

Abstract
Atypical haemolytic uraemic syndrome (aHUS) is associated with (genetic) alterations in alternative complement pathway. Nevertheless, comprehensive evidence that the complement system in aHUS patients is more prone to activation is still lacking. Therefore, we performed a thorough analysis of complement activation in acute phase and in remission of this disease. Complement activation patterns of the aHUS patients in acute phase and in remission were compared to those of healthy controls. Background levels of complement activation products C3b/c, C3bBbP and terminal complement complex (TCC) were measured using enzyme-linked immunosorbent assay (ELISA) in ethylenediamine tetraacetic acid (EDTA) plasma. In vitro-triggered complement activation in serum samples was studied using zymosan-coating and pathway-specific assay. Furthermore, efficiencies of the C3b/c, C3bBbP and TCC generation in fluid phase during spontaneous activation were analysed. Patients with acute aHUS showed elevated levels of C3b/c (P < 0·01), C3bBbP (P < 0·0001) and TCC (P < 0·0001) in EDTA plasma, while values of patients in remission were normal, compared to those of healthy controls. Using data from a single aHUS patient with complement factor B mutation we illustrated normalization of complement activation during aHUS recovery. Serum samples from patients in remission showed normal in vitro patterns of complement activation and demonstrated normal kinetics of complement activation in the fluid phase. Our data indicate that while aHUS patients have clearly activated complement in acute phase of the disease, this is not the case in remission of aHUS. This knowledge provides important insight into complement regulation in aHUS and may have an impact on monitoring of these patients, particularly when using complement inhibition therapy.
Keywords: biomarkers, clinical immunology, complement activation, haemolytic uraemic syndrome
Introduction
Atypical haemolytic uraemic syndrome (aHUS) is a severe renal illness with up to 50% of cases progressing to end-stage renal disease (ESRD) and up to 25% of lethal outcomes in the acute phase of aHUS 1. Complement dysregulation, leading to glomerular endothelial cell damage is considered to be a central element in aHUS pathogenesis and complement inhibition therapy has been reported to be successful and has recently been approved for aHUS treatment 2–4.
The complement system, a part of the innate immune system, can be activated via three pathways: the classical, the lectin and the alternative. These pathways converge at the cleavage and activation of the central complement component C3. This results in formation of the C3b fragment, which binds zymogen complement factor B (CFB) and, in association with properdin, forms the alternative pathway C3 convertase (C3bBbP). C3bBbP cleaves and activates more C3 molecules, leading to formation of the C5b-9 terminal complement complex (TCC) and release of the potent anaphylatoxins C3a and C5a. To protect healthy host tissue, the C3 convertase activity is controlled by complement inhibitors, such as complement factor I (CFI), complement factor H (CFH) and membrane co-factor protein (CD46/MCP). As a result of C3 convertase inactivation, various C3 degradation products are formed, such as iC3b, C3c and C3dg 5,6.
Currently, alternative complement pathway deficiencies are identified in 50–60% of aHUS patients. Genetic variants affecting CFH, CFI, MCP, C3 and CFB as well as autoantibodies against CFH (anti-CFH) are associated with aHUS pathogenesis. Aberrations affecting genes outside the complement cascade, encoding thrombomodulin (THBD) and diacylglycerol kinase ε (DGKE), were also reported 7–20. Aetiological analysis of patients with aHUS is critically important, especially in renal transplantation, which is frequently required in this patient group. For example, patients who carry mutations in genes encoding CFH, CFI or C3 are at higher risk of disease recurrence in the graft (40–90%), whereas such probability is lower for the aHUS patients carrying MCP mutations and anti-CFH autoantibodies 21–24. Functional analysis of complement activation in aHUS patients remains very limited. The C3 and C4 levels, which are often used in clinical practice, often show normal values. Recently, a single study reported that C5a and TCC are higher in aHUS than in thrombotic thrombocytopenic purpura (associated not with complement aberrations, but with ADAMTS13 deficiency) 25. However, it is still not clear whether complement system in aHUS patients is more prone to activation than in healthy individuals.
In this study, for the first time we performed a thorough analysis of complement activation in aHUS patients in acute phase and in remission of the disease. Monitoring of complement activation dynamics in ethylenediamine tetraacetic acid (EDTA) plasma and serum samples may provide a simple and important tool in assessment of complement activation in aHUS patients, particularly when complement inhibition therapy is used.
