

Abstract
Natural killer (NK) cell-mediated antibody-dependent cellular cytotoxicity (ADCC) has been linked to protection from HIV infection and slower progression towards AIDS. However, antibody-dependent activation of NK cells results in phenotypical alterations similar to those observed on NK cells from individuals with progressive HIV infection. Activation of NK cells induces matrix metalloproteinase (MMP)-mediated cleavage of cell surface CD16. In the present study we assessed the phenotype and functional profile of NK cells exhibiting post-activation MMP-mediated CD16 cleavage. We found that NK cells achieving the highest levels of activation during stimulation exhibit the most profound decreases in CD16 expression. Further, we observed that educated KIR3DL1+ NK cells from human leucocyte antigen (HLA)-Bw4-carrying donors exhibit larger decreases in CD16 expression post-activation than the KIR3DL1− NK cell subset containing cells educated via other inhibitory receptor/ligand combinations and non-educated NK cells. Lastly, we assessed the ex-vivo expression of CD16 on educated KIR3DL1+ NK cells and the KIR3DL1− NK cell subset from HLA-Bw4-carrying HIV-uninfected and HIV-infected donors. Suggestive of in-vivo activation of KIR3DL1+ NK cells during HIV infection, CD16 expression was higher on KIR3DL1+ than KIR3DL1− NK cells in uninfected donors but similar on both subsets in HIV-infected donors. These results are discussed in the context of how they may assist with understanding HIV disease progression and the design of immunotherapies that utilize antibody-dependent NK cell responses.
Keywords: AIDS, KIR3DL1, NK cell education
Introduction
A prophylactic vaccine is needed to curtail the HIV epidemic. Recently, much research has been directed towards understanding the potential of non-neutralizing antibodies to prevent HIV infection. Interest in the protective potential of non-neutralizing antibody effector functions has been stimulated by two major observations. First, the ability of the passively transferred b12 broadly neutralizing antibody to protect macaques from simian/HIV (SHIV) infection is reduced if the constant region (Fc) of the antibody is modified in a manner that abrogates binding to Fc receptors on cells of the innate immune system1. Secondly, the recent RV144 vaccine trial in Thailand provided a modest level of protection from HIV infection, despite not inducing robust cytotoxic T lymphocyte (CTL) responses or broadly neutralizing antibodies2,3. A correlate of protection analysis of this trial revealed that the presence of antibodies capable of mediating antibody-dependent cellular cytotoxicity (ADCC) corresponded with protection when vaccinated individuals also had low levels of immunoglobulin (Ig)A antibodies that could inhibit IgG binding and induction of ADCC4,5. Taken together, these data suggest that protection from HIV infection might be achievable by vaccine induced non-neutralizing antibodies. This notion is not surprising in the context of additional data suggesting a role for ADCC in preventing HIV transmission from mothers to breast-fed infants6, or a study linking anti-viral ADCC to the ability of a live-attenuated SIV vaccine to protect macaques from infection with pathogenic SIV7.
In addition to reports linking ADCC to protection from HIV infection, numerous studies have suggested a role for ADCC in slowing progression to AIDS. Antibodies capable of mediating ADCC are found at higher levels in HIV-infected individuals that naturally control HIV replication8. Individuals with slow progressing HIV infections also tend to exhibit ADCC-mediating antibodies against a broader range of viral epitopes9. Furthermore, a study in SIV-infected macaques linked sustained plasma ADCC activity to slower disease progression10. While these studies focused on the ability of anti-viral antibodies to mediate ADCC, other investigators have examined the ability of effector cells, such as natural killer (NK) cells, from HIV-infected individuals to mediate ADCC. In general, the ability of effector cells to mediate ADCC is reduced in HIV-infected patients11–13. More specifically, the ability of NK cells to mediate ADCC has been linked to disease progression status12. This decreased potential to mediate ADCC has been linked to the expression levels of the low-affinity receptor for the constant region of IgG, CD1612. These data have been interpreted previously as NK cell ADCC competency associating with slower progression to AIDS. It should be noted, however, that a large body of literature now exists demonstrating that activation of NK cells results in decreased CD16 surface expression and functional anergy14–17. Thus, activation of NK cells to mediate ADCC might itself induce phenotypical and functional alterations in NK cells that are similar to those associated with progression to AIDS. ADCC may represent a ‘double-edged sword’ that has the potential to protect from HIV infection and disease progression, but also carries the potential to induce functional and phenotypical alterations in NK cells that contribute to disease progression.
Decreased cell surface expression of CD16 on NK cells is observed following antibody-dependent and -independent stimulations14,16,17. The cells that exhibit post-activation decreases in CD16 expression appear to be those that were activated during stimulation, as expression of the CD107a degranulation marker is found on NK cells with reduced CD16 expression14. Furthermore, post-activation decreases in CD16 expression are induced by matrix metalloproteinases (MMP) that are produced in NK cells upon activation. The ADAM17 MMP has been implicated in cleaving CD16 from the surface of NK cells activated by cytokines, as well as antibody-dependent and -independent stimuli17.
Although much has been established about post-activation MMP-mediated cleavage of CD16 from NK cells, many questions remain about the identity of the NK cells losing CD16 surface expression. Given that cleavage of CD16 appears to occur on activated NK cells14, we hypothesized that the degree of NK cell activation would be predictive of the amount of CD16 cleavage induced. As stronger cumulative activating signalling is required to induce NK cell interferon (IFN)-γ production than is required to induce expression of the CD107a degranulation marker18, we predicted that IFN-γ+ CD107a+/− post-activation NK cells would express lower levels of CD16 expression than IFN-γ− CD107a+ NK cells, which would exhibit lower CD16 expression levels than IFN-γ− CD107a− NK cells. Lastly, we also predicted that the heightened function of educated NK cells, defined as those expressing the inhibitory killer immunoglobulin-like receptor 3DL1 (KIR3DL1) that recognizes a self major histocompatibility complex class I (MHC-I or HLA-I) molecule containing the human leucocyte antigen (HLA)-Bw4 ligand, would result in lower levels of CD16 expression post-activation than those exhibited by the KIR3DL1− NK cell subset, which contains non-educated NK cells and NK cells educated through alternative inhibitory receptor/ligand combinations19–21. We utilized two previously described assays to measure antibody-dependent activation of NK cells16, and now present data illustrating a role for the degree of NK cell activation on post-activation CD16 cleavage.
