

Abstract
The aim of this study was to investigate the expression of the interleukin (IL)-36 axis in patients with primary Sjögren’s syndrome (pSS). Blood and minor labial salivary glands (MSG) biopsies were obtained from 35 pSS and 20 non-Sjögren’s syndrome patients (nSS) patients. Serum IL-36α was assayed by enzyme-linked immunosorbent assay (ELISA). IL-36α, IL-36R, IL-36RA, IL-38, IL-22, IL-17, IL-23p19 and expression in MSGs was assessed by reverse transcription–polymerase chain reaction (RT–PCR), and tissue IL-36α and IL-38 expression was also investigated by immunohistochemistry (IHC). αβ and γδ T cells and CD68+ cells isolated from MSGs were also studied by flow cytometry and confocal microscopy analysis. IL-36α was over-expressed significantly in the serum and in the salivary glands of pSS. Salivary gland IL-36α expression was correlated with the expression levels of IL-17, IL-22 and IL-23p19. IL-38, that acts as inhibitor of IL-36α, was also up-regulated in pSS. αβ+ CD3+ T cells and CD68+ cells were the major source of IL-36α in minor salivary glands of pSS. γδ T cells were not significantly expanded in the salivary glands of pSS but produced more IL-17, as their percentage correlated with the focus score. Higher expression of IL-36α and IL-36R was also demonstrated in γδ T cells isolated from pSS compared to controls. In this study we demonstrate that a significant increase in circulating and tissue levels of IL-36α occurs in pSS patients.
Keywords: IL-36a, IL36RA, IL-38, Sjögren’s syndrome, γδ T cells
Introduction
Interleukin (IL)-36α, IL-36β, IL-36γ and IL-36Ra, collectively called IL-36 cytokines, are novel members of the IL-1 family of cytokines 1 involved in the development of human diseases such as psoriasis 2,3. IL-36Ra and IL-36 agonists are highly expressed in the psoriatic skin 4, and anti-tumour necrosis factor (TNF) treatment results in decreased expression of the IL-36 agonists and IL-36Ra, with an improved clinical outcome 4. In psoriasis, IL-36 seems to exert a proinflammatory role by mediating the cross-talk between dendritic (DCs) and skin tissue cells, the recruitment of inflammatory cells and the expansion of IL-17A-producing γδ T cells 3. Although IL-36α was also found to be up-regulated in joints of patients with psoriatic and rheumatoid arthritis 2, it seems to have no proinflammatory effect in this context. In fact, in several experimental arthritis models, IL-36γ, IL-36Ra and IL-36R did not correlate with arthritis severity and treatment with neutralizing antibodies against IL-36R did not affect arthritis severity in collagen-induced arthritis 5,6.
Primary Sjögren’s syndrome (pSS) is an autoimmune disorder in which multiple activation of the innate immune system and of B and T cells has been implicated in disease pathogenesis 7. In particular, activation of the pathogenic IL-23/IL-17/IL-22 axis has been demonstrated recently to be one of the key mediators of primary pSS pathogenesis 8,9. IL-36 receptor (IL-36R) signalling appears to be absolutely crucial for the control of this axis 10,11.
Despite the demonstrated relevance of the IL-23/IL-17/IL-22 axis, the role of IL-36 and, in particular, the immunological behaviour of γδ T cells in pSS, have not so far been defined.
In the results included in this study, we show that IL-36, but not IL-36R and IL-36 receptor antagonist (IL-36RA), is strongly over-expressed in the salivary glands and in the serum of pSS patients and correlated with the degree of tissue inflammation. In addition, we show that γδ T cells strongly express the IL-36R and produce high levels of IL-36 and IL-17, providing the first evidence of a putative proinflammatory role of IL-17+ IL-36+ γδ T lymphocytes in the inflammatory responses of the disease.
Methods
Patients
Blood and minor labial salivary glands (MSG) biopsies were obtained from 35 patients (30 female, mean age 43 ± 12 years, mean disease duration 23 ± 18 months) fulfilling the American–European Consensus Group criteria for pSS 12 and 20 patients (15 female, mean age 48 ± 9 years, mean disease duration 28 ± 14 months) displaying subjective complaints of dry mouth or eyes, who did not meet the American–European Consensus Group criteria for pSS (nSS) and were considered as the control group. All the nSS subjects had negative biopsy at histological assessment. Disease activity was assessed by using the EULAR Sjögren’s Syndrome Disease Activity Index (ESSDAI) 13. All the patients and controls gave their informed consent and the study was approved by the ethical committee of the University of Palermo. Serological evaluations, which included tests for the presence of anti-nuclear antibodies (positive in 85% of pSS), anti-SSA/Ro/anti-SSB/La (positive in 71% of pSS), levels of CRP (14 ± 6 mg/l in pSS versus 3 ± 1·8 in controls, P < 0·05) and erythrocyte sedimentation rate (ESR) (44 ± 12 mm/h in pSS versus 12 ± 6 in controls, P < 0·05) were performed. Multiple minor labial salivary glands biopsies were obtained from all the pSS patients and controls and placed into formalin fixative and RNAlater for immunohistochemistry and reverse transcription–polymerase chain reaction (RT–PCR) analyses, respectively. Ten matched biopsies were also placed in RPMI for isolation of salivary gland mononuclear cells (SGMC) and flow cytometry analysis.