Materials and methods
Study population
The research population consisted of two groups of aHUS patients in acute phase (n = 6) and in remission (n = 11) of the disease. Acute phase was defined as the presence of haemolytic anaemia, thrombocytopenia and acute renal failure. Patients in remission had their last aHUS episode more than 1 year ago. In addition, from a single aHUS patient with CFB p.Lys323Glu mutation, samples were taken during two acute aHUS episodes, in the following convalescence periods during plasmapheresis (PF) treatment and in remission phase at the time of the study. The patients were referred to the Pediatric Nephrology Department of the Radboud University Medical Center. The control group (n = 19) consisted of healthy adult volunteers. For the controls, the following exclusion criteria were applicable: fever, bacterial/viral infection in the previous 2 weeks, chronic illness, inborn or acquired immune disorders and immunosuppressive medication. The study was approved by the institutional review board and was performed in accordance with the appropriate version of the Declaration of Helsinki. Informed consent of all patients and/or their parents as well as of controls was obtained before analysis.
Genetic analysis
Genomic DNA was isolated from peripheral blood leucocytes as described by Miller et al. 26. Coding fragments of CFH, CFI, MCP, C3, CFB, THBD and DGKE genes were amplified from genomic DNA by means of polymerase chain reaction (PCR). Primer sequences are available upon request. The obtained PCR products included DNA sequences of the individual exons, flanked by the splice donor site and the splice acceptor site. The amplimers were subjected to double-stranded DNA sequence analysis on an ABI 3130 xl GeneticAnalyzer (Applied Biosystems, San Francisco, CA, USA). Sequence analyses were performed using Sequencher version 4·8 software (Gene Codes, Ann Arbor, MI USA). Anti-CFH autoantibodies were detected as described previously 27.
Sample collection
EDTA blood and serum samples of patients and controls were collected and processed within 1 h after sampling. Samples were aliquoted and stored at −80°C. To avoid in vitro complement activation after sampling, EDTA plasma samples were used to compare complement activation in acute phase and in remission of aHUS. Serum samples were used in the in vitro complement activation experiments.
Complement activation analyses
The levels of complement activation products – C3b and its degraded form C3c (C3b/c), the alternative complement pathway convertase C3bBbP and the fluid phase terminal sC5b-9 complement complex (TCC) in EDTA plasma – were quantified using enzyme-linked immunosorbent assay (ELISA), as described in detail previously by Bergseth et al. 28. International complement standard 2 (ICS 2) was used for quantification of activation products in complement activation units per ml (CAU/ml) 28.
Zymosan-induced activation of serum was performed in 96-well plates coated using 2 μg/well of zymosan A from Saccharomyces cerevisiae (Sigma-Aldrich, St Louis, MO, USA). Complement activation and detection of C3b and TCC deposition on the solid phase was performed as described previously 29.
To analyse the classical, lectin and alternative pathway activation separately, the Wieslab® Complement system Screenkit (Euro Diagnostica, Malmö, Sweden) was used according to the manufacturer’s protocol.
In kinetic activation analysis, serum samples were diluted 1 : 2 in Dulbecco’s phosphate-buffered saline with MgCl2 and CaCl2 (Sigma-Aldrich) and incubated at 37°C with gentle agitation; samples were collected at 0, 10, 20, 30 and 60 min of incubation and immediately placed on ice. To stop the complement activation, EDTA (Sigma-Aldrich) was added to a final concentration of 20 mM. Complement activation products were quantified as described above for EDTA plasma samples.
Statistical analysis
Statistical analysis was performed using the unpaired two-tailed t-test. Statistically significant differences with P < 0·05 are indicated.
Results
Genetic background of the aHUS patients
For this study, samples from two groups of patients in acute phase (n = 6) and in remission (n = 11) were available. The patients in both groups were screened for the presence of pathogenic genetic variations in genes encoding CFH, CFI, MCP, C3, CFB, THBD and DGKE; furthermore, the presence of anti-CFH autoantibodies was analysed. In the acute-phase aHUS group, two of the patients carried heterozygous genetic variants in C3 or MCP and one had anti-CFH autoantibodies, while three patients had aHUS of undefined aetiology (Table 1). In the aHUS remission group six patients carried heterozygous changes in CFH, CFI, C3 or THBD; in five patients no genetic aberrations or anti-CFH autoantibodies were found (Table 2). Familial and idiopathic aHUS patients were present in both groups (Tables 1 and 2).
Table 1.
