

Abstract
The transcriptional activity of genes largely depends on the accessibility of specific chromatin regions to transcriptional regulators. This process is controlled by diverse post-transcriptional modifications of the histone amino termini of which reversible acetylation plays a vital role. Histone acetyltransferases (HATs) are responsible for the addition of acetyl groups and histone deacetylases (HDACs) catalyse the reverse reaction. In general, though not exclusively, histone acetylation is associated with a positive regulation of transcription, whereas histone deacetylation is correlated with transcriptional silencing. The elucidation of unequivocal links between aberrant action of HDACs and tumorigenesis lies at the base of key scientific importance of these enzymes. In particular, the potential benefit of HDAC inhibition has been confirmed in various tumour cell lines, demonstrating antiproliferative, differentiating and pro-apoptotic effects. Consequently, the dynamic quest for HDAC inhibitors (HDIs) as a new class of anticancer drugs was set off, resulting in a number of compounds that are currently evaluated in clinical trials. Ironically, the knowledge with respect to the expression pattern and function of individual HDAC isoenzymes remains largely elusive. In the present review, we provide an update of the current knowledge on the involvement of HDACs in the regulation of fundamental cellular processes in the liver, being the main site for drug metabolism within the body. Focus lies on the involvement of HDACs in the regulation of growth of normal and transformed hepatocytes and the transdifferentiation process of stellate cells. Furthermore, extrapolation of our present knowledge on HDAC functionality towards innovative treatment of malignant and non-malignant, hyperproliferative and inflammatory disorders is discussed.
Keywords: histone deacetylases, histone deacetylase inhibitors, protein acetylation, cell cycle, hepatocytes, hepatoma, transdifferentiation, hepatic stellate cells
Introduction
-
Histone Deacetylases
Classification
Histone-dependent mode of action
Non-histone targets
-
Cell cycle-related histone and non-histone substrates of histone deacetylases
Retinoblastoma
Protein 53 transcription factor
Cyclin-dependent kinase inhibitor p21Cip1
Proliferating cell nuclear antigen
-
Inhibition of histone deacetylases in liver cells
Physiological condition: effects of histone deacetylase inhibitors on primary hepatocytes
-
Pathophysiological condition
Liver malignancy – hepatocellular carcinoma
-
Liver fibrosis
Histone deacetylase inhibitors as modulators of the hepatic stellate cell myofibroblastic phenotype
The role of histone deacetylases in the regulation of pro-fibrogenic and pro-inflammatory cascades
Conclusions
Introduction
Regulatory pathways of many cellular activities, including cell proliferation, merge on the fundamental process of transcription. Indeed, transcription of genetic information enclosed in DNA and subsequent translation allow the cell to generate the necessary factors, i.e. proteins, in response to various intra- and extracellular stimuli. One of the key mechanisms determining transcriptional activity of genes is the acetylation of histones. The latter are highly conserved low molecular weight proteins encountered in the nucleus of eukaryotic cells and in the nucleoid of some prokaryotic species. Six main histone classes can be distinguished, namely H1, H2A, H2B, H3, H4 and prokaryotic archeal histones [1, 2]. Two copies of H2A, H2B, H3 and H4 compose the core of the nucleosome structure, which is a basic unit of eukaryotic chromatin. Winded around the histone centre is a 146bp DNA strand representing the second component of the nucleosome [3]. The high lysine/arginine content of the histones endows them with strong basic properties, which facilitate the interaction with the negatively charged DNA. The introduction of acetyl groups by histone acetyltransferases (HATs) reduces the affinity of the histone cores to the DNA strands. As a result a relaxed chromatin structure is formed, enabling initiation of transcription. On the contrary, histone deacetylation, which is a virtue of histone deacetylase (HDAC), reconstitutes a dense chromatin configuration, being impermissive for transcriptional factors [4]. Thus, generally, yet not exclusively, histone hyperacetylation is associated with transcriptional activation, whereas histone hypoacetylation leads to transcriptional repression [5–7]. As many cellular activities find their outcome at the transcriptional level, both HATs and HDACs can be perceived as master regulators of cellular homeostasis.
Histone deacetylases
Classification
Taunton and colleagues identified the first mammalian HDAC isoenzyme, being HDAC-1 [8]. Since then, the existence of 17 other enzymes with HDAC activity has been reported. They are divided in four major classes based on phylogenetic analysis and homology to yeast HDACs. Class I consists of HDAC-1, -2, -3 and -8 whereas class II is represented by HDAC-4, -5, -6, -7, -9 and 10. HDAC-11 constitutes the single representative of class IV [9]. Those enzymes belong to a group of zinc-dependent aminohydrolases owing to the presence of a Zn2+ in their catalytic centre. Class III HDACs, also called sirtuins, constitute a distinct group of HDACs requiring NAD+ for their catalytic activity and will not be discussed in the current review [10, 11]. Whereas class I HDACs are rather small proteins with predominantly nuclear localization, class II enzymes are large and capable of translocating between cellular compartments [9, 12].
Histone-dependent mode of action
HDAC-1 and HDAC-2 as well as other HDACs do not act autonomously but create multi-subunit protein complexes [13]. Because they lack intrinsic DNA-binding activity, HDACs require DNA-binding proteins to localize specific genes destined for deacetylation-mediated transcriptional repression. At the same time, other protein components of the complex act as cofactors, augmenting the enzymatic activity of HDACs [13]. In fact, three HDAC1/2-containing corepressor complexes have been identified: (i) Sin3, (ii) the nucleosome remodeling and deacetylation complex (NuRD) and (iii) the corepressor of RE1-silencing transcription factor (coREST) [14, 15]. Another member of class I HDACs, namely HDAC-3, resides in a complex with the nuclear receptor corepressor (NCoR) and silencing mediator for retinoid and thyroid receptors (SMRT) [16, 17]. These HDAC-3 containing complexes also take part in interactions occurring within the HDAC family. SMRT/NCoR corepressors not only bind to class IIa HDACs but are also a prerequisite for their enzymatic activity [18, 19].
HDAC-dependent deacetylation of histones affects gene transcription by at least two parallel mechanisms (Fig. 1). As described before, histone deacetylation enhances the electrostatic interaction between the histone core of nucleosomes and DNA, leading to chromatin condensation [4]. Apart from altering the physical properties of chromatin, removal of acetyl groups modifies the array of post-translational modifications of amino acids in histones’ amino termini. These chemical modifications, including phosphorylation, acetylation, methylation, ubiquitination and sumoylation, are thought to create a specific code, designated as the histone code [2]. Addition or removal of a functional group on one amino acid residue may influence the status of neighbouring amino acid residues, not only in the same histone tail but also in tails of other histones, or even in other nucleosomes. As a result, the context of the encrypted message may be changed [13, 20]. This secret molecular language of chemical marks is deciphered by nuclear proteins such as transcription factors or chromatin modifying enzymes. These are endowed with specific recognition modules with bromo- and chromodomains that recognize acetylated sites or amino acids in methylated form, respectively [21, 22]. Depending on the nature of the recruited protein, a specific action will be initiated. Thus, although we are used to associate histone deacetylation with chromatin inactivation, the factual effect on transcription not only depends on the net electric charge of all modifications on histone tails but also on the type of proteins, i.e. silencers or activators of transcription, recruited in response to acetyl group displacement. As a consequence HDACs may either positively or negatively affect gene expression. The latter was confirmed by the observations made in HDAC inhibitor (HDI)-treated systems, where a similar number of genes was found to be either up-regulated or down-regulated upon the treatment [23–25]. Furthermore, several independent groups reported the requirement of HDAC activity in the activation of transcription in cytokine-inducible systems such as interferon-controlled genes. (For an extensive review of this topic we refer to [26, 27].)
Figure 1.
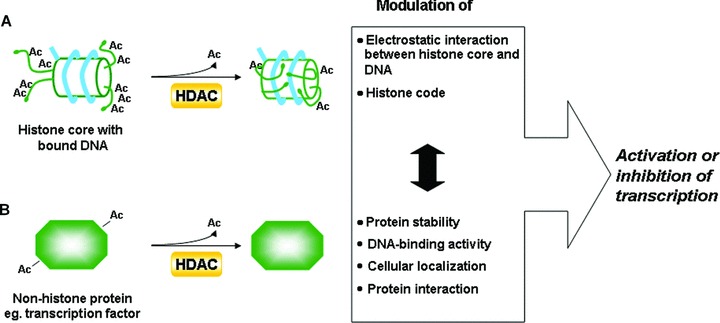
Mechanisms by which HDACs may regulate the process of transcription. (A) Histone-related pathway: HDACs deacetylate histones leading to the increase of chromatin compaction and alternations in the histone code. (B) Non-histone related pathway: HDACs deacetylate non-histone protein targets affecting diverse aspects of protein physiology. These pathways interconnect with each other (which is indicated by a black double-sided arrow), e.g. modification of the histone code could result in the recruitment of a specific transcriptional regulator. If the latter is deacetylated by one of the HDACs its DNA-binding activity, cellular localization or half-life may be affected. Consequently, the final decision whether the transcription of a particular gene will be initiated is a sum of all actions, some being transcription stimulating, others transcription inhibiting.
Non-histone targets
Recent studies indicate that histones are not the sole target of HDACs. In fact, phylogenetic analysis of bacterial HDACs revealed that in the progress of evolution these enzymes emerged before the histone proteins, strongly suggesting that other protein type(s) were the primary HDACs’ substrates [9, 11]. In addition, a number of cytoplasmic and nuclear proteins, including transcription factors, DNA repair enzymes, cell-cycle regulators, molecular chaperones as well as structural proteins were found to be reversibly acetylated [28]. Acetylation/deacetylation may modify the stability of the protein involved, its localization, DNA-binding ability and its interactions with other proteins [29, 30]. Recently, a novel nomenclature was proposed for some of the chromatin-modifying enzymes, replacing the historical term of HATs by the more general one, of, ‘lysine (K)-acetyltransferases’ (KATs) [31]. Rethinking of our perception of KATs and HDACs seems necessary as their significance raises from transcriptome-related enzymes to proteome-controlling enzymes and finally to masters of the acetylome [29].