Materials and methods
Participants
Whole blood was collected from eight HIV-uninfected donors by forearm venipuncture into vacuettes containing sodium heparin anti-coagulant (Greiner Bio-One, Frickenhausen, Germany). These samples served as a source of effector cells for the HIV+ plasma and anti-CD16 NK cell activation assays. Six of these donors were typed with sequence-based typing at the HLA-B locus by the Victorian Transplantation and Immunogenetics Service (Parkville, Australia), and were shown to carry HLA-B alleles containing the HLA-Bw4 public epitope. Plasma from an HIV-infected individual was obtained from a client of the Melbourne Sexual Health Centre. As this donor has been shown previously to carry anti-HIV antibodies capable of activating NK cells, this plasma was used as a source of antibodies for the HIV+ plasma NK cell activation assay16. To complete NK cell phenotype studies, peripheral blood mononuclear cells (PBMC) were obtained from 25 HIV-uninfected controls and 25 HIV-infected donors, enrolled into the Canadian Cohort of HIV-infected Slow Progressors (SP). SP had a median CD4 count of 655 cells/mm3 (range = 370–1190) and a median viral load (VL) of 562 RNA copies/ml of plasma (range = <40–25 170). These subjects were typed for HLA-I alleles using sequence-based typing kits from Atria Genetics, Inc. (South San Francisco, CA, USA). Assign 3·5+ software was used to interpret sequence information for allele assignment (Conexio Genetics, Perth, Australia). KIR3DL/S1 generic genotyping was performed by polymerase chain reaction (PCR) using two pairs of primers specific for amplification of either KIR3DL1 or KIR3DS1 alleles, as described previously22. KIR3DL1 allotyping was performed by sequencing KIR3DL1 exons, as described previously23. Single nucleotide polymorphisms (SNP) corresponding to the KIR3DL1 alleles were identified by aligning the sequenced DNA to a reference consensus sequence consisting of KIR3DL1 cDNA sequences. Informed consent was obtained from all donors prior to the collection of biological samples. The ethics committees of all participating institutions approved the conducted studies.
HIV+ plasma NK cell activation assay
Activation of NK cells by antibodies within HIV+ plasma was assessed utilizing a previously described intracellular cytokine staining assay16. Briefly, 150 μl of whole blood from HIV-uninfected donors was mixed with 50 μl of HIV-infected plasma in the presence of 1 μg/ml of HIV-1bal gp120 (NIH AIDS Reagent Program, Bethesda, MD, USA), 5 μg/ml brefeldin A (Sigma, St Louis, MO, USA) and 6 μg/ml monensin (BD Biosciences, San Jose, CA, USA) for 5 h at 37oC. Control conditions contained all reagents in experimental conditions, with the exception of HIV gp120. Following incubation fluorochrome-conjugated antibodies against cell surface antigens [peridinin chlorophyll (Per-CP)-conjugated anti-CD3 (BD Biosciences), phycoerythrin-cyanin 7 (PE-Cy7)-conjugated anti-CD56 (BD Biosciences), PE-conjugated anti-KIR3DL1 (BD Biosciences), allophycocyanin (APC)-Cy7-conjugated anti-CD16 (In Vitro Technologies, Melbourne, Australia) and APC-conjugated anti-CD107a (BD Biosciences)] were added to the whole blood. After lysing red blood cells using lysis buffer (BD Biosciences) the remaining white blood cells were permeabilized and stained with Alexa Fluor 700-conjugated anti-IFN-γ (BD Biosciences). Finally, cells were fixed in formaldehyde and acquired using a BD FACS Canto II flow cytometer. Data were analysed using FlowJo version 9·2 software (TreeStar Inc., Ashland, OR, USA).
Anti-CD16 NK cell activation assay
As described previously16, NK cells in whole blood were activated by the addition of the 3G8 anti-CD16 antibody clone. Briefly, 150 μl of whole blood from HIV-uninfected donors was incubated with fluorescein isothiocyanate (FITC)-conjugated anti-CD16 antibody (BD Biosciences) or no antibody in the presence of 5 μg/ml brefeldin A, and 6 μg/ml monensin for 5 h at 37oC. Following incubation, fluorochrome-conjugated antibodies against cell surface antigens [Per-CP-conjugated anti-CD3 (BD Biosciences), PE-Cy7-conjugated anti-CD56 (BD Biosciences), PE-conjugated anti-KIR3DL1 (BD Biosciences) and APC-conjugated anti-CD107a (BD Biosciences)] were added to the whole blood. Additionally, at this step FITC-conjugated anti-CD16 (BD Biosciences) was added to the control well to assess the CD16 expression on non-activated NK cells. Following lysis of red blood cells the remaining white blood cells were permeabilized and stained with Alexa Fluor 700-conjugated anti-IFN-γ (BD Biosciences). Finally, cells were fixed in formaldehyde and acquired using a BD FACS Canto II flow cytometer. Data were analysed using FlowJo version 9·2 software (TreeStar Inc.).
