

Abstract
Therapy for Crohn’s disease (CD) with thiopurines is limited by systemic side effects. A novel formulation of fixed-dose, delayed-release 6-mercaptopurine (DR-6MP) was developed, with local effect on the gut immune system and minimal absorption. The aim of this study was to evaluate the safety and efficacy of DR-6MP in patients with moderately severe CD compared to systemically delivered 6-mercaptopurine (Purinethol). Seventy CD patients were enrolled into a 12-week, double-blind controlled trial. The primary end-point was the percentage of subjects with clinical remission [Crohn’s Disease Activity Index (CDAI) < 150] or clinical response (100-point CDAI reduction). Twenty-six (56·5%) and 13 (54·2%) subjects from the DR-6MP and Purinethol cohorts, respectively, completed the study. DR-6MP had similar efficacy to Purinethol following 12 weeks of treatment. However, the time to maximal clinical response was 8 weeks for DR-6MP versus 12 weeks for Purinethol. A higher proportion of patients on DR-6MP showed clinical remission at week 8. A greater improvement in Inflammatory Bowel Disease Questionnaire (IBDQ) score was noted in the DR-6MP group. DR-6MP led to a decrease of CD62+ expression on T cells, implying a reduction of lymphocyte adhesion to site of inflammation. DR-6MP was safer than Purinethol, with significantly fewer adverse events (AEs). There was no evidence of drug-induced leucopenia in the DR-6MP group; the proportion of subjects who developed hepatotoxicity was lower for the DR-6MP. Non-absorbable DR-6MP is safe and biologically active in the gut. It is clinically effective, exerting a systemic immune response with low systemic bioavailability and a low incidence of side effects.
Keywords: Crohn’s disease, gut immune system, 6-mercaptopurine, oral therapy
Introduction
The purine analogues, azathioprine (AZA) and 6-mercaptopurine (6-MP), have been the mainstay of long-term therapy for inflammatory bowel disease (IBD) patients for many years 1. Their role as steroid-sparing agents and in the maintenance of remission is well recognized, and with the advent of metabolite testing their use has been refined 1.
AZA is a prodrug that is converted non-enzymatically into 6-MP, which is metabolized further to 6-thioguanine (6-TG). 6-TG has an anti-proliferative activity via strand breakage through displacement of purine nucleotide incorporation into DNA 2,3. Several months of administration are required before the anti-inflammatory effect of thiopurines in IBD becomes apparent 3. The latency of clinical efficacy of thiopurines is not understood fully, but may be related to the observation that only prolonged administration of the drug leads to contraction of antigen-specific T cell memory clones 4. A role for azathioprine and its metabolites in the control of T cell apoptosis by modulation of Rac1 activation upon CD28 co-stimulation was described. Azathioprine and its metabolites induced apoptosis of T cells from patients with CD via co-stimulation with CD28 and blockade of Rac1. These findings support that 6-Thio-GTP derivates may be useful as potent immunosuppressive agents 5.
An updated meta-analysis and a recent controlled trial have questioned whether thiopurines offer advantage over placebo for induction of remission or clinical improvement in active CD 6,7. Adverse events (AEs) were more common in patients receiving thiopurines. AZA therapy was inferior to infliximab for induction of steroid-free remission. However, a combination of AZA and infliximab was superior to infliximab alone for induction of steroid-free remission 5.
Therapy for Crohn’s disease (CD) with thiopurines is limited by systemic side effects 8–10. AEs leading to discontinuation or dose-reduction of thiopurine therapy occur in 9–28% of patients 8–10. Common AEs reported include allergic reactions, leucopenia, pancreatitis and nausea 6. In a recent meta-analysis, it was shown that 10% of patients in the anti-metabolite group withdrew due to AEs compared to 5% of placebo patients 6. Serious AEs were reported in 14% of patients receiving azathioprine compared to 4% of placebo patients. Side effects are associated with low tolerability and low compliance 11.
Oral immune therapy uses an inherent ability of the gastrointestinal immune system to modulate the systemic immune response 11. The effect is based on activation of gut-associated lymphoid tissue cells leading to systemic immune modulation 12,13. As this mode of therapy does not induce generalized immune suppression, it does not expose the patient to systemic malignancies 12,14.
A novel formulation of low-dose, delayed-release 6-mercaptopurine (DR-6MP) was developed with local delivery to the terminal ileum and no systemic absorption.
The aim of initial pharmacokinetic (PK) and proof-of-concept open-label (POC) studies was to evaluate the safety and efficacy of DR-6MP in patients with CD and to compare its pharmacokinetic profile to 6-MP (Purinethol). The aim of the Phase IIa, multi-centre, randomized, double-blind study was to evaluate the biological activity and clinical efficacy and safety of DR-6MP versus Purinethol in patients with moderately active CD. We hypothesized that a local effect of DR-6MP in the gut would have the potential to modulate the systemic immune system in that it would be at least equally effective and with fewer systemic side effects than 6-MP.
Patients and methods
Phase I pharmacokinetic (PK) trial
In a cross-over trial, 12 CD patients in remission received one dose (40 mg) of DR-6MP versus 100 mg of Purinethol, and Cmax, Tmax and area under the curve (AUC) were measured for 24 h.
Phase I: POC trial
In a randomized, parallel-group, open-label, 12-week study, 13 patients with moderate CD [Crohn’s Disease Activity Index (CDAI) 220–400] received 40 mg DR-6MP (n = 10) or 100–150 mg Purinethol (n = 3). The primary end-point was clinical remission. This POC trial was conducted for the evaluation of the potential systemic immune effect of the non-absorbable DR-6MP. Patients were followed by CDAI score, fluorescence activated cell sorter (FACS) analysis and interferon (IFN)-γ enzyme-linked immunospot (ELISPOT) assay. Results are presented only for the six subjects who completed the study and provided both baseline and week 12 data.
Phase IIA clinical trial
Study design and patients
Patients were enrolled into a multi-centre, randomized, double-blind, double-dummy, two-arm, 12-week study. The study was conducted to determine if the initial promising results of the pilot POC study would be repeatable in a larger, more rigorous controlled study, and if a higher DR-6MP dose (80 mg) could elicit a more robust clinical/immunological effect. Eligible patients were aged 18–75 years who had been diagnosed with CD according to validated criteria, with a CDAI of 220–450 at inclusion. Patients who had received treatment with immunomodulators (within 4 weeks of baseline) or anti-tumour necrosis factor (TNF) (within 6 weeks), or with an immediate need for surgery, severe comorbidity, documented infection, renal or liver failure, contraindication to thiopurines according to labelling recommendations, malignancy, history of drug abuse or predictable poor compliance, were excluded. Patients who had at least one of the following screening laboratory parameters were also excluded: leucocyte count < 3500 mm3; platelet count < 100 000 mm3; haemoglobin < 8·5 g/dl; serum albumin < 2·5 g/dl; and liver enzymes or serum bilirubin > ×2 upper limit of normal. Pregnant women were also ineligible. Patient participation in the study was terminated if at any of the study visits the absolute neutrophil count was < 1000 mm3 or alanine transaminase/aspartate aminotransferase (ALT/AST) > ×5 upper limit of normal. The study was performed at 11 centres in Israel. The institutional review board at each centre approved the protocol, as well as the Central Review Board of the Israeli Ministry of Health, and all patients provided written informed consent. This trial was registered with ClinicalTrials.gov, number NCT01094613.