Histology and immunohistochemistry
Paraffin-embedded sections of 5-μm thickness were stained with haematoxylin and eosin (H&E) for the histological evaluation of the presence of lymphocytic infiltrates and/or foci. A focus was defined as an aggregate of ≥ 50 lymphocytes and the focus score was reported as the number of foci per 4 mm2 of tissue. Immunohistochemistry was performed on 5-μm-thick paraffin-embedded sections from salivary glands and from lymph nodes used as positive controls, as described previously 14. Lymph nodes were obtained from patients with lymphoadenomegaly undergoing biopsy for diagnostic purposes suspecting lymphoproliferative diseases. Only lymph nodes showing reactive lymphoid hyperplasia were used for immunohistochemistry (IHC) experiments. The primary antibodies rabbit anti-human IL-36α and rabbit anti-human IL-38 (Table 1) were added and incubated for 1 h at room temperature. Isotype-matched irrelevant antibodies were used as a negative control [rabbit immunoglobulin (Ig)G polyclonal antibody (ab27472); AbCam, Cambridge, UK]. The number of positive cells was determined by counting the reactive cells on microphotographs obtained from three randomly selected high-power microscopic fields (original magnification ×400). To specifically address the nature of IL-36-producing cells, double-staining for CD3/IL-36α, CD19/IL-36α and CD68/IL-36α and triple staining for CD138/CD78/IL-36α (Table 1) was performed on paraffin-embedded sections of salivary glands and the sections were treated with fluorescein isothiocyanate (FITC)-, Rhodamine Redor Cy-5-conjugated anti-mouse or anti-rabbit antibodies (Invitrogen, Carlsbad, CA, USA) plus RNasi (200 ng/ml) and counterstained using Toto-3 iodide (642/660; Invitrogen) or 4’,6-diamidino-2-phenylindole (DAPI) (Life Technologies, Paisley, UK). Confocal analysis was used to acquire fluorescence staining.
Table 1.
List of primers and antibodies
| Primers and sequences and antibodies | |||
|---|---|---|---|
| IL-36 | Hs00205367_m1 | IL-36α | Rabbit anti-human (1 : 100 dilution), (Sigma, St Louis, MO, USA) |
| IL-36R | Hs00909276_m1 | γδ | PE anti-human γδ (Becton Dickinson, San Jose, CA, USA) |
| IL-36RA | Hs00893626_m1 | IL-17 | PerCP/Cy5.5 anti-human IL-17A Antibody (Biolegend, San Diego, CA, USA) |
| IL-38 | Hs00544662_g1 | IFN-γ | FITC anti-human IFN-γ antibody (Biolegend, San Diego, CA, USA) |
| IL-17 | Hs00174383_m1 | IL-36α | Mouse anti-human IL-36α (clone 162122, R&D, Minneapolis, MN, USA) |
| IL-23p19 | Hs00372324_m1 | Anti-mouse FITC | Rabbit anti-mouse FITC-conjugated (Sigma, St Louis, MO, USA) |
| IL-22 | Hs01574154_m1 | IL-36R | Rabbit anti-human IL-36R (Novus biologicals, Littleton, CO, USA) |
| CD138 | Mouse anti-human (Dako, Glostrup, DK) (1 : 100 dilution) | Anti-rabbit PerCP | Goat anti-rabbit PerCP-conjugated (Sigma, St Louis, MO, USA) |
| CD68 | Mouse anti-human (Dako, Glostrup, DK) (1 : 100 dilution) | CD68 | Mouse anti-human CD68 PE-conjugated (Biolegend, San Diego, CA, USA) |
| CD78 | Mouse anti-human (Dako, Glostrup, DK) (1 : 100 dilution) | CD3 | Mouse anti-human CD4 PE-conjugated (Biolegend, San Diego, CA, USA) |
| CD19 | Mouse anti-human (Dako, Glostrup, Denmark) (1 : 100 dilution) | IL-38 | Rabbit anti-human (1 : 50 dilution) Novus Biological (Littleton, CO, USA) |
| CD3 | Mouse anti-human (Dako, Glostrup, Denmark) (1 : 100 dilution) | CD38 | PE anti-human CD38 (Biolegend, San Diego, CA, USA) |
| CD45 | Mouse anti-human APC Cy7-conjugated (Biolegend, San Diego, CA, USA) | ||
| 7-AAD | Biolegend, San Diego, CA, USA | ||
| CD138 | Mouse anti-human APC-conjugated (Biolegend, San Diego, CA, USA) | ||
7-AAD = 7-aminoactinomycin D; APC = allophycocyanin; PE = phycoerythrin; PerCP = peridinin chlorophyll; FITC = fluorescein isothiocyanate; IL = interleukin; IFN = interferon.