Phenotypical staining to identify KIR3DL1+/− NK cells expressing CD16
PBMC were obtained from a total of 50 individuals, including healthy uninfected donors (n = 25) and HIV-infected individuals (n = 25), enrolled into the Canadian Cohort of HIV Infected SP. PBMC (106 per subject) were cell surface-stained with anti-CD3 (PerCP), anti-CD56 (APC), anti-KIR3DL1 (PE) (clone DX9) and anti-CD16-Pacific blue (all from BioLegend, San Diego, CA, USA) for 30 min in the dark at room temperature (RT). Stained cells were then washed with RPMI supplemented with 10% fetal bovine serum, fixed with 1% paraformaldehyde solution (PFA) (Fisher Scientific, Loughborough, UK), and 300 000–400 000 events were acquired using a FACSCanto flow cytometer (BD Biosciences) to assess the median fluorescence intensity (MFI) of CD16 cell surface expression of CD3−, CD56+, KIR3DL1+/− NK cells. Data were analysed using FlowJo version 9·4 software (TreeStar Inc.).
Inhibition of MMP
To assess the impact of MMPs on the surface expression of CD16 following stimulation, both the HIV+ plasma and anti-CD16 NK cell activation assays were conducted under conditions where the GM6001 MMP-inhibiting drug (50 μM) (Sigma) or an equivalent volume of the DMSO (Sigma) drug vehicle were added during stimulation. The GM6001 drug was used at a concentration of 50 μM.
Statistics
Statistical analyses were conducted using GraphPad Prism version 4·0. Differences between paired data for two and more than two groups were analysed using Wilcoxon’s matched-pairs test and the Friedman test followed by Dunn’s post-hoc tests, respectively. Differences between data sets were considered to be significantly different at P < 0·05. Data throughout the paper are represented in the [median (range)] format.
Results
CD16 expression following NK cell activation
As a prelude to investigating the role of education and degree of NK cell activation on the MMP-mediated cleavage of CD16, we first assessed the ability of both the HIV+ plasma and anti-CD16 NK cell activation assays to stimulate NK cells and decrease CD16 expression in an MMP-dependent manner. Robust NK cell activation, defined as IFN-γ production and/or CD107a expression on CD3−CD56dim NK cells, was observed in all eight whole blood donors following activation of NK cells in both the HIV+ plasma [17·9% (7·2–32·8%)] and anti-CD16 activation assays [40·8% (28·6–70%)] (Fig. 1a,b). As both assays induced NK cell activation, we assessed if the observed activation corresponded to the decreased surface expression of CD16 that has been demonstrated previously. Both the HIV+ plasma [1751 (1029–3195) versus 575 (332–1593), P < 0·01] and anti-CD16 [4031 (1371–7575) versus 728 (428–1171), P < 0·01] NK cell activation assays induced substantial CD16 down-regulation, as measured by median fluorescence intensity (MFI) (Fig. 1c). Finally, to confirm that the observed CD16 down-regulation was due to MMP-mediated cleavage, we activated NK cells in both assays in the presence of the MMP-inhibitor GM6001 (50 μM) or an equivalent volume of the DMSO drug vehicle. Post-activation CD16 surface expression was maintained at a higher level upon activation in the presence of GM6001, compared to DMSO (Fig. 1d). These data show that activation of NK cells through anti-HIV antibodies or anti-CD16 antibody triggers cleavage of CD16 from the surface of NK cells in an MMP-dependent manner.
Figure 1.

Activation of natural killer (NK) cells in the anti-CD16 and HIV+ plasma assays results in matrix metalloproteinase (MMP)-mediated cleavage of CD16 from the NK cell surface. (a) Contour plots demonstrate the gating strategy implemented. Initially, gating was upon the lymphocyte population (top, left), followed by the CD3−CD56dim NK cell population (top, right). The NK cell population was assessed for activation, represented by interferon (IFN)-γ production and CD107a expression, in the control and activation conditions of both the HIV+ plasma and anti-CD16 assays (bottom). (b) Graphs depict the relative number of NK cells activated to produce IFN-γ and/or express CD107a in the control and stimulated conditions of the anti-CD16 (right) and HIV+ plasma (left) assays. (c) Contour plots depict the expression of CD16 on the control (left) and activated (right) conditions in both the HIV+ plasma (bottom) and anti-CD16 (top) NK cell activation assays. Graphs illustrate the relative CD16 median fluorescence intensity (MFI) on NK cells pre- and post-activation in the HIV+ plasma (bottom) and anti-CD16 (top) NK cell activation assay. (d) Contour plots portray the expression of CD16 on non-activated NK cells (left), as well as NK cells stimulated in the presence of dimethylsuphoxide (DMSO) drug vehicle (middle) or MMP-inhibitor GM6001 (50 μM) (right).
Influence of degree of NK cell activation on post-activation CD16 expression
Given that the utilized assays were able to detect NK cell activation and the resulting MMP-mediated CD16 cleavage, we next assessed the impact that the degree of NK cell activation has on the level of MMP-mediated CD16 cleavage. It has been demonstrated previously that activated NK cells are likely to exhibit reduced CD16 expression14, but it is currently not known how the degree to which an NK cell becomes activated impacts upon the post-activation level of CD16 expression. Previous investigations have demonstrated that NK cells require stronger activation to produce IFN-γ than to express the CD107a degranulation marker18. If our hypothesis, that the NK cells most likely to exhibit reduced CD16 expression were those with the highest potential to respond, is correct, it would be expected that CD107a+/− IFN-γ+ NK cells would express lower levels of CD16 than CD107a+ IFN-γ− NK cells which, in turn, should have lower CD16 expression levels than CD107a− IFN-γ− NK cells.