Two protocols for steroid therapy were used in the study. The first were those subjects who could enter the study using adjunctive treatment (either 5-ASA compounds or low-dose chronic steroids, i.e. up to 6 mg budesonide or 15 mg prednisone or chronic antibiotics) if the CDAI criteria (220–450) were still met. These subjects were included as they were active CD patients in spite of the adjunctive treatment; evaluation of these patients would be whether the addition of either DR-6MP or Purinethol to their regular treatment elicited clinical remission or response. The second type of steroid use permitted in the study was that if someone needed steroid rescue, based on site investigator determination, this was allowed, beginning from week 2 until week 6, at a dose of 40–60 mg starting dose and titrating downwards, so that at the final visit the subject was steroid-free.
Randomization
Patients were randomized in a 2 : 1 randomization scheme: 80 mg DR-6MP (test drug) versus 1–1·5 mg/kg/daily Purinethol (reference drug). Randomization was performed centrally with computer-derived permutation tables. Patients were assigned unique study numbers correlated with the site number and the subject-specific randomized blinded treatment assignment; the study numbers were sent to the investigator after receipt and validation of the inclusion form by the statistical centre.
Procedures
Patients randomized to the DR-6MP group were prescribed 80 mg DR-6MP 15, administered as 2 × 40 mg tablets, once daily, at night. Patients randomized to the Purinethol group were prescribed Purinethol at the initial starting dose of 1 mg/kg/day (generally 50 or 75 mg) Purinethol (DSM; Gates Pharmaceuticals, A division of Teva Pharmaceuticals USA), administered once daily in the morning. Following safety review of the weeks 1 and 2 laboratory data, Purinethol patients were subsequently titrated to the optimal dose of 1·5 mg/kg/day (75, 100, 125 or 150 mg) at week 4 and were maintained at that dose for the remainder of the study, in the absence of AEs/laboratory abnormalities. Following the baseline visit, the investigators received copies of the patients’ laboratory test results but were blinded to the measurements of white blood count (WBC) and differential as well as liver function tests (ALT, AST, direct and total bilirubin), as it was presumed that these specific parameters would be affected only in the Purinethol arm, but not in the DR-6MP arm. Complete unblinded laboratory test results were reviewed in real time by an independent investigator (assigned as the central Study Safety Physician), who was responsible for any drug dose changes. Adjustments of dosage (for the Purinethol arm only, as per the titration paradigm) or temporary or permanent cessation of therapy (both arms) were based on the laboratory test review and decision of the Study Safety Physician, while the site Principle Investigators (PIs) and patients remained blinded, due to the unique drug card design employed in the study. Thiopurine methyltransferase phenotyping/genotyping was not performed. The commonly tested mutations are rare in the Israeli population 16.
Medical history and current medications were recorded at study inclusion. Disease activity was measured by the CDAI. Patients were seen every 2 weeks after baseline until week 8, with an additional safety visit at week 1 and a final evaluation at week 12. Physical examination and laboratory tests [C-reactive protein (CRP), erythrocyte sedimentation rate (ESR) blood counts, liver function tests] were performed at baseline and at each bi-weekly visit. The Inflammatory Bowel Disease Questionnaire (IBDQ) was completed at baseline and at week 12. The IBDQ measures disease-specific quality of life, and higher scores indicate better quality of life. Monitoring for AEs was performed to week 12; any serious AEs within 30 days after study completion were also recorded.
End-points
The primary efficacy outcome was the proportion of subjects with clinical response at week 12. Clinical response was defined as reduction in CDAI score by 100 points from baseline or remission (CDAI score < 150), even if achieved with reduction of CDAI score of fewer than 100 points from baseline.
Prespecified secondary outcomes were time to clinical response; the proportion of subjects in clinical remission (CDAI score < 150) at weeks 2, 4, 6, 8 and 12; time to induction of remission; the proportion of subjects achieving clinical remission or response without steroid rescue therapy at weeks 4, 6, 8 and 12, IBDQ score at week 12 relative to baseline;, mucosal healing as determined by colonoscopy at week 12 relative to baseline (subset of subjects); systemic immunological improvement: CRP, ESR change from baseline at weeks 2, 4, 6, 8 and 12; and FACS analysis: immunological markers (all subjects) measured at week 12 relative to baseline and IFN-γ ELISPOT: (subset of subjects) measured at week 12 relative to baseline.
Safety evaluations included comparison of the incidence, frequency and severity of AEs in each treatment group (DR-6MP versus Purinethol) and comparison of the changes from baseline within each treatment group of clinically significant laboratory values, specifically ALT, AST (hepatoxicity) and WBC (leucopenia).
Intent-to-treat (ITT) population
The ITT population included all randomized/enrolled patients who received a subject study number, signed the informed consent form (ICF) and received at least one dose of study medication, with the last observation carried forward (LOCF) following that administration. The ITT population included 64 subjects (40 subjects in the DR-6MP 80 mg cohort and 24 subjects in the Purinethol cohort). Safety population was defined as the ITT population.
The primary efficacy end-point was also analysed in the population of randomized/enrolled patients who received a subject study number, signed the ICF and provided week 12 CDAI data (ITT CDAI completers population), which included 28 subjects in the DR-6MP treatment arm and 13 subjects in the Purinethol treatment arm.
Of 135 planned patients, only 70 patients were enrolled, 64 of whom were analysable. The reduction in enrolment was due to early closure of the recruitment based on an internal business decision of the Sponsor.
FACS analysis
FACS analysis was performed as described 12, with the following modifications. Cells were suspended in 100 μl FACS buffer [1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS)] and incubated with the following surface anti-human antibodies: CD4-fluorescein isothiocyanate (FITC), CD25-phycoerythrin (PE), CD8-eFlour450, CD56-FITC, CD3-allophycocyanin (APC), CD62-PE and CD127-APC (eBiosciences, San Diego, CA, USA) for 30 min. Tubes with intracellular staining for forkhead box protein 2 (FoxP3) antibody (APC; eBiosciences) were fixed and permeabilized according to the manufacturer’s instructions (eBiosciences) for 40 min and then incubated for 30 min with FoxP3 antibody. Cells were washed twice with FACS buffer and incubated at 4°C until analysis. Cell phenotyping was performed by a LSR-II (Becton Dickinson, Franklin Lakes, NJ, USA) and analysed by FACS-DIVA software. Live cells only were counted, and background fluorescence from non-antibody-treated lymphocytes and isotype control was subtracted.