RNA extraction from salivary gland biopsies and quantitative TaqMan RT–PCR
Soon after removal, salivary gland biopsies were also stored in RNAlater® solution (Applied Biosystems, Foster City, CA, USA). RT–PCR was performed as described previously 14. Master mix and Taqman® gene expression assays for glyceraldehyde 3-phosphate dehydrogenase (GAPDH control) and target genes IL-36A (Hs00205367_m1), IL-36R (Hs00909276_m1), IL-36RA (Hs01104220_g1) and IL-38 (Hs00544662_g1) were obtained from Applied Biosystems (Foster City, CA, USA). Data were quantified using sds 2·1 software and normalized using GAPDH as endogenous control. Relative changes in gene expression between nSS and pSS samples were determined using the ΔΔCt method. Levels of the target transcript were normalized to a GAPDH endogenous control, constantly expressed in both groups (ΔCt). For ΔΔCt values, additional subtractions were performed between pSS (n = 35) and nSS (n = 20) ΔCt values. Final values were expressed as fold of induction.
Isolation of minor salivary gland mononuclear cells and flow cytometry
Minor SGMCs were obtained as described previously from 10 pSS and 10 nSS patients. Cell viability (trypan blue dye exclusion) was always > 95%. Four-colour flow cytometry analysis was performed using a fluorescence activated cell sorter (FACS)Calibur (BD Biosciences, San Jose, CA, USA). At least 50 000 cells (events), gated on the lymphocyte or monocyte/macrophage regions, were acquired for each sample and data are presented as a percentage of total alive cells in the CD45 channel.
Enzyme-linked immunosorbent assay (ELISA) for serum IL-36α
Serum IL-36α was assayed by commercial ELISA (Cusabio Biotech, Wuhan, China).
Statistical analysis
Parametric and non-parametric statistical analysis was performed calculating the mean ± standard error of the mean (s.e.m.) and median, respectively. For comparison of parametric and non-parametric data, the t-test and Mann–Whitney rank-sum test were used where appropriate. Spearman’s correlation analysis was utilized to quantify the expression associations between the genes of interest. Data are expressed as mean ± s.e.m. P-values less than 0·05 were considered significant.
Results
IL-36α is over-expressed in pSS patients
The IL-36 axis was evaluated in MSG of pSS and nSS patients. A significantly increased expression of IL-36 α (Fig. 1a), but not of IL-36R (Fig. 1b), was found in pSS salivary gland tissue, as detected by RT–PCR, and correlated with those of IL-17, IL-22 and IL-23p19 (Supporting information, Fig. S1a–c; r2 = 0·45, r2 = 0·66, r2 = 0·73, respectively; P < 0·05). IL-36α expression was also correlated with the focus score (Fig. 1e). IL-36RA and IL-38, both inhibitors of IL-36α, showed different behaviour, IL-36RA being significantly down-regulated (Fig. 1c), and IL-38 strongly over-expressed (Fig. 1d). Because of the high expression in MSG, we next analysed IL-36α levels in the serum of pSS patients. Serum IL-36α levels were significantly higher in pSS compared to controls (mean ± s.e.m.: 439 ± 165 versus 88 ± 47; P < 0·0001) (Fig. 1f). pSS patients segregate into two groups: those with notably elevated IL-36α (> 550 pg/ml) and those with little or no IL-36α. Interestingly, pSS patients with higher IL-36α serum levels manifest distinct profiles, showing higher disease activity (Fig. 1g) and higher levels of mRNA for IL-36 (Fig. 1h), IL-36R and IL-38 (data not shown) compared to those with lower levels.
Figure 1.
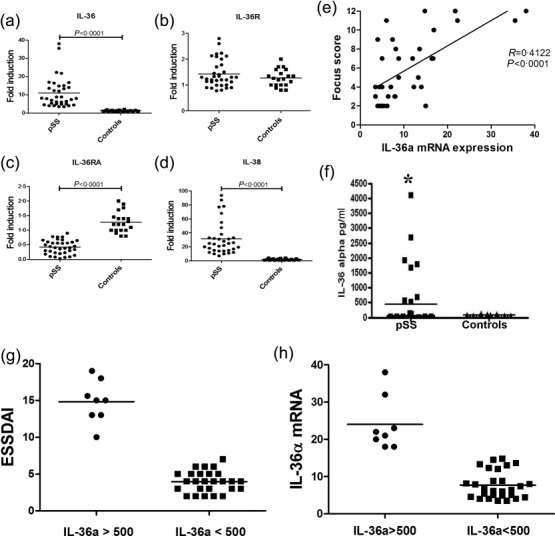
Interleukin (IL)-36α axis in primary Sjögren’s syndrome (pSS) patients as assessed using reverse transcription–polymerase chain reaction (RT–PCR). (a–d) Relative m-RNA quantification of IL-36α (a), IL-36R (b), IL-36RA (c) and IL-38 (d) was assessed by quantitative reverse transcription–polymerase chain reaction (qRT–PCR) in minor salivary glands obtained from 35 pSS patients and 20 control subjects. IL-36α (a) and IL-38 (d) showed a significant over-expression in pSS compared to controls. Conversely, IL-36R expression was not modulated between patients and controls (b), IL-36 RA being down-regulated significantly (c). (e) Correlation between IL-36α m-RNA expression and the lymphocytic focus score. The r2 (r2 = 0.4122) and P (P < 0·0001) values were determined with Spearman’s correlation coefficient. (f) IL-36α levels detected in the serum of pSS and controls by enzyme-linked immunosorbent assay (ELISA) (*P < 0·0001). (g) IL-36α serum levels according to EULAR Sjögren’s Syndrome Disease Activity Index (ESSDAI) scores.