Initially, we assessed whether activated NK cells belong predominantly to the CD16dim/− NK cell population. Assessments of post-activation CD16+ and CD16 dim/− NK cells in both the HIV+ plasma [1·9% (0·9–7·1%) versus 33·6% (10·7–48·5%), P < 0·01] and anti-CD16 [11·6% (1·7–40·5%) versus 56·1% (33·2–73·2%), P < 0·01] NK cell activation assays revealed that responding NK cells (i.e. IFN-γ+ and/or CD107a+) were detected more frequently in the CD16dim/− NK cell gate compared to the CD16+ gate (Fig. 2a). To assess if the degree of activation determined the level of post-activation CD16 expression, we compared NK cells with IFN-γ+ CD107a+, IFN-γ+ CD107a−, IFN-γ− CD107a+ and IFN-γ− CD107a− functional profiles for their MFI of CD16 expression. Friedman tests revealed significant differences between these populations in both the HIV+ plasma (P < 0·0001) and anti-CD16 (P < 0·0001) NK cell activation assays. In the HIV+ plasma NK cell activation assay the level of CD16 expression was significantly lower in IFN-γ+ CD107a+ [64·4 (0–104)] and IFN-γ+ CD107a− [108 (23·8–210)] NK cells than in IFN-γ−CD107a− [835 (433–1735)] NK cells (P < 0·001 and P < 0·01, respectively), whereas CD16 MFI in IFN-γ− CD107a+ [199 (83·9–359)] NK cells did not differ significantly from that in IFN-γ− CD107a− NK cells (P > 0·05) (Fig. 2b). Further, IFN-γ+ CD107a+ NK cells exhibited significantly lower CD16 MFI than IFN-γ− CD107a+ NK cells (P < 0·05) (Fig. 2b). Similarly, in the anti-CD16 NK cell activation assay the level of CD16 expression was significantly lower in IFN-γ+ CD107a+ [384·5 (215–487)] and IFN-γ+ CD107a− [429 (242–559)] NK cells than in IFN-γ− CD107a− [1184 (793–1952)] NK cells (P < 0·001 and P < 0·05, respectively), whereas CD16 MFI in IFN-γ− CD107a+ [589·5 (310–1042)] NK cells did not differ significantly from that in IFN-γ− CD107a− NK cells (P > 0·05) (Fig. 2b). Further, IFN-γ+ CD107a+ NK cells exhibited significantly lower CD16 MFI than IFN-γ− CD107a+ NK cells (P < 0·05) (Fig. 2b). Collectively, these data demonstrate that activated NK cells exhibit decreased CD16 expression. Furthermore, the level to which CD16 expression is decreased reflects the degree to which stimulation activated the NK cell.
Figure 2.
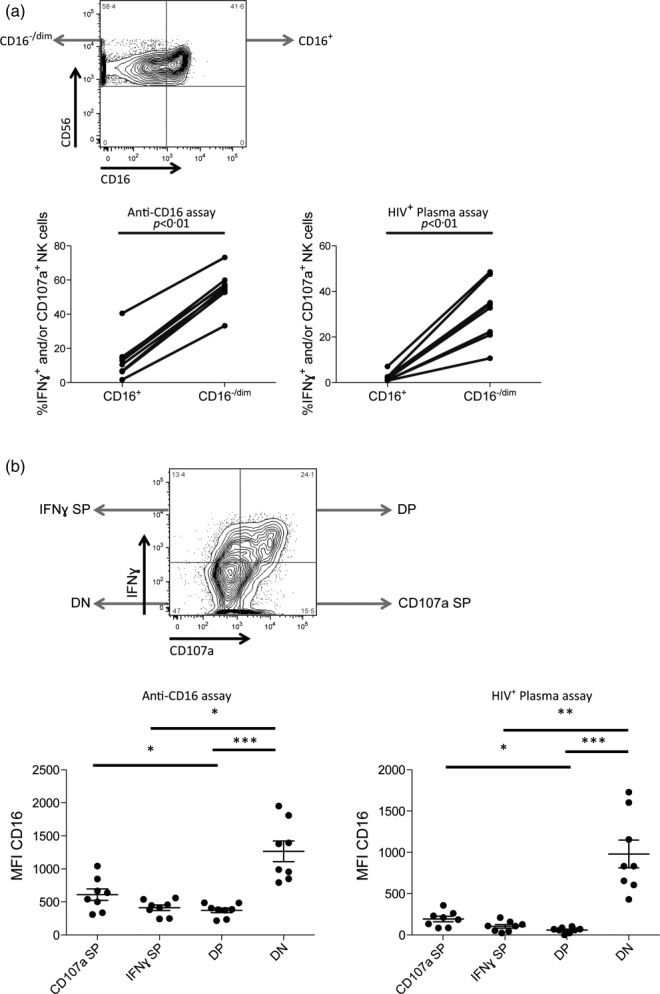
Functional profile of natural killer (NK) cells exhibiting reduced cell surface CD16 expression. (a) The fluorescence activated cell sorter (FACS) plot illustrates gating to identify CD16dim/− and CD16+ NK cells (top). Graphs (bottom) illustrate the relative activation [i.e. interferon (IFN)-γ production and/or CD107a expression] of the CD16dim/− and CD16+ NK cell populations after activation in the HIV+ plasma (right) and anti-CD16 (left) NK cell activation assays. (b) The FACS plot illustrates the gating utilized to identify the IFN-γ+ CD107a+ [i.e., double positive (DP)], IFN-γ+ CD107a− [i.e., IFNγ single positive (SP)], IFN-γ− CD107a+ [i.e., CD107a SP] and IFN-γ− CD107a− [i.e., double negative (DN)] functional profiles. Graphs depict the relative CD16 median fluorescence intensity (MFI) on NK cells exhibiting the IFN-γ+ CD107a+ [i.e., DP], IFN-γ+ CD107a− [i.e., IFNγ SP], IFN-γ− CD107a+ [i.e., CD107a SP] and IFN-γ− CD107a− [i.e., DN] functional profiles after stimulation in the HIV+ plasma (right) and anti-CD16 (left) NK cell activation assays. Lines above graphs represent statistically significant post-hoc comparisons. *P < 0·05; **P < 0·01; ***P < 0·001.