Subject-specific, antigen-directed IFN-γ ELISPOT assays
IFN-γ spot-forming cells (SFC) were identified using a modified subject-specific, antigen-directed ELISPOT assay (Mabtech, Nacka, Sweden), as described previously 17. Filtration plates (96-well), coated with high protein binding hydrophobic polyvinylidene disulphide (PVDF) membrane, were used (Millipore Corp., Bedford, MA, USA). The plates were coated with 1-D1K anti-IFN-γ coating antibody (15 mg/ml; Mabtech) for 24 h at 40C. Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll gradient separation of 20 ml blood samples, collected in acid citrate dextrose tubes and processed within 1 h. PBMC were washed twice in RPMI-1640 with 10% fetal bovine serum (FBS). Cells were cultured in 96-well plates (1 × 105 cells/well) with RPMI-1640 and 10% FBS. Plates were incubated for 48 h at 37oC with 5% CO2. The plates were washed and 100 μl biotinylated antibody (7-B6-1-biotin; Mabtech) at a concentration of 1 μg/ml in filtered PBS with 0·5% FBS was added. Plates were incubated for 3 h at room temperature. Following washing, 100 μl of streptavidin–alkaline phosphatase was added, and the plates were incubated for 90 min at room temperature. The plates were washed and substrate (BioRad, Richmond, CA, USA) was added for 30 min, until reddish-purple spots appeared. Using a dissection microscope, dark spots, reflecting IFN-γ-secreting clones, were counted independently by two investigators. The results were expressed as means of triplicate IFN-γ-secreting cells per 105 PBMC, after subtraction of the mean spots from wells without the study drug.
The immunology tests described above were performed exclusively at Hadassah, Ein Kerem, Jerusalem, and the safety laboratory tests from the 11 centres were conducted at a central laboratory (AML Laboratories, Herzliyah, Israel).
Statistical analysis
All measured variables and derived parameters were listed individually by treatment group and subject number. All key data were summarized in tables using appropriate summary statistics by treatment group. Continuous parameters were summarized using n, mean, minimum, maximum, standard deviation (s.d.) and 95% confidence interval (CI). Categorical parameters were presented by their frequency counts and percentages and, if appropriate, 95% CI. All tests applied were two-tailed, and a P-value of 5% or less was considered statistically significant. The data were analysed using the sas® 9·1 software package (SAS Institute, Cary, NC, USA).
Results
Phase I PK trial
Figure 1 shows that DR-6MP is not absorbed significantly. Cmax, AUC and Tmax were 82·1 ± 28·7 ng/ml, 216·1 ± 73·8 ng/ml/h and 1·9 ± 1·1 h, respectively, for Purinethol (n = 11) versus 6·1 ng/ml, 10·2 ng/ml/h and 9 h, respectively, for DR-6MP, demonstrating negligible systemic bioavailability and delayed release of the study drug. Quantifiable data for the DR-6MP arm were detected for only one patient, with the remaining the patients showing negligible detection levels or no absorption at all. This study was performed using a 40 mg single dose.
Figure 1.
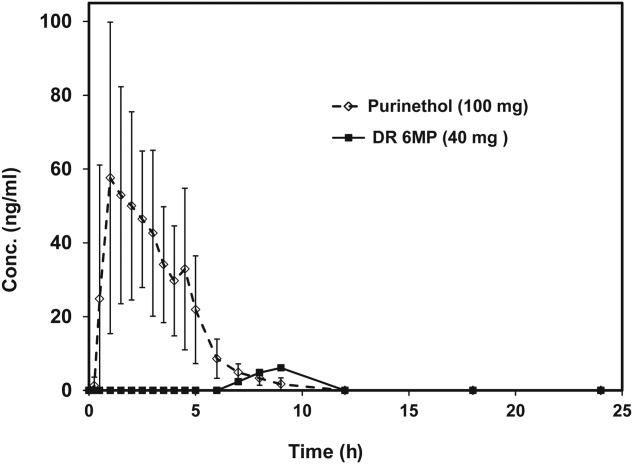
Plasma levels of delayed-release 6-mercaptopurine (DR-6MP) 40 mg in the pharmacokinetic (PK) trial.
Dissimilarity of dose administered between the two treatments, i.e. 40 mg (DR-6MP) versus 100 mg (Purinethol), was performed as the issue was not to compare the bioavailability of like doses, but to compare the bioavailability of the clinical doses to be used, with each arm representing what was presumed to be the clinical dose for that treatment.
Administration of a single dose of both drugs significantly increased systemic immunological regulatory cells 24 h after ingestion relative to baseline: CD4+CD25+FoxP3+ mean of 1·13% (P = 0·008) and 1·43% (P = 0·006) for DR-6MP and Purinethol, respectively, although there were no statistical differences between treatments observed.
Phase I POC trial
Administration of DR-6MP in active CD patients resulted in remission at 12 weeks in three of 10 versus one of three patients for Purinethol. The data presented below are provided for the six subjects (n = 5 for DR-6MP, n = 1 for Purinethol) who completed the study and provided both baseline and week 12 data points. Reduction in week 12 mean CDAI score from baseline was comparable: DR-6MP (285 ± 63–116 ± 55) versus Purinethol (264–120). In two DR-6MP patients, remission was achieved as early as 2–4 weeks. No definitive treatment-related AEs were noted for DR-6MP. Table1 shows the effect of treatment on IFN-γ ELISPOT assay.
Table 1.
Interferon (IFN)-γ enzyme-linked immunospot (ELISPOT) assay results at baseline and at week 12 for a subgroup of patients in the proof-of-concept (POC) trial
| Patient no. | Baseline | Week 12 |
|---|---|---|
| 1 (DR-6MP) | 5·3 | 2·4 |
| 4 (DR-6MP) | 4·4 | 1·0 |
| 05 (DR-6MP) | 4·0 | 1·2 |
| 7 (DR-6MP) | 5·0 | 1·4 |
| 22 (DR-6MP) | 4·0 | 1·4 |
| 10 (Purinethol) | 5·0 | 1·3 |
Results presented as IFN-γ-secreting cells per 105 peripheral blood mononuclear cells (PBMC).