Cell cycle-related histone and non-histone substrates of histone deacetylases
The role of HDACs in cellular proliferation was emphasized by the discovery that their anomalous action constitutes a cornerstone of carcinogenesis, a process which results from a disrupted balance between cell proliferation and apoptosis, in favour of unlimited growth [10, 28, 29, 32]. Global loss of histone H4 acetylation is a common trait of cancer cells, indicating disruption of the acetylome’s homeostasis [33]. The latter may occur due to aberrant recruitment to specific loci or altered expression of individual HDAC isoenzymes. Alternatively, disruption of KAT activity may be the cause. Abnormal targeting of HDACs to promoters of genes involved in the control of cell growth and differentiation by PML-RAR-α, PZLF-RAR-α and AML1-ETO chimeric oncoproteins results in leukaemias [34]. On the other hand, overexpression of distinct HDACs was encountered in a number of tumour specimens: (i) HDAC-1 in prostate, gastric and colon tumours, (ii) HDAC-2 in colorectal, cervical and gastric cancer and (iii) HDAC-3 in colon cancer [10]. These observations triggered the development of new anticancer drugs interfering with HDAC enzymatic activity.
The discovery of the first mammalian HDAC coincided in time with the identification of the first HDI generation represented by sodium butyrate (NaB) and trichostatin A (TSA) [35, 36]. Currently, there is an abundance of structurally diverse HDI molecules available. Several of them have even reached advanced clinical trials and recently the first HDI, for the treatment of cutaneous T-cell lymphoma, marketed under the name Vorinostat®, was approved by the Food and Drug Administration agency (for detailed reviews see [10, 29, 37]).
As demonstrated by cDNA microarray studies, inhibition of HDACs alters the expression of merely 2% to 10% of all genes, with as many genes up-regulated as down-regulated [23–25]. One therefore would expect only negligible consequences for the cell’s physiology. In cancer cells, however, chemical down-regulation of the HDAC activity as well in vitro as in vivo inhibits cell growth and induces apoptosis. In addition, the action of HDIs proved to be cancer cell-selective, leaving normal, healthy cells unharmed. Recently, additional anti-metastatic, anti-angiogenic and immunomodulatory properties of HDIs were reported [10, 30, 32, 38, 39].
In the following paragraph, the importance of HDAC enzymes in the regulation of cell cycle proteins such as retinoblastoma (Rb), protein 53 (p53), protein 21 Cip1 (p21Cip1) and proliferating cell nuclear antigen (PCNA) will be discussed in depth.
Retinoblastoma
Rb is an archetypal tumour suppressor, characterized by a distinctive binding region in the form of a pocket, which enables specific protein–protein interaction (e.g. with E2F transcription factors) [40, 41]. The Rb protein plays a crucial role in the regulation of the mid-late G1 restriction point. It exerts a growth-inhibitory function by a dual approach. Via a pocket domain-mediated binding, Rb sequesters E2F and inhibits its transcriptional activity. The latter controls the expression of a panel of proteins having a central role in DNA replication and cell cycle regulatory activities, i.e. DNA polymerase α, PCNA, and cyclin A and E [42]. Rb also recruits HDAC-1 and directs it to E2F-responsive promoters [43]. Different proteins act as a bridge between HDAC-1 and Rb, including the Jumonji domain 2 A (JMJD2A) and Rb-binding protein 1 (RBBP1) [44, 45]. HDAC-1-mediated histone deacetylation in the region of E2F-target promoters results in their transcriptional repression and hinders G1/S-phase transition. Upon mitogenic stimulation, cyclin-dependent kinase (cdk)/cyclin complexes phosphorylate the Rb protein and abrogate the HDAC-1/Rb/E2F interaction. Liberated E2F can then activate its target gene promoters, allowing initiation of the S-phase and subsequent cell cycle progression [28]. Additionally, the Rb and E2F proteins as such can be directly subjected to reversible acetylation. In the case of E2F acetylation, highly conserved acetylation sites lie next to its DNA-binding domain, resulting in the enhancement of its DNA-binding activity, activation potential and protein half-life. HDAC-1 can deacetylate E2F1 via an indirect Rb-mediated mechanism [28]. In the case of Rb acetylation, it was found that not only the efficiency of phosphorylation by the cdk/cyclin complex was lowered, but also the binding affinity of MDM2 ubiquitin ligase was increased [28]. Yet, the biological relevance of these observations remains elusive.
Protein 53 transcription factor
Another master regulator of cell cycle progression, of which the activity is modulated by reversible acetylation, is p53 (Fig. 2). As nicely portrayed by Shu and colleagues, p53 is a key tumour suppressor protein at the crossroads of cellular stress response pathways [46]. In unstressed cells, p53 is expressed at a steady, low level and undergoes constant turnover due to polyubiquitination and concomitant proteolytic breakdown. p53 ubiquitination is catalysed by a group of E3-ubiquitin ligases including MDM2, Pirh2, COP1, CHIP and ARF-BP enzymes [47, 48]. A myriad of stress stimuli, such as DNA damage, hypoxia and oncogene activation, prompts the stabilization and subsequent accumulation of the p53 protein in the nucleus [41]. Activated p53 binds to its p53-responsive elements in the genome, initiating a suitable transcriptional program in response to the stress-inducing trigger. The biological outcome may involve growth arrest, DNA repair, cellular senescence or apoptosis [49]. Numerous studies indicate that post-translational modifications play a significant role in modulating the p53 activity. In addition to ubiqitination, various kinases, including ATM, ATR and DNA-PK, phosphorylate p53. The addition of phosphate groups takes place at the N-terminus of the protein, which is engaged in MDM2 binding. As a result, the interaction between p53 and ubiquitinase is disrupted. Early in vitro studies suggested that acetylation of specific lysine residues in the C-terminal domain, which could become the anchors for poly-ubiquitin chains, could prevent the proteosomal degradation of the p53 protein and hence increase its half-life [50]. Conversely, recruitment of HDAC-1 either by the metastasis associated protein 2 (MTA2) or the HDAC-1-containing Sin3 complex mediated by MDM2, and subsequent removal of acetyl groups, restores accessibility of lysines to ubiquitin ligase [28, 51]. Unfortunately, this rather plausible mechanism of modulating p53 protein stability by acetylation could not be confirmed in the subsequent in vivo studies [52, 53]. On the other hand, strong evidence exists that site-specific acetylation of p53 protein in the region of the DNA-binding domain regulates the affinity of this transcription factor towards its target promoters and hence shapes the downstream events. For instance, the acetylation of K320 stimulates the activation of cell cycle-restraining genes such as cdk inhibitor p21Cip1, whereas K373 acetylation provokes activation of pro-apoptotic genes such as Bcl-2-associated X protein (Bax). The position of the acetylated lysine is also not random but rather determined by the type of the acetylation-inducing stimulus such as the type of DNA damage (Fig. 2) [54]. Also, the HDAC-1 ‘sister’ enzyme, HDAC-2 was recently implicated in the negative regulation of p53 activity [55]. By using HDAC-2-targeting short hairpin RNAs, Harms and Chen could demonstrate that HDAC-2 knock-down in MCF7 cell line inhibited cellular proliferation and induced cellular senescence. These effects were partially p53-dependent as demonstrated by activation of major p53 target genes such as p21Cip1 and MDM2. Interestingly, neither increased stability nor enhanced acetylation of the p53 protein could be detected. Instead, HDAC-2 silencing appeared to improve the DNA-binding ability of the transcription factor. The mechanism by which HDAC-2 interferes with the DNA-binding activity of p53 however, remains relatively unknown [55].
Figure 2.
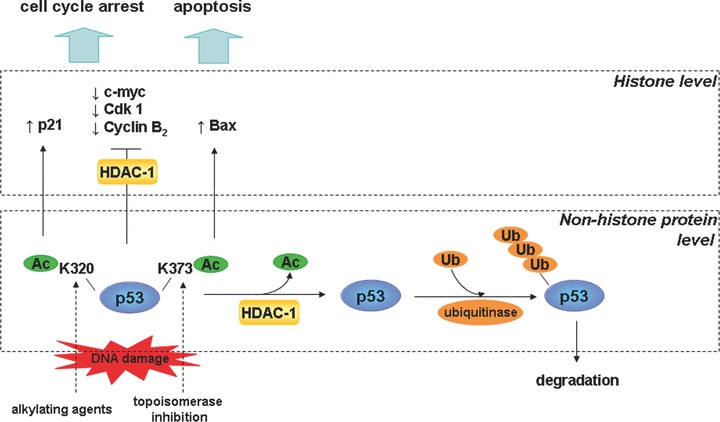
Different levels of HDAC-mediated control in the p53 pathway. In unstressed cells, HDACs may facilitate p53 turnover by removal of acetyl groups from lysines in p53. Those amino acids become ‘visible’ to ubiquitin ligases that are now able to attach ubiquitin particles destining p53 protein to proteosomal degradation. In stressed cells, p53 becomes acetylated, increasing its half-life and leading to accumulation of p53 protein in the cell. The nature of stress stimuli determines the acetylation site and subsequently the type of p53-controled transcriptional response. Although some genes are induced, others are repressed via HDAC-mediated histone deacetylation in the region of corresponding promoters.
Reversible acetylation of histones also constitutes one of the mechanisms by which p53 regulates the transcription of its target genes. Although some genes are up-regulated upon p53 activation (p21Cip1, Gadd45, 14–3-3σ), others are repressed [48, 56]. In the latter instance, HDAC-1/Sin3 acts as a p53 corepressor by restraining the production of c-Myc proto-oncogene transcripts and promoting a G1-phase cell cycle arrest [57]. A study by Imbriano and colleagues indicated that p53 also down-regulates the expression of genes lacking a p53-regulatory sequence [58]. Instead, the promoter regions of these genes contain NF-Y-binding boxes (CCAAT). Following DNA damage, p53 recruits, in concert with the NF-Y transcription factor HDACs and represses cdk1 and cyclin B2 transcription. The ensuing outcome is a G2-phase cell cycle arrest. Chromatin immunoprecipitation experiments revealed the identity of the recruited HDACs. Although HDAC-1 was the first to associate with the investigated promoters, HDAC-4 and HDAC-5 followed with a temporal delay, possibly due to the time required for their translocation from the cytosol to the nucleus [58].