Clinical and genetic data available for patients in acute phase of atypical haemolytic uraemic syndrome (aHUS)
| Patient no. | aHUS pathogenic change | Gender | Age at onset/time of study* | Treatment | Outcome |
|---|---|---|---|---|---|
| P1a† | C3: p.Arg161Trp 15,16 | Male | 1st episode: 6 years;2nd episode: 12 years | Plasma therapy | Remission |
| P2a | None | Female | 7 years | Plasma therapy | Remission |
| P3a | Anti-CFH autoantibodies 19 | Female | 11 years | Plasma therapy, haemodialysis | Remission |
| P4a | MCP: p.Asp271_Ser272del 10 | Male | 1st episode: 4 years;2nd episode: 8 years | Plasma therapy | Remission |
| P5a† | None | Male | 1st episode: 7 months;2nd episode: 2 years | Plasma therapy, eculizumab | Remission under eculizumab |
| P6a | None | Male | 5 years | Plasma therapy, CVVHDF | Remission |
Age at onset of the latest episode for these patients is equal to the age at the time of the study. First episodes of patients P1a, P4a and P5a resolved in (almost) complete remission without need of a renal transplantation.
Familial aHUS. CVVHDF = continuous veno-venous haemodiafiltration; CFH = complement factor H; MCP = membrane co-factor protein.
Table 2.
Clinical and genetic data available for patients in remission phase of atypical haemolytic uraemic syndrome (aHUS)
| Patient no. | aHUS pathogenic change | Gender | Age at first onset/time of study | Treatment | Outcome | Transplantation history |
|---|---|---|---|---|---|---|
| P1r* | CFH: p.Arg1206Cys 8 | Male | 22 years/27 years | Plasma therapy, haemodialysis | Remission | None |
| P2r*† | CFH: p.Arg1206Cys | Female | 21 years/48 years | Plasma therapy, haemodialysis | ESRD | aHUS in graft |
| P3r | CFI: p.Arg474Stop 9 | Female | 49 years/57 years | Plasma therapy, haemodialysis | ESRD | No recurrence |
| P4r* | C3: p.Arg161Trp 15,16 | Male | 52 years/59 years | Plasma therapy, haemodialysis | ESRD | No recurrence |
| P5r* | C3: p.Arg161Trp | Female | 23 years/35 years | Plasma therapy | ESRD | No recurrence |
| P6r | THBD: p.Ala43Thr 18 | Female | 2 months/5 years | Antibiotics, CVVH | Remission | None |
| P7r | None | Male | 24 years/29 years | Haemodialysis | ESRD | None, on the waiting list for transplantation |
| P8r | None | Female | 3 years/23 years | Haemodialysis | ESRD | aHUS in graft |
| P9r | None | Male | 1 years/13 years | Peritoneal dialysis | ESRD | aHUS in graft |
| P10r | None | Female | 15 years/24 years | Haemodialysis | ESRD | No recurrence |
| P11r§ | None | Male | 8 years/17 years | Blood transfusions | ESRD | No recurrence |
Familial aHUS.
Mother of P1r.
Familial steroid-resistant nephrotic syndrome. Full spectrum aHUS developed during cyclosporin treatment. CFH = complement factor H; CFI = complement factor I; CVVH = continuous veno-venous haemofiltration; ESRD = end stage renal disease; THBD = thrombomodulin.
Complement activation in acute phase and in remission of aHUS
First, we analysed complement activation in acute phase and in remission of aHUS. To this end, we measured concentrations of complement activation products C3b/c, C3bBbP and TCC in EDTA plasma of aHUS patients and compared the values to those of healthy controls (n = 19). These assays reflect activation of the alternative complement pathway, which is implicated in aHUS pathogenesis. In the acute phase of the disease, we observed significantly elevated levels of C3b/c, C3bBbP and TCC (Fig. 1a). In four of the six patients in the acute phase all three activation products were elevated; in the other two patients two of the three activation product were elevated, indicating ongoing complement activation in all these patients (Table 3). The values of C3bBbP and TCC were increased in all six patients, including the three patients with normal serum C3 levels in the acute phase. Conversely, patients in remission showed C3b/c, C3bBbP and TCC values that were completely comparable to those of healthy controls (Fig. 1b).
Figure 1.