Phase IIA clinical trial
The aim of this trial was to determine the clinical effect of DR-6MP and its non-inferiority to its comparator, Purinethol. Seventy subjects were enrolled overall (46 randomized to the DR-6MP group and 24 to the Purinethol group), 64 of whom were analysable and comprise the ITT population. Six subjects from the DR-6MP cohort were excluded from the analysis. Two subjects (4·3%) were excluded because they received DR-6MP 40 mg (enrolled under an earlier protocol version, evaluating both low dose (40 mg) and high dose (80 mg) DR-6MP versus Purinethol. The protocol was amended subsequently to one test dose of 80 mg versus Purinethol). Four subjects (8·7%) were excluded because they never began treatment (i.e. after screening/randomization, three patients withdrew consent and one had a flare-up necessitating hospitalization) (Fig. 2).
Figure 2.
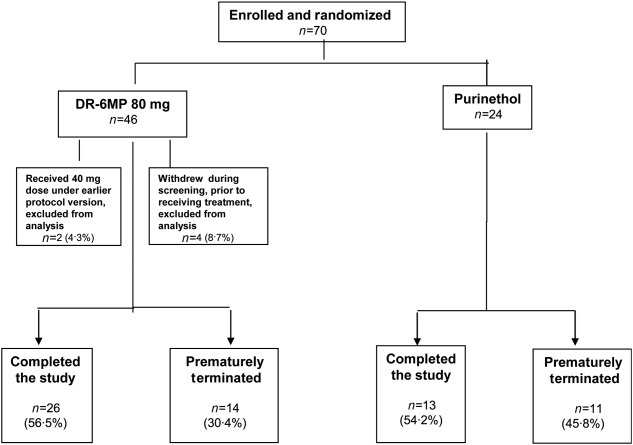
Disposition of subjects the Phase IIa trial.
The two protocols for steroid therapy used in this study affected a subgroup of patients (n = 15) whose data were also analysed in the study. In this subgroup, subjects who were on combination steroids/antibiotics and DR-6MP showed a statistically significant reduction in CDAI at week 12; this was not seen for the Purinethol arm (week 12%CDAI change, DR-6MP versus Purinethol: −36.1%, P = 0·03 versus −18·6%, P = 0·5, respectively), indicating that DR-6MP was effective both as monotherapy and combination therapy. The steroid rescue option was recommended for only two subjects in the study, one in the DR-6MP arm (who exercised the option) and one in the Purinethol arm (who did not exercise the option). Both subjects completed the study. The fact that such an insignificant number of subjects required steroid rescue indicates that the clinical remission and/or response observed in the study overall was due to thiopurine use, whether administered systemically or via local delivery.
Demographics and baseline characteristics
All study subjects were Caucasian, except for one subject from the DR-6MP cohort who was of Ethiopean descent. Nineteen subjects (47·5%) from the DR-6MP cohort and 15 subjects (62·5%) from the Purinethol cohort were female. The average age of the study subjects at screening was 35·5 ± 11·4 years (range = 18–55) in the DR-6MP cohort and 33·7 ± 12·5 years (range = 19–64) in the Purinethol cohort. The number of years since CD diagnosis was 7·9 ± 8·2 years (range 0–26) for subjects in the DR-6MP cohort and 5·5 ± 5·7 (range = 0–22) for subjects in the Purinethol cohort.
Efficacy
As the study was terminated prior to enrolment of the planned number of patients (135), the study population was too small to reach statistical significance in most parameters; nevertheless, trends could be determined and statistical significance was evident in some parameters. Analyses were conducted on ITT (LOCF), ITT (week 12 completers) and PP populations.
Because more patients were included in the ITT set it is more robust, and therefore the data for the ITT population are presented below, except where noted. The clinical efficacy of DR-6MP after 12 weeks of treatment was similar to that of Purinethol. CDAI score decreased to a similar extent in both cohorts (Table2) and a similar proportion of subjects achieved clinical response and remission (Fig. 3). Patients treated with DR-6MP had a significantly greater decrease in CDAI score at week 8 (Table3), reaching a clinical response 4 weeks earlier than patients treated with Purinethol. The time to maximal clinical response was 8 weeks for DR-6MP versus 12 weeks for Purinethol (Fig. 4). A higher proportion of patients in the DR-6MP cohort had clinical response and clinical remission at week 8, compared with those treated with Purinethol (48·3 versus 21·4% Ptrend = 0·0915; 34·5 versus 0% P = 0·0121, respectively; Fig. 5). IBDQ score improved significantly between baseline and week 12 in both cohorts, with a greater change noted in the DR-6MP group. For the DR-6MP cohort, the IBDQ increased from 117·5 at baseline to 154·7 at week 12, an increase of 37·2 (P < 0·0001), while for the Purinethol group, the IBDQ increased from 127·3 at baseline to 152·5 at week 12, an increase of 25·2 (P = 0·0264 between the two differences).
Table 2.
Crohn’s Disease Activity Index (CDAI) score change from baseline at week 12, Phase IIa trial [intent-to treat (ITT) population]
| CDAI score | |||||
|---|---|---|---|---|---|
| DR-6MP 80 mg n = 40 | Purinethol n = 24 | ||||
| n | Mean ± s.d. | n | Mean ± s.d. | P-valuea | |
| Change at week 12 (P-valuea) | 28 | −99·8 ± 89·6 (<0·0001) | 13 | −94·2 ± 83·9 (0·0034) | 0·6969 |
| % Change at week 12 (P-valuea) | 28 | −36·4 ± 32·6 (<0·0001) | 13 | −35·8 ± 31·2 (0·0034) | 0·9003 |
DR-6MP = delayed-release 6-mercaptopurine; s.d. = standard deviation.
P-value by signed-rank test for the statistical significance of CDAI change within treatment. **P-value by non-parametric Wilcoxon test for the statistical significance of the difference in CDAI change between treatments. ***P-value by analysis of covariance (ancova) adjusted for baseline CDAI, age, gender and baseline weight for the statistical significance of the difference in CDAI change between treatments.
Figure 3.
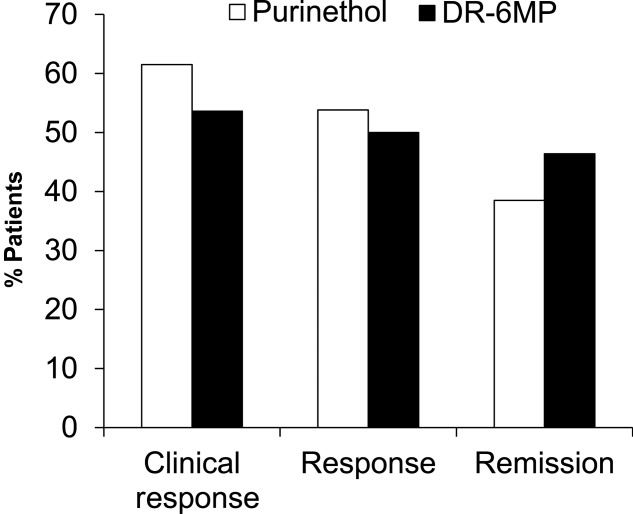
Proportion of responders at week 12 [intent-to treat Crohn’s Disease Activity Index (ITT CDAI) completers population] in the Phase IIa trial. Clinical response = CDAI decrease of at least 100 points or CDAI < 150. Response = CDAI drcrease of at least 100 points. Clinical remission – CDAI < 150. The population analysed included all subjects who provided CDAI data.