Cyclin-dependent kinase inhibitor p21Cip1
One of the most renowned target genes of the p53 pathway is p21Cip1[59, 60]. Its protein product negatively regulates cell cycle progression by inhibiting the activity of several cdk/cyclin complexes and blocking DNA replication [61].
The first evidence of HDACs’ participation in the regulation of p21Cip1 expression was delivered by Lagger and colleagues [62]. They demonstrated that targeted disruption of HDAC-1 alleles during mouse embryogenesis leads to severe proliferation defects associated with abnormal protein and mRNA levels of p21Cip1 and p27 (a related cdk inhibitor). Analysis of core histones with respect to their post-translational modifications’ status revealed an increased acetylation in the regions of corresponding promoters [62]. Recently, similar observations were made by another group using HDAC-1-directed RNA interference to knock-down the expression of the enzyme in mouse preimplantation embryos [63]. Noteworthy, both groups detected elevated expression of HDAC-2 and -3 enzymes following HDAC-1 elimination. However, neither higher HDAC-2 nor HDAC-3 activity was able to counterbalance the loss of HDAC-1. In the line with former findings, an alternative approach uncovered reduced p21Cip1 mRNA expression in a transgenic mice overexpressing human HDAC-1 isoenzyme [64, 65]. These data underscore the unique role of HDAC-1 in controlling the transcriptional status of p21Cip1 gene.
Although the p21Cip1 gene promoter contains several p53-binding sites, p53 is not always directly responsible or even obligatory for the transcriptional induction of p21Cip1 gene. Other transcription factors, namely SP-1 and SP-3, may be in command. Depending on the circumstances, SP-1 can choose between two interaction partners, either p53 or HDAC-1. Cooperation between p53 and SP-1 results in the p21Cip1 induction, whereas interaction with HDAC-1 has the opposite effect [66]. Recently, also other members of the HDAC family, namely HDAC-3 and -4, were implicated in the repression of p21Cip1 by a similar SP-1 involving mode of action [67]. The finding that HDAC-4 interacts with SP-1 at the p21Cip1 promoter is particularly interesting as this HDAC is thought to be enzymatically inactive. Yet, upon silencing of HDAC-4, hyperacetylation of histones at the former promoter was detected, followed by the induction of p21Cip1. This phenomenon could be ascribed to the fact that class IIa HDACs often ‘borrow’ deacetylase activity of class I members [18, 19]. Indeed, this assumption recently was proven by Wilson et al.[68] showing that HDAC-4 could repress the p21Cip1 expression by cooperation with the HDAC-3-NCoR/SMRT corepressor complex [68]. Dormant HDAC-4 may act as a sort of assembly platform connecting transcription factors (e.g. SP-1 and SP-3) to enzymatically active HDAC isoenzymes.
Proliferating cell nuclear antigen
An additional example of the involvement of HDAC enzymes in cell cycle control is the regulation of DNA replication, the latter being an intrinsic element of every cell division. It requires a plethora of factors, including PCNA (also known as processivity factor for DNA polymerase γ). Milutinovic and colleagues found that PCNA physically interacts with HDAC-1. Co-localization of HDAC-1 with PCNA, the resident protein of the DNA replication fork, suggests that this enzyme might participate in the reproduction of the histone acetylation pattern of the parental cells upon DNA replication. Because high fidelity of this process is crucial for maintaining a proper chromatin structure, HDAC-1 could be perceived as a guardian of the chromosomal stability [69]. Alternatively, Naryzhny and Lee proposed a model in which HDAC-1 and p300 acetyltransferase cooperate in the PCNA function/location switch [70]. They identified three PCNA isoforms based on their acetylation status: (i) highly acetylated PCNA was detected in nuclear extract, chromatin and nuclear matrix fractions; (ii) moderately acetylated PCNA protein was found in all cellular fractions tested, including cytoplasm and (iii) deacetylated-PCNA mainly was present in the nuclear fraction extract. Interestingly, the ratio of PCNA subspecies seemed to be cell cycle-regulated: whereas cells arrested in G0-phase exhibited low levels of highly acetylated and deacetylated PCNA isoforms, the quantity of the latter increased upon progression towards the S-phase of cell cycle. Because deacetylated PCNA exhibited a lower binding affinity to β- and γ-DNA polymerases, it probably could be involved in the termination of DNA replication whereas highly and moderately acetylated PCNA varieties might regulate earlier stages of the process [70].
Inhibition of histone deacetylases in liver cells
Hyperproliferation, uncontrolled inflammation and a distorted balance between cell survival and apoptosis could underlie a multitude of pathologies. Consequently, HDIs are perceived as promising drug candidates in the treatment of a wide range of diseases (reviewed by [71]), including fibroproliferative disorders, such as liver fibrosis, systemic sclerosis, pulmonary fibrosis and renal fibrosis. HDI-based treatment could also involve diseases of dysregulated immunity, i.e. autoimmune disorders (for review see [38, 72–74]). This is, however, not the end of the list. As demonstrated by recent research, HDIs may also be valuable tools in the prevention of neuronal degeneration as occurring in, e.g. Huntington’s disease [75–77]. It has also been suggested that HDIs could be helpful in combating infections with protozoan parasites [78] or with viruses, including HIV [79–81].
Physiological condition: effects of histone deacetylase inhibitors on primary hepatocytes
The majority of somatic cells in the mature organism stops dividing and enters a state of quiescence, i.e. G0-phase of the cell cycle. Some of these cells will irreversibly lose their ability to proliferate and finally progress into a state of senescence, whereas others retain their capacity to divide upon stimulation [82]. Hepatocytes which represent the predominant cell type in the liver (∼80% of the organ mass) belong to the latter group of cells that are semi-permanently withdrawn from the cell cycle [83]. These parenchymal cells perform multiple metabolic and secretory functions including protein synthesis, storage and transformation of carbohydrates, synthesis of cholesterol, bile salts and phospholipids, metabolism of endogenous substances and, most importantly, biotransformation of xenobiotics [84]. In fact, the majority of drugs undergoes biotransformation in the liver. In particular oral drugs are absorbed by the gut and transported via portal circulation directly to the liver.
Due to the high proliferative capacity of the hepatocytes, the liver is among the few internal organs that are capable of natural regeneration of lost tissue. This capability is imperative for its survival. In the case of acute liver injury, hepatocyte proliferation is aimed at replacing necrotic and apoptotic cells [85]. Basically, all types of cells in the liver, namely hepatic stellate cells (HSCs), Kupffer cells, Pit cells, bile duct epithelial cells and fenestrated endothelial cells, contribute to the regeneration of liver parenchyma, not only by their own proliferation, but also by stimulation of hepatocyte proliferation [86].
The in vivo regenerative response in the liver is manifested by a semi-synchronous cell-cycle re-entry of surviving hepatocytes, accompanied by an induction of a number of differentiation-promoting pathways [87, 88]. These allow preservation of metabolic homeostasis during the regeneration of the organ. In vitro, the proliferation and differentiation programmes are inversely related. Thus, in sharp contrast to the in vivo situation, cultured primary hepatocytes are unable to redifferentiate upon cell division and lose their liver-specific functions [89]. Therefore, currently available in vitro models based on primary hepatocytes fail to effectively reproduce the in vivo status of the cells for longer periods, restricting their applicability in, for instance, long-term pharmaco-toxicological tests of new chemical entities and implying the use of animal-based tests.
Our previous research clearly shows that HDI therapy may be beneficial for cultured primary rat hepatocytes. Exposure of hepatocytes to HDIs allows overcoming the isolation procedure-induced cell cycle re-entry of the cells [90]. As a consequence, mitogen-stimulated primary hepatocytes cease to proliferate in the presence of TSA. The specific timing of cell cycle arrest is dictated by the onset of exposure to the HDI involved [90]. We found that hepatocytes exposed to TSA from cell seeding onwards respond with an early S-phase cell cycle arrest, as demonstrated by block of DNA replication and lack of the S/G2/M-phase marker cdk1 (Fig. 3). When HDI exposure is initiated already during the isolation process of the cells, namely when hepatocytes are still in the state of considerable quiescence, neither the proto-oncogene c-jun, nor cyclin D1 could be detected, pointing towards an early G1-phase cell cycle arrest [90]. Interestingly, ω-carboxypenthyl p-dimethylaminobenzamide hydroxamate (4-Me2N-BAVAH), a metabolically more stable structural TSA analogue, is a more potent inhibitor of hepatocyte proliferation. Indeed, we found that 4-Me2N-BAVAH could already promote G1-cell cycle arrest when exposed from the time of plating onwards. Unexpectedly, p21Cip1, controlling the progression through the G1-phase and as such a potential mediator of the observed cell cycle arrest, was not induced. Instead, diminished activation of transcription factor NF-κβ seemed to underlie the transcriptional repression of the cyclin D1, although, the exact mechanism remains to be elucidated [91]. HDACs regulate NF-κβ signalling pathways at multiple levels. For instance, both HDAC-1 and HDAC-3 have been implicated in the transcriptional silencing of NF-κβ-controlled genes [92–94]. In addition, HDAC-3 directly deacetylates the transcription factor promoting its export from the nucleus and sequestration by its inhibitor, Iκβ[95]. Inhibition of deacetylase activity would augment the NF-κβ function by relieving HDAC-mediated repression. Nevertheless, a study in mouse epithelial JB6 cells revealed that HDIs down regulated cyclin D1 expression by diminishing NF-κβ-DNA binding [96]. Another study could demonstrate that TSA prevented the phosphorylation of the inhibitor Iκβ and its subsequent degradation, resulting in a decreased NF-κβ activation [97]. Whether similar mechanisms are responsible for the inhibition of NF-κβ activation in primary hepatocytes upon exposure to HDIs is still to be determined.
Figure 3.