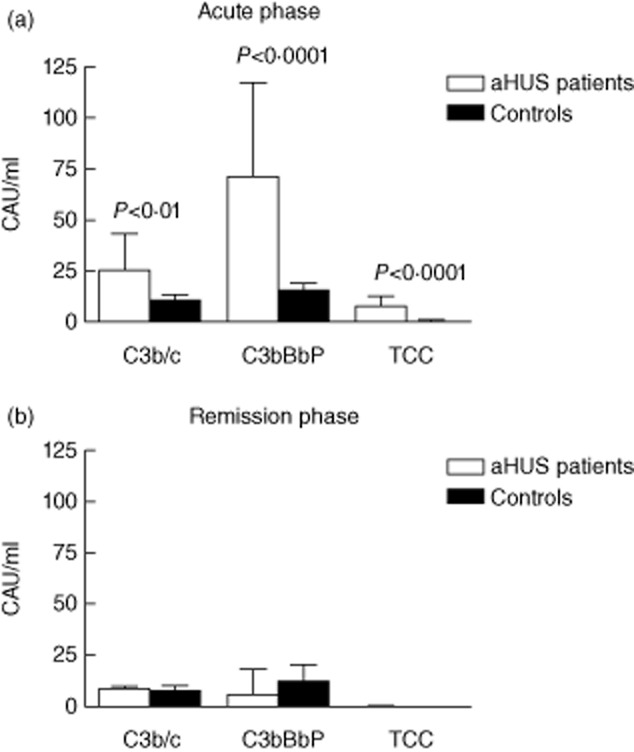
Plasma complement activation products in patients with atypical haemolytic uraemic syndrome (aHUS) in acute phase and in remission. Ethylenediamine tetraacetic acid (EDTA) plasma samples of six aHUS patients in acute phase (a) and of 11 patients in remission phase (b) were analysed for the levels of C3b/c, C3bBbP and terminal complement complex (TCC). The values were compared to those of 19 healthy controls. Data were quantified using international complement standard 2 (ICS 2) in complement activation units per ml (CAU/ml) and are presented as mean ± standard deviation. Values that were statistically different from those of healthy controls (P < 0·05) are indicated.
Table 3.
Complement assessment of patients in the acute phase of atypical haemolytic uraemic syndrome (aHUS)
| Patient no. | C3 700–1500* | C4 100–400* | C3b/c (<15·8 )† | C3bBbP (<23·5)† | TCC (<2·2)† |
|---|---|---|---|---|---|
| P1a | 1110 | 323 | 57·82 | 148·7 | 7·85 |
| P2a | 1130 | 300 | 18·15 | 29·3 | 15·16 |
| P3a | 639 | 292 | 12·25 | 46·59 | 5·39 |
| P4a | 657 | 164 | 34·25 | 67·48 | 2·56 |
| P5a | 927 | 205 | 11·28 | 34·74 | 4·84 |
| P6a | 620 | 250 | 19·63 | 99·95 | 10·94 |
Normal serum concentration range (mg/l) defined by the standard clinical protocol.
Normal concentration range (CAU/ml) defined by this study as mean ± 2 standard deviations of control values (n = 19). Values outside the normal range are indicated in bold type. TCC = terminal complement complex.
From a single female patient carrying a well-characterized CFB p.Lys323Glu mutation, samples taken during two acute aHUS episodes, during PF treatments and in remission at the time of the study were available (Table 4) 17. Increased concentrations of all three complement activation markers (first episode) and C3b/c and C3bBbP (second episode) were detected in acute phase. Complement activation levels decreased considerably during PF treatments and were completely normalized in remission.
Table 4.
Complement assessment of a single female patient during two acute atypical haemolytic uraemic syndrome (aHUS) episodes, in convalescence periods after five (first episode) and 10 (second episode) plasmapheresis (PF) sessions and in remission at the time of the study. The patient recovered without need of a renal transplant and carries pathogenic p.Lys323Glu mutation in CFB 17
| Disease phase | Age (years) | C3b/c (<15·8 )* | C3bBbP (<23·5)* | TCC (<2·2)* |
|---|---|---|---|---|
| First acute episode | 2·5 | 155·17 | 61·58 | 2·88 |
| After 5 PF sessions | 2·5 | 21·57 | 19·87 | 1·3 |
| Second acute episode | 3·5 | 116·16 | 115·49 | 0·74 |
| After 10 PF sessions | 3·5 | 22·82 | 10·82 | 0·67 |
| Remission phase | 21 | 10·33 | 6·00 | 0·46 |
Normal concentration range (CAU/ml) defined by this study as mean ± 2 standard deviations of control values (n = 19). Values outside the normal range are indicated in bold type. TCC = terminal complement complex.
Together, our data indicate that in acute aHUS episodes complement activation is taking place, but returns to the level of healthy controls during remission.
Induced complement activation in serum samples of aHUS patients in acute phase
Ongoing complement activation in the acute phase of aHUS may lead to complement consumption, as seen partially from the C3 serum levels (Table 3). This may cause diminished capacity for complement activation. Next, we tested this hypothesis using a pathway-specific activation assay, used widely in clinical laboratories (Fig. 2).
Figure 2.

Pathway-specific activation of serum samples from acute atypical haemolytic uraemic syndrome (aHUS) patients. Specific activation of the classical (CP), the lectin (LP) and the alternative (AP) pathways of complement was measured in six acute-phase patients and in 19 controls with TCC deposition as readout. Data are presented as percentage of activation of the positive control provided with the assay. Low alternative pathway activation value in a sample of patient P6a is indicated.
