while covered to shield them from light, for 1 h at room temperature prior to standard flow cytometry staining protocol. For ex vivo staining, mice were intracardially perfused, and their CP tissues were extracted into small Petri dishes containing 10 μM DAF-2 DA. Dishes were covered and incubated for 1 h at room temperature, prior to tissue handling for flow cytometry.
Statistical analysis
Data were analyzed using the Student's t-test to compare between two groups. One-way ANOVA was used to compare several groups. The Newman–Keuls post hoc test was used for follow-up pairwise comparison of groups after the null hypothesis was rejected (P < 0.05). Results are presented as mean ± s.e.m. In the graphs, y-axis error bars represent s.e.m. Statistical calculations were performed using standard functions of Microsoft Excel and Prism 5.0 software (GraphPad Software).
Acknowledgments
We thank Dr. Shelley Schwarzbaum for editing the manuscript. This research was supported by a European Research Council (E.R.C.) advanced grant (232835), an EU Seventh Framework Program (FP7) grant (279017), and the Israel Science Foundation (ISF) Legacy-Bio-Med program (1899-08). M.S. holds The Maurice and Ilse Katz Professorial Chair in Neuroimmunology.
Author contributions
KB and AK, in equal contribution and under the mentoring of MS, conceived the general ideas of this study, performed all of the experiments, analyzed the data, and prepared it for presentation. ZP assisted in imaging flow cytometry experiments and subsequent data analysis. KB, AK, and MS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure Legends
Review Process File
References
- Aliyev A, Seyidova D, Rzayev N, Obrenovich ME, Lamb BT, Chen SG, Smith MA, Perry G, de la Torre JC, Aliev G. Is nitric oxide a key target in the pathogenesis of brain lesions during the development of Alzheimer's disease? Neurol Res. 2004;26:547–553. doi: 10.1179/01610425017613. [DOI] [PubMed] [Google Scholar]
- Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–2882. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal-Price A, Matthias A, Brown GC. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J Neurochem. 2002;80:73–80. doi: 10.1046/j.0022-3042.2001.00675.x. [DOI] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, Amit I, Schwartz M. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346:89–93. doi: 10.1126/science.1252945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci USA. 2008;105:15558–15563. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Britschgi M, Wyss-Coray T. Immune cells may fend off Alzheimer's disease. Nat Med. 2007;13:408–409. doi: 10.1038/nm0407-408. [DOI] [PubMed] [Google Scholar]
- Brown GC. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans. 2007;35:1119–1121. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, Castro A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci USA. 2010;107:12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci USA. 2006;103:11784–11789. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. Eur J Neuorsci. 2007;26:413–416. doi: 10.1111/j.1460-9568.2007.05652.x. [DOI] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AJ, Heales SJ. Nitric oxide and neurological disorders. Mol Aspects Med. 2005;26:67–96. doi: 10.1016/j.mam.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Emerich DF, Skinner SJ, Borlongan CV, Vasconcellos AV, Thanos CG. The choroid plexus in the rise, fall and repair of the brain. BioEssays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007;127:514–525. doi: 10.1038/sj.jid.5700701. [DOI] [PubMed] [Google Scholar]
- Falcao AM, Marques F, Novais A, Sousa N, Palha JA, Sousa JC. The path from the choroid plexus to the subventricular zone: go with the flow! Front Cell Neurosci. 2012;6:34. doi: 10.3389/fncel.2012.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra P, Fernandez AP, Castro-Blanco S, Encinas JM, Serrano J, Bentura ML, Munoz P, Martinez-Murillo R, Rodrigo J. Expression of nitric oxide system in clinically evaluated cases of Alzheimer's disease. Neurobiol Dis. 2004;15:287–305. doi: 10.1016/j.nbd.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Fernandez AP, Pozo-Rodrigalvarez A, Serrano J, Martinez-Murillo R. Nitric oxide: target for therapeutic strategies in Alzheimer's disease. Curr Pharm Des. 2010;16:2837–2850. doi: 10.2174/138161210793176590. [DOI] [PubMed] [Google Scholar]