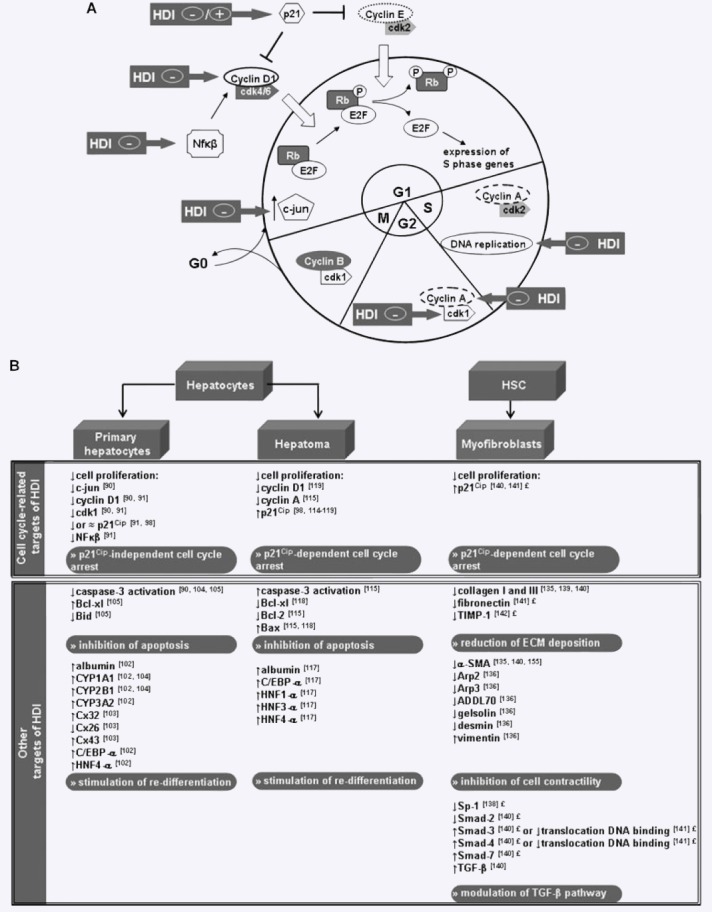
(A) Schematic representation of the cell cycle and cell cycle-related targets of HDIs described in liver cells. Positive or negative effect of HDIs on the expression/activity of cell cycle-specific proteins or events is indicated by a ‘+’ or ‘−’ sign respectively. (B) Summary of biological effects of HDIs in cultures of primary hepatocytes, hepatoma cell lines and myofibroblast-like cells. Symbols; ↓- down-regulation, ↑- up-regulation, ≈ no change of expression/activity, ‘£’-findings described in myofibroblasts of non-hepatic origin. Abbreviations not appearing in the text of the current review are: B-cell CLL/lymphoma 2 (Bcl-2), BH3-interacting domain death agonist (Bid), Bcl-2-associated X protein (Bax), cytochrome P450 (CYP), connexin (Cx) and hepatocyte nuclear factor (HNF).
Our finding, namely a p21Cip1-independent cell cycle arrest in response to exposure of primary hepatocytes to HDIs, was confirmed by another group [98]. Indeed, normal human skin fibroblasts and primary neuronal cells did not demonstrate p21Cip1 up-regulation upon HDI exposure. The latter findings may suggest that p21Cip1-independent cell cycle arrest is a common feature of HDAC inhibition in healthy, (primary) cells [23, 99]. Nevertheless, there is a subtle indication that HDACs control p21Cip1 expression in hepatic tissue. Indeed, forced expression of human HDAC-1 diminishes the expression of p21Cip1 mRNA in mouse liver [64, 65]. Unfortunately, the authors did not clarify whether this phenomenon was of a primary or secondary nature. Accordingly, the following question was not answered; was an increased HDAC-1 activity at the p21Cip1 gene promoter responsible for observed decrease of p21Cip1 mRNA or rather the diminished production of its upstream regulator, i.e. p53? Furthermore, the expression of p21Cip1 protein in response to HDAC-1 overexpression was not investigated [64, 65].
Recently, new light was shed on the role of HDAC-1 in the control of liver proliferation by Wang et al.[100]. They showed that in the quiescent liver of aged mice HDAC-1 protein was elevated and interacted with the negative regulator of liver proliferation, namely CCAAT-enhancer-binding protein (C/EBP)-α. The latter guided the HDAC-1 enzyme to E2F-responsive gene promoters enabling deacetylation of local histones and subsequent transcriptional silencing of the corresponding genes. The expression of proliferation-specific transcription factors, i.e. Forkhead box M1 B (FoxM1B) and c-myc, was inhibited in the former manner reflecting a diminished ability of the liver to regenerate during the aging process. Furthermore, the observed increase of the HDAC-1 protein was cyclin D3-dependent and resulted from an intensified translation of HDAC-1 mRNA [100]. Surprisingly, HDAC-1 was also elevated in the liver of young mice undergoing partial hepatectomy (PH) [64, 65, 101]. Nonetheless, as opposed to the previous study, a related transcription factor, namely C/EBP-β acted as a primary HDAC-1 interaction partner instead of C/EBP-α. Following PH, the HDAC-1-C/EBP-β complex repressed the transcription of the former C/EBP isoform, and by doing so abolished the C/EBP-α-mediated block of liver proliferation [101]. A substantial role of HDAC-1 in this process was underlined by the fact that depletion of the enzyme by siRNA technology prevented cyclin D1 and PCNA induction upon PH, whereas C/EBP-α protein was expressed at high levels [101].
Our research group has shown that apart from cell proliferation, HDACs also modulate other aspects of hepatocyte physiology. As such, exposure of cultured adult rat hepatocytes to TSA improved both their xenobiotic biotransformation capacities and albumin secretion, and their gap junctional intercellular communication [102–104]. Additionally, an increased survival rate of hepatocytes was seen in conventional monolayer cultures by delaying activation of programmed cell death pathways [104, 105].
Pathophysiological condition
Liver malignancy-hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is one of the most frequent and one of the five deadliest malignancies on a global scale [106]. Viral infection with hepatitis B and/or C, as well as chronic exposure to toxic compounds, lead to uncontrolled inflammation with consequent expansion of liver fibrosis/cirrhosis, creating favourable conditions for HCC development [107]. In current clinical practise, treatment options are limited to surgical resection and liver transplantation. Due to the scarcity of healthy liver transplants, only a limited number of patients can be treated. Fifty to 80% of the patients undergoing surgery will suffer from recurrence within 5 years from resection [108]. There is, however, a growing understanding of the molecular events that participate in hepatocarcinogenesis. They may include telomere shortening, evoking chromosomal instability, loss of cell cycle checkpoints (p53, Rb, IGF2R), activation of proliferation and survival promoting pathways (PI3K, ERK, Wnt/β-catenin, NF-κβ) as well as inhibition of apoptosis (imbalance in the pro- and anti-apoptotic members of the Bcl-2 family) [109–112]. Recently, a direct link between HDAC activity and HCC incidence was discovered. It was indeed found, that chronic hepatitis B virus patients, being carrier of the specific HDAC-10 sequence variant, HDAC10–589C>T, run a higher risk of HCC development. Additionally, this specific allele demonstrates higher transcriptional activity probably suggesting that increased HDAC-10 expression could be one of the contributors to the carcinogenesis process [113].
Although HDIs constitute a large group of structurally and chemically diverse compounds, their effects on tumour cells are surprisingly uniform. In contrast to primary non-transformed cells, HDIs-mediated cell cycle arrest in cancer cell lines bears the emblem of p21Cip1 induction. As early as in 1998, the elevated expression of p21Cip1 upon exposure of human hepatoma cell lines to the weak HDI NaB was detected [114]. In the following years, several other groups reported p21Cip1 induction in human and rodent hepatoma cells using distinct HDIs such as, TSA, valproate, ITF2357 (hydroxamic acid derivative), 4-phenylbutyrate and a bioconjugate of butyrate with hyaluronan [98, 115–119]. The reported p21Cip1 up-regulation was associated with cyclin D1 or cyclin A gene repression, indicating a G0/G1- or G2/M-phase cell cycle arrest, respectively (Fig. 3) [115, 119]. HDIs activate the expression of p21Cip1 by enhancing histone acetylation and promoting co-activator recruitment in the region of the p21Cip1 gene promoter. A number of studies indicate a key role of the SP-1 transcription factor in p21Cip1 activation, autonomous of the p53 pathway [61, 120]. It was suggested that the exposure to HDIs discharges the HDAC-1 enzyme from SP-1 binding sites at the p21Cip1 gene promoter. As a consequence, repression is relieved and transcription is initiated [121]. Dispensability of p53 in this process was illustrated using the PLC/PRF/5 HCC cell line expressing mutant p53, and the Hep3B HCC cell line which lacks production p53. These cell lines were not impaired in their p21Cip1-induction ability upon HDI exposure [114, 116, 119, 122]. Nevertheless, the p53 involvement in HDI-mediated p21Cip1 gene transcription cannot be completely ruled out. As such a site-specific p53 acetylation as a consequence of HDAC inhibition followed by p21Cip1 induction was described [123]. A general conclusion is that the p21Cip1 induction pathway, in response to HDI treatment, is dependent on both the cell type and the functionality of p53.
Liver fibrosis
Liver fibrosis is a disease arising as a result of dysregulated repair processes in reaction to liver insults [124]. It is believed that HSCs are major perpetrators of the pathological changes. HSCs belong to the non-parenchymal subpopulation in the liver. In normal, healthy liver, these cells occupy the space of Disse, embracing the hepatic sinusoids with their long protrusions. In their quiescent state they are known to store retinoids within the numerous cytoplasmic lipid droplets [125]. Upon liver injury, HSCs undergo a complete metamorphosis during a process commonly referred to as ‘transdifferentiation’. HSCs become ‘activated’ and adopt a distinct myofibroblastic phenotype [126]. The latter is characterized by a higher proliferative capacity, fibrogenic and pro-inflammatory properties and enhanced contractility. The cell morphology is also transformed and is accompanied by a loss of lipid droplets [127]. HSC-derived myofibroblasts constitute a prominent cell type in the pathogenesis of liver cirrhosis. It must be stressed that also other cells, including portal fibroblasts, biliary epithelial cells and bone marrow-derived fibrocytes, may give rise to myofibroblast-like cells [125, 128, 129]. A common feature of the whole myofibroblast population, independent of cell origin, is the expression of the myogenic marker α-smooth muscle actin (α-SMA) (Fig. 4) [86].
Figure 4.