Table 3.
Crohn’s Disease Activity Index (CDAI) score change from baseline at week 8, Phase IIa trial [intent-to-treat (ITT) population]
| CDAI score | ||||||
|---|---|---|---|---|---|---|
| DR-6MP 80 mg n = 40 | Purinethol n = 24 | |||||
| n | Mean ± s.d. | n | Mean ± s.d. | P-valuea | P-valuea | |
| Change at week 8 (P-valuea) | 29 | −103·5 ± 74·8 (<0·0001) | 14 | −42·2 ± 71·0 (0·0494) | 0·0570 | 0·0424 |
| % Change at week 8 (P-valuea) | 29 | −36·7 ± 24·5 (<0·0001) | 14 | −12·0 ± 28·8 (0·1040) | 0·0269 | 0·0130 |
DR-6MP = delayed-release 6-mercaptopurine; s.d. = standard deviation.
P-value by signed-rank test for the statistical significance of CDAI change within treatment. **P-value by non-parametric Wilcoxon test for the statistical significance of the difference in CDAI change between treatments. ***P-value by analysis of covariance (ancova) adjusted for baseline CDAI, age, gender and baseline weight for the statistical significance of the difference in CDAI change between treatments.
Figure 4.
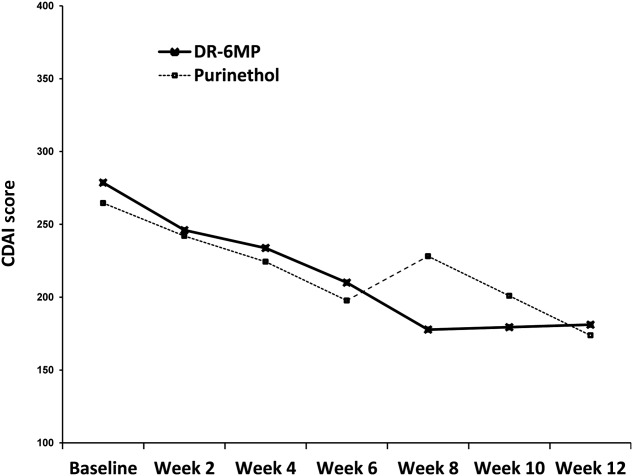
Crohn’s Disease Activity Index (CDAI) score by week [intent-to treat (ITT) population] in the Phase IIa trial.
Figure 5.
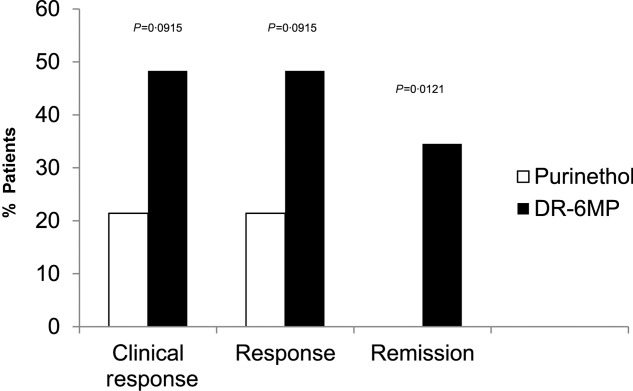
Proportion of responders at week 8 [intent-to treat Crohn’s Disease Activity Index (ITT CDAI) completers population] completers population), Phase IIa trial. Clinical response = CDAI decrease of at least 100 points or CDAI < 150. Response = CDAI drcrease of at least 100 points. Clinical remission – CDAI < 150. The population analysed included all subjects who provided CDAI data.
Reductions in general immune systemic markers CRP and ESR are a measure of efficacy. As a systemically acting drug, Purinethol was expected to have an effect on immune systemic markers; it remained to be seen if locally delivered DR-6MP could exert a comparable systemic effect. The reduction in CRP and ESR at week 12 relative to baseline was slightly greater for the Purinethol group compared to DR-6MP (CRP change −13·3 versus −9·9, and ESR change −12·7 versus −10, respectively); P = Not significant (n.s.)?
DR-6MP induced a different immunological profile than that of Purinethol from baseline to end of study (Fig. 6). DR-6MP 80 mg led to a decrease of CD62+ expression on peripheral T cells as measured by FACS analysis, implying a reduction of lymphocyte adhesion to the site of inflammation. In contrast, Purinethol led to an increase in CD62+ expression. DR-6MP led to a decrease in CD4+CD25+FoxP3+ and CD3+CD56+ expression, while Purinethol led to an increase in expression of these parameters. Both treatments led to an increase in CD4+/CD8+ ratio, and to a decrease in CD4+CD25+ cells. Treatment with DR-6MP for 12 weeks led to a decrease in IFN-γ-positive T cell clones reacting against the subject colon-derived proteins; this was not seen for Purinethol (Fig. 7). However, as both pre- and post-colonoscopy data were needed for the evaluation, data were available for only six patients in the DR-6MP group versus one patient in the Purinethol arm.
Figure 6.

Change in FACS immunology parameters from baseline to week 12 by treatment, Phase IIa trial [intent-to treat (ITT) population].
Figure 7.
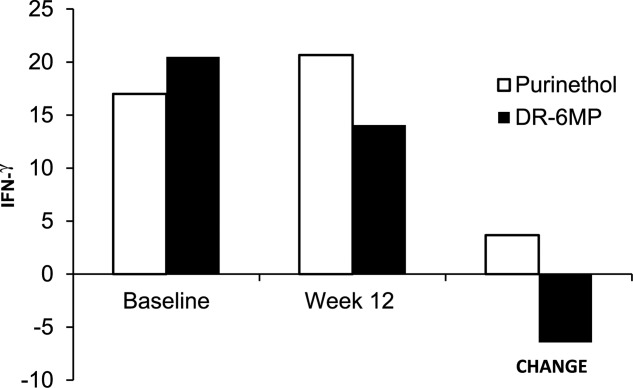
Change in interferon (IFN)-gamma enzyme-linked immunospot (ELISPOT) assay from baseline to week 12 by treatment, Phase IIa trial. Analysis included a subgroup of patients with available ELISPOT assays [for delayed-release 6-mercaptopurine (DR-6MP), n = 6; for Purinethol, n = 1]. Subset of patients = only patients who had undergone a colonoscopy and provided biopsy samples at both baseline and week 12 could be analysed.