Our results indicate that the classical and lectin pathway activation profiles are not different between the acute phase patients and healthy individuals. In the alternative pathway experiment, only one of the acute phase patients (P6a) demonstrated activation outside the normal range.
Induced complement activation in serum samples of aHUS patients in remission
Next, we analysed whether, when triggered, serum samples of aHUS patients in remission are able to reach higher complement activation levels than the samples of healthy controls. To this end, we performed complement activation on the zymosan-coated surface. Zymosan is able to directly activate the lectin and the alternative complement pathways and also activates the classical pathway, when anti-zymosan antibodies are present in the serum 30. We quantified C3b and TCC deposition on the zymosan-coated surface in response to the activation of the serum samples. No significant differences between remission phase aHUS samples and control samples were observed by using zymosan-induced complement activation (Fig. 3a).
Figure 3.
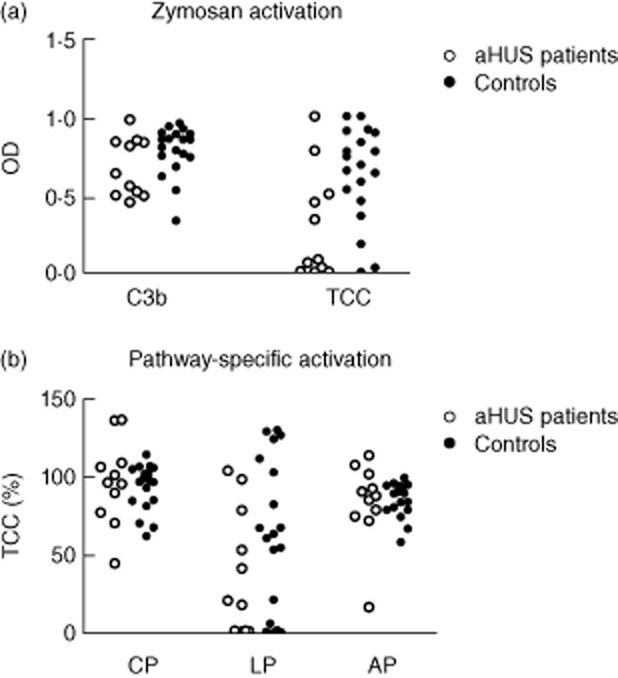
Induced complement activation of serum samples from atypical haemolytic uraemic syndrome (aHUS) patients in remission. (a) Serum samples of 11 patients in remission phase of aHUS and of 19 healthy controls were analysed for complement activation in zymosan-coated wells, using C3b and TCC deposition as readout. Results are given as absorbance units of optical density (OD). (b) Specific activation of the classical (CP), the lectin (LP) and the alternative (AP) pathway of complement was measured with terminal complement complex (TCC) deposition as readout. Data are presented as percentage of activation of the positive control provided with the assay.
Furthermore, we performed pathway-specific analysis to quantify activation of the classical, lectin and alternative pathways separately (Fig. 3b). This analysis also did not reveal significant differences between complement activation in aHUS patients in remission and in healthy controls.
These data indicate that complement activation in serum samples of aHUS patients in remission is not triggered to a higher level than in the samples of healthy controls.
Kinetics of complement activation in serum samples of aHUS patients in remission
So far, no differences in complement activation between patients in remission and controls were observed. Finally, we analysed whether serum samples of aHUS patients in remission have faster activation kinetics compared to the samples of healthy controls. Thus, we studied kinetics of spontaneous complement activation in fluid phase. Serum samples were incubated at 37°C to induce spontaneous alternative pathway complement activation; concentrations of C3b/c, C3bBbP and TCC were then analysed at various time-points (Fig. 4). No statistically significant differences were observed between patients and controls.
Figure 4.