Post-activation CD16 expression on educated NK cells
The observation that the degree of NK cell activation influences the post-activation level of CD16 expression is consistent with our hypothesis that NK cells with the highest potential to mediate antibody-dependent responses are the most susceptible to MMP-mediated CD16 cleavage. The potential of NK cells to mediate both antibody-dependent and -independent functions is determined during the ontological process of education, where NK cells expressing inhibitory receptors that recognize self HLA-I ligands acquire the ability to respond upon receipt of a cumulative activating signal19–21. Previous work from our group and others has shown educated NK cells to be more primed to mediate antibody-dependent activation than non-educated NK cells19–21. As such, the observation of lower CD16 expression on the most highly activated NK cells suggests that decreased CD16 expression occurs most prominently on educated NK cells. To test this possibility, we evaluated the pre- and post-activation levels of CD16 on the KIR3DL1+ and the KIR3DL1− NK cell subsets from six donors known to carry the HLA-Bw4 ligand. KIR3DL1 expression in these donors was detected using the DX9 antibody clone, which recognizes inhibitory KIR3DL1 to the exception of activating KIR3DS1 isoforms24. Assessment of the activation of the educated KIR3DL1+ subset and the KIR3DL1− subset revealed that the KIR3DL1+ NK cells exhibited higher activation in both the HIV+ plasma [25·4% (13·2–51·9%) versus 15·3% (7·2–29·1%), P < 0·05] and anti-CD16 [49% (34·5–81·6%) versus 37·2% (23·9–68·1%), P < 0·05] NK cell activation assays (Fig. 3a). Further, KIR3DL1+ NK cells, compared to KIR3DL1− NK cells, exhibited a significantly larger decrease in CD16 expression post-activation in the HIV+ plasma [80% (46–90%) versus 67% (34–72%), P < 0·05] and anti-CD16 [87% (73–92%) versus 80·5% (59–89%), P < 0·05] NK cell activation assays (Fig. 3b). These data support the hypothesis that MMP-mediated CD16 cleavage preferentially targets those NK cells with the highest potential to mediate antibody-dependent activation.
Figure 3.
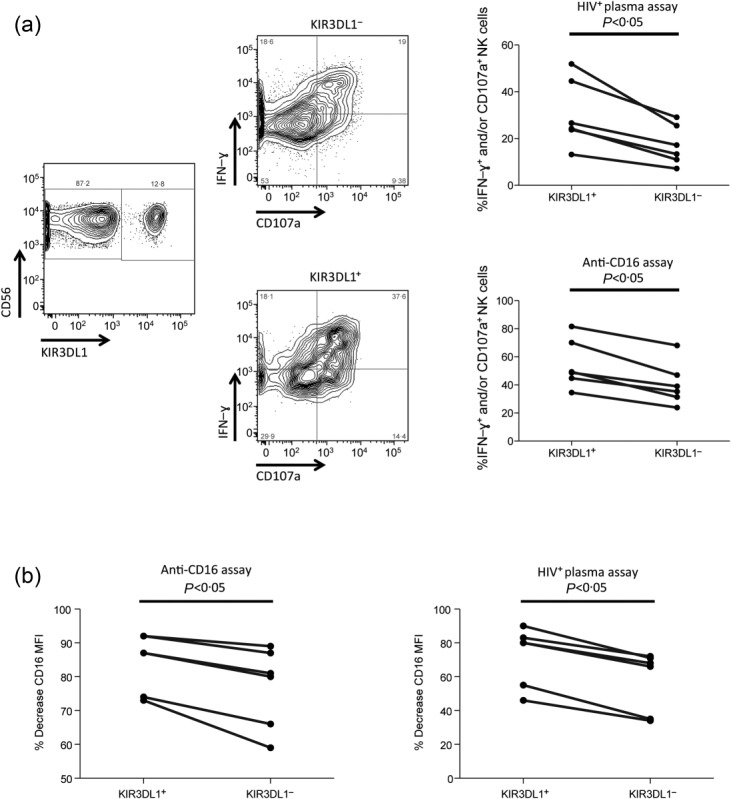
Influence of natural killer (NK) cell education on NK cell antibody-dependent function and post-activation CD16 cleavage. (a) Fluorescence activated cell sorter (FACS) plots depict the gating utilized to identify KIR3DL1+ and KIR3DL1− NK cells, as well as the ability of these NK cell subsets to produce interferon (IFN)-γ+ and express CD107a. Graphs portray the relative activation (i.e. IFN-γ+ and/or CD107a+) of KIR3DL1+ and KIR3DL1− NK cells after activation in the HIV+ plasma (top) and anti-CD16 (bottom) NK cell activation assays. (b) Graphs depict the percentage decrease in CD16 median fluorescence intensity (MFI) on KIR3DL1+ and KIR3DL1− NK cell populations after activation in the anti-CD16 (left) and HIV+ plasma (right) NK cell activation assays.