Immunohistochemical analysis of IL36α and IL-38 expression in pSS salivary glands
We next evaluated the protein expression and tissue distribution of IL-36α and IL-38 in the MSG. A significantly higher expression of IL-36α was observed in pSS compared to controls. In control subjects, IL-36α expression was rarely detected and observed essentially among oval- or fan-shaped cells with bi-lobed or multi-lobed nuclei located closely to ducts, morphologically resembling plasma cells (Fig. 2a). Conversely, in pSS the expression of IL-36α was observed essentially in inflammatory infiltrates among lymphoid cells, cells displaying dendritic morphology and cells resembling plasma cells (Fig. 2b–e). No significant acinar and/or ductal epithelial expression of IL-36α was observed in patients and in controls (Figs 2a–e and 4b). The semi-quantitative analysis of IL-36α-expressing cells revealed a significant positive correlation between the number of IL-36α+ cells and the ratio between the numbers of IL-36α+ cells and the total of infiltrating mononuclear cells (Fig. 2g). We next evaluated the expression of IL-38 in pSS patients and controls. As shown in Fig. 2h–k,m, IL-38 was also up-regulated in the salivary glands of pSS, expressed mainly among acinar epithelial cells and infiltrating mononuclear cells (Fig. 2i–k). In control subjects, basal expression of IL-38 was also observed in the context of ductal epithelial cells (Fig. 2h). In order to study the cellular source of IL-36α among infiltrating mononuclear cells, flow cytometry analysis of isolated SGMCs and confocal microscopy analysis of paraffin-embedded sections was also performed. As shown in Fig. 3a,b, T cell receptor (TCR)-αβ+ cells and macrophages were the major source of IL-36α in SGMC, with plasma cells accounting for only a small percentage of IL-36α+ cells (the gating strategy is shown in Supporting information, Fig. S2). According to previous reports 15–17, CD3+ cells (Fig. 4a–c) and CD68+ macrophages (Fig. 4d,) were confirmed by confocal microscopy analysis to express IL-36α in the salivary glands of pSS. Conversely, only a small percentage of B cells (Fig. 4f–h) and plasma cells (Fig. 4i,j) expressed IL-36α.
Figure 2.
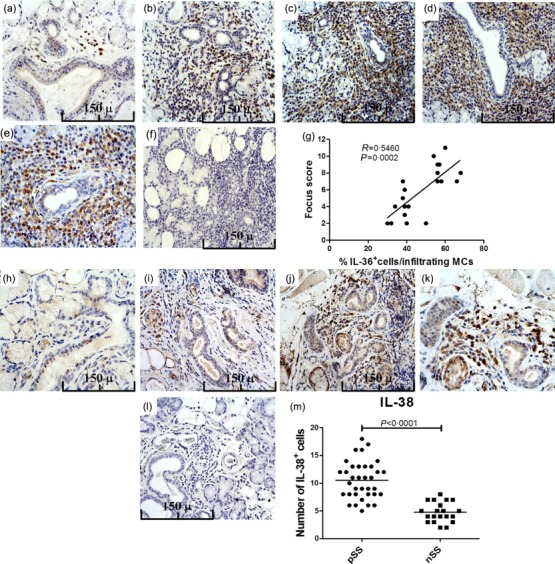
Interleukin (IL)-36α and IL-38 expression in primary Sjögren’s syndrome (pSS) patients’ minor salivary glands as assessed by immunohistochemistry. Representative microphotographs showing IL-36α immunostainings in 20 control subjects and 35 pSS patients. (a) IL-36α expression in minor salivary glands of controls. (b–e) IL-36α expression in the minor salivary glands of pSS patients with different focus score: focus scores 2 (b), 3 (c) and 4 (d,e). (f) Representative microphotographs showing minor salivary gland sections of a patient with pSS and focus score 4 stained with control isotype antibody. (g) Correlation of IL-36α+ cells with the focus scores. Number of IL-36α-expressing cells was correlated with the ratio between the number of IL-36+ cells and the number of infiltrating mononuclear cells in minor salivary glands of pSS. The r2 (r2 = 0·5460) and P (P = 0·0002) values were determined with Spearman’s correlation coefficient. (h–k) Representative microphotographs showing IL-38 immunostainings in 20 control subjects and 35 pSS patients. (h) In control subjects IL-38 was observed exclusively among ductal epithelial cells. (i–k) In pSS patients a strong expression of IL-38 was observed among acinar epithelial cells and infiltrating mononuclear cells. (l) Representative microphotographs showing minor salivary gland sections of a patient with pSS and focus score 4 stained with control isotype antibody. (m) Quantification of IL-38+ cells in pSS and non-Sjögren’s syndrome patients (nSS). (a–d,f,h–j,l) Original magnification ×250; (e,k) original magnification ×400.