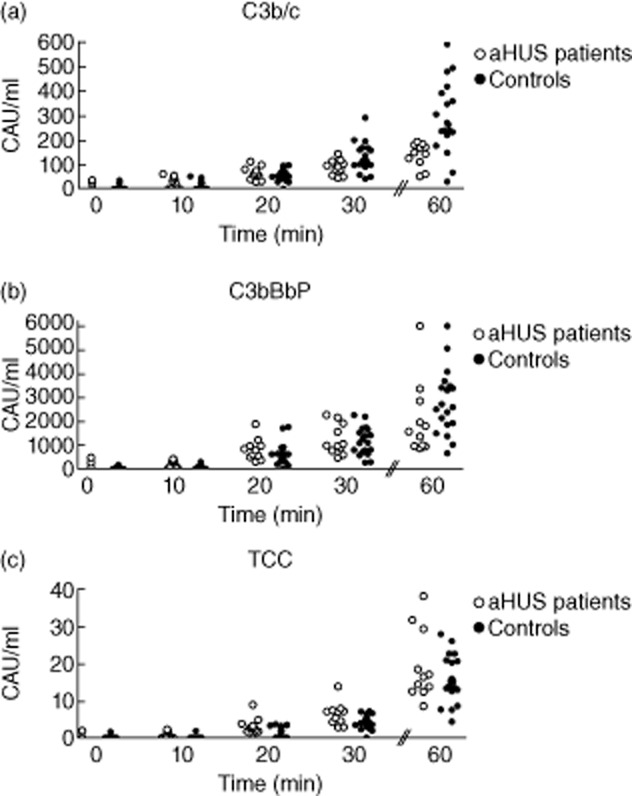
In vitro kinetics of spontaneous complement activation in the serum samples of atypical haemolytic uraemic syndrome (aHUS) patients in remission. Samples of 11 aHUS patients in remission and of 19 healthy controls were incubated at 37°C with gentle agitation. Samples were collected at 0, 10, 20, 30 and 60 min of incubation and levels of C3b/c (a), C3bBbP (b) and terminal complement complex (TCC) (c) were analysed. Data were quantified using international complement standard 2 (ICS 2) and are presented as complement activation units per ml (CAU/ml).
These results indicate that there is no difference in complement activation kinetics between aHUS patients in remission and healthy controls.
Discussion
Complement dysregulation is an established hallmark of aHUS. To the best of our knowledge, in this study we present for the first time a comprehensive analysis of complement regulation in the blood of aHUS patients in acute phase and in remission.
We observed a clear activation of the alternative complement pathway in the acute phase group, as revealed by significant increases in C3b/c, C3bBbP and TCC in EDTA plasma. However, the levels of these complement activation markers in aHUS patients in remission were normal. Moreover, when analysed in a single aHUS patient with CFB mutation, complement activation was detected clearly in both aHUS episodes, but not in remission.
Therefore we suggest that, in remission, complement activation in blood of aHUS patients normalizes to the levels of healthy individuals. As our experiments included a limited number of patients, studies in larger cohorts should be performed in the future.
The levels of complement activation markers measured in this study reflect complement activation in the fluid phase. The sC5b-9 (fluid phase TCC) does not normally shed from the cell surface, as when first inserted into the membrane there is a firm attachment. However, activation might take place at the surface but not close enough for the TCC to be inserted, thereby generating a fluid phase complex, as seen typically by artificial surfaces such as haemodialysis membrane 31,32.
Our data do not exclude the possibility of ongoing complement activation on the cell surface in remission, that could be detected experimentally using sheep erythrocytes or endothelial cells and that was outside the scope of this study.
We found that in a single aHUS patient with CFB mutation, complement activation was detected clearly in both aHUS episodes, but not in remission. Previously, complement activation in a single aHUS patient with CFH mutation p.Ser1191Leu was monitored before and after infusion of fresh frozen plasma 33. Only a limited effect of plasma infusion on complement activation was observed using a haemolytic alternative pathway assay and no difference was observed in a pathway-specific screen. Assays used in this previous study reflect a resultant of several factors, including the amount of native components present, which does not necessarily reflect the degree of ongoing activation. The latter is reflected by the activation product assays we have used. Consistent with these previous data, all but one patient in our acute-phase group demonstrated normal activation in a pathway-specific assay (Fig. 2).
Further, we analysed whether complement activation in serum samples of remission patients can be triggered more efficiently than in samples of healthy controls. Our data indicate that both zymosan-induced activation and pathway-specific activation of aHUS serum samples give results similar to those of the control group. The results of kinetic analysis in the fluid phase also appeared to be comparable in aHUS remission patients and in controls.
These findings are indeed intriguing, as six of 11 patients carry aHUS-predisposing genetic aberrations, which are expected to cause more efficient complement activation. Patients P1r and P2r carry p.Arg1206Cys substitution in the CFH gene. Previously, the p.Arg1206Ala variant was characterized in functional studies using a recombinant CFH19-20 fragment 8. In this work, impaired binding of CFH19-20 fragment carrying p.Arg1206Ala to C3b/d and mouse glomerular endothelium was shown. We expect the p.Arg1206Cys to have a similar effect on protein function. Patients P4r and P5r carry the C3 variant p.Arg161Trp that was reported previously by us and others, and has a negative impact on CFH and MCP binding and increases the affinity to CFB 15,16. Furthermore, thrombomodulin, a component of the coagulation system, was reported previously to regulate inactivation of C3b in vitro. The p.Ala43Thr change (P6r) was shown to affect this function negatively 18. Particularly interesting is the CFI mutation in patient P3r, which leads to a stop codon in the serine protease domain that is important for C3b and C4b inactivation.