Relative ex-vivo expression of CD16 on KIR3DL1+ and KIR3DL1− NK cells from HLA-Bw4-carrying HIV-infected and uninfected donors
Given that educated KIR3DL1+ NK cells exhibit a functional advantage in the HIV+ plasma assay and are thus more susceptible to activation-induced CD16 cleavage, we next assessed whether HIV infection influenced the relative expression of CD16 on the KIR3DL1+ and the KIR3DL1− CD56+ NK cells from carriers of Bw4 antigens. If educated KIR3DL1+ NK cells are becoming activated preferentially during HIV infection, it would be predicted that the relative CD16 expression between KIR3DL1+ and KIR3DL1− NK cells would be altered in HIV-infected individuals, compared to uninfected donors. An initial screen of the expression of CD16 on KIR3DL1+ and KIR3DL1− NK cells from HLA-Bw4-carrying HIV-uninfected donors revealed a significantly higher CD16 MFI on KIR3DL1+ compared to KIR3DL1− NK cells [2242 (246–18187) versus 1133 (0–19394), P < 0·05] (Fig. 4). In comparison, HLA-Bw4-carrying HIV-infected individuals exhibited similar CD16 expression levels on KIR3DL1+ and KIR3DL1− NK cells [941 (290–4703) versus 941 (259–3635), P > 0·05] (Fig. 4). These data suggest that educated KIR3DL1+ NK cells are becoming activated during HIV infection and thus losing their CD16 expression. This observation might be of relevance in explaining the lack of functional advantage upon antibody-dependent activation for KIR3DL1+ educated NK cells from HIV-infected donors20.
Figure 4.
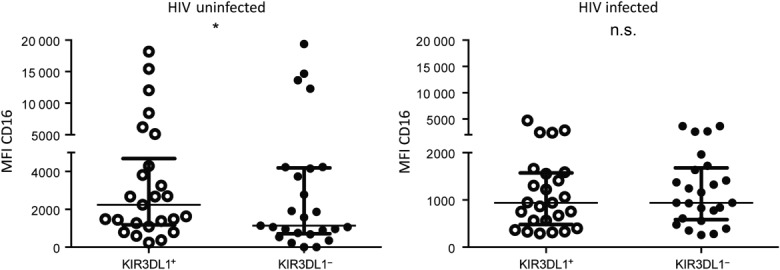
Ex-vivo expression of CD16 on KIR3DL1+ or KIR3DL1− natural killer (NK) cells (CD3−CD56+) from Bw4 carriers. Graphs depict the relative expression of CD16, measured as median fluorescence intensity, on KIR3DL1+ and KIR3DL1− NK cells from HIV-uninfected (left) and HIV-infected (right) donors. Symbols above the graphs represent the significance of Wilcoxon’s matched-pairs tests. *P < 0·05; n.s. = not significant.
Discussion
To our knowledge, the data within this paper provide several novel observations. First, this is the initial demonstration that the degree of post-activation MMP-mediated CD16 cleavage is proportional to the level of activation achieved by the NK cell during stimulation. Secondly, this is the first evidence that educated NK cells experience more CD16 cleavage post-activation. Lastly, this is the first evidence that CD16 expression is reduced on educated NK cells during HIV infection. These observations are consistent with previous literature demonstrating the involvement of MMPs in the cleavage of CD16 from the surface of activated NK cells14,16,17. Our data have ramifications for understanding HIV disease progression and will be instructive for future considerations of utilizing therapies that elicit NK cell responses.
Numerous studies have revealed the ability of effector cells to mediate ADCC decreases in HIV-infected patients11–13,25. In particular, the potential of NK cells to respond to antibody-dependent stimuli is decreased in individuals progressing towards AIDS12. Although the reasons for the decreased potential of NK cells from HIV-infected individuals to mediate ADCC are unknown, it has been suggested that MMP-induced decreased CD16 expression on NK cells from HIV-infected patients could be a major contributing factor. Ex-vivo treatment of PBMC from HIV-infected individuals with an MMP inhibitor restores CD16 expression on NK cells and increases the ability of these cells to respond to antibody-dependent activation26. Corroborating a role for decreased CD16 expression in the diminishing potential of NK cells to mediate antibody-dependent function in HIV infection, it has been demonstrated that the antibody-dependent function of NK cells from HIV infected individuals correlates with CD16 expression levels12. Lastly, some investigations suggest that despite anti-retroviral therapies that suppress viral replication, NK cells from HIV-infected patients exhibit reduced CD16 expression27.
Building upon these previous studies, the work presented in this paper demonstrates that MMP-mediated cleavage of CD16 from activated NK cells occurs in a manner that is dependent upon the level of activation an NK cell exhibits upon stimulation. As expected from this observation, educated KIR3DL1+ NK cells from HLA-Bw4-carrying donors, which exhibit more robust activation upon antibody-dependent stimulation than KIR3DL1− NK cells from these individuals, exhibited more post-activation CD16 cleavage than the KIR3DL1− NK cell subset. Given this observation, we hypothesize that reduced NK cell-mediated antibody-dependent functions in HIV infection are a result of activation-induced reduced CD16 expression on the KIR3DL1+ NK subset from Bw4 carriers. Our group has demonstrated previously that educated KIR3DL1+ NK cells from HLA-Bw4-carrying HIV-infected individuals, unlike KIR3DL1+ NK cells from HLA-Bw4-carrying uninfected donors, no longer exhibit higher antibody-dependent function than the KIR3DL1− subset20. The data presented in Fig. 4 are suggestive of CD16 expression being reduced on KIR3DL1+ NK cells from HIV-infected donors. A preferential decrease of CD16 expression on educated NK cells might provide a mechanism to explain deficient antibody-dependent NK cell functionality in HIV infection, as well as the loss of the functional advantage of educated KIR3DL1+ NK cells upon antibody-dependent activation20.