Schematic representation of HSC activation. Liver damage (caused by viral infection, metabolic disease, drugs, toxins, cholestasis) leads to the transdifferentiation of quiescent HSCs into activated myofibroblast-like cells under the influence of different mediators secreted by resident liver cells as well as non-liver resident cells. The most striking phenotypical changes are the loss of lipid droplets, fibrogenesis and the increase in contractile capacity. During resolution of liver damage, it is not yet clear what the fate of the activated stellate cells is: re-differentiation into quiescent HSCs or apoptosis of the activated myofibroblast-like cells. Abbreviations not appearing in the text of the current review are: platelet derived growth factor (PDGF), endothelin 1 (ET-1), reactive oxygen species (ROS) and monocyte chemotactic protein 1 (MCP-1). The figure has been adapted from [127].
Damage to liver parenchyma induces a regenerative response that is characterized by three phases, namely initiation, perpetuation and resolution [127]. (i) During the initiation phase, injured hepatocytes, HSCs, bile duct cells and activated immune system cells secrete a plethora of pro-inflammatory mediators, rendering the surviving liver cells responsive towards cytokines and growth factors. (ii) In the perpetuation phase, the activated cells synthesize and excrete growth-promoting stimuli in a paracrine and autocrine manner. These enhance the signalling spiral aimed at promoting hepatocyte proliferation and extracellular matrix (ECM) deposition by HSCs/myofibroblasts to replace the damaged tissue. (iii) It is speculated that upon restoration of proper liver architecture, activated HSCs may reverse to a quiescent state or undergo spontaneous apoptosis, accompanied by degradation of the ECM surplus. These latter events characterize the final stage of the regeneration process, i.e. the resolution [127]. In contrast, repetitive or persistent liver damage may eradicate the resolution phase. Liver cells, predominantly HSCs, become entrapped in a vicious inflammatory circle of cell activation, proliferation and ECM overproduction, being the hallmark of liver fibrosis. Paradoxically, the regenerative capabilities of hepatocytes become compromised in the former setting. Funakoshi et al.[130] have shown that cyclin D1 levels decline in hepatocytes whereas the expression of cdk inhibitors such as p15 and p16, increase in the progression of liver fibrosis [130]. Simultaneously, the chief profibrogenic cytokine transforming growth factor (TGF)-β, produced by activated HSCs, is known to have detrimental effects on hepatocyte survival [131]. Impaired proliferation combined with diminished viability of the hepatocytes result in substitution of functional liver parenchyma by a collagen-rich matrix. Threads of newly synthesized collagen force themselves between groups of liver cells, creating nodules of surviving hepatocytes which attempt to regenerate. Over time, progressive parenchymal remodelling leads to deformation of the lobular organization of the liver which actually defines liver cirrhosis [132]. Abnormal cirrhotic architecture severely diminishes blood flow in the liver, resulting in portal hypertension and hepatocellular insufficiency. In these circumstances, hepatocytes are not able to perform their usual biochemical tasks, bringing about grave consequences for the functioning of the whole organism. Moreover, there exists a well-documented positive correlation between the occurrence of liver cirrhosis and subsequent HCC development [133].
Histone deacetylase inhibitors as modulators of the hepatic stellate cell myofibroblastic phenotype
Activated HSCs are master regulators of ECM production and turn-over, and thus appear attractive targets for antifibrotic therapy. Exposure of primary rat HSCs to TSA or NaB was shown to hinder their transdifferentiation, which otherwise occurs spontaneously when cultured on a plastic substrate. Most notably, HDI treatment had an impact on several key features of the myofibroblastic phenotype, namely on cell morphology, proliferation, ECM production and cell contractility. These changes were accompanied by histone H4 hyperacetylation. The effects of HDAC inhibition were less pronounced when full HSCs activation already had occurred [134, 135]. It is also worth mentioning that the TSA concentration that was effective in blocking HSCs transformation was significantly lower than that inhibiting hepatocyte proliferation (100 nM versus 1 μM). Consequently, at this low concentration, HDIs are effective in blocking HSCs transdifferentiation; however, they do not compromise the ability of hepatocytes to regenerate. Further analysis of TSA-exposed HSCs revealed several HDI sensitive genes implicated in actin cytoskeleton formation, such as nucleating proteins Arp2 and Arp3, capping proteins ADDL70 and gelsolin, in addition to type III intermediate filaments (Fig. 3). TSA-induced alterations of actin dynamics preserved the actin cytoskeleton typical for quiescent HSCs. As a consequence, the migration ability of HSCs was severely affected [136].
The authenticity of the above described findings was confirmed in other in vitro systems. The disproportionate deposition of ECM leading to fibrotic changes is not a unique feature of the fibrotic liver, but rather a common component of a group of disorders which may assault eye, kidney, lung and heart. The main perpetrators in these cases are fibroblasts that undergo a transdifferentiation process. The latter is analogous to that of HSCs in the damaged liver and also gives rise to myofibroblast-like cells [137]. The myofibroblastic phenotype may be mimicked in vitro by stimulating cultured fibroblasts with TGF-β. Several studies describe that HDIs are able to inhibit TGF-β-induced transformation of human, murine and rat skin fibroblasts in culture [138–140]. A common feature of HDAC inhibition is the reduced production of collagen I. Other effects of exposure of TGF-β-stimulated fibroblasts or fibroblasts isolated from systemic sclerosis patients to HDIs included diminished expression of fibronectin, α-SMA and induction of the cdk inhibitor p21Cip1[140, 141]. Additionally, Young and colleagues reported that TSA had a negative impact on the TGF-β mediated expression of TIMP-1, tissue inhibitor of matrix metalloproteinases (MMPs). Interestingly, valproic acid did not block the TGF-β-induced expression of TIMP, demonstrating HDAC specificity in this process [142]. Under normal conditions, matrix metalloproteinases orchestrate ECM turnover. They break down the key components of ECM, maintaining a balanced ECM production/degradation ratio. However, in pathological situations, although abundant in the extracellular environment, MMPs remain latent due to elevated levels of TIMPs [132]. An up-regulated expression of TIMP-1 and TIMP-2 is believed to be an intrinsic element of progressive fibrosis, further augmenting the effect of ECM overproduction [143]. The ability of TSA to inhibit TIMP-1 expression has more than one implication. Apart from the direct effect on of the ECM degradation rate, TIMP-1 also acts as an anti-apoptotic factor for activated HSCs [144]. Lowering TIMP levels by TSA may thus accelerate fibrosis regression by promoting ECM proteolysis and elimination of profibrogenic cells. However, a cell-type specific response should be anticipated and experiments on HSCs should be carried out to demonstrate the importance of these original findings.
The role of histone deacetylases in the regulation of pro-fibrogenic and pro-inflammatory cascades
The mechanism by which HDIs modulates the transdifferentiation process of myofibroblast-like cells, including activated HSCs, remains a subject of speculation, although a few recent studies may provide a hint. An evident candidate appears to be the TGF-β signalling pathway, because TGF-β controlled gene promoters include collagen I and III, α-SMA, TIMP-1 and MMP-1 [129, 145]. The canonical TGF-β signal transduction pathway is initiated upon molecular bridging of specific receptors by TGF-β ligand and subsequent recruitment of cytosolic mediators, the Smads proteins. The first to associate with the activated receptors are Smad-2 and Smad-3, i.e. receptor-activated Smads (R-Smads). They are further joined by another family member, Smad-4. This multi-subunit complex translocates to the nucleus to exert its transcription-related effects. Two additional, though inhibitory Smads (I-Smads), Smad-6 and Smad-7, are responsible for counteracting this process [129, 145]. This simplistic illustration of the TGF-β pathway becomes much more complex if one considers the diversity of proteins that associate and interact with Smads and the non-Smad pathways induced by TGF-β. Although the Smads possess intrinsic DNA-binding ability, they must cooperate with other proteins to increase the binding affinity and selectivity towards their target gene promoters [146]. Furthermore, the activated R-Smad/Smad-4 complex may select interaction partners with either gene transcription-inducing or repressive properties. These mulitprotein factors contain the chromatin modulating enzymes, KATs or HDACs, respectively, and compete for binding to Smad proteins. The transcriptional outcome will be determined by the ratio of rivalling transcriptional coactivators and corepressors [147, 148].
Thus far, several Smad-linked corepressors with deacetylase activity have been identified, but only few studies have demonstrated actual recruitment of these HDAC complexes by Smads to TGF-β-responsive genes [149]. These include TGIF, Ski and SnoN, Evi-1 and PIASy [150–152]. Only few of them (i.e. TGIF and PIASy) can directly interact with HDACs. Nonetheless, the most common mode of action involves the recruitment of HDAC-containing complexes, such as mSin3 (TGIF, Ski and SnoN), CtBP (TIGF and Evi-1) or NcoR (Ski and SnoN) [147, 150–152]. The chromatin-condensing properties of HDACs will act as a silencing tool of Smads for TGF-β-controlled genes. Interestingly, HDACs not only regulate the transcriptional activity of Smad-responsive genes, but also of Smads themselves. Although there is no actual evidence on reversible acetylation of R-Smads or Smad-4, the former mechanism seems to regulate the stability of I-Smad, Smad-7 [28]. In this regard, the acetylation of specific lysine residues, which alternatively may be used as ubiquitination sites, protects Smad-7 from proteolytic degradation. Conversely, HDAC-mediated deacetylation of these lysines decreases the half-life of the protein [153]. These observations support the notion that HDACs are involved in the regulation of TGF-β signalling at multiple levels and hence, might occupy a central position in TGF-β driven myofibroblastic differentiation.