Although weight is measured as part of the vital signs and is included typically as a safety parameter, in the case of CD patients, where weight loss is one of the characteristic features of the disease, a change in the expected weight loss can be considered as a parameter of clinical efficacy. Weight and body mass index (BMI) increased in the DR-6MP treatment arm during the 12 weeks of treatment while both parameters decreased in the Purinethol arm, with a statistically significant difference between treatments observed at week 8. This can also be explained by less nausea with improved appetite.
Safety
Overall, DR-6MP 80 mg was safe and well tolerated. DR-6MP was safer than Purinethol, with a significantly lower proportion of subjects with AEs (67·5 versus 95·8%, P = 0·0079). Most AEs were transient and of mild or moderate severity. In addition, the most common drug-related AEs were gastrointestinal disorders, which can be expected for this drug. However, the proportion of subjects reporting drug-related gastrointestinal disorders was lower for DR-6MP than for Purinethol (20 versus 33·3%, respectively). The proportion of subjects with drug-related nausea (10 versus 12·5%), abdominal pain (five versus 12·5%), decreased appetite (2·5 versus 12·5%) and upper abdominal pain (2·5 versus 8·3%) was higher in the Purinethol group compared with the DR-6MP treatment arm. The proportion of subjects who withdrew from the study due to AEs was similar. The percentage of patients after 12 weeks of treatment who had WBC within the normal range was higher in the DR-6MP 80 mg cohort compared with the Purinethol group (Fig. 8). At the end of 12 weeks, 87% of patients on DR-6MP 80 mg had normal levels of WBC compared with only 69·2% in the Purinethol group. The proportion of subjects who developed drug-induced hepatotoxicity (i.e. increase in LFTs necessitating drug dose reduction or study termination) was lower for the DR-6MP group compared to the Purinethol group (2·5 versus 8·3%). Moreover, two subjects who had to stop Purinethol treatment due to hepatoxicity were treated with DR-6MP on a ‘compassionate care’ basis at their individual sites; both subjects stabilized, with normal LFTs for nearly 7 months on DR-6MP.
Figure 8.
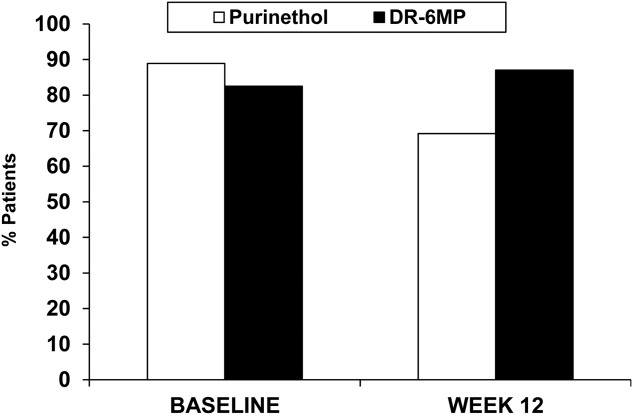
Patients (%) with white blood cell (WBC) result within normal range at baseline and week 12, Phase IIa trial [intent-to treat (ITT)] completers population] population).
A comparable percentage of pancreatitis events occurred in both treatment arms [two (5%) in the DR-6MP group and one (4·2%) in the Purinethol group], supporting the notion that DR-6MP is biologically active systemically, even though it is negligibly absorbed. Unlike hepatotoxicity or leucopenia, however, which is dose-dependent, pancreatitis can be an idiosyncratic allergic reaction to even minute amounts of drug in susceptible patients.
Discussion
Our study shows that non-absorbable DR-6MP was biologically active in the gut, exerting a systemic clinical and immune effect. DR-6MP exerted a clinical effect that was non-inferior and, in some measures, superior to that of the comparator, Purinethol. Treatment with DR-6MP 80 mg was safe, with fewer drug-related AEs reported, including fewer drug-related gastrointestinal disorders, less hepatotoxicity and no leucopenia compared to Purinethol. DR-6MP showed an earlier onset of clinical efficacy compared with that of Purinethol. Overall, the data support the premise that DR-6MP is not absorbed systemically and has a local effect in the gut that affects the systemic immune system.
Exerting an effect at the level of the gut-associated lymphoid system suggests that oral administration of DR-6MP may overcome some of the obstacles encountered with the absorbable formulation of the drug. These may include the severe immune suppression and long-term effects of immune suppression endured by patients with chronic disorders who use these agents for long periods of time.
Although corticosteroids are effective for induction of remission of CD, many patients relapse when steroids are withdrawn or become steroid-dependent 5. The success of thiopurines and methotrexate as a treatment for rheumatoid arthritis and other immune-mediated disorders led to their evaluation in patients with refractory CD 6,18. Thiopurines maintain remission and modify the disease course in IBD 19. None the less, a recent trial showed that universal administration of azathioprine within 6 months of CD diagnosis was no more effective in reducing time to clinical remission than conventional management (i.e. azathioprine added only as needed, as in cases of steroid dependency or poor response, chronic active disease with frequent flares or perianal disease) 6,7. However, thiopurine treatment is limited by a high rate of side effects and intolerance, which can lead to drug withdrawal in approximately 17% of patients 19.
Oral immune therapy uses the inherent ability of the gut immune system to alter different subsets of lymphocytes systemically 13,20. This method is not associated with generalized immune suppression and therefore is not expected to expose patients to infections or malignancy, nor to any side effects in the gut. Manipulation of a proinflammatory environment via a local effect on the gut immune system represents a therapeutic strategy for restoration of immune homeostasis and tolerance 21. Several orally administered immune modulatory agents are being developed as a means to alter the systemic immune system via their effects at the gut level 12,14. These products were shown to exert diverse effects on the systemic immune system when administered orally compared with parenteral administration 14,22. Oral immune therapy can be achieved by immune modulatory agents or disease-associated antigens 9. It was suggested to exert its effect at the level of the mesenteric lymph nodes and/or the gut-associated lymphoid tissue 12–14,23–28.
The results of the present trial show different effects on the T cell profile between the non- absorbable and absorbable formulations of 6-mercaptopurine, which can be explained by different mechanisms of action for the DR-6MP versus Purinethol.
The initial Phase I PK trial described here shows that a DR-6MP 40-mg single dose is not absorbed. Based on the negligible levels detected following single dosing, it is reasonably extrapolated that 80 mg for 12 weeks would have a similar effect, although this would have to be demonstrated in a steady-state PK study of the higher dose. The administration of a dose of 40 mg DR-6MP for 12 weeks was biologically active, as shown by the Phase I POC trial.