Figure 4.
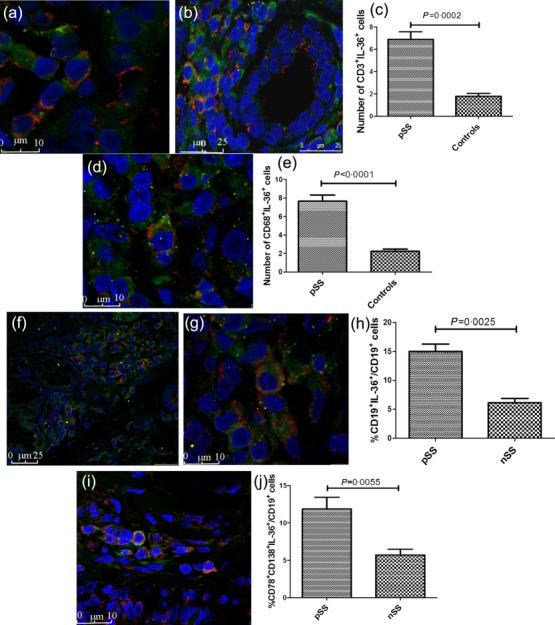
Confocal analysis of cells expression of interleukin (IL)-36α in minor labial salivary glands (MSGs). (a,b) Representative images of confocal analysis of IL-36α and CD3 co-localization in salivary glands of pSS patients. Salivary gland (SG) epithelial cells do not show IL-36α epithelial staining (b). Nuclei were counterstained with toto-3 (blue). (c) Number of CD3+IL-36+ cells in primary Sjögren’s syndrome (pSS) patients and controls. (d) Representative image of confocal analysis of IL-36α and CD68 co-localization in salivary glands of pSS patients. Nuclei were counterstained with toto-3 (blue). (d) Quantification of CD68+IL-36α+ cells in pSS patients and controls. (f–g) Representative images of confocal analysis of IL-36α and CD19 co-localization in salivary glands of pSS patients. Nuclei were counterstained with toto-3 (blue). (h) Quantification of CD19+IL-36α+/CD19+ cells in pSS patients and controls. (i) Representative image of confocal analysis of IL-36α, CD78 and CD138 co-localization in salivary glands of pSS patients. (i) Percentages of CD38+IL-36+ cells in pSS and non-Sjögren’s syndrome patients (nSS) as assessed by flow cytometry. Data are shown as mean ± standard error of the mean.
Figure 3.
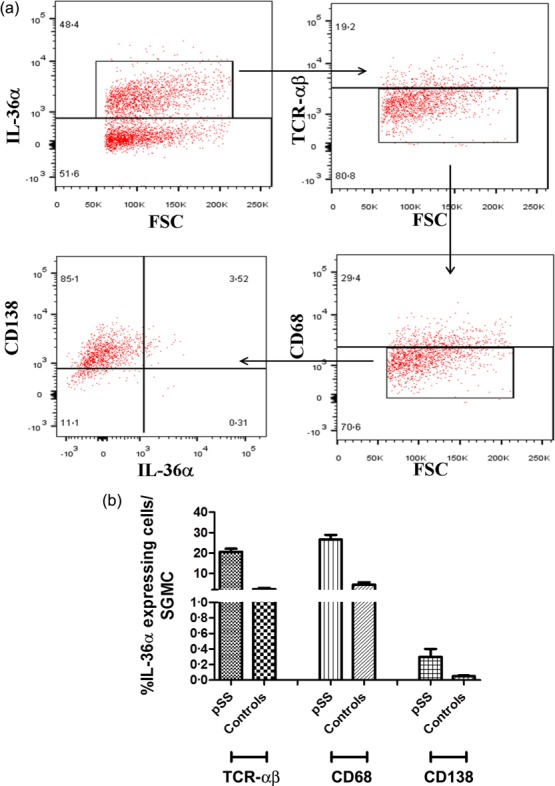
Interleukin (IL)-36α is expressed by T cells and macrophages in the salivary glands of primary Sjögren’s syndrome (pSS) patients. Salivary glands mononuclear cells were isolated from biopsies obtained from 10 pSS patients and 10 controls. (a) Representative dot-plots showing IL-36α expression by T cell receptor (TCR)-αβ+, CD68+ and CD138 cells. (b) Percentages of IL-36α-producing cells among TCRαβ+, CD68+ and CD138 cells in pSS patients and controls.