At present no studies have been carried out to demonstrate an effect of HDIs on the TGF-β-mediated events in primary HSCs in vivo or in vitro. Several studies, however addressed this issue by using skin fibroblasts. Rombouts et al. demonstrated in rat skin fibroblasts that, apart from its pronounced effects on collagen I and α-SMA mRNA levels, TSA also alters the transcription of TGF-β as well as of its downstream mediators in activated rat skin fibroblasts [140]. The TSA-induced TGF-β expression was paralleled by the up-regulation of Smads-3, -4 and -7 whereas the Smad-2 mRNA level decreased. It is difficult to interpret those observations because HDACs regulate the TGF-β pathway at multiple levels. Increased TGF-β production together with up-regulation of Smad-3 and -4 indicate that TSA treatment could amplify the profibrogenic cascade. On the other hand, down-regulation of Smad-2 could limit the formation rate of transcriptionally active Smad complexes. Similarly, elevated Smad-7 could constrain signal transduction within the pathway. Nevertheless, judging from the reduced expression of fibrosis-related proteins (collagen I and III, α-SMA), one could expect an antifibrogenic net outcome of HDI treatment. In a study by Rishikof et al.[139], the HDI phenylbutyrate was used to inhibit TGF-β mediated effects in human lung fibroblasts. Although type I collagen expression was decreased, the responsible mechanism in these cells seemed to be Smad independent. A mechanism including a phenylbutyrate-dependent stimulation of cAMP production and histone acetylation was proposed [139]. A similar observation was made by Ghosh et al.[138] in human foreskin fibroblasts where TSA treatment did not directly modulate Smad activity. There, exposure of TGF-β activated fibroblasts to TSA resulted in diminished SP-1 levels (Fig. 3) [138]. As SP-1-Smad interaction is essential for the activation of the transcription from the collagen I gene promoter in response to TGF-β signals, the SP-1 levels might suggest an important response of the cells to TSA exposure [154]. Glenisson and colleagues took these analyses one step further. They explored the contribution of individual HDAC isotypes to TGF-β-induced myofibroblastic differentiation in primary human skin fibroblasts [155]. By comparing the effect of TSA treatment with the result of silencing of a single HDAC in TGF-β treated cells, they found that knocking-down expression of HDAC-4, and in lesser extent HDAC-6 and -8, mimics the effects of TSA-induced HDAC inhibition with respect to the down-regulation of α-SMA. Interestingly, HDAC-2 and HDAC-7 knock-down potentiated the production of α-SMA gene transcripts. This study indicates that different members of the HDAC family govern the TGF-β-induced α-SMA transcription in human skin fibroblasts. It also suggests that during fibrogenesis this process might be HDAC-4 dependent. Therefore, HDAC-4-specific inhibitors could perhaps ameliorate liver condition by inhibiting the transdifferentiation process. Thus far, neither in vivo studies nor studies in primary HSCs were carried out.
Conclusions
The observations discussed in this paper accentuate the multiple and diverse roles of the HDAC family in the control of cellular homeostasis. Consequently, these enzymes may present attractive pharmacological targets to modulate disease processes. However, finding the equilibrium between blocking ‘pernicious’ and promoting ‘beneficial’ HDAC(s) seems to be a prerequisite. It is clear that the function of HDACs varies with cell type, signalling pathway and target gene promoter. Additionally, HDACs may regulate the same processes by a dual mechanism, namely by operating at the level of chromatin and/or at the protein level (Fig. 2). Furthermore, HDACs are debarred of DNA-binding ability and require multiple associates in order to localize target genes. It is therefore of key importance to identify the cell- and process-specific functions of each HDAC isoenzyme and their potential interaction partners. A total shutdown of the deacetylation machinery obtained by a general HDI, such as TSA, and subsequent apoptosis is desired when cancer cells are targeted. Adverse effects by inhibition of all HDACs in healthy primary cells, particularly during long-term treatment, might occur. For instance, in the TGF-β signalling pathway, driving the fibrogenic cascade in HSCs, HDACs play opposite roles. Inhibition of HDAC activity by TSA prevents the manifestation of myofibroblastic features (i.e. collagen production, α-SMA expression) and induces the inhibitors of Smad signalling (TGIFs and Smad-7). The same inhibitors, TGIF as well as Ski and SnoN, require HDAC activity to repress the transcription on target gene promoters involved in non-benign processes in HSCs but also in other liver cells.
To the best of our knowledge, no in vivo studies with HDIs in animal models of liver fibrosis have been published. It would, however, be tempting to speculate, based on the in vitro studies with HSCs and fibroblasts that HDIs could be of therapeutic use as antifibrotics. The current HDIs that entered pre-clinical and/or clinical trials for their use in the treatment of cancer, i.e. suberoylanilide hydroxamic acid or valproic acid in breast cancer, have according to the readily available scientific literature not been tested for their in vivo antifibrogenic capacity.
In summary, HDIs have an enormous potential as pharmacological agents for the treatment of various diseases and as modulators of the phenotype of primary cells in vitro. To fully benefit from the advantageous effects of these agents, however, we need to fathom the enzymes they are targeting. With the advent of new technologies, such as RNA interference, elucidation of their specific roles can be awaited in the near future.
References
- 1.Sandman K, Reeve JN. Archaeal histones and the origin of the histone fold. Curr Opin Microbiol. 2006;9:520–5. doi: 10.1016/j.mib.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Luger K. Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev. 2003;13:127–35. doi: 10.1016/s0959-437x(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 4.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224–9. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Ruijter AJ, van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–49. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene. 2007;26:5528–40. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- 7.Legube G, Trouche D. Regulating histone acetyltransferases and deacetylases. EMBO Rep. 2003;4:944–7. doi: 10.1038/sj.embor.embor941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–11. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 9.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 11.Hildmann C, Riester D, Schwienhorst A. Histone deacetylases–an important class of cellular regulators with a variety of functions. Appl Microbiol Biotechnol. 2007;75:487–97. doi: 10.1007/s00253-007-0911-2. [DOI] [PubMed] [Google Scholar]
- 12.Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- 13.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–8. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 14.Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26:5433–8. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- 15.Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem Biol. 2002;9:3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 16.Gallinari P, Di Marco S, Jones P, et al. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 17.Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26:5439–49. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- 18.Fischle W, Dequiedt F, Hendzel MJ, et al. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 19.Fischle W, Kiermer V, Dequiedt F, et al. The emerging role of class II histone deacetylases. Biochem Cell Biol. 2001;79:337–48. [PubMed] [Google Scholar]
- 20.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code. Curr Opin Genet Dev. 2005;15:163–76. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 21.de la Cruz CC, Kirmizis A, Simon MD, et al. The polycomb group protein SUZ12 regulates histone H3 lysine 9 methylation and HP1 alpha distribution. Chromosome Res. 2007;15:299–314. doi: 10.1007/s10577-007-1126-1. [DOI] [PubMed] [Google Scholar]
- 22.Santos-Rosa H, Caldas C. Chromatin modifier enzymes, the histone code and cancer. Eur J Cancer. 2005;41:2381–402. doi: 10.1016/j.ejca.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Glaser KB, Staver MJ, Waring JF, et al. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther. 2003;2:151–63. [PubMed] [Google Scholar]
- 24.Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci USA. 2004;101:540–5. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–53. [PMC free article] [PubMed] [Google Scholar]
- 26.Nusinzon I, Horvath CM. Histone deacetylases as transcriptional activators. Sci STKE. 2005;2005 doi: 10.1126/stke.2962005re11. ?Role reversal in inducible gene regulation.: re11. [DOI] [PubMed] [Google Scholar]
- 27.Smith CL. A shifting paradigm: histone deacetylases and transcriptional activation. Bioessays. 2008;30:15–24. doi: 10.1002/bies.20687. [DOI] [PubMed] [Google Scholar]
- 28.Glozak MA, Sengupta N, Zhang X, et al. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 29.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 30.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 31.Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–6. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 32.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 33.Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 34.Kramer OH, Gottlicher M, Heinzel T. Histone deacetylase as a therapeutic target. Trends Endocrinol Metab. 2001;12:294–300. doi: 10.1016/s1043-2760(01)00438-6. [DOI] [PubMed] [Google Scholar]
- 35.Riggs MG, Whittaker RG, Neumann JR, et al. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268:462–4. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida M, Kijima M, Akita M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–9. [PubMed] [Google Scholar]
- 37.Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol. 2007;74:659–71. doi: 10.1016/j.bcp.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 38.Huang L. Targeting histone deacetylases for the treatment of cancer and inflammatory diseases. J Cell Physiol. 2006;209:611–6. doi: 10.1002/jcp.20781. [DOI] [PubMed] [Google Scholar]
- 39.Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol. 2008;615:261–98. doi: 10.1007/978-1-4020-6554-5_13. [DOI] [PubMed] [Google Scholar]
- 40.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 41.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 42.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 43.Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–73. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- 44.Gray SG, Iglesias AH, Lizcano F, et al. Functional characterization of JMJD2A, a histone deacetylase- and retinoblastoma-binding protein. J Biol Chem. 2005;280:28507–18. doi: 10.1074/jbc.M413687200. [DOI] [PubMed] [Google Scholar]