The Phase IIA clinical trial showed that the clinical efficacy of DR-6MP 80 mg after 12 weeks of treatment was similar to that of Purinethol. The smaller number of patients enrolled in the current trial, relative to the original statistical plan, did not enable the study to reach statistical significance for several of the parameters tested. Nevertheless, positive trends were evident. CDAI score decreased to a similar extent in both cohorts and a similar proportion of subjects achieved clinical response and remission. At week 8, a higher proportion of patients in the DR-6MP cohort had a clinical response and clinical remission, as well as a significantly greater decrease in mean CDAI score, compared with those treated with Purinethol. Therefore, the maximal clinical effect was achieved much earlier with DR-6MP; the DR-6MP group reached its maximal effect at week 8, which then continued to week 12. The Purinethol group reached its maximal effect at week 12. As a corollary to the clinical efficacy finding of CDAI, ‘quality of life’ was evaluated using the IBDQ. IBDQ score improved significantly between baseline and week 12 in both cohorts.
Several of the subjects entered the study with active CD while on adjunctive treatments (5-ASA or steroids/chronic antibiotics) and remained on them concomitantly with the randomly assigned DR-6MP or Purinethol for the duration of the study. In both these subsets, a significant CDAI decrease from baseline to week 12 was seen only in the DR-6MP treatment arm (5-ASA % change: −38·5, P = 0·039; steroids/antibiotics % change: −36·1, P = 0·031), indicating that DR-6MP may be efficacious both as monotherapy or as combination therapy. A statistically significant decrease in CDAI score (% change −22·9, P = 0·0078) was observed between baseline and week 12 in the subset of patients who had previously failed thiopurine treatment and were assigned non-randomly to the DR-6MP, indicating that even patients for whom Purinethol or azathioprine are not viable alternatives could benefit from DR-6MP treatment. However, the lack of a placebo group in this trial does not enable final conclusions.
A similar decrease in general systemic immune markers CRP and ESR at week 12 relative to baseline was observed for both treatment arms, with a slightly greater decrease noted in the Purinethol arm. This result was expected following Purinethol treatment, but also occurred following locally delivered DR-6MP with negligible systemic absorption. These results may suggest that DR-6MP initially works locally in the intestinal mucosa affecting regulatory T cells (Tregs), and then later by expressing itself systemically.
The proportion of subjects reporting AEs was statistically significantly lower with DR-6MP than with Purinethol. The proportion of subjects reporting drug-related gastrointestinal disorders was lower for the DR-6MP. There was no evidence of drug-induced leucopenia in the DR-6MP group; the percentage of patients after 12 weeks of treatment who had white blood cells within the normal range was higher in the DR-6MP cohort. The proportion of patients who developed hepatotoxicity was lower for the DR-6MP group compared to the Purinethol arm.
The suggested mechanism of action of DR-6MP at the level of the gut immune system while not being systemically absorbed has two additional advantages. The effect is not dose-dependent, and therefore does not require dose adjustments to body weight. The lack of absorption potentially omits the need to adjust the dose based on TPMT genetic variants.
The pathogenic changes of CD depend on migration of circulating leucocytes into intestinal tissues 29. CD is characterized by immune dysregulation and leucocyte recruitment into the gastrointestinal tract. Cell adhesion molecules (CAM) mediate the extravasation of leucocytes and their accumulation in inflamed intestinal mucosa 30. DR-6MP induced a different immunological profile than that of Purinethol from baseline to the end of the trial. DR-6MP led to a decrease in CD4+CD25+FoxP3+ and CD3+CD56 expression, while Purinethol led to an increase in expression of these parameters. Both treatments led to an increase in CD4+/CD8+ ratio and to a decrease in CD4+CD25+ cells. Treatment with DR-6MP for 12 weeks led to a decrease in IFN-γ-positive T cell clones reacting against the subject colon-derived proteins.
Leucocyte rolling and adhesion are regulated by inducible selectins on vascular endothelia 29. CD62 is an adhesive protein designated as an endothelial P-selectin. CD62+ expresses on the surface membrane of all leucocytes and plays a role in early adhesive rolling interactions between white cells and the vascular wall. CD62 was tested in histologically uninvolved tissues adjacent to inflammation in CD. Compared with the normal gut, there was a several-fold increase of P-selectin immunoreactivity on veins, venules and capillaries in the highly inflamed gut 29. DR-6MP led to a decrease of CD62+ expression on peripheral T cells as measured by FACS analysis, implying a reduction of lymphocyte adhesion to the site of inflammation. In contrast, Purinethol led to an increase in CD62+ expression. It is therefore hypothesized that while the Purinethol exerts its effects via a suppressive effect on the immune system as an anti-metabolite, the DR-6MP works via a different type of mechanism to affect the systemic immune system.
It should be noted that, in the present study, Purinethol was used as the reference for induction of remission although it is used typically as maintenance treatment. Nevertheless, within 8 weeks, DR-6MP was shown to be effective for induction of remission, while exhibiting a lower level of AEs and a beneficial effect on the immune system. As such, DR-6MP should be tested against reference drugs used mainly for induction of remission.
In summary, a non-absorbable DR-6MP formulation is safe and biologically active in the gut. It exerts a systemic immune response and is clinically effective, with low systemic bioavailability and a lower incidence of side effects. Larger cohorts of patients are required for final conclusions. The data show that DR-6MP can alter the systemic immune system and supports its potential use as an immune modulatory agent for a greater number of disorders in which immune modulation is required, but is currently limited due to the immune suppression induced by drugs such as Purinethol. Moreover, the type of immune modulation promoted by DR-6MP systemically may be of greater benefit compared with other immune suppression agents.
Acknowledgments
The study was designed and supported by Teva.
Disclosure
Teva took part in the manuscript preparation. Y. I. is a consultant for Teva.