γδ+/IL-17+IL-36+T cells are expanded in the salivary glands of pSS patients and correlated with the focus score
Because of the role of IL-36α in modulating the function of γδ T cells in psoriasis 3 and the absence of studies regarding their immunological behaviour in pSS, we next analysed these innate cells in patients and controls using flow cytometry (the gating strategy is shown in Supporting information, Fig. S2). No significant expansion of γδ T cells was observed in pSS compared to controls (3·8 ± 0·7 versus 3·1 ± 0·8; P > 0·05) (data not shown); γδ T cells from pSS patients, however, produced higher amounts of IL-17 compared to controls (4·27 ± 0·3 versus 0·73 ± 0·04; P < 0·005) (Fig. 5a–c). Although numerically not expanded compared to controls, the percentages of γδ T cells in pSS was correlated significantly with the focus score (Fig. 5j). Conversely, no difference in γδ+/IFN-γ+ T cell levels in pSS and controls [0·34 ± 0·22 versus 0·21 ± 0·11, P = not significant (n.s.), data not shown] was observed. Notably, a significantly higher percentage of γδ+ T cells from pSS patients co-expressed IL-17 and IL-36α (3·5 ± 0·5% versus 0·45 ± 0·01%, P < 0·001) (Fig. 5d–f) and the IL-36R (4·07 ± 0·7% in pSS versus 0·81 ± 0·4% in nSS; P < 0·005) (Fig. 5g–i), suggesting a possible autocrine stimulatory loop.
Figure 5.
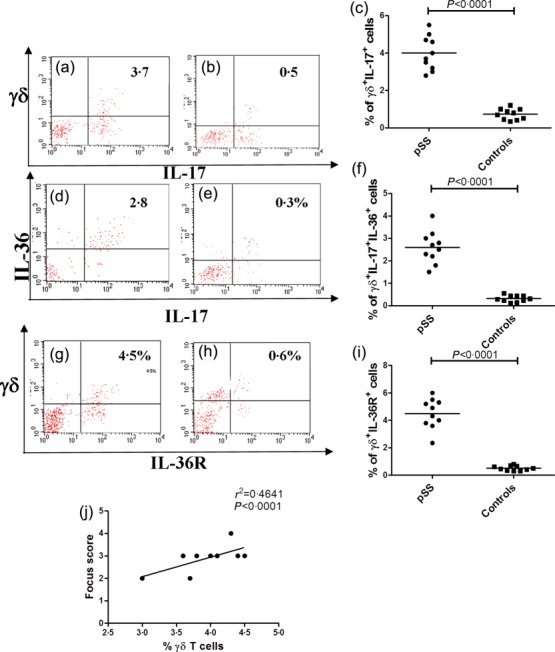
γδ T cells in primary Sjögren’s syndrome (pSS) salivary glands as assessed by flow cytometry. Salivary gland mononuclear cells were isolated from biopsies obtained from 10 pSS patients and 10 controls. (a) representative dot-plot showing the percentage of IL-17+ γδ T cells among salivary gland mononuclear cells of pSS patients (a) and control subjects (b). (c) Percentages of γδ+IL-17+ cells in pSS and controls. (d,e) Representative dot-plot showing the percentage of IL-36+IL-17+ γδ T cells among salivary gland mononuclear cells of pSS patients (d) and non-Sjögren’s syndrome patients (nSS) subjects (e). (f) Percentages of γδ+IL-17+IL-36+ cells in pSS patients and controls. (g,h) Representative dot plot showing the percentage of IL-36R+ γδ T cells among salivary gland mononuclear cells of pSS patients (g) and controls (h). i: percentages of γδ+IL-36R+ cells in pSS patients and controls. (j) Correlation between the percentages of γδ+ T cells and the lymphocytic focus score.
Discussion
In this study we demonstrate for the first time, to our knowledge, that the IL-36 axis is modulated in pSS and probably involved in the control of the IL-23/IL-17/IL-22 axis, as demonstrated previously in other autoimmune diseases 18,19, and suggest a role for γδ+/IL-17+/IL-36α+ in the pathogenesis of pSS.
IL-36α is a member of the IL-1 cytokine family, expressed mainly in keratinocytes, bronchial epithelium, brain tissue, monocytes/macrophages αβ and γδ T lymphocytes 1. IL-36α signals through a heterodimeric complex consisting of the IL-1 receptor accessory protein 10. Ligation of IL-36α with its cognate receptor seems to induce proinflammatory responses, being potentially involved in the pathogenesis of systemic autoimmune diseases such as rheumatoid arthritis and psoriasis 2,3. In particular, IL-36α has been proposed as a central regulator of human immune responses acting in the interface between innate and adaptive immunity by expanding both IL-17+ γδ Τ cells and IL-17+ αβ T cells 3,17,20. The immune effects of IL-36α seem to be antagonized by two different IL-36R ligands, IL-36RA and IL-38. Ligation of IL-36RA and/or of IL-38 with the IL-36R may, in fact, reverse the proinflammatory properties of IL-36α 21.