- 45.Lai A, Kennedy BK, Barbie DA, et al. RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol Cell Biol. 2001;21:2918–32. doi: 10.1128/MCB.21.8.2918-2932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shu KX, Li B, Wu LX. The p53 network: p53 and its downstream genes. Colloids Surf B Biointerfaces. 2007;55:10–8. doi: 10.1016/j.colsurfb.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Juven-Gershon T, Oren M. Mdm2: the ups and downs. Mol Med. 1999;5:71–83. [PMC free article] [PubMed] [Google Scholar]
- 48.Liu G, Chen X. Regulation of the p53 transcriptional activity. J Cell Biochem. 2006;97:448–58. doi: 10.1002/jcb.20700. [DOI] [PubMed] [Google Scholar]
- 49.Tokino T, Nakamura Y. The role of p53-target genes in human cancer. Crit Rev Oncol Hematol. 2000;33:1–6. doi: 10.1016/s1040-8428(99)00051-7. [DOI] [PubMed] [Google Scholar]
- 50.Ito A, Kawaguchi Y, Lai CH, et al. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–45. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo J, Su F, Chen D, et al. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–81. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 52.Krummel KA, Lee CJ, Toledo F, et al. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci USA. 2005;102:10188–93. doi: 10.1073/pnas.0503068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng L, Lin T, Uranishi H, et al. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25:5389–95. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knights CD, Catania J, Di Giovanni S, et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–44. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harms KL, Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007;67:3145–52. doi: 10.1158/0008-5472.CAN-06-4397. [DOI] [PubMed] [Google Scholar]
- 56.Murphy M, Ahn J, Walker KK, et al. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999;13:2490–501. doi: 10.1101/gad.13.19.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho JS, Ma W, Mao DY, et al. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol. 2005;25:7423–31. doi: 10.1128/MCB.25.17.7423-7431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Imbriano C, Gurtner A, Cocchiarella F, et al. Direct p53 transcriptional repression: in vivo analysis of CCAAT-containing G2/M promoters. Mol Cell Biol. 2005;25:3737–51. doi: 10.1128/MCB.25.9.3737-3751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.el-Deiry WS. p21/p53, cellular growth control and genomic integrity. Curr Top Microbiol Immunol. 1998;227:121–37. doi: 10.1007/978-3-642-71941-7_6. [DOI] [PubMed] [Google Scholar]
- 60.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 61.Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367–74. doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 62.Lagger G, O’Carroll D, Rembold M, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–81. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma P, Schultz RM. Histone deacetylase 1 (HDAC1) regulates histone acetylation, development, and gene expression in preimplantation mouse embryos. Dev Biol. 2008;319:110–20. doi: 10.1016/j.ydbio.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang AG, Kim SU, Lee SH, et al. Histone deacetylase 1 contributes to cell cycle and apoptosis. Biol Pharm Bull. 2005;28:1966–70. doi: 10.1248/bpb.28.1966. [DOI] [PubMed] [Google Scholar]
- 65.Wang AG, Seo SB, Moon HB, et al. Hepatic steatosis in transgenic mice overexpressing human histone deacetylase 1. Biochem Biophys Res Commun. 2005;330:461–6. doi: 10.1016/j.bbrc.2005.02.179. [DOI] [PubMed] [Google Scholar]
- 66.Lagger G, Doetzlhofer A, Schuettengruber B, et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell Biol. 2003;23:2669–79. doi: 10.1128/MCB.23.8.2669-2679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang W, Tan D, Wang X, et al. Histone deacetylase 3 represses p15(INK4b) and p21(WAF1/cip1) transcription by interacting with Sp1. Biochem Biophys Res Commun. 2006;339:165–71. doi: 10.1016/j.bbrc.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 68.Wilson AJ, Byun DS, Nasser S, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19:4062–75. doi: 10.1091/mbc.E08-02-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Milutinovic S, Zhuang Q, Szyf M. Proliferating cell nuclear antigen associates with histone deacetylase activity, integrating DNA replication and chromatin modification. J Biol Chem. 2002;277:20974–8. doi: 10.1074/jbc.M202504200. [DOI] [PubMed] [Google Scholar]
- 70.Naryzhny SN, Lee H. The post-translational modifications of proliferating cell nuclear antigen: acetylation, not phosphorylation, plays an important role in the regulation of its function. J Biol Chem. 2004;279:20194–9. doi: 10.1074/jbc.M312850200. [DOI] [PubMed] [Google Scholar]
- 71.Elaut G, Rogiers V, Vanhaecke T. The pharmaceutical potential of histone deacetylase inhibitors. Curr Pharm Des. 2007;13:2584–620. doi: 10.2174/138161207781663064. [DOI] [PubMed] [Google Scholar]
- 72.Adcock IM, Ito K, Barnes PJ. Histone deacetylation: an important mechanism in inflammatory lung diseases. Copd. 2005;2:445–55. doi: 10.1080/15412550500346683. [DOI] [PubMed] [Google Scholar]
- 73.Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25:552–63. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- 74.Blanchard F, Chipoy C. Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases. Drug Discov Today. 2005;10:197–204. doi: 10.1016/S1359-6446(04)03309-4. [DOI] [PubMed] [Google Scholar]
- 75.Anne-Laurence B, Caroline R, Irina P, et al. Chromatin acetylation status in the manifestation of neurodegenerative diseases: HDAC inhibitors as therapeutic tools. Subcell Biochem. 2007;41:263–93. [PubMed] [Google Scholar]
- 76.Sadri-Vakili G, Cha JH. Histone deacetylase inhibitors: a novel therapeutic approach to Huntington’s disease (complex mechanism of neuronal death) Curr Alzheimer Res. 2006;3:403–8. doi: 10.2174/156720506778249407. [DOI] [PubMed] [Google Scholar]
- 77.Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–50. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andrews KT, Tran TN, Lucke AJ, et al. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob Agents Chemother. 2008;52:1454–61. doi: 10.1128/AAC.00757-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Demonte D, Quivy V, Colette Y, et al. Administration of HDAC inhibitors to reactivate HIV-1 expression in latent cellular reservoirs: implications for the development of therapeutic strategies. Biochem Pharmacol. 2004;68:1231–8. doi: 10.1016/j.bcp.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 80.Varier RA, Kundu TK. Chromatin modifications (acetylation/ deacetylation/ methylation) as new targets for HIV therapy. Curr Pharm Des. 2006;12:1975–93. doi: 10.2174/138161206777442092. [DOI] [PubMed] [Google Scholar]
- 81.Quivy V, De Walque S, Van Lint C. Chromatin-associated regulation of HIV-1 transcription: implications for the development of therapeutic strategies. Subcell Biochem. 2007;41:371–96. [PubMed] [Google Scholar]
- 82.Vermeulen K, Berneman ZN, Van Bockstaele DR. Cell cycle and apoptosis. Cell Prolif. 2003;36:165–75. doi: 10.1046/j.1365-2184.2003.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marcos R, Monteiro RA, Rocha E. Design-based stereological estimation of hepatocyte number, by combining the smooth optical fractionator and immunocytochemistry with anti-carcinoembryonic antigen polyclonal antibodies. Liver Int. 2006;26:116–24. doi: 10.1111/j.1478-3231.2005.01201.x. [DOI] [PubMed] [Google Scholar]
- 84.Elaut G, Henkens T, Papeleu P, et al. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr Drug Metab. 2006;7:629–60. doi: 10.2174/138920006778017759. [DOI] [PubMed] [Google Scholar]
- 85.Zimmermann A. Regulation of liver regeneration. Nephrol Dial Transplant. 2004;19:iv6–10. doi: 10.1093/ndt/gfh1034. [DOI] [PubMed] [Google Scholar]
- 86.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brenner DA. Signal transduction during liver regeneration. J Gastroenterol Hepatol. 1998;13:S93–5. [PubMed] [Google Scholar]
- 88.Taub R. Liver regeneration 4: transcriptional control of liver regeneration. FASEB J. 1996;10:413–27. [PubMed] [Google Scholar]
- 89.Glaise D, Ilyin GP, Loyer P, et al. Cell cycle gene regulation in reversibly differentiated new human hepatoma cell lines. Cell Growth Differ. 1998;9:165–76. [PubMed] [Google Scholar]
- 90.Papeleu P, Loyer P, Vanhaecke T, et al. Trichostatin A induces differential cell cycle arrests but does not induce apoptosis in primary cultures of mitogen-stimulated rat hepatocytes. J Hepatol. 2003;39:374–82. doi: 10.1016/s0168-8278(03)00288-5. [DOI] [PubMed] [Google Scholar]
- 91.Papeleu P, Wullaert A, Elaut G, et al. Inhibition of NF-kappaB activation by the histone deacetylase inhibitor 4-Me2N-BAVAH induces an early G1 cell cycle arrest in primary hepatocytes. Cell Prolif. 2007;40:640–55. doi: 10.1111/j.1365-2184.2007.00466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hoberg JE, Yeung F, Mayo MW. SMRT derepression by the IkappaB kinase alpha: a prerequisite to NF-kappaB transcription and survival. Mol Cell. 2004;16:245–55. doi: 10.1016/j.molcel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 93.Graham B, Gibson SB. The two faces of NFkappaB in cell survival responses. Cell Cycle. 2005;4:1342–5. doi: 10.4161/cc.4.10.2047. [DOI] [PubMed] [Google Scholar]
- 94.Shetty S, Graham BA, Brown JG, et al. Transcription factor NF-kappaB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol Cell Biol. 2005;25:5404–16. doi: 10.1128/MCB.25.13.5404-5416.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–30. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 96.Hu J, Colburn NH. Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res. 2005;3:100–9. doi: 10.1158/1541-7786.MCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 97.Larsen L, Tonnesen M, Ronn SG, et al. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia. 2007;50:779–89. doi: 10.1007/s00125-006-0562-3. [DOI] [PubMed] [Google Scholar]
- 98.Chiba T, Yokosuka O, Arai M, et al. Identification of genes up-regulated by histone deacetylase inhibition with cDNA microarray and exploration of epigenetic alterations on hepatoma cells. J Hepatol. 2004;41:436–45. doi: 10.1016/j.jhep.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 99.Boutillier AL, Trinh E, Loeffler JP. Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J Neurochem. 2003;84:814–28. doi: 10.1046/j.1471-4159.2003.01581.x. [DOI] [PubMed] [Google Scholar]