The absence of a phenotype in remission-phase patients might be in line with the relatively mild nature of often heterozygous aHUS changes. Penetrance (the chance to acquire aHUS) is estimated to be approximately 60% for aHUS variants. It is suggested that next to a genetic variant or anti-CFH autoantibodies, the presence of aHUS-predisposing single nucleotide polymorphisms and external stimuli, such as infections, surgeries or use of medication, is needed for development of aHUS 34.
In this study, low serum C3 levels were found only in three acute-phase patients, indicating complement activation. Moreover, only one patient showed aberrant alternative pathway activation in a pathway-specific activation assay. However, the activation markers, which were elevated in all patients, may provide a more promising tool for the detection of complement activation in aHUS.
The fact that the samples from patients in remission of aHUS display normal complement activation pattern in blood has an important clinical implication. This indicates that in remission phase aHUS patients are likely to have the same low complement activation levels as those of healthy controls. Therefore, these assays may be used in the early detection of relapse of aHUS in the future. Recently, the complement component C5 inhibitor eculizumab (Soliris®) was approved for the treatment of aHUS 2–4. Patients in the acute phase of aHUS who undergo treatment with complement inhibition therapy can be monitored for the levels of complement activation markers (C3b/c, C3bBbP and TCC). The value of these markers in patients undergoing eculizumab treatment will be studied in the near future, which may lead to the development of individualized therapy based on the levels of complement activation.
In conclusion, we demonstrate that complement activation can be shown clearly in the blood of aHUS patients during acute episodes of aHUS and that levels of complement activation markers in the fluid phase return completely to normal during remission. No change in complement activation profile compared with controls was seen when complement was triggered in vitro in the serum samples of aHUS patients in remission. These assays may be extremely helpful in the diagnostic setting to monitor complement dysregulation during aHUS attacks, especially when using complement inhibition therapy.
Acknowledgments
The authors thank patients, their parents and healthy volunteers for participation in this study. Furthermore, we would like to thank G. Bergseth, A. Pharo, J. K. Lindstad and N. Postriganova for technical assistance. Clinical data were provided by Dr M. Kömhoff and Dr M. Seelen (University Medical Centre Groningen, the Netherlands); Dr S. Konings (Catharina Ziekenhuis, the Netherlands); Dr J. van de Wetering (Erasmus MC, the Netherlands); Dr H. van Hamersvelt (Radboud University Medical Center, the Netherlands); Dr M. Groeneveld (MC Haaglanden, the Netherlands); Dr J. Vande Walle (Ghent University Hospital, Belgium); Dr F. Boereboom (Dianet Utrecht, the Netherlands); Dr R. van Etten (Amphia Ziekenhuis, the Netherlands); Dr J. Groothoff (Academic Medical Center, the Netherlands); and Dr A. Bökenkamp (VU University Medical Center Amsterdam, the Netherlands). This work was supported financially by the Dutch Kidney Foundation (KFB 11·007, IP 10·22, KBSO 11·019, C09·2313) and ERA-EDTA (ERA STF 138–2013).
Disclosure
The authors have no financial conflicts of interest related to this study.
Author contributions
Research idea and study design by E. V., T. E. M. and L. v.d. H.; data acquisition by E. V., T. v.d. V. and D. W.; data analysis/interpretation by E. V., N. v.d. K, T. E. M. and L. v.d. H.; statistical analysis by E. V.