Aside from providing a potential explanation for the diminished antibody-dependent functionality of NK cells during HIV infection, the work presented raises questions concerning the role of antibody-dependent NK cell functions in slowing HIV disease progression. Numerous investigations have linked anti-viral ADCC to slower progression towards AIDS8–10. This notion, however, is difficult to reconcile with the data presented in this paper, as well as data published by several other groups, demonstrating that antibody-dependent activation of NK cells induces phenotypical alterations that are reflective of alterations associated with HIV disease progression12,14–17,25,27. In addition to studies investigating alterations in NK cell phenotype during HIV infection, soluble CD16 is detected in the sera of HIV-infected donors and correlates with disease progression28. This observation suggests that the activation-induced cleavage of CD16 may result in a serological marker that associates with HIV disease progression, further suggesting a linkage between chronic NK cell activation and HIV disease progression. Given these outstanding issues, more research is needed to clarify how antibody-dependent NK cell responses might contribute to slower HIV disease progression. We predict that ADCC during acute infection might play an important role by setting the viral load set point at low enough levels that persistent high levels of antibody-dependent NK cell activation are not needed during chronic infection. As such, NK cells from individuals that control HIV replication are more likely to express surface CD16 than those from donors with more progressed infections12. It is worth noting that we observed a decreased expression of CD16 on KIR3DL1+ NK cells in HIV infected donors with slowly progressing disease (Fig. 4). Given that HIV infected individuals with more progressed infections exhibit more profound decreases in CD16 expressing NK cells than HIV controllers12, this diminished CD16 expression on KIR3DL1+ NK cells would be expected to be more robust in rapidly progressing HIV-infected patients.
Although the data presented in this paper have several potential ramifications for understanding HIV disease progression, they might also be of interest for designing immunotherapies for eradicating HIV infection. Recently there has been much interest in curing HIV infection by utilizing drugs to ‘shock’ HIV out of hiding and utilizing vaccine-induced immune responses to clear latently infected reactivated cells29. ADCC has been suggested as a potential immune response for utilization30. Given our data suggesting that NK cells can be quickly stripped of CD16 expression upon activation, as well as other data suggesting reduced CD16 expression even after successful viral suppression through anti-retroviral therapy27, it might be reasonable to consider therapeutic application of MMP-inhibiting drug along with ADCC-competent anti-viral antibodies to clear latently infected reactivated cells. The MMP-inhibiting drug would have the potential to increase the CD16 expression and functional potential of NK cells, which would be able to utilize the anti-viral antibodies successfully to target reactivated latently infected cells. Furthermore, the presence of the MMP-inhibiting drug should prevent CD16 cleavage after activation of NK cells by anti-viral ADCC16.
Recently, there has been much interest in utilizing antibody-dependent NK cell functions to target HIV. This interest has been generated by a large body of literature demonstrating an association between ADCC and prevention of infection or slower progression towards AIDS1,4–10. The data presented in the current paper, however, demonstrate that antibody-dependent activation of NK cells phenotypically alters NK cells in a manner similar to that observed in progressive HIV infection. Given these incongruent observations, future research is required to understand the conditions under which antibody-dependent NK cell responses might provide protection against HIV disease progression. Similarly, investigating NK cell activation-induced phenotypical alterations in other chronic viral infections or malignancies would lead to a greater understanding of the mechanisms of pathogenesis and protection of these other conditions. Research leading to a thorough understanding of activation-induced alterations in NK cell phenotypes would be instrumental for designing antibody-based therapies and prophylactics.
Acknowledgments
We acknowledge Rachel Bouchard and Stephanie Matte for co-ordinating the cohorts of HIV-uninfected and HIV-infected slow progressors utilized for NK cell phenotyping studies. The following physicians are acknowledged for recruiting and following HIV-infected slow progressors: Dr Danielle Rouleau, Dr Colin Kovacs, Dr Mona Loutfy, Dr Marianne Harris, Dr Benoit Trottier, Dr Jean-Pierre Routy, Dr Anita Rachlis, Dr Pierre Cote, Dr Marian Klien, Dr Bertrand Lebouche, Dr Alan Piche, Dr Kenneth Logue and Dr Edward Ralph. Finally, we would like to acknowledge the study participants. This work was supported by programme grant number 510448 from the National Health and Medical Research Council (NHMRC), as well as grants from the Canadian Institutes for Health Research (CIHR) numbers HOP-123800, MOP-111155 and THA-118628 and the Fonds de la Recherche du Québec Santé (FRQ-S) AIDS and Infectious Diseases Network. G. I. is the recipient of a Canadian HIV Trials Network postdoctoral fellowship. N. F. B. is a member of the Research Institute of the McGill University Health Centre, an institution funded in part by the FRQ-S. M. S. P. is the recipient of a CIHR postdoctoral fellowship.
Author contributions
The study was designed by M. S. P. and S. J. K. C.-C. T., G. I. and M. S. P. performed experiments. Clinical specimens were provided by J. B., C. T. and S. J. K. C.-C. T., G. I., N. F. B., S. J. K. and M. S. P. completed data analyses and manuscript preparation.
Disclosures
The authors declare no conflicts of interest.