- 100.Wang GL, Salisbury E, Shi X, et al. HDAC1 cooperates with C/EBPalpha in the inhibition of liver proliferation in old mice. J Biol Chem. 2008;283:26169–78. doi: 10.1074/jbc.M803544200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang GL, Salisbury E, Shi X, et al. HDAC1 promotes liver proliferation in young mice via interactions with C/EBPbeta. J Biol Chem. 2008;283:26179–87. doi: 10.1074/jbc.M803545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Henkens T, Papeleu P, Elaut G, et al. Trichostatin A, a critical factor in maintaining the functional differentiation of primary cultured rat hepatocytes. Toxicol Appl Pharmacol. 2007;218:64–71. doi: 10.1016/j.taap.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 103.Vinken M, Henkens T, Vanhaecke T, et al. Trichostatin a enhances gap junctional intercellular communication in primary cultures of adult rat hepatocytes. Toxicol Sci. 2006;91:484–92. doi: 10.1093/toxsci/kfj152. [DOI] [PubMed] [Google Scholar]
- 104.Fraczek J, Deleu S, Lukaszuk A, et al. Screening of amide analogues of Trichostatin A in cultures of primary rat hepatocytes: search for potent and safe HDAC inhibitors. Invest New Drugs. 2008;27:338–46. doi: 10.1007/s10637-008-9180-x. [DOI] [PubMed] [Google Scholar]
- 105.Vanhaecke T, Henkens T, Kass GE, et al. Effect of the histone deacetylase inhibitor trichostatin A on spontaneous apoptosis in various types of adult rat hepatocyte cultures. Biochem Pharmacol. 2004;68:753–60. doi: 10.1016/j.bcp.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 106.Parkin DM, Bray F, Ferlay J, et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 107.Schafer DF, Sorrell MF. Hepatocellular carcinoma. Lancet. 1999;353:1253–7. doi: 10.1016/S0140-6736(98)09148-X. [DOI] [PubMed] [Google Scholar]
- 108.Lau WY, Lai EC. Hepatocellular carcinoma: current management and recent advances. Hepatobiliary Pancreat Dis Int. 2008;7:237–57. [PubMed] [Google Scholar]
- 109.Xu XR, Huang J, Xu ZG, et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc Natl Acad Sci USA. 2001;98:15089–94. doi: 10.1073/pnas.241522398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen X, Cheung ST, So S, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–39. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fabregat I, Roncero C, Fernandez M. Survival and apoptosis: a dysregulated balance in liver cancer. Liver Int. 2007;27:155–62. doi: 10.1111/j.1478-3231.2006.01409.x. [DOI] [PubMed] [Google Scholar]
- 112.Tannapfel A, Wittekind C. Genes involved in hepatocellular carcinoma: deregulation in cell cycling and apoptosis. Virchows Arch. 2002;440:345–52. doi: 10.1007/s00428-002-0617-x. [DOI] [PubMed] [Google Scholar]
- 113.Park BL, Kim YJ, Cheong HS, et al. HDAC10 promoter polymorphism associated with development of HCC among chronic HBV patients. Biochem Biophys Res Commun. 2007;363:776–81. doi: 10.1016/j.bbrc.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 114.Yamamoto H, Fujimoto J, Okamoto E, et al. Suppression of growth of hepatocellular carcinoma by sodium butyrate in vitro and in vivo. Int J Cancer. 1998;76:897–902. doi: 10.1002/(sici)1097-0215(19980610)76:6<897::aid-ijc21>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 115.Herold C, Ganslmayer M, Ocker M, et al. The histone-deacetylase inhibitor Trichostatin A blocks proliferation and triggers apoptotic programs in hepatoma cells. J Hepatol. 2002;36:233–40. doi: 10.1016/s0168-8278(01)00257-4. [DOI] [PubMed] [Google Scholar]
- 116.Svechnikova I, Gray SG, Kundrotiene J, et al. Apoptosis and tumor remission in liver tumor xenografts by 4-phenylbutyrate. Int J Oncol. 2003;22:579–88. [PubMed] [Google Scholar]
- 117.Yamashita Y, Shimada M, Harimoto N, et al. Histone deacetylase inhibitor trichostatin A induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. Int J Cancer. 2003;103:572–6. doi: 10.1002/ijc.10699. [DOI] [PubMed] [Google Scholar]
- 118.Armeanu S, Pathil A, Venturelli S, et al. Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J Hepatol. 2005;42:210–7. doi: 10.1016/j.jhep.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 119.Coradini D, Speranza A. Histone deacetylase inhibitors for treatment of hepatocellular carcinoma. Acta Pharmacol Sin. 2005;26:1025–33. doi: 10.1111/j.1745-7254.2005.00195.x. [DOI] [PubMed] [Google Scholar]
- 120.Richon VM, Sandhoff TW, Rifkind RA, et al. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA. 2000;97:10014–9. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gui CY, Ngo L, Xu WS, et al. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci USA. 2004;101:1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chiba T, Yokosuka O, Fukai K, et al. Cell growth inhibition and gene expression induced by the histone deacetylase inhibitor, trichostatin A, on human hepatoma cells. Oncology. 2004;66:481–91. doi: 10.1159/000079503. [DOI] [PubMed] [Google Scholar]
- 123.Zhao Y, Lu S, Wu L, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1) Mol Cell Biol. 2006;26:2782–90. doi: 10.1128/MCB.26.7.2782-2790.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–9. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guyot C, Lepreux S, Combe C, et al. Hepatic fibrosis and cirrhosis: the (myo)fibroblastic cell subpopulations involved. Int J Biochem Cell Biol. 2006;38:135–51. doi: 10.1016/j.biocel.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 126.Mann DA, Smart DE. Transcriptional regulation of hepatic stellate cell activation. Gut. 2002;50:891–6. doi: 10.1136/gut.50.6.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Forbes SJ, Russo FP, Rey V, et al. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology. 2004;126:955–63. doi: 10.1053/j.gastro.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 129.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–48. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Funakoshi F, Masaki T, Kita Y, et al. Proliferative capability of hepatocytes and expression of G1-related cell cycle molecules in the development of liver cirrhosis in rats. Int J Mol Med. 2004;13:779–87. [PubMed] [Google Scholar]
- 131.Gressner AM, Lahme B, Mannherz HG, et al. TGF-beta-mediated hepatocellular apoptosis by rat and human hepatoma cells and primary rat hepatocytes. J Hepatol. 1997;26:1079–92. doi: 10.1016/s0168-8278(97)80117-1. [DOI] [PubMed] [Google Scholar]
- 132.Muddu AK, Guha IN, Elsharkawy AM, et al. Resolving fibrosis in the diseased liver: translating the scientific promise to the clinic. Int J Biochem Cell Biol. 2007;39:695–714. doi: 10.1016/j.biocel.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 133.Okuda H. Hepatocellular carcinoma development in cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21:161–73. doi: 10.1016/j.bpg.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 134.Niki T, Rombouts K, De Bleser P, et al. A histone deacetylase inhibitor, trichostatin A, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology. 1999;29:858–67. doi: 10.1002/hep.510290328. [DOI] [PubMed] [Google Scholar]
- 135.Rombouts K, Niki T, Wielant A, et al. Trichostatin A, lead compound for development of antifibrogenic drugs. Acta Gastroenterol Belg. 2001;64:239–46. [PubMed] [Google Scholar]
- 136.Rombouts K, Knittel T, Machesky L, et al. Actin filament formation, reorganization and migration are impaired in hepatic stellate cells under influence of trichostatin A, a histone deacetylase inhibitor. J Hepatol. 2002;37:788–96. doi: 10.1016/s0168-8278(02)00275-1. [DOI] [PubMed] [Google Scholar]
- 137.Derk CT, Jimenez SA. Systemic sclerosis: current views of its pathogenesis. Autoimmun Rev. 2003;2:181–91. doi: 10.1016/s1568-9972(03)00005-3. [DOI] [PubMed] [Google Scholar]
- 138.Ghosh AK, Mori Y, Dowling E, et al. Trichostatin A blocks TGF-beta-induced collagen gene expression in skin fibroblasts: involvement of Sp1. Biochem Biophys Res Commun. 2007;354:420–6. doi: 10.1016/j.bbrc.2006.12.204. [DOI] [PubMed] [Google Scholar]
- 139.Rishikof DC, Ricupero DA, Liu H, et al. Phenylbutyrate decreases type I collagen production in human lung fibroblasts. J Cell Biochem. 2004;91:740–8. doi: 10.1002/jcb.10742. [DOI] [PubMed] [Google Scholar]
- 140.Rombouts K, Niki T, Greenwel P, et al. Trichostatin A, a histone deacetylase inhibitor, suppresses collagen synthesis and prevents TGF-beta(1)-induced fibrogenesis in skin fibroblasts. Exp Cell Res. 2002;278:184–97. doi: 10.1006/excr.2002.5577. [DOI] [PubMed] [Google Scholar]
- 141.Huber LC, Distler JH, Moritz F, et al. Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum. 2007;56:2755–64. doi: 10.1002/art.22759. [DOI] [PubMed] [Google Scholar]
- 142.Young DA, Billingham O, Sampieri CL, et al. Differential effects of histone deacetylase inhibitors on phorbol ester- and TGF-beta1 induced murine tissue inhibitor of metalloproteinases-1 gene expression. FEBS J. 2005;272:1912–26. doi: 10.1111/j.1742-4658.2005.04622.x. [DOI] [PubMed] [Google Scholar]
- 143.Iredale JP, Benyon RC, Arthur MJ, et al. Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology. 1996;24:176–84. doi: 10.1002/hep.510240129. [DOI] [PubMed] [Google Scholar]
- 144.Murphy FR, Issa R, Zhou X, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277:11069–76. doi: 10.1074/jbc.M111490200. [DOI] [PubMed] [Google Scholar]
- 145.Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007;13:3056–62. doi: 10.3748/wjg.v13.i22.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Itoh S, ten Dijke P. Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol. 2007;19:176–84. doi: 10.1016/j.ceb.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 147.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 148.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–54. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.van Grunsven LA, Verstappen G, Huylebroeck D, et al. Smads and chromatin modulation. Cytokine Growth Factor Rev. 2005;16:495–512. doi: 10.1016/j.cytogfr.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 150.Izutsu K, Kurokawa M, Imai Y, et al. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling. Blood. 2001;97:2815–22. doi: 10.1182/blood.v97.9.2815. [DOI] [PubMed] [Google Scholar]
- 151.Long J, Matsuura I, He D, et al. Repression of Smad transcriptional activity by PIASy, an inhibitor of activated STAT. Proc Natl Acad Sci USA. 2003;100:9791–6. doi: 10.1073/pnas.1733973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wotton D, Knoepfler PS, Laherty CD, et al. The Smad transcriptional corepressor TGIF recruits mSin3. Cell Growth Differ. 2001;12:457–63. [PubMed] [Google Scholar]
- 153.Simonsson M, Heldin CH, Ericsson J, et al. The balance between acetylation and deacetylation controls Smad7 stability. J Biol Chem. 2005;280:21797–803. doi: 10.1074/jbc.M503134200. [DOI] [PubMed] [Google Scholar]
- 154.Poncelet AC, Schnaper HW. Sp1 and Smad proteins cooperate to mediate transforming growth factor-beta 1-induced alpha 2(I) collagen expression in human glomerular mesangial cells. J Biol Chem. 2001;276:6983–92. doi: 10.1074/jbc.M006442200. [DOI] [PubMed] [Google Scholar]
- 155.Glenisson W, Castronovo V, Waltregny D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim Biophys Acta. 2007;1773:1572–82. doi: 10.1016/j.bbamcr.2007.05.016. [DOI] [PubMed] [Google Scholar]