In our study, levels of IL-36α were increased significantly in the serum and salivary glands of pSS patients. Serum levels of IL-36α were increased significantly in pSS patients and correlated with disease activity, suggesting a potential role of IL-36α dosage in monitoring the activation of the immune system in pSS. Salivary gland IL-36α was correlated significantly with the level of tissue inflammation and detected, in particular, in the infiltrating cells only, being virtually absent among ductal and acinar epithelial cells. Using flow cytometry and confocal microscopy analysis, these cells were confirmed to be, to a larger extent, αβ+ CD3+ T lymphocytes and CD68+ macrophages. Plasma cells were also confirmed, however, to be a tissue source of IL-36α in both pSS and controls. Tissue IL-36α levels also correlated with the expression of IL-17, IL-22 and IL-23p19. Despite the high levels of IL-36α observed, IL-36R was not up-regulated in pSS, suggesting that the physiological basal expression of IL-36R might be not increased further under inflammatory stimuli. Several studies support the dual regulation of the two known inhibitors of IL-36α, IL-36RA and IL-38 in autoimmune diseases (reviewed in 22). In our study, IL-36RA and IL-38 were modulated differently in pSS. IL-36RA was, in fact, down-regulated significantly, suggesting an imbalance in IL-36 response in pSS. Conversely, IL-38 was up-regulated significantly suggesting, in our opinion, the occurrence of an attempt by the immune system to counteract the imbalanced activation of IL-36.
γδ T cells represent a small subset of T cells that possess a distinct TCR on their surface and are considered to be located at the border between the more evolutionarily primitive innate immune system and the more evolved adaptive immune system 23. Similarly to αβ T cells, γδ T cells have been demonstrated to be polarized into T helper type 17 (Th17) (producing only IL-17), Th1/17 (producing both IFN-γ and IL-17) and Th22 (producing only IL-22) populations, with distinct cytokines required for their initial polarization and later maintenance 23. These data indicate that γδ T cells participate in both the regulation and the propagation of murine lupus.
IL-36α has been proved to promote the expansion and the production of IL-17 by γδ+ T cells in psoriatic skin 3. In this study we provide the first demonstration that, in pSS, γδ+ T cells isolated from the salivary glands, although not significantly expanded, are correlated with the lymphocytic focus score. γδ+ T cells were only a small percentage of infiltrating T cells in pSS, but they appeared to be activated because of their expression of high levels of IL-36R and the production of higher levels of IL-17 and IL-36α. The absence of IFN-γ production suggests the presence of specific and coordinate regulation of effector γδ+ T cells functions. γδ+ T cells seem to participate in both the regulation and the propagation of systemic autoimmunity 24 to be the major IL-17 producers in the skin of patients with psoriasis and to promote adaptive immune response by promoting a Th1 response 25. Our findings may suggest that activated IL-36R+IL-17+ γδ+ T cells might be a specific feature of pSS contributing to adaptive and immune responses in pSS and highlight the importance of innate sources of IL-17 in the pathogenesis of disease.
In conclusion, in this study we demonstrate that a significant increase in circulating and tissue levels of IL-36α occurs in pSS. Further studies, however, are required to define more clearly the role of IL-36α in pSS pathogenesis and in the modulation of γδ+ T cell immune functions.
Acknowledgments
This work was supported by a grant from Ministero della Università e della Ricerca Scientifica from Italy.
Disclosure
The authors declare no conflicts of interest.
Supporting Information
Additional Supporting information may be found in the online version of this article at the publisher’s Web site:
Fig. S1. Correlation between proinflammatory cytokines and IL-36α. (b) Correlation between interleukin (IL)-17 and IL-36α m-RNA expression. (b) Correlation between IL-22 and IL-36α m-RNA expression. (c) Correlation between IL-23p19 and IL-36α m-RNA expression. The r2 and P-values were determined with Spearman’s correlation coefficient.
Fig. S2. Gating strategy for flow cytometry analysis. In this sample gating, cells were first gated for lymphocytes [side-scatter (SSC)-A versus forward scatter (FSC)-A] and the lymphocyte gate analysed further for CD45 expression and for uptake of 7-aminoactinomycin D (7AAD) to determine live versus dead cells.