References
- Westra D, Wetzels JF, Volokhina EB, van den Heuvel LP, van de Kar NC. A new era in the diagnosis and treatment of atypical haemolytic uraemic syndrome. Neth J Med. 2012;70:121–129. [PubMed] [Google Scholar]
- Waters AM, Licht C. aHUS caused by complement dysregulation: new therapies on the horizon. Pediatr Nephrol. 2011;26:41–57. doi: 10.1007/s00467-010-1556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EK, Goodship TH, Kavanagh D. Complement therapy in atypical haemolytic uraemic syndrome (aHUS) Mol Immunol. 2013;56:199–212. doi: 10.1016/j.molimm.2013.05.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic–uremic syndrome. N Engl J Med. 2013;368:2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- Harboe M, Thorgersen EB, Mollnes TE. Advances in assay of complement function and activation. Adv Drug Deliv Rev. 2011;63:976–987. doi: 10.1016/j.addr.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Stahl AL, Vaziri-Sani F, Heinen S, et al. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008;111:5307–5315. doi: 10.1182/blood-2007-08-106153. [DOI] [PubMed] [Google Scholar]
- Lehtinen MJ, Rops AL, Isenman DE, van der Vlag J, Jokiranta TS. Mutations of factor H impair regulation of surface-bound C3b by three mechanisms in atypical hemolytic uremic syndrome. J Biol Chem. 2009;284:15650–15658. doi: 10.1074/jbc.M900814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41:e84. doi: 10.1136/jmg.2004.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2003;100:12966–12971. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noris M, Brioschi S, Caprioli J, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. 2003;362:1542–1547. doi: 10.1016/S0140-6736(03)14742-3. [DOI] [PubMed] [Google Scholar]
- Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14:703–712. doi: 10.1093/hmg/ddi066. [DOI] [PubMed] [Google Scholar]
- Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–1279. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeaux-Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112:4948–4952. doi: 10.1182/blood-2008-01-133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumenina LT, Frimat M, Miller EC, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. 2012;119:4182–4191. doi: 10.1182/blood-2011-10-383281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volokhina E, Westra D, Xue X, Gros P, van de Kar N, van den Heuvel L. Novel C3 mutation p.Lys65Gln in aHUS affects complement factor H binding. Pediatr Nephrol. 2012;27:1519–1524. doi: 10.1007/s00467-012-2183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240–245. doi: 10.1073/pnas.0603420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic–uremic syndrome. N Engl J Med. 2009;361:345–357. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood. 2008;111:1512–1514. doi: 10.1182/blood-2007-09-109876. [DOI] [PubMed] [Google Scholar]
- Quaggin SE. DGKE and atypical HUS. Nat Genet. 2013;45:475–476. doi: 10.1038/ng.2622. [DOI] [PubMed] [Google Scholar]
- Noris M, Remuzzi G. Atypical hemolytic–uremic syndrome. N Engl J Med. 2009;361:1676–1687. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- Kavanagh D, Goodship TH. Atypical hemolytic uremic syndrome. Curr Opin Hematol. 2010;17:432–438. doi: 10.1097/MOH.0b013e32833cae86. [DOI] [PubMed] [Google Scholar]
- Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60. doi: 10.1186/1750-1172-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragon-Durey MA, Sethi SK, Bagga A, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21:2180–2187. doi: 10.1681/ASN.2010030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataland SR, Holers VM, Geyer S, Yang S, Wu HM. Biomarkers of the alternative pathway and terminal complement activity at presentation confirms the clinical diagnosis of aHUS and differentiates aHUS from TTP. Blood. 2014;123:3733–3738. doi: 10.1182/blood-2013-12-547067. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra D, Volokhina E, van der Heijden E, et al. Genetic disorders in complement (regulating) genes in patients with atypical haemolytic uraemic syndrome (aHUS) Nephrol Dial Transplant. 2010;25:2195–2202. doi: 10.1093/ndt/gfq010. [DOI] [PubMed] [Google Scholar]
- Bergseth G, Ludviksen JK, Kirschfink M, Giclas PC, Nilsson B, Mollnes TE. An international serum standard for application in assays to detect human complement activation products. Mol Immunol. 2013;56:232–239. doi: 10.1016/j.molimm.2013.05.221. [DOI] [PubMed] [Google Scholar]
- Harboe M, Garred P, Lindstad JK, et al. The role of properdin in zymosan- and Escherichia coli-induced complement activation. J Immunol. 2012;189:2606–2613. doi: 10.4049/jimmunol.1200269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MC, Pensky J, Naff GB. Inhibition of zymosan-induced alternative complement pathway activation by concanavalin A. Infect Immun. 1982;38:1279–1284. doi: 10.1128/iai.38.3.1279-1284.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. The role of complement in biomaterial-induced inflammation. Mol Immunol. 2007;44:82–94. doi: 10.1016/j.molimm.2006.06.020. [DOI] [PubMed] [Google Scholar]
- DeAngelis RA, Reis ES, Ricklin D, Lambris JD. Targeted complement inhibition as a promising strategy for preventing inflammatory complications in hemodialysis. Immunobiology. 2012;217:1097–1105. doi: 10.1016/j.imbio.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen S, Pluthero FG, van Eimeren V, Quaggin SE, Licht C. Monitoring and modeling treatment of atypical hemolytic uremic syndrome. Mol Immunol. 2013;54:84–88. doi: 10.1016/j.molimm.2012.10.044. [DOI] [PubMed] [Google Scholar]
- Kavanagh D, Goodship TH. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15–20. doi: 10.1182/asheducation-2011.1.15. [DOI] [PubMed] [Google Scholar]