References
- Waters OR, Lawrance IC. Understanding the use of immunosuppressive agents in the clinical management of IBD. Curr Drug Targets. 2011;12:1364–71. doi: 10.2174/138945011796150343. [DOI] [PubMed] [Google Scholar]
- Bokkerink JP, Stet EH, De Abreu RA. 6-Mercaptopurine: cytotoxicity and biochemical pharmacology in human malignant T-lymphoblasts. Biochem Pharmacol. 1993;45:1455–63. doi: 10.1016/0006-2952(93)90045-x. [DOI] [PubMed] [Google Scholar]
- Ebbesen MS, Nersting J, Jacobsen JH, et al. Incorporation of 6-thioguanine nucleotides into DNA during maintenance therapy of childhood acute lymphoblastic leukemia-the influence of thiopurine methyltransferase genotypes. J Clin Pharmacol. 2013;53:670–4. doi: 10.1002/jcph.81. [DOI] [PubMed] [Google Scholar]
- Ben-Horin S, Goldstein I, Fudim E, et al. Early preservation of effector functions followed by eventual T cell memory depletion: a model for the delayed onset of the effect of thiopurines. Gut. 2009;58:396–403. doi: 10.1136/gut.2008.157339. [DOI] [PubMed] [Google Scholar]
- Tiede I, Fritz G, Strand S, et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest. 2003;111:1133–45. doi: 10.1172/JCI16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chande N, Tsoulis DJ, MacDonald JK, et al. Azathioprine or 6-mercaptopurine for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. 2013;4:CD000545. doi: 10.1002/14651858.CD000545.pub4. [DOI] [PubMed] [Google Scholar]
- Cosnes J, Bourrier A, Laharie D, et al. Early administration of azathioprine vs conventional management of Crohn’s disease: a randomized controlled trial. Gastroenterology. 2013;145:758–65. doi: 10.1053/j.gastro.2013.04.048. , e2; quiz e14–5. [DOI] [PubMed] [Google Scholar]
- Hanauer SB, Korelitz BI, Rutgeerts P. Postoperative maintenance of Crohn’s disease remission with 6-mercaptopurine, mesalamine, or placebo: a 2-year trial. Gastroenterology. 2004;127:723–9. doi: 10.1053/j.gastro.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Mate-Jimenez J, Hermida C, Cantero-Perona J, et al. 6-mercaptopurine or methotrexate added to prednisone induces and maintains remission in steroid-dependent inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2000;12:1227–33. doi: 10.1097/00042737-200012110-00010. [DOI] [PubMed] [Google Scholar]
- Markowitz J, Grancher K, Kohn N, et al. A multicenter trial of 6-mercaptopurine and prednisone in children with newly diagnosed Crohn’s disease. Gastroenterology. 2000;119:895–902. doi: 10.1053/gast.2000.18144. [DOI] [PubMed] [Google Scholar]
- Gilissen LP, Wong DR, Engels LG, et al. Therapeutic drug monitoring of thiopurine metabolites in adult thiopurine tolerant IBD patients on maintenance therapy. J Crohns Colitis. 2012;6:698–707. doi: 10.1016/j.crohns.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Ilan Y, et al. Oral tolerance: can we make it work? Hum Immunol. 2009;70:768–76. doi: 10.1016/j.humimm.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Mizrahi M, Ilan Y. The gut mucosa as a site for induction of regulatory T-cells. Curr Pharm Des. 2009;15:1191–202. doi: 10.2174/138161209787846784. [DOI] [PubMed] [Google Scholar]
- Weiner HL, da Cunha AP, Quintana F. Oral tolerance. Immunol Rev. 2011;241:241–59. doi: 10.1111/j.1600-065X.2011.01017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastida G, Nos P, Aguas M, et al. The effects of thiopurine therapy on health-related quality of life in inflammatory bowel disease patients. BMC Gastroenterol. 2010;10:26. doi: 10.1186/1471-230X-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasirer Y, Mevorach R, Renbaum P, et al. Thiopurine S-methyltransferase (TPMT) activity is better determined by biochemical assay versus genotyping in the Jewish population. Dig Dis Sci. 2014;59:1207–12. doi: 10.1007/s10620-013-3008-z. [DOI] [PubMed] [Google Scholar]
- Margalit M, Israeli E, Shibolet O, et al. A double-blind clinical trial for treatment of Crohn’s disease by oral administration of Alequel, a mixture of autologous colon-extracted proteins: a patient-tailored approach. Am J Gastroenterol. 2006;101:561–8. doi: 10.1111/j.1572-0241.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Feagan BG, Lichtenstein GR, et al. Medical management of mild to moderate Crohn’s disease: evidence-based treatment algorithms for induction and maintenance of remission. Aliment Pharmacol Ther. 2007;26:987–1003. doi: 10.1111/j.1365-2036.2007.03455.x. [DOI] [PubMed] [Google Scholar]
- Kennedy NA, Rhatigan E, Arnott ID. A trial of mercaptopurine is a safe strategy in patients with inflammatory bowel disease intolerant to azathioprine: an observational study, systematic review and meta-analysis. Aliment Pharmacol Ther. 2013;38:1255–66. doi: 10.1111/apt.12511. [DOI] [PubMed] [Google Scholar]
- Castro-Sanchez P, Martin-Villa JM, et al. Gut immune system and oral tolerance. Br J Nutr. 2013;109(Suppl. 2):S3–11. doi: 10.1017/S0007114512005223. [DOI] [PubMed] [Google Scholar]
- Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. 2007;204:33–9. doi: 10.1084/jem.20061531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Maron R, Tukpah AM. Mucosal anti-CD3 monoclonal antibody attenuates collagen-induced arthritis that is associated with induction of LAP+ regulatory T cells and is enhanced by administration of an emulsome-based Th2-skewing adjuvant. J Immunol. 2010;185:3401–7. doi: 10.4049/jimmunol.1000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cunha AP, Weiner HL, et al. Induction of immunological tolerance by oral anti-CD3. Clin Dev Immunol. 2012;2012:425021. doi: 10.1155/2012/425021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassani B, Villablanca EJ, Quintana FJ. Gut-tropic T cells that express integrin alpha4beta7 and CCR9 are required for induction of oral immune tolerance in mice. Gastroenterology. 2011;141:2109–18. doi: 10.1053/j.gastro.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peron JP, Yang K, Chen ML, et al. Oral tolerance reduces Th17 cells as well as the overall inflammation in the central nervous system of EAE mice. J Neuroimmunol. 2010;227:10–7. doi: 10.1016/j.jneuroim.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Ochi H, Abraham M, Ishikawa H, et al. New immunosuppressive approaches: oral administration of CD3-specific antibody to treat autoimmunity. J Neurol Sci. 2008;274:9–12. doi: 10.1016/j.jns.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ochi H, Chen ML, et al. Inhibition of autoimmune diabetes by oral administration of anti-CD3 monoclonal antibody. Diabetes. 2007;56:2103–9. doi: 10.2337/db06-1632. [DOI] [PubMed] [Google Scholar]
- Israeli E, Ilan Y, et al. Oral administration of Alequel, a mixture of autologous colon-extracted proteins for the treatment of Crohn’s disease. Therap Adv Gastroenterol. 2010;3:23–30. doi: 10.1177/1756283X09351733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurmann GM, Bishop AE, Facer P. Increased expression of cell adhesion molecule P-selectin in active inflammatory bowel disease. Gut. 1995;36:411–8. doi: 10.1136/gut.36.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazen D, Jendoubi-Ayed S, Aleya WB, et al. Polymorphism in ICAM-1, PECAM-1, E-selectin, and L-selectin genes in Tunisian patients with inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2009;21:167–75. doi: 10.1097/MEG.0b013e32830e6fc8. [DOI] [PubMed] [Google Scholar]