References
- van de Veerdonk FL, Netea MG. New insights in the immunobiology of IL-1 family members. Front Immunol. 2013;4:167. doi: 10.3389/fimmu.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S, Derer A, Messbacher ME, et al. The novel cytokine interleukin-36α is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis. 2013;72:1569–74. doi: 10.1136/annrheumdis-2012-202264. [DOI] [PubMed] [Google Scholar]
- Tortola L, Rosenwald E, Abel B, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012;122:3965–76. doi: 10.1172/JCI63451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston A, Xing X, Guzman AM, et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol. 2011;186:2613–22. doi: 10.4049/jimmunol.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamacchia C, Palmer G, Rodriguez E, et al. The severity of experimental arthritis is independent of IL-36 receptor signaling. Arthritis Res Ther. 2013;15:R38. doi: 10.1186/ar4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derer A, Groetsch B, Harre U, et al. Blockade of IL-36 receptor signaling does not prevent from TNF-induced arthritis. PLoS One. 2014;9:e101954. doi: 10.1371/journal.pone.0101954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary Sjögren’s syndrome. Nat Rev Rheumatol. 2013;9:544–56. doi: 10.1038/nrrheum.2013.110. [DOI] [PubMed] [Google Scholar]
- Ciccia F, Guggino G, Rizzo A, et al. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann Rheum Dis. 2012;71:295–301. doi: 10.1136/ard.2011.154013. [DOI] [PubMed] [Google Scholar]
- Nguyen CQ, Hu MH, Li Y, Stewart C, Peck AB. Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjögren’s syndrome: findings in humans and mice. Arthritis Rheum. 2008;58:734–43. doi: 10.1002/art.23214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresnigt MS, van de Veerdonk FL. Biology of IL-36 cytokines and their role in disease. Semin Immunol. 2013;25:458–65. doi: 10.1016/j.smim.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Gresnigt MS, Rösler B, Jacobs CW, et al. The IL-36 receptor pathway regulates Aspergillus fumigatus-induced Th1 and Th17 responses. Eur J Immunol. 2013;43:416–26. doi: 10.1002/eji.201242711. [DOI] [PubMed] [Google Scholar]
- Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American–European Consensus Group. Ann Rheum Dis. 2002;61:554–8. doi: 10.1136/ard.61.6.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seror R, Ravaud P, Bowman SJ, et al. EULAR Sjogren’s syndrome disease activity index: development of a consensus systemic disease activity index for primary Sjogren’s syndrome. Ann Rheum Dis. 2010;69:1103–9. doi: 10.1136/ard.2009.110619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia F, Alessandro R, Rodolico V, et al. IL-34 is overexpressed in the inflamed salivary glands of patients with Sjogren’s syndrome and is associated with the local expansion of pro-inflammatory CD14brightCD16+ monocytes. Rheumatology (Oxf) 2013;52:1009–17. doi: 10.1093/rheumatology/kes435. [DOI] [PubMed] [Google Scholar]
- Smith DE, Renshaw BR, Ketchem RR, Kubin M, Garka KE, Sims JE. Four new members expand the interleukin-1 superfamily. J Biol Chem. 2000;275:1169–75. doi: 10.1074/jbc.275.2.1169. [DOI] [PubMed] [Google Scholar]
- Barksby HE, Nile CJ, Jaedicke KM, Taylor JJ, Preshaw PM. Differential expression of immunoregulatory genes in monocytes in response to Porphyromonas gingivalis and Escherichia coli lipopolysaccharide. Clin Exp Immunol. 2009;156::479–87. doi: 10.1111/j.1365-2249.2009.03920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne S, Palmer G, Lamacchia C, et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood. 2011;118:5813–23. doi: 10.1182/blood-2011-05-356873. [DOI] [PubMed] [Google Scholar]
- Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev. 2013;52:116–32. doi: 10.1111/imr.12027. [DOI] [PubMed] [Google Scholar]
- Vigne S, Palmer G, Martin P, et al. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood. 2012;120:3478–87. doi: 10.1182/blood-2012-06-439026. [DOI] [PubMed] [Google Scholar]
- van de Veerdonk FL, Stoeckman AK, Wu G, et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc Natl Acad Sci USA. 2012;109:3001–5. doi: 10.1073/pnas.1121534109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Peng X, Li Y, Li M. Role of IL-38 and its related cytokines in inflammation. Mediat Inflamm. 2015;2015:807976. doi: 10.1155/2015/807976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S, Shi C, Budd RC. γδ T cells promote a Th1 response during coxsackievirus B3 infection in vivo: role of Fas and Fas ligand. J Virol. 2002;76:6487–94. doi: 10.1128/JVI.76.13.6487-6494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Shen X, Ding C, et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Correlation between proinflammatory cytokines and IL-36α. (b) Correlation between interleukin (IL)-17 and IL-36α m-RNA expression. (b) Correlation between IL-22 and IL-36α m-RNA expression. (c) Correlation between IL-23p19 and IL-36α m-RNA expression. The r2 and P-values were determined with Spearman’s correlation coefficient.
Fig. S2. Gating strategy for flow cytometry analysis. In this sample gating, cells were first gated for lymphocytes [side-scatter (SSC)-A versus forward scatter (FSC)-A] and the lymphocyte gate analysed further for CD45 expression and for uptake of 7-aminoactinomycin D (7AAD) to determine live versus dead cells.