

Abstract
DNA repair is a fundamental cellular function, indispensable for cell survival, especially in conditions of exposure to environmental or pharmacological effectors of DNA damage. The regulation of this function requires a flexible machinery to orchestrate the reversal of harmful DNA lesions by making use of existing proteins as well as inducible gene products. The accumulation of evidence for the involvement of ubiquitin-proteasome system (UPS) in DNA repair pathways, that is reviewed here, has expanded its role from a cellular waste disposal basket to a multi-dimensional regulatory system. This review is the first of two that attempt to illustrate the nature and interactions of all different DNA repair pathways where UPS is demonstrated to be involved, with special focus on cancer- and chemotherapy-related DNA-damage repair. In this first review, we will be presenting the proteolytic and non-proteolytic roles of UPS in the post-translational regulation of DNA repair proteins, while the second review will focus on the UPS-dependent transcriptional response of DNA repair after DNA damage and stress.
Keywords: DNA repair, ubiquitin-proteasome system, SUMO, NEDD8, MGMT, mismatch repair, base-excision repair, nucleotide-excision repair, double-strand break repair, Fanconi anaemia pathway, post-replication repair
Introduction
-
Proteolytic roles of UPS in DNA repair
MGMT repair pathway
MMR pathway
BER pathway
NER pathway
DSB repair pathway
PRR pathway
FA pathway
-
Non-proteolytic roles of UPS in DNA repair
FA pathway
BER pathway
DSB repair pathway
PRR pathway
NER pathway
Conclusive remarks
Introduction
One of the most important cellular processes for survival against environmental and endogenous exposure to DNA damage is the mobilisation of a co-ordinated network of DNA repair pathways. Depending on the nature of the induced DNA lesions, there are five main DNA repair pathways: direct or reversion repair by O6-methylguanine DNA methyltransferase (MGMT), mismatch repair (MMR), base-excision repair (BER), nucleotide-excision repair (NER) and double-strand break (DSB) repair, which involves homologous recombination (HR) and non-homologous end joining (NHEJ) repair sub-pathways [1–6]. Additionally, there exist a DNA replication block bypassing repair mechanism termed post-replication repair (PRR), consisting of both error-prone translesion synthesis (TLS) and error-free damage avoidance [7], as well as a DNA-crosslink repair pathway, combining HR and TLS, the Fanconi anaemia pathway (FA) [8].
The emergence of DNA repair deregulation as a key aspect of carcinogenesis and cancer progression has been previously described and attributed, at least partially, to generation of genomic instability at vulnerable sites, based on their structure and characteristics [9]. Aberrantly proliferating precancerous cells are subjected to increased oncogene-induced DNA replication stress, which renders them susceptible to DSB formation, with subsequent activation of the DNA-damage checkpoint and p53-dependent apoptosis. In the context of cancer progression, these cells might be subject to selective pressure for inactivation of such key checkpoint genes, as is p53 [9, 10], which would ultimately lead to the evasion of normal DNA-damage response events, as observed in cancerous phenotypes.
The ubiquitin-proteasome system (UPS) is long-established to be a cellular tool for the marking and proteolytic degradation of proteins involved in a wide variety of structural and functional roles inside the cell. The UPS includes the ‘ubiquitously’ expressed 76-amino acid protein ubiquitin (Ub), the multi-subunit protein organelle 26S proteasome, consisting of one 20S catalytic and two 19S regulatory subunits, and finally, a three-step enzymatic cascade of Ub-activating (E1), Ub-conjugating (E2) and Ub-ligase (E3) enzymes that attach Ub to the target protein [11–14]. In an expanding outlook of the ubiquity protein family there are several Ub-like proteins, sharing similarities in both structure and activation process, mainly represented by the small ubiquitin-like modifier (SUMO) protein and neural precursor cell expressed, developmentally down-regulated protein 8 (NEDD8) [15].
The step from the identification of UPS as a principal tool for intracellular proteolysis to the correlation of UPS dysfunction with the onset of disease has recently been illustrated for neurodegenerative disorders and malignancies [16, 17]. Regarding the former, Parkinson’s disease could serve as a paradigm, where the combined effect of environmental (such as dieldrin) and genetic factors (such as a-synuclein mutations) was shown to be responsible for the apoptotic death of dopaminergic neuronal cells via impairment of proteasome activity [18]. Overall, there seems to be a complex interplay between mitochondrial dysfunction, oxidative stress and aggregate formations, which are both the cause and result of aberrant inhibition of the protein degradation process [19].
In the cancer paradigm, the aberrant turnover of many oncoproteins and tumour suppressors for whom UPS is known to be the regulator, seems to have profound implications in oncogenesis in various types of cancers, such as renal cell carcinoma (VHL – HIF1α pathway), colorectal cancer (adenomatous polyposis coli (APC) –β-catenin pathway), cervical cancer (p53 targeting for degradation, mediated by E6 HPV proteins with E3 Ub ligase activity), breast cancer (E3 ligase activity of BRCA1 protein), glioblastoma (EGFR amplification as a result of lack of down-regulation by CBL E3 ligase). Moreover, hyperfunction or hypofunction of UPS in terms of protein degradation has been implicated in the deregulation of a number of cell cycle regulatory proteins in cancer, such as cyclin E and p27 [17, 20].
In the issue of cancer cell-related resistance to chemotherapeutic drugs, there is striking evidence from recent literature that both Ub family members as signalling molecules and the proteasome, either as a full entity (26S) or through its constituent subunits (20S, 19S) with distinct roles, proteolytic and non-proteolytic, are strong regulators of the DNA repair machinery. The complexity of UPS-DNA repair connection includes post-translational as well as transcriptional modifications of many repair proteins (Fig. 1). The DNA repair processes are reviewed here in the context of post-translational modifications induced by UPS (Fig. 2, Table 1), in an effort to offer a perspective of better understanding the profile of underlying mechanisms but also discover the utility and contribution of proteasome inhibitors to the reversal of DNA repair, which is an important matter in relation to therapeutic outcome, in terms of their expanding use in the treatment of malignancies, alone or in combination with other antineoplastic drugs, notably DNA-damaging agents.
Figure 1.
Modes of UPS involvement in regulation of DNA repair. *Abbreviations: O6meG, O6-methylated guanine; MGMT, O6-methylguanine-DNA methyltransferase; DR, direct repair; hMSH2, human MutS homologue 2; hMSH6, human MutS homologue 6; hMLH1, human MutL homologue 1; hPMS2, human post-meiotic segregation increased 2 protein; MMR, mismatch-repair; NEDD8, neural precursor cell expressed, developmentally down-regulated 8; EXO I, human exonuclease 1; SSBs, single-strand breaks; DSBs, double-strand breaks; TDG, thymine-DNA glycosylase; APE, apurinic endonuclease; Lig 3, DNA-ligase 3; FEN1, flap structure-specific endonuclease 1; BER, base-excision repair; NER, nucleotide-excision repair; GGR, global genomic repair; TCR, transcription-coupled repair; CSN, COP9 signalosome; SUMO, small ubiquitin-like modifier; UNG2, uracil-DNA glycosylase 2; PARP-1, poly-ADP-ribose polymerase; XRCC1, X-ray repair complementing defective repair in Chinese hamster cells 1; XPC, Xeroderma pigmentosum complementation group C; HR23, homologue of Rad23; DDB1, damage-specific DNA binding protein 1; DDB2, damage-specific DNA binding protein 2; NEDD8, neural precursor cell expressed developmentally down-regulated 8; XPG, Xeroderma pigmentosum complementation group G; XPF, Xeroderma pigmentosum complementation group F; ERCC1, excision repair cross-complementing rodent repair deficiency complementation group 1; Lig 1, DNA-ligase 1; HR, homologous recombination; NHEJ, non-homologous end joining; XRCC4, X-ray repair complementing defective repair in Chinese hamster cells 4; Lig 4, DNA-ligase 4; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; PCNA, proliferating cell nuclear antigen protein; DSBR, double-strand break repair; MRN, Mre11-Rad50-Nbs1 complex; RPA, replication protein A; BRCA 1,2, breast cancer 1,2 genes; XRCC2, X-ray repair complementing defective repair in Chinese hamster cells 2; XRCC3; X-ray repair complementing defective repair in Chinese hamster cells 3; XRCC4, X-ray repair complementing defective repair in Chinese hamster cells 4; PRR, post-replication repair; TLS, translesion DNA synthesis; Pol η, DNA polymerase η; Pol ζ, DNA polymerase ζ; Pol γ, ɛ, DNA polymerases γ, ɛ; FANCC, Fanconi anaemia, complementation group C; FANCD2, Fanconi anaemia complementation group D2; FANCI, Fanconi anaemia complementation group I.