- Finkelstein A, Kunis G, Seksenyan A, Ronen A, Berkutzki T, Azoulay D, Koronyo-Hamaoui M, Schwartz M. Abnormal changes in NKT cells, the IGF-1 axis, and liver pathology in an animal model of ALS. PLoS One. 2011;6:e22374. doi: 10.1371/journal.pone.0022374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George TC, Fanning SL, Fitzgerald-Bocarsly P, Medeiros RB, Highfill S, Shimizu Y, Hall BE, Frost K, Basiji D, Ortyn WE, Morrissey PJ, Lynch DH. Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J Immunol Methods. 2006;311:117–129. doi: 10.1016/j.jim.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Javed H, Khan MM, Ahmad A, Vaibhav K, Ahmad ME, Khan A, Ashafaq M, Islam F, Siddiqui MS, Safhi MM, Islam F. Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type. Neuroscience. 2012;210:340–352. doi: 10.1016/j.neuroscience.2012.02.046. [DOI] [PubMed] [Google Scholar]
- Johanson C, McMillan P, Tavares R, Spangenberger A, Duncan J, Silverberg G, Stopa E. Homeostatic capabilities of the choroid plexus epithelium in Alzheimer's disease. Cerebrospinal Fluid Res. 2004;1:3. doi: 10.1186/1743-8454-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE, Stopa EG, McMillan PN. The blood-cerebrospinal fluid barrier: structure and functional significance. Methods Mol Biol. 2011;686:101–131. doi: 10.1007/978-1-60761-938-3_4. [DOI] [PubMed] [Google Scholar]
- Katsuyama K, Shichiri M, Marumo F, Hirata Y. NO inhibits cytokine-induced iNOS expression and NF-kappaB activation by interfering with phosphorylation and degradation of IkappaB-alpha. Arterioscler Thromb Vasc Biol. 1998;18:1796–1802. doi: 10.1161/01.atv.18.11.1796. [DOI] [PubMed] [Google Scholar]
- Kojima H, Sakurai K, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, Hirata Y, Nagano T. Development of a fluorescent indicator for nitric oxide based on the fluorescein chromophore. Chem Pharm Bull. 1998;46:373–375. doi: 10.1248/cpb.46.373. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, Konig S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011;71:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, Schwartz M. IFN-gamma-dependent activation of the brain's choroid plexus for CNS immune surveillance and repair. Brain. 2013;136:3427–3440. doi: 10.1093/brain/awt259. [DOI] [PubMed] [Google Scholar]
- Kunis G, Baruch K, Miller O, Schwartz M. Immunization with a Myelin-Derived Antigen Activates the Brain's Choroid Plexus for Recruitment of Immunoregulatory Cells to the CNS and Attenuates Disease Progression in a Mouse Model of ALS. J Neurosci. 2015;35:6381–6393. doi: 10.1523/JNEUROSCI.3644-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lin X. Positive and negative signaling components involved in TNFalpha-induced NF-kappaB activation. Cytokine. 2008;41:1–8. doi: 10.1016/j.cyto.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Lin AY, Szmydynger-Chodobska J, Rahman MP, Mayer B, Monfils PR, Johanson CE, Lim YP, Corsetti S, Chodobski A. Immunohistochemical localization of nitric oxide synthase in rat anterior choroidal artery, stromal blood microvessels, and choroid plexus epithelial cells. Cell Tissue Res. 1996;285:411–418. doi: 10.1007/s004410050657. [DOI] [PubMed] [Google Scholar]
- Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann N Y Acad Sci. 2002;962:318–331. doi: 10.1111/j.1749-6632.2002.tb04077.x. [DOI] [PubMed] [Google Scholar]
- Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- Maguire O, Collins C, O'Loughlin K, Miecznikowski J, Minderman H. Quantifying nuclear p65 as a parameter for NF-kappaB activation: correlation between ImageStream cytometry, microscopy, and Western blot. Cytometry A. 2011;79:461–469. doi: 10.1002/cyto.a.21068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Sousa N, Palha JA. Blood-brain-barriers in aging and in Alzheimer's disease. Mol Neurodegener. 2013;8:38. doi: 10.1186/1750-1326-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HE, Stamler JS. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res. 1996;24:2236–2242. doi: 10.1093/nar/24.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schlevogt B, Kierdorf K, Bottcher C, Erny D, Kummer MP, Quinn M, Bruck W, Bechmann I, Heneka MT, Priller J, Prinz M. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neuorsci. 2001;13:529–538. doi: 10.1046/j.1460-9568.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J, Vodovotz Y, Kipiani K, Beal MF. Protection from Alzheimer's-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202:1163–1169. doi: 10.1084/jem.20051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng HB, Libby P, Liao JK. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J Biol Chem. 1995;270:14214–14219. doi: 10.1074/jbc.270.23.14214. [DOI] [PubMed] [Google Scholar]
- Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14:1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- Qureshi GA, Baig S, Bednar I, Sodersten P, Forsberg G, Siden A. Increased cerebrospinal fluid concentration of nitrite in Parkinson's disease. NeuroReport. 1995;6:1642–1644. doi: 10.1097/00001756-199508000-00013. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- Raposo C, Graubardt N, Cohen M, Eitan C, London A, Berkutzki T, Schwartz M. CNS repair requires both effector and regulatory T cells with distinct temporal and spatial profiles. J Neurosci. 2014;34:10141–10155. doi: 10.1523/JNEUROSCI.0076-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Baruch K. Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: boosting autoimmunity to fight-off chronic neuroinflammation. J Autoimmun. 2014a;54:8–14. doi: 10.1016/j.jaut.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J. 2014b;33:7–22. doi: 10.1002/embj.201386609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim KW, Klein E, Kalchenko V, Bendel P, Lira SA, Jung S, Schwartz M. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38:555–569. doi: 10.1016/j.immuni.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Schwartz M. CNS sterile injury: just another wound healing? Trends Mol Med. 2013;19:135–143. doi: 10.1016/j.molmed.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmydynger-Chodobska J, Strazielle N, Zink BJ, Ghersi-Egea JF, Chodobski A. The role of the choroid plexus in neutrophil invasion after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1503–1516. doi: 10.1038/jcbfm.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmydynger-Chodobska J, Strazielle N, Gandy JR, Keefe TH, Zink BJ, Ghersi-Egea JF, Chodobski A. Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J Cereb Blood Flow Metab. 2012;32:93–104. doi: 10.1038/jcbfm.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taskiran D, Sagduyu A, Yuceyar N, Kutay FZ, Pogun S. Increased cerebrospinal fluid and serum nitrite and nitrate levels in amyotrophic lateral sclerosis. Int J Neurosci. 2000;101:65–72. doi: 10.3109/00207450008986493. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neurosci Lett. 1999;260:204–206. doi: 10.1016/s0304-3940(98)00986-0. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi A, Olivas AD, Noble-Haeusslein LJ. Inflammation and spinal cord injury: infiltrating leukocytes as determinants of injury and repair processes. Clin Neurosci Res. 2006;6:283–292. doi: 10.1016/j.cnr.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaknin I, Kunis G, Miller O, Butovsky O, Bukshpan S, Beers DR, Henkel JS, Yoles E, Appel SH, Schwartz M. Excess circulating alternatively activated myeloid (M2) cells accelerate ALS progression while inhibiting experimental autoimmune encephalomyelitis. PLoS One. 2011;6:e26921. doi: 10.1371/journal.pone.0026921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas T, Ugalde C, Spuch C, Antequera D, Moran MJ, Martin MA, Ferrer I, Bermejo-Pareja F, Carro E. Abeta accumulation in choroid plexus is associated with mitochondrial-induced apoptosis. Neurobiol Aging. 2010;31:1569–1581. doi: 10.1016/j.neurobiolaging.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Wang CX, Nuttin B, Heremans H, Dom R, Gybels J. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. J Neuroimmunol. 1996;69:151–156. doi: 10.1016/0165-5728(96)00080-x. [DOI] [PubMed] [Google Scholar]
- Xu YZ, Nygard M, Kristensson K, Bentivoglio M. Regulation of cytokine signaling and T-cell recruitment in the aging mouse brain in response to central inflammatory challenge. Brain Behav Immun. 2010;24:138–152. doi: 10.1016/j.bbi.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Xu PX, Wang SW, Yu XL, Su YJ, Wang T, Zhou WW, Zhang H, Wang YJ, Liu RT. Rutin improves spatial memory in Alzheimer's disease transgenic mice by reducing Abeta oligomer level and attenuating oxidative stress and neuroinflammation. Behav Brain Res. 2014;264:173–180. doi: 10.1016/j.bbr.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Zaremba J, Losy J. Early TNF-alpha levels correlate with ischaemic stroke severity. Acta Neurol Scand. 2001;104:288–295. doi: 10.1034/j.1600-0404.2001.00053.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure Legends
Review Process File