References
- Hessell AJ, Hangartner L, Hunter M, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–4. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- Montefiori DC, Karnasuta C, Huang Y, et al. Magnitude and breadth of the neutralizing antibody response in the RV144 and Vax003 HIV-1 vaccine efficacy trials. J Infect Dis. 2012;206:431–41. doi: 10.1093/infdis/jis367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–20. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- Haynes BF, Gilbert PB, McElrath MJ, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366:1275–86. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaras GD, Ferrari G, Shen X, et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc Natl Acad Sci USA. 2013;110:9019–24. doi: 10.1073/pnas.1301456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuka J, Nduati R, Odem-Davis K, Peterson D, Overbaugh J. HIV-specific antibodies capable of ADCC are common in breastmilk and are associated with reduced risk of transmission in women with high viral loads. PLOS Pathog. 2012;8:e1002739. doi: 10.1371/journal.ppat.1002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert MD, Harvey JD, Lauer WA, et al. ADCC develops over time during persistent infection with live-attenuated SIV and is associated with complete protection against SIV(mac)251 challenge. PLOS Pathog. 2012;8:e1002890. doi: 10.1371/journal.ppat.1002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambotte O, Ferrari G, Moog C, et al. Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS. 2009;23:897–906. doi: 10.1097/QAD.0b013e328329f97d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren LH, Chung AW, Isitman G, et al. Specific antibody-dependent cellular cytotoxicity responses associated with slow progression of HIV infection. Immunology. 2013;138:116–23. doi: 10.1111/imm.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks ND, Kinsey N, Clements J, Hildreth JE. Sustained antibody-dependent cell-mediated cytotoxicity (ADCC) in SIV-infected macaques correlates with delayed progression to AIDS. AIDS Res Hum Retroviruses. 2002;18:1197–205. doi: 10.1089/08892220260387940. [DOI] [PubMed] [Google Scholar]
- Brenner BG, Gryllis C, Wainberg MA. Role of antibody-dependent cellular cytotoxicity and lymphokine-activated killer cells in AIDS and related diseases. J Leukoc Biol. 1991;50:628–40. doi: 10.1002/jlb.50.6.628. [DOI] [PubMed] [Google Scholar]
- Jia M, Li D, He X, et al. Impaired natural killer cell-induced antibody-dependent cell-mediated cytotoxicity is associated with human immunodeficiency virus-1 disease progression. Clin Exp Immunol. 2013;171:107–16. doi: 10.1111/j.1365-2249.2012.04672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler DS, Stanley SD, Nastala CA, et al. Alterations in antibody-dependent cellular cytotoxicity during the course of HIV-1 infection. Humoral and cellular defects. J Immunol. 1990;144:3375–84. [PubMed] [Google Scholar]
- Grzywacz B, Kataria N, Verneris MR. CD56(dim)CD16(+) NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia. 2007;21:356–9. doi: 10.1038/sj.leu.2404499. [DOI] [PubMed] [Google Scholar]
- Jewett A, Cavalcanti M, Giorgi J, Bonavida B. Concomitant killing in vitro of both gp120-coated CD4+ peripheral T lymphocytes and natural killer cells in the antibody-dependent cellular cytotoxicity (ADCC) system. J Immunol. 1997;158:5492–500. [PubMed] [Google Scholar]
- Parsons MS, Tang CC, Jegaskanda S, et al. Anti-HIV antibody-dependent activation of NK cells impairs NKp46 expression. J Immunol. 2014;192:308–15. doi: 10.4049/jimmunol.1301247. [DOI] [PubMed] [Google Scholar]
- Romee R, Foley B, Lenvik T, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17) Blood. 2013;121:3599–608. doi: 10.1182/blood-2012-04-425397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115:2167–76. doi: 10.1182/blood-2009-08-238469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfossi N, Andre P, Guia S, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–42. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Parsons MS, Wren L, Isitman G, et al. HIV infection abrogates the functional advantage of natural killer cells educated through KIR3DL1/HLA-Bw4 interactions to mediate anti-HIV antibody-dependent cellular cytotoxicity. J Virol. 2012;86:4488–95. doi: 10.1128/JVI.06112-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MS, Zipperlen K, Gallant M, Grant M. Killer cell immunoglobulin-like receptor 3DL1 licenses CD16-mediated effector functions of natural killer cells. J Leukoc Biol. 2010;88:905–12. doi: 10.1189/jlb.1009687. [DOI] [PubMed] [Google Scholar]
- Boulet S, Sharafi S, Simic N, et al. Increased proportion of KIR3DS1 homozygotes in HIV-exposed uninfected individuals. AIDS. 2008;22:595–9. doi: 10.1097/QAD.0b013e3282f56b23. [DOI] [PubMed] [Google Scholar]
- Boulet S, Kleyman M, Kim JY, et al. A combined genotype of KIR3DL1 high expressing alleles and HLA-B*57 is associated with a reduced risk of HIV infection. AIDS. 2008;22:1487–91. doi: 10.1097/QAD.0b013e3282ffde7e. [DOI] [PubMed] [Google Scholar]
- Trundley A, Frebel H, Jones D, Chang C, Trowsdale J. Allelic expression patterns of KIR3DS1 and 3DL1 using the Z27 and DX9 antibodies. Eur J Immunol. 2007;37:780–7. doi: 10.1002/eji.200636773. [DOI] [PubMed] [Google Scholar]
- Hu PF, Hultin LE, Hultin P, et al. Natural killer cell immunodeficiency in HIV disease is manifest by profoundly decreased numbers of CD16+CD56+ cells and expansion of a population of CD16dimCD56- cells with low lytic activity. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10:331–40. [PubMed] [Google Scholar]
- Liu Q, Sun Y, Rihn S, et al. Matrix metalloprotease inhibitors restore impaired NK cell-mediated antibody-dependent cellular cytotoxicity in human immunodeficiency virus type 1 infection. J Virol. 2009;83:8705–12. doi: 10.1128/JVI.02666-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtfuss GF, Cheng WJ, Farsakoglu Y, et al. Virologically suppressed HIV patients show activation of NK cells and persistent innate immune activation. J Immunol. 2012;189:1491–9. doi: 10.4049/jimmunol.1200458. [DOI] [PubMed] [Google Scholar]
- Khayat D, Soubrane C, Andrieu JM, et al. Changes of soluble CD16 levels in serum of HIV-infected patients: correlation with clinical and biologic prognostic factors. J Infect Dis. 1990;161:430–5. doi: 10.1093/infdis/161.3.430. [DOI] [PubMed] [Google Scholar]
- Autran B, Descours B, Bacchus C. Immune control of HIV-1 reservoirs. Curr Opin HIV AIDS. 2013;8:204–10. doi: 10.1097/COH.0b013e32835fe6d2. [DOI] [PubMed] [Google Scholar]
- Kent SJ, Reece JC, Petravic J, et al. The search for an HIV cure: tackling latent infection. Lancet Infect Dis. 2013;13:614–21. doi: 10.1016/S1473-3099(13)70043-4. [DOI] [PubMed] [Google Scholar]