Figure 2.
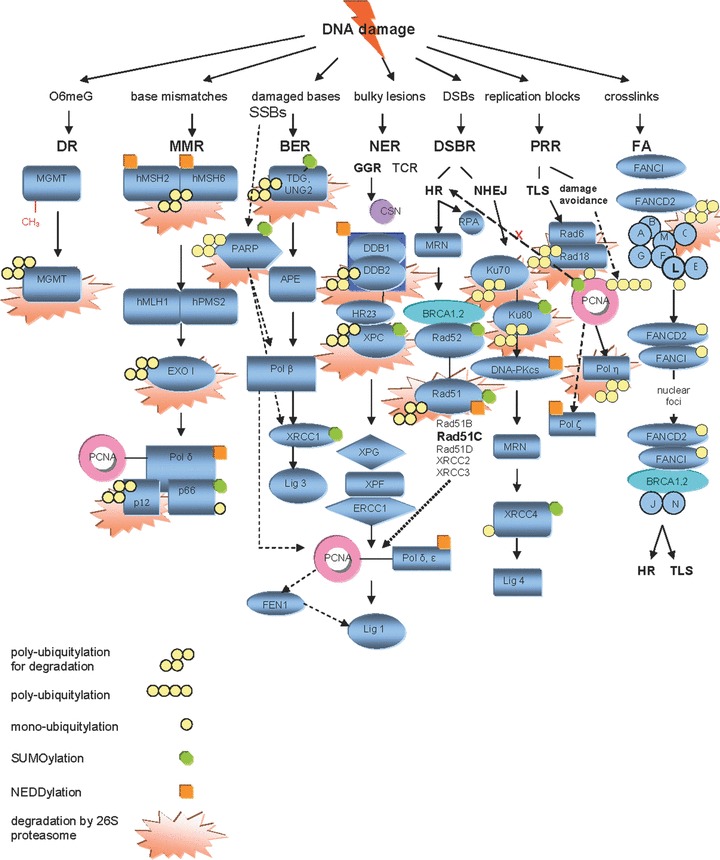
Post-translational modifications of DNA repair by UPS. The regulatory roles of UPS exerted on DNA repair on the post-translational level include (A) signalling for chromatin recruitment (FANCD2-FANCI, XRCC4 mono-ubiquitylation), (B) stabilisation and sustain of activity (XPC, Rad52 SUMOylation), (C) facilitation of DNA unbinding and turnover (TDG, XPC SUMOylation), (D) choice of sub-pathway (PCNA mono-, poly-ubiquitination, SUMOylation) and (E) degradation of DNA repair proteins considered to be proteolytic targets of the proteasome, in a way that both their levels and availability in DNA repair complexes are modified after completion of repair (MGMT, hMutSa complex, hEXO1b, TDG, UNG2, PARP-1, XPC, Rad51, Ku70, Ku80, Rad18, FANCC). There is both proteolytic and non-proteolytic contribution of UPS to this regulation of DNA repair proteins. The first is affected via the attachment of several Ub molecules and subsequent recognition and degradation by the proteasome, while the second is orchestrated through conjugation of the repair protein with a single Ub or Ub-like modifier (mainly SUMO and NEDD8).
Table 1.
Ubiquitin family- and proteasome-target proteins involved in DNA repair (post-translational regulation)
| Target protein | DNA repair pathway | Modifier | Result |
|---|---|---|---|
| MGMT | DR | Poly-ubiquitin & 26S proteasome | Degradation |
| hMutSa complex (MSH2/MSH6) | MMR | Poly-ubiquitin & 26S proteasome | Degradation |
| NEDD8 | Unknown | ||
| hEXO1b | MMR | Poly-ubiquitin & 26S proteasome | Degradation |
| TDG | BER | Poly-ubiquitin & 26S proteasome | Degradation |
| SUMO | Release from DNA | ||
| UNG2 | BER | Poly-ubiquitin & 26S proteasome | Degradation |
| PARP-1 | BER | Poly-ubiquitin | Degradation |
| 26S proteasome | |||
| Ubc9/SUMO | Deceleration of DNA synthesis for DNA repair prior to replication | ||
| XRCC1 | BER | SUMO | Unknown |
| XPC | NER | Poly-ubiquitin | Increased DNA-binding |
| 26S proteasome | Degradation | ||
| SUMO | Stabilisation | ||
| HR23 | NER | 26S proteasome | Binding to 26S proteasome, inhibition of XPC |
| 19S proteasome | Multi-ubiquitylation | ||
| DDB2 | NER | Poly-ubiquitin & 26S proteasome | Degradation |
| Rad51 | HR | Poly-ubiquitin & 26S proteasome | Degradation |
| 19S proteasome (DSS1) | Rad51 focus formation, binding at DNA-damage site | ||
| SUMO | Nuclear trafficking | ||
| NEDD8 | Unknown | ||
| Rad52 | HR | SUMO | Protection from degradation, accumulation after DNA damage |
| Ku70 | NHEJ | Poly-ubiquitin & 26S proteasome | Degradation |
| Ku80 | NHEJ | Poly-ubiquitin & 26S proteasome | Degradation |
| SUMO | Unknown | ||
| XRCC4 | NHEJ | Mono-ubiquitin | Lig 4 stabilisation, damage foci & DNA binding, interaction with NHEJ proteins |
| SUMO | Warrants further examination | ||
| DNA PKcs | NHEJ | NEDD8 | Unknown |
| PCNA | TLS | Mono-ubiquitin | Recruitment of TLS polymerases |
| SUMO | HR inhibition, Pol ζ recruitment | ||
| Poly-ubiquitin | Switch to damage avoidance sub-pathway | ||
| Rad18 | TLS | Poly-ubiquitin & 26S proteasome | Degradation |
| Mono-ubiquitin | Equilibration between nuclear and cytoplasmic protein levels | ||
| Pol η | TLS | Poly-ubiquitin & 26S proteasome | Degradation |
| Pol δ, ɛ, ζ | NEDD8 | Unknown | |
| p12 subunit of Pol δ | MMR, BER, NER, HR | Poly-ubiquitin & 26S proteasome | Degradation |
| p66 subunit of Pol δ | MMR, BER, NER, HR | Mono-ubiquitin | Conformation change (affecting the overall structure & function of the polymerase complex) |
| Poly-ubiquitin | |||
| SUMO | |||
| FANCC | FA | Poly-ubiquitin & 26S proteasome | Degradation |
| FANCD2 | FA | Mono-ubiquitin | Interaction with FANCI, chromatin targeting |
| FANCI | FA | Mono-ubiquitin | Interaction with FANCD2, chromatin targeting |
Abbreviations: MGMT, O6-methylguanine-DNA methyltransferase; DR, direct repair; MSH2, MutS homologue 2; MSH6, MutS homologue 6; MMR, mismatch-repair; NEDD8, neural precursor cell expressed developmentally down-regulated 8; hEXO1b, human exonuclease 1b; TDG, thymine-DNA glycosylase; BER, base-excision repair; SUMO, small ubiquitin-like modifier; UNG2, uracil-DNA glycosylase 2; PARP-1, poly-ADP-ribose polymerase; Ubc9, ubiquitin-conjugating enzyme 9; XRCC1, X-ray repair complementing defective repair in Chinese hamster cells 1; XPC, Xeroderma pigmentosum, complementation group C; NER, nucleotide-excision repair; HR23, homologue of Rad23; DDB2, damage-specific DNA binding protein 2; HR, homologous recombination; NHEJ, non-homologous end joining; XRCC4, X-ray repair complementing defective repair in Chinese hamster cells 4; Lig 4, DNA-ligase 4; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; PCNA, proliferating cell nuclear antigen protein; TLS, translesion DNA synthesis; Pol η, DNA polymerase η; Pol δ, ɛ, ζ, DNA polymerases δ, ɛ, ζ; FANCC, Fanconi anaemia, complementation group C; FANCD2, Fanconi anaemia complementation group D2; FANCI, Fanconi anaemia complementation group I.
Proteolytic roles of UPS in DNA repair
The attachment of a single or multiple Ub molecules, together with the exact mode and location of interactions between the chain of Ubs and the lysine residues of the target proteins determines different fates for the latter. Modulation through mono-ubiquitylation or poly-ubiquitination at Lys63 (K63) is implicated in non-proteolytic functions of Ub, ranging from membrane transport to regulation of transcription. Poly-ubiquitination at Lys48 (K48) is involved in directing to proteasomal degradation, after the formation of at least four Ub adducts [21].
MGMT repair pathway
Ubiquitination and sequential proteolytic degradation is an important post-translational modification for the pathway of direct repair of O6-alkylation lesions by human MGMT, taking place once the protein becomes irreversibly inactivated after the single-step transfer of a methyl group in its active centre [22, 23]. The proposed underlying molecular explanation lies in the generation of a conformational change in the protein, caused by the alkylation-transfer reaction, which facilitates the conjunction of MGMT with a Ub ligase, thus targeting MGMT for degradation. This regulatory procedure offers the cell an effective way of removing inactive proteins that, not only have completed their purpose, but also may delay or burden the initial DNA repair process against the damaging action of alkylating agents [24].
MMR pathway
The UPS is also involved in the degradation of the hMutSα complex, a heterodimer of hMSH2 and hMSH6 proteins of the MMR pathway. Its functional duty is to recognize the DNA mispairs or loops as the first step of a repair process. This is followed by the recruitment of a second heterodimer complex, hMutLα (hMLH1/hPMS2), promoting, along with hMutSα, the excision of the lesion by exonuclease I (hEXO1b) and the DNA strand gap repair synthesis by DNA polymerase δ[2]. Despite the fact that proteasome inhibitor-induced stabilisation of hMutSα proteins is not accompanied by increased MMR activity, low hMutSα protein levels in cells with high degradation rate of hMutSα proteins is a limiting factor for MMR activity, suggesting a regulatory proteolytic role for UPS in MMR pathway [25].
Exonuclease I was also found to be poly-ubiquitinated and degraded by the proteasome at the S phase under conditions of inhibition of DNA replication, and this process was facilitated by phosphorylation induction as a result of stalled replication forks [26].
BER pathway
The BER pathway is, at least partially, regulated by proteolysis involving the UPS. DNA glycosylases, which are responsible for the initial step of recognition and removal of damaged or incorrect bases [2], are subjected to cell cycle-dependent ubiquitination and proteasome-mediated degradation. This was proven for thymine-DNA glycosylase (TDG) and uracil-DNA glycosylase 2 (UNG2), two uracil DNA glycosylases (UDGs). TDG is eliminated during G1-S phases and until G2, as opposed to UNG2 that follows a reverse pattern, a condition essential for cell cycle progression and cell proliferation, as the two biochemically redundant UDGs control non-redundant cell cycle stage-specific pathways for uracil repair; while UNG2 is active during DNA replication, TDG functions in non-replicating DNA. Thus, the UPS appears to be a focal point in the co-ordination of redundant BER pathways [27].
Poly-ADP ribose polymerase (PARP) is another key repair protein involved in BER that was recently associated with UPS-dependent post-translational modification. PARP binds at sites of single-strand breaks (SSBs) and its increased activity, following auto-poly(ADP)-ribosylation, facilitates the replacement of the damaged base by direct interaction and poly(ADP)-ribosylation of other BER proteins such as X-ray repair complementing defective repair in Chinese hamster cells 1 (XRCC1), DNA ligase III and DNA polymerase β[2]. PARP-1 is also known to be cleaved by caspases 3 and/or 7 during apoptosis. Cleavage of PARP-1 by caspases inactivates the enzyme, which is the prerequisite of apoptosis. Poly-ubiquitylation of full length PARP-1 was clearly shown to occur both in vitro and in vivo, and it is inhibited by activated DNA and NAD+. The requirement of proteasome inhibitor to observe poly-ubiquitylation of PARP-1 in vivo suggests that poly-ubiquitylated PARP-1 is susceptible to degradation by the 26 S proteasome. Therefore, it seems that the DNA-activated enzyme is protected from proteolysis to enable its role in nuclear pathways such as DNA repair. Additionally, ubiquitylation of PARP-1 was unable to protect PARP-1 from caspase-3 catalyzed cleavage [28]. Moreover, the proteolytic post-translational modification of PARP by UPS is crucial for DNA repair in general, given the regulatory role of PARP, exerted through potential poly(ADP)-ribosylation of various DNA repair involved proteins, including MSH6 (MMR pathway), Xeroderma Pigmentosum A (a damage recognition factor of NER pathway), DNA-protein kinase (DNA-PK, NHEJ pathway) and Ku70 (NHEJ pathway) [2].
NER pathway
Regarding the influence of UPS on NER pathway, there is a complex group of interactions between NER proteins, Ub and subunits of 26 S proteasome, establishing both proteolytic and non-proteolytic roles for UPS. The NER pathway is responsible for the removal of UV-induced DNA lesions and other bulky adducts, such as intrastrand crosslinks, large chemical adducts generated from exposure to aflatoxin, benzopyrene and other genotoxic agents [2]. It involves about 30 proteins and consists of two distinct sub-pathways, the global genomic repair (GGR) and the transcription-coupled repair (TCR) [2].
The first evidence for a proteolytic role in NER came from studies in Rad4, the yeast Xeroderma Pigmentosum group C (XPC) homologue, which was suggested to be a degradation target for 26S proteasome, based on the observation of faster repair in yeast mutants of 19S proteasome subunits and after overexpression of Rad4 [29]. XPC-HR23 heterodimer is part of the damage recognition process of human GGR [2]. Rad4 protein was finally shown, by Western analysis, to be ubiquitylated and degraded by the proteasome following exposure of cells to UV [30]. Additionally, a negative role of proteolysis was supported as interpretation of increased NER in 19S proteasome mutants, with Rad23 (another important NER protein, analysed below) contribution being crucial for Rad4 stability and repair [29, 30]. The proposed model distinguishes a regulatory inhibition of multi-ubiquitination and 26S proteasome-related proteolysis of Rad4 upon DNA damage, which is followed by the proteolytic degradation of Rad4 after the completion of DNA repair [31, 32]. This switch mechanism is regulated by the co-existence and interactions (intra- and intermolecular) of one Ub-like (UbL) amino-terminal domain and two carboxy-terminal ubiquitin-associated (UBA) domains on Rad23, the yeast homologue of human HR23[33, 34]. HR23 is expressed in two homologues, HR23A and HR23B, which are functionally equivalent in NER, despite the predominance of XPC/HR23B complexes in normal cells [35]. Both HR23A and HR23B were found to interact with the S5a subunit of 26S proteasome [36]. UbL domain of HR23 binds the 26S proteasome while UBAs inhibit the formation of multi-Ub chains on Rad4 surface [37, 38]. Furthermore, the UBA-2 domain allows Rad23 to interact with the proteasome without facing destruction that would otherwise render Rad23 a short-lived protein [39]. The proper Rad23 activity additionally requires the recognition of the delivered proteolytic substrates by Rpn10, a proteasome-associated multi-Ub chain binding protein [40, 41]. The proteasome-dependent shut-off mechanism after the completion of repair ensures that the accumulated NER proteins are not implicated in improper incision of DNA structures and consequent genomic instability, during normal conditions [31]. More recent studies have elucidated that it is the Ub-ligase activity of UV-damaged DNA binding component (DDB2) complex that stimulates the dissociation of the negative regulator COP9 signalosome (CSN) through neddylation (NEDD8) and leads to poly-ubiquitylation of both XPC and DDB2. Thereafter, poly-Ub DDB2 disengages from DNA, heading for proteolytic degradation, while poly-Ub XPC demonstrates enhanced DNA-binding properties and recruits proteins involved in the DNA unwinding, excision of the damage and resynthesis [42–44]. Degradation of XPC by 26S proteasome does occur, both early after UV-damage and later after completion of repair and this is necessary for recruiting XPG, a NER protein involved in the excision of the lesion. As it were shown from previous data, XPC-hHR23B and XPG cannot simultaneously exist in the repair complex and the entry of XPG into the complex coincides with XPC-hHR23B leaving the complex [45]. This explains why inhibition of XPC degradation in the case of mutation at K655, which is the same potential site for ubiquitylation and SUMOylation described below, abolishes XPG recruitment and subsequent repair [46]. This proteolytic contribution of UPS to NER was also confirmed by the finding of diminished NER repair function because of attenuation of UV-induced XPC recruitment to DNA-damaged sites after proteasome inhibition [47].
The involvement of NER in the repair of formaldehyde-induced superbulky DNA-protein crosslinks (DPCs) has expanded the question concerning the regulatory role of UPS. Indeed it was shown, with the use of an oligodeoxyribonucleotide carrying a DPC, that protease digestion significantly improved repair and this was also observed in plasmid-based in vivo assays for both transcribed and non-transcribed regions. In contrast, the process was attenuated in XPG-deficient cells or after proteasome inhibition. Consequently, it is evident that proteolysis by 26S proteasome is the first step in the removal of DPC poly-peptide chain, prior to NER processing of the adduct [48].
DSB repair pathway
The regulation of repair of DNA DSBs could not evade the indiscriminate impact of UPS on DNA repair pathways. In favour of proteolysis involvement in DSB repair is the recruitment of subunits of 19S and 20S proteasome to DSBs in vitro and the association of deletion of its encoding gene with growth defects and hypersensitivity to DNA-damaging agents. A scenario of UPS-mediated degradation of DSB repair proteins following completion of DNA repair is speculated [49], in agreement with a consistent role of the BRCA1/BARD1 complex in promoting poly-ubiquitylation by its Ub-ligase activity [49, 50].
Furthermore, the proteolytic activity of UPS seems to determine the choice of type of HR repair as demonstrated by the shift of repair process from gene conversion (GC), which is an error-free sub-pathway of HR, to error-prone single-strand annealing (SSA), observed in mammalian cells after proteasome inhibition. In the same study, the authors are also implying a role for BRCA2 in mediating the proteasome-DSB repair machinery interaction via interacting with the proteasome subunits RPN3 and RPN7 [51]. Further evidence highlights the suppression of HR by proteasome inhibition via a Rad51-dependent way [52, 53]. The Rad51 nucleoprotein filament, interacting with Rad52, is involved in the DNA strand exchange with the undamaged homologous DNA molecule and its assembly at the site of damage is facilitated by different Rad51 paralogues, such as Rad51B, Rad51C, Rad51D, XRCC2 and XRCC3 [2]. RNAi-mediated knockdown of Rad51C was observed to lead to decreased levels of Rad51, and this effect appears to be increased following DNA damage compared to normal conditions [52]. In both cases, depletion of Rad51 resulted from Ub-mediated proteasomal degradation because in the presence of a proteasome-specific inhibitor, accumulation of ubiquitinated-Rad51 was detected. Therefore, proteasome-mediated degradation plays a role in removing Rad51 from breaks following DNA repair, and Rad51C is involved in the regulation of this activity [52]. On the other hand, the suppression of proteasome-dependent promotion of early HR after proteasome inhibition is likely explained by the abolishment of formation of Rad51 foci. In specific, after the recognition of a DSB, a 3’ single-stranded DNA tail (ssDNA) is generated. Replication protein A (RPA), which is a signalling intermediate for HR, binds to the ssDNA and allows the recruitment of the the Rad51 nucleoprotein filament. Proteasomal inhibition inhibits the formation of Rad51 foci, without interfering with generation of the ssDNA tail or the RPA foci [53, 54].
The emergence of proteolytic involvement of UPS in DSB repair is further supported by the finding that treatment of cells with a proteasome inhibitor dramatically reduced the percentage of cells forming phospho-Nbs1 foci following exposure to the DNA-damaging agent etoposide (ETOP) [55]. Nbs1 forms a part of the Mre11-Rad50-Nbs1 (MRN) complex, which is the main processor of ETOP-induced DSBs at G0/G1 phase, when NHEJ is the predominant form of repair. In contrast, proteasome inhibition did not alter the percentage of ETOP-treated cells in S/G2 phase containing RPA. It is important to clarify that following etoposide treatment, cells form foci containing either RPA or the MRN complex, but not both [55]. Consequently, these results are consistent with a cell cycle- and proteasome-dependent formation of ETOP-induced RPA or MRN complex repair foci [55].
NHEJ repair proteins are further modulated by UPS proteolytic properties. The heterodimeric repair protein complex Ku70/Ku80 is the first recognition and damage-binding step of NHEJ repair [2]. Ku70 is subjected to ubiquitination and subsequent degradation by the proteasome upon apoptotic stress, demonstrated by increased levels of Ku70 after inhibition of proteasome activity. Ubiquitylated Ku70 is inhibitory for the formation of Ku70/Ku80 complex (Ku), given that these two proteins are known to stabilise each other [56]. A more recent study clarifies that both Ku proteins are degraded in response to DSBs in an Ub-mediated manner. In the proposed model, binding of the Ku heterodimer to DNA induces a signal recognized by an E3 Ub-ligase, which then poly-ubiquitylates Ku80. This leads to either a conformational change in Ku releasing it from DNA or, more likely, to the recruitment of an additional factor that actively dissociates Ku80 from DNA. After dissociation, Ku80 is recognized and degraded by the proteasome [57]. Strikingly, K48-linked poly-ubiquitylation, but not proteasomal degradation, is required for the efficient removal of Ku80 from DNA. Furthermore, NHEJ completion and removal of Ku80 from DNA are independent from one another [57]. This is in line with the observation that NHEJ is not significantly influenced by proteasome inhibition, contrarily to the substantial effect exerted on HR, mentioned above [53].
PRR pathway
The PRR-UPS connection is a complicated story too. There are intriguing implications of a proteolytic role of UPS, based on an epistasis analysis study of mutated genes encoding either catalytic subunits or the maturase of 20S proteasome and PRR-essential repair proteins like Rad18, with respect to UV sensitivity. It was observed that UV-sensitivity of yeast with mutated maturase or β2 or β5 20S subunits does not exceed the sensitivity corresponding to mutated Rad18. Similarly, it was shown that TLS error-prone DNA polymerase zeta (Polζ) determines the UV-induced mutagenesis in maturase-mutant strains and that deletion of the former dramatically diminishes the frequency of mutations. In contrast, the presence of more error-free Polη in mutated 20S proteasome strains raises UV-sensitivity and mutagenicity, suggesting a proteolytic role in rearrangement of the replication fork in response to DNA damage [58]. The same authors describe of a proposed model in which proteasome activity negatively regulates the Rad6/Rad18 (E2/E3 complex) dependent, mono-ubiquitination-driven switch of proliferating cell nuclear antigen (PCNA) to TLS polymerases Polζ and Polη, whereas on the other hand promotes SUMO-dependent error-prone TLS by Polζ. PCNA is the replication processivity factor responsible for initiation and choice of PRR sub-pathway, depending on the recruitment of different TLS post-replicative DNA polymerases [59].
The proteolytic involvement of UPS in PRR became more evident after the recent finding that Polη is post-translationally controlled by ubiquitination-mediated proteolytic degradation, the transient inhibition of which is believed to be a contributing mechanism for stabilisation of Polη after UV-damage [60]. Poly-ubiquitination of Rad18 is another process indicative of proteasome-dependent proteolysis involvement in PRR function. Poly-ubiquitinated Rad18 bands, undetected under normal human cell culture conditions, were clearly detected when proteasome inhibitors were added to the culture. Additionally, poly-ubiquitination of Rad18 was reconstructed in an in vitro system consisting of purified E1, E2 (Rad6), Rad18 and Ub, indicating that Rad18 poly-ubiquitination occurs through auto-ubiquitination. Poly-ubiquitinated Rad18 was degraded in vitro by adding a proteasome extract [61].
Finally, there is an exciting recent finding that substantiates the 26S proteasome proteolytic involvement in PRR: the blockage of UV- and cisplatin-induced TLS by proteasome inhibition in various human cancer cells, irrespective of cell origin, histological type or p53 status, in a cancer-specific way leaving normal cell lines unaffected. The mechanism by which proteasome inhibitors inhibit error-prone TLS remains to be elucidated [62].
The contribution of DNA polymeraseδ to DNA replication and DNA repair, as the last effector of repair synthesis, after damage removal, in many repair pathways (BER, NER, MMR, HR), is further enriched by a UPS-related modification: degradation of its fourth subunit, p12. This degradation is rapidly induced after DNA damage or replication stress but blocked after proteasome inhibition [63].
FA pathway
UPS post-translationally affects the FA pathway, a replication stress response pathway named after the respective autosomal recessive disorder, which is characterized by a unique hypersensitivity to DNA-crosslinking agents [8]. This was directly demonstrated for Fanconi anaemia, complementation group C (FANCC), a member of the FA protein group that was found to be expressed in a cell cycle-dependent manner, with the lowest levels seen in cells being at the G1/S transition and the highest levels in the M-phase. The important part is that inhibitors of proteasome function blocked this cycle-dependent regulation, leading to an increase in FANCC protein level in a direct way, independent of other effects on cell cycle process (such as G2/M arrest) [64].
Non-proteolytic roles of UPS in DNA repair
FA pathway
The FA pathway is an example of non-proteolytic regulation of DNA repair as well. Consisting of at least 12 proteins, the FA pathway is activated after DNA damage and the forming nuclear core complex (FANCA, B, C, E, F, G, L, M) causes the mono-ubiquitylation of Fanconi anaemia complementation group D2 (FANCD2) protein by acting as a Ub-ligase, thus facilitating the interaction with chromatin, HR and TLS proteins [8, 43]. Mono-ubiquitylation of FANCD2 is reversed by USP1, a member of the protease family of de-ubiquitylating enzymes (DUBs) after the completion of repair [43, 65]. It was also shown that proteasome inhibition blocks both basal and DNA damage-induced (irradiation, UV, DNA crosslinking agents, hydroxyurea) mono-ubiquitylation and nuclear foci formation of FANCD2 [54]. When depletion of 19S and 20S proteasome subunits was tested under the conditions mentioned above, it became clear that the proteasome proteolytic function is required for the activation of the FA pathway; nevertheless, inhibition of FANCD2 mono-ubiquitination requires a more profound proteasome inhibition than inhibition of FANCD2 foci formation and the implicated mechanisms do not involve USP1 activation, ATR (upsteam DNA-damage response kinase) inhibition, decreased expression of the FA core complex (E3) or E2 enzymes [54]. The authors seem to believe to a dual, both proteolytic and non-proteolytic involvement of the proteasome in the FA pathway regulation, anticipating the identification of new proteolytic targets required for the formation of the DNA repair (FANCD2, BRCA1, Rad51) foci as well as a new FANCD2 deubiquitinating enzyme [54].
A further FA protein, Fanconi anaemia complementation group I (FANCI), was recently identified as a target of mono-ubiquitination induced by DNA damage. It has many properties analogous to FANCD2 and most strikingly, co-localizes and interacts with FANCD2 in a mutually dependent, Ub-related manner, suggesting the existence of a FANCD2-FANCI (ID) subcomplex in which both partners have to be mono-ubiquitinated to form nuclear foci [66, 67].
BER pathway
Non-proteolytic post-translational modifications are also part of BER regulation. This is the case for TDG, which becomes conjugated with SUMO-1 and SUMO-2/3 proteins after the hydrolysis of mispaired U or T base, opposite G [68]. This reversible SUMOylation reduces the DNA-binding affinity of the enzyme to the cleaved abasic site, resulting not only in the efficient progression of abasic site processing by apurinic and apyrimidinic endonuclease (APE) but also in the release and turnover of TDG for subsequent rounds of recognition of base mispairs [43, 68].
The non-proteolytic modulation of BER by SUMO is further substantiated in the light of evidence showing that Ubc9, a SUMO E2-ligase, interacted with PARP in a yeast-two-hybrid system. The explanation of this modification is unknown but is speculated to involve deceleration of DNA synthesis for DNA repair prior to replication and a possible role in apoptosis [69, 70].
Finally, XRCC1, a molecular scaffold protein that coordinates the assembly of BER complexes at the damaged sites, was also found to be a SUMO substrate, however, the effect of sumoylation on its function is unknown [69, 71].
DSB repair pathway
The documentation of a non-proteolytic link between DSB repair and UPS, based on the examination of DSB repair efficiency in conditions of modified 19S proteasome subunits was proved to be a difficult venture. The importance of 19S participation in DSB repair was justified by the observation of growth defects and hypersensitivity to drugs generating DSBs in yeast with mutated groups of genes encoding a 19S subunit (Sem1) and proteins of either HR or NHEJ. Sem1 recruitment to an artificially induced DSB was found to be dependent on both HR (Rad52) and NHEJ (DNA pol4) proteins [49]. The role of proteasome at DSBs is speculated to be the proteolytic degradation of one or more components of the DSB repair machinery following the completion of repair [49]. Moreover, the human homologue for Sem1, DSS1, is required for DNA-damage-induced Rad51 nuclear focus formation and was implicated in a possible involvement in the BRCA2-Rad51 complex binding at sites of damage [72].
However, the observation that Sem1 deletion reduces proteolytic activity of proteasome without interfering with Pre2 recruitment (encoding a 20S proteasome subunit) to the DSB [73], favours a proteolytic explanation for post-translational regulation of HR by UPS. The supporters of the latter could also argue on the grounds of a recent genetic analysis study of a human cDNA library in yeast, screening for new proteins implicated in HR. Among other findings, it was shown that in Rad52-deficient strains, the catalytic proteasome subunits (the complete β2 and the partially deleted α3, β8) greatly increased resistance to the alkylating agent methylmethane sulphonate (MMS) [74].
Nevertheless, it seems that non-proteolytic modification of HR by UPS cannot be excluded, as earlier studies have reported a non-covalent association of SUMO-1 and Ubc9 (a SUMO E2 conjugating enzyme) with human Rad51, demonstrated by yeast two-hybrid and co-immunoprecipitation experiments [75–77]. SUMOylation was also observed in yeast Rad52 upon DNA damage and triggered by Mre11-Rad50-Xrs2 (MRX) complex-governed DSBs. Although sumoylation-defective Rad52 is largely recombination proficient, mutant analysis revealed that the SUMO modification sustains Rad52 activity and concomitantly protects the protein against accelerated proteasomal degradation [78]. The clarification of this non-proteolytic modification of HR by UPS, achieved in another study, is focused on SUMOylated Rad52 accumulation following exposure to MMS and on partial defect observed in MMS-induced interchromosomal HR in SUMOylation-defective mutant Rad52 cells [79].
With respect to NHEJ, a non-degradative role of UPS is justified in light of X-ray repair complementing defective repair in Chinese hamster cells 4 (XRCC4) mono-ubiquitination. XRCC4-DNA ligase IV complex is a target of the activated by Ku heterodimer DNA-PK, and its role is the link of broken DNA ends [2]. After DNA damage, XRCC4 is (further) phosphorylated, and ubiquitinated, exerting a stabilizing role for ligase IV. However, proteasome inhibition does not lead to accumulation of XRCC4 protein levels, supporting a non-proteolytic role for UPS in this aspect. Moreover, the calculation of several potential SUMOylation sites within the protein offers another proof of its non-proteolytic regulation via SUMOylation (in addition to mono-ubiquitination and phosphorylation) [80]. XRCC4 transient SUMOylation was shown to be crucial for the protein’s nuclear translocation to complete NHEJ events; a mutated XRCC4 protein (with compromised ability to become SUMOylated) was radiation sensitive and failed to complete NHEJ-dependent V(D)J recombination of B- and T-cell receptors, whereas genetic fusion of the SUMO sequence to the C-terminus of the mutant protein restores nuclear localization and radiation resistance [69, 81]. Another NHEJ protein, Ku80, was found to be a SUMO-substrate in an in vitro expression cloning experiment that identified 40 human SUMO1 substrates. Both SUMO1 and SUMO2 conjugates of the protein were identified, although SUMO1 modification was much stronger. Ku80 also appeared poly-SUMOylated [71].
PRR pathway
The literature highlights many descriptions of non-proteolytic interactions between PRR and UPS, involving different modifications of PCNA by SUMOylation, mono- and poly-ubiquitylation, playing a key role in the cellular control of management of the replication fork in response to replication-blocking DNA lesions. During normal conditions, PCNA is SUMOylated and consequently inhibited from the initiation of inappropriate HR via the recruitment of Srs helicase. Upon DNA damage, PCNA is subjected to mono-ubiquitylation, which promotes the recruitment of TLS error-prone DNA polymerases for bypassing the DNA-damaged site, or to further poly-ubiquitylation that favours the error-free damage avoidance PRR sub-pathway. The mono-ubiquitylation of PCNA is controlled by USP1, a deubiquitylating enzyme, possibly contributing to the release of TLS polymerases after the completion of lesion bypass, facilitating the re-engagement of normal replicative polymerases [82]. The Ub-polymerase interaction is another important factor that regulates the access of TLS polymerases to stalled replication forks in vivo, as documented by the lower levels of replication foci of mutated pol η and pol ι, defective in interacting with poly-Ub and Ub-PCNA, in response to DNA damage [83]. Ub is central to Rad18 regulation as well. Rad6-dependent mono-ubiquitination of human Rad18 occurs by auto-ubiquitination and is assumed to serve as an indirect regulatory mechanism for PRR through equilibration of levels between nucleic non-Ub Rad18 and cytoplasmic reservoir of mono-Ub Rad18 protein [61].
NER pathway
Non-proteolytic regulation also plays a central role in NER function. XPC is subjected to SUMOylation and ubiquitylation following DNA damage, in a DDB2 and XPA-dependent way. Both modifications stabilize the protein, enabling increased binding to DNA, and initiation of a new round of damage-recognition by hypothetically facilitating its disengagement from NER complex, respectively. Therefore, it is evident that XPC levels are non-proteolytically controlled through alternating interactions with Ub and SUMO [84].
The finding that inhibition of 19S proteasomal ATPases positively affects NER activity, which, however, remained unchanged by blocking of protein degradation by the proteasome, has led to consider 19S proteasome subunit as a rearranging, disassembling factor for NER protein complexes at DNA-damaged sites, through its chaperone-like activity [85]. This negative role of 19S was further shown to be independent of proteolysis, as yeast strains with mutations in 19S regulatory subunits promoted partial recovery of NER in vivo in rad23 deletion mutants. Therefore, it was suggested that Rad23 not only recruits the 19S proteasome subcomplex but also attenuates its negative in vivo effect and enhances NER through its UBA-mediated interaction with other members of the nucleotide-excision repairosome [86]. More recent studies proceed further by proposing a dual non-proteolytic model for NER regulation by UPS. In this model, NER is divided into two pathways. Pathway I, involving pre-existing proteins, consists of Rad23 interaction with 19S proteasome subunit through its UbL domain. Pathway II, concerning de novo protein synthesis, is controlled through ubiquitylation of Rad4 by a novel Ub-ligase, followed by Rad4 degradation, which, however, is not a prerequisite for the efficiency of this axis [87, 88]. The supporting data are based on the cycloheximide-selective reduction of NER in Rad23-defective yeast strains (suggesting pathway II), contrary to the cycloheximide independent repair impairment in Rad7-containing E3 ligase mutated strains (suggesting pathway I). Rad7 was identified as part of a new E3 Ub-ligase that ubiquitinates Rad4 following exposure to UV-radiation. Additionally, although Rad4 protein levels were reduced in Rad23-defective cells, stabilisation of Rad4 in proteolytic-defective mutants had no effect on the NER-defective phenotype of a rad23-defective strain. The interpretation of these results suggest that the reduced levels of Rad4 in Rad23-deleted cells are not as a result of increased proteolysis of Rad4 in these cells, but rather as a result of reduced production of Rad4 transcript [88].
In contrast, another research team has demonstrated that inhibition of 19S proteasome subunit function by overexpression of 19S regulatory complex hSug1 or its mutant protein hSug1mk positively modulates NER in human cells [47]. It is important to keep in mind that a critical view of contradicting information, based on the differences of methods and cellular systems used and of NER function between yeast and human, is demanded.
The DNA polymeraseδ is also non-proteolytically modified by UPS. Apart from p12 subunit ubiquitination and degradation by the proteasome, p66 subunit was found to be modified by Ub and SUMO-3 [89]. However, in contrast to the results seen with p12, the treatment of cells with a proteasome inhibitor does not significantly increase the levels of p66 and does not affect either the levels or the proportions of mono- or poly-ubiquitinated species, suggesting that p66 is not targeted to the proteasome by an Ub-dependent mechanism [89].
Additionally, to all the information given above on post-translational modifications of DNA repair proteins by Ub and SUMO, the role of NEDD8 in DNA repair (apart from the one described above, exerted in NER) seems to concern a more direct level of regulation of DNA repair proteins. This was supported by the finding that NEDD8 interacts with MSH2, MSH6, DNA PKcs, Rad51 homologue 3, DNA polymerasesδ, ɛ, ζ, RNA polymerase II, topoisomerases Top 2α and 2β, poly-ubiquitin and subunits of both 19S and 20S proteasome, in proteomic analyses with the use of affinity purification and tandem mass spectrometry [90, 91]. Although NEDD8 has been primarily reported to function as a regulator of Ub-protein ligases and secondly as a decoy for proteins to undergo proteasomal degradation [15], further studies are expected to elucidate the significance and the order of the interactions mentioned above, in the context of the whole succession of downstream events in DNA repair pathways.
Conclusive remarks
It is without doubt that the complexity and variability of the interconnection between UPS and DNA repair is really enormous. We have attempted to present an illustrative picture of UPS function in cancer biology regarding the influence exerted on every separate DNA repair pathway by Ub family members and the proteasome within the context of post-translational modifications, more depictive of the range of interrelating networks rather than presenting in-depth details of actual interactions. Moreover, the role in DNA repair of important signalling proteins with an established interaction with UPS, such as p53 [92, 93] and the tumour suppressors BRCA-1 [50] and BRCA-2 [94], was underscored, despite the existence of gross evidence indicating that many DNA repair proteins function as p53- or/and BRCA-target genes or conjugates [95–98]. As a result of the existence of multiple levels of regulation and the numerous factors participating, it is inevitable that more questions are raised than answered. Further studies are expected to elucidate the exact sequence of molecular events involving the mobilisation of UPS in discrete ways on the purpose of DNA repair regulation. However, one thing is certain: protein degradation, once synonymous with UPS, is only the tip of the iceberg in regard with this multi-functional cellular tool.
Acknowledgments
The authors declare no conflict of interest related to this article.
References
- 1.Hansen WK, Kelley MR. Review of mammalian DNA repair and translational implications. J Pharmacol Exp Ther. 2000;295:1–9. [PubMed] [Google Scholar]
- 2.Christmann M, Tomicic MT, Roos WP, et al. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 3.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes. Mutat Res. 2005;577:275–83. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Pourquier P. La réparation de l’ ADN, cible potentielle d’un développement thérapeutique en cancérologie. Bull Cancer. 2006:124–44. ; hors série: [Google Scholar]
- 5.Sánchez-Pérez I. DNA inhibitors in cancer treatment. Clin Transl Oncol. 2006;8:642–6. doi: 10.1007/s12094-006-0034-8. [DOI] [PubMed] [Google Scholar]
- 6.Damia G, D’Incalci M. Targetting DNA repair as a promising approach in cancer therapy. Eur J Cancer. 2007;43:1791–801. doi: 10.1016/j.ejca.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Barbour L, Xiao W. Regulation of alternative replication bypass pathways at stalled replication forks and its effects on genome stability: a yeast model. Mutat Res. 2003;532:137–55. doi: 10.1016/j.mrfmmm.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy RD, D’ Andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19:2925–40. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 9.Tsantoulis PK, Kotsinas A, Sfikakis PP, et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27:3256–64. doi: 10.1038/sj.onc.1210989. [DOI] [PubMed] [Google Scholar]
- 10.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 11.Baumeister W, Walz J, Zuhl F, et al. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–80. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 12.Ciechanover A, Orian A, Schwartz AL. The ubiquitin-mediated proteolytic pathway: mode of action and clinical implications. J Cell Biochem Suppl. 2000;34:40–51. doi: 10.1002/(sici)1097-4644(2000)77:34+<40::aid-jcb9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Adams J. The proteasome: structure, function, and role in the cell. Cancer Treat Rev. 2003;29(Suppl 1):S3–9. doi: 10.1016/s0305-7372(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 14.Behuliak M, Celec P, Gardlik R, et al. Ubiquitin – the kiss of death goes Nobel. Will you be quitting. Bratisl Lek Listy. 2005;106:93–100. [PubMed] [Google Scholar]
- 15.Hermann J, Lerman LO, Lerman A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ Res. 2007;100:1276–91. doi: 10.1161/01.RES.0000264500.11888.f0. [DOI] [PubMed] [Google Scholar]
- 16.Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005;12:1178–90. doi: 10.1038/sj.cdd.4401692. [DOI] [PubMed] [Google Scholar]
- 17.Reinstein E, Ciechanover A. Narrative review: protein degradation and human diseases: the ubiquitin connection. Ann Intern Med. 2006;145:676–84. doi: 10.7326/0003-4819-145-9-200611070-00010. [DOI] [PubMed] [Google Scholar]
- 18.Sun F, Anantharam V, Latchoumycandane C, et al. Dieldrin induces ubiquitin-proteasome dysfunction in alpha-synuclein overexpressing dopaminergic neuronal cells and enhances susceptibility to apoptotic cell death. J Pharmacol Exp Ther. 2005;315:69–79. doi: 10.1124/jpet.105.084632. [DOI] [PubMed] [Google Scholar]
- 19.Sun F, Kanthasamy A, Anantharam V, et al. Environmental neurotoxic chemicals-induced ubiquitin proteasome system dysfunction in the pathogenesis and progression of Parkinson’s disease. Pharmacol Ther. 2007;114:327–44. doi: 10.1016/j.pharmthera.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol. 2005;23:4776–89. doi: 10.1200/JCO.2005.05.081. [DOI] [PubMed] [Google Scholar]
- 21.Haglund K, Dikic I. Ubiquitylation and cell signalling. EMBO J. 2005;24:3353–9. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pegg AE, Wiest L, Mummert C, et al. Use of antibodies to human O6-alkylguanine-DNA alkyltransferase to study the content of this protein in cells treated with O6-benzylguanine or N-methyl-N’-nitro-N-nitrosoguanidine. Carcinogenesis. 1991;12:1679–83. doi: 10.1093/carcin/12.9.1679. [DOI] [PubMed] [Google Scholar]
- 23.Srivenugopal KS, Yuan XH, Friedman HS, et al. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry. 1996;35:1328–34. doi: 10.1021/bi9518205. [DOI] [PubMed] [Google Scholar]
- 24.Xu-Welliver M, Pegg AE. Degradation of the alkylated form of the DNA repair protein, O6-alkylguanine-DNA alkyltransferase. Carcinogenesis. 2002;23:823–30. doi: 10.1093/carcin/23.5.823. [DOI] [PubMed] [Google Scholar]
- 25.Hernandez-Pigeon H, Laurent G, Humbert O, et al. Degradation of mismatch repair hMutSα heterodimer by the ubiquitin-proteasome pathway. FEBS Lett. 2004;562:40–4. doi: 10.1016/S0014-5793(04)00181-4. [DOI] [PubMed] [Google Scholar]
- 26.El-Shemerly M, Janscak P, Hess D, et al. Degradation of human exonuclease 1b upon DNA synthesis inhibition. Cancer Res. 2005;65:3604–9. doi: 10.1158/0008-5472.CAN-04-4069. [DOI] [PubMed] [Google Scholar]
- 27.Hardeland U, Kunz C, Focke F, et al. Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 2007;35:3859–67. doi: 10.1093/nar/gkm337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang T, Simbulan-Rosenthal CM, Smulson ME, et al. Polyubiquitylation of PARP-1 through ubiquitin K48 is modulated by activated DNA, NAD+, and dipeptides. J Cell Biochem. 2008;104:318–28. doi: 10.1002/jcb.21624. [DOI] [PubMed] [Google Scholar]
- 29.Lommel L, Chen L, Madura K, et al. The 26S proteasome negatively regulates the level of overall genomic nucleotide excision repair. Nucleic Acids Res. 2000;28:4839–45. doi: 10.1093/nar/28.24.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lommel L, Ortolan T, Chen L, et al. Proteolysis of a nucleotide excision repair protein by the 26S proteasome. Curr Genet. 2002;42:9–20. doi: 10.1007/s00294-002-0332-9. [DOI] [PubMed] [Google Scholar]
- 31.Sweder K, Madura K. Regulation of repair by the 26S proteasome. J Biomed Biotechnol. 2002;2:94–105. doi: 10.1155/S1110724302205033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ng JM, Vermeulen W, van der Horst GT, et al. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 2003;17:1630–45. doi: 10.1101/gad.260003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madura K. The ubiquitin-associated (UBA) domain. Cell Cycle. 2002;1:235–44. [PubMed] [Google Scholar]
- 34.Goh AM, Walters KJ, Elsasser S, et al. Components of the ubiquitin-proteasome pathway compete for surfaces on Rad23 family proteins. BMC Biochem. 2008;9:4–13. doi: 10.1186/1471-2091-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okuda Y, Nishi R, Ng JM, et al. Relative levels of the two mammalian Rad23 homologs determine composition and stability of the xeroderma pigmentosum group C protein complex. DNA Repair. 2004;3:1285–95. doi: 10.1016/j.dnarep.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 36.Hiyama H, Yokoi M, Masutani C, et al. Interaction of hHR23 with S5a. The ubiquitin-like domain of hHR23 mediates interaction with S5a subunit of 26S proteasome. J Biol Chem. 1999;274:28019–25. doi: 10.1074/jbc.274.39.28019. [DOI] [PubMed] [Google Scholar]
- 37.Chen L, Shinde U, Ortolan TG, et al. Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition if multi-ubiquitin chain assembly. EMBO Rep. 2001;2:933–8. doi: 10.1093/embo-reports/kve203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raasi S, Pickart CM. Rad23-ubiquitin associated domains (UBA) inhibit 26S proteasome-catalysed proteolysis by sequestering lysine 48-linked polyubiquitin chains. J Biol Chem. 2003;278:8951–9. doi: 10.1074/jbc.m212841200. [DOI] [PubMed] [Google Scholar]
- 39.Heessen S, Masucci MG, Dantuma NP. The UBA2 domain functions as an intrinsic stabilization signal that protects Rad23 from proteasomal degradation. Mol Cell. 2005;18:225–35. doi: 10.1016/j.molcel.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 40.Lambertson D, Chen L, Madura K. Pleiotropic defects caused by loss of the proteasome-interacting factors Rad23 and Rpn10 of Saccharomyces cerevisiae. Genetics. 1999;153:69–79. doi: 10.1093/genetics/153.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen L, Madura K. Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol Cell Biol. 2002;22:4902–13. doi: 10.1128/MCB.22.13.4902-4913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Mahdy LA, Zhu Q, Wang QE, et al. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem. 2006;281:13404–11. doi: 10.1074/jbc.M511834200. [DOI] [PubMed] [Google Scholar]
- 43.Huang TT, D’ Andrea AD. Regulation of DNA repair by ubiquitylation. Nat Rev Mol Cell Biol. 2006;7:323–34. doi: 10.1038/nrm1908. [DOI] [PubMed] [Google Scholar]
- 44.Bergink S, Jaspers NG, Vermeulen W. Regulation of UV-induced DNA damage response by ubiquitylation. DNA Repair. 2007;6:1231–42. doi: 10.1016/j.dnarep.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Wakasugi M, Sancar A. Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc Natl Acad Sci USA. 1998;95:6669–74. doi: 10.1073/pnas.95.12.6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang QE, Praetorius-Ibba M, Zhu Q, et al. Ubiquitylation-independent degradation of Xeroderma pigmentosum group C protein is required for efficient nucleotide excision repair. Nucleic Acids Res. 2007;35:5338–50. doi: 10.1093/nar/gkm550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang QE, Wani MA, Chen J, et al. Cellular ubiquitination and proteasomal functions positively modulate mammalian nucleotide excision repair. Mol Carcinog. 2005;42:53–64. doi: 10.1002/mc.20065. [DOI] [PubMed] [Google Scholar]
- 48.Baker DJ, Wuenschell G, Xia L, et al. Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J Biol Chem. 2007;282:22592–604. doi: 10.1074/jbc.M702856200. [DOI] [PubMed] [Google Scholar]
- 49.Krogan NJ, Lam MH, Fillingham J, et al. Proteasome involvement in the repair of DNA double-strand breaks. Mol Cell. 2004;16:1027–34. doi: 10.1016/j.molcel.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 50.Starita LM, Parvin JD. The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr Opin Cell Biol. 2003;15:345–50. doi: 10.1016/s0955-0674(03)00042-5. [DOI] [PubMed] [Google Scholar]
- 51.Gudmundsdottir K, Lord CJ, Ashworth A. The proteasome is involved in determining differential utilization of double-strand break repair pathways. Oncogene. 2007;26:7601–6. doi: 10.1038/sj.onc.1210579. [DOI] [PubMed] [Google Scholar]
- 52.Bennett BT, Knight KL. Cellular localization of human Rad51C and regulation of ubiquitin-mediated proteolysis of Rad51. J Cell Biochem. 2005;96:1095–109. doi: 10.1002/jcb.20640. [DOI] [PubMed] [Google Scholar]
- 53.Murakawa Y, Sonoda E, Barber LJ, et al. Inhibitors of the proteasome suppress homologous DNA recombination in mammalian cells. Cancer Res. 2007;67:8536–43. doi: 10.1158/0008-5472.CAN-07-1166. [DOI] [PubMed] [Google Scholar]
- 54.Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res. 2007;67:7395–405. doi: 10.1158/0008-5472.CAN-07-1015. [DOI] [PubMed] [Google Scholar]
- 55.Robison JG, Dixon K, Bissler JJ. Cell cycle- and proteasome-dependent formation of etoposide-induced replication protein A (RPA) or Mre11/Rad50/Nbs1 (MRN) complex repair foci. Cell Cycle. 2007;6:2399–407. doi: 10.4161/cc.6.19.4772. [DOI] [PubMed] [Google Scholar]
- 56.Gama V, Yoshida T, Gomez JA, et al. Involvement of the ubiquitin in decreasing Ku70 levels in response to drug-induced apoptosis. Exp Cell Res. 2006;312:488–99. doi: 10.1016/j.yexcr.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 57.Postow L, Ghenoiu C, Woo EM, et al. Ku80 removal from DNA through double strand break-induced ubiquitylation. J Cell Biol. 2008;182:467–79. doi: 10.1083/jcb.200802146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Podlaska A, McIntyre J, Skoneczna A, et al. The link between 20S proteasome activity and post-replication DNA repair in Saccharomyces cerevisiae. Mol Microbiol. 2003;49:1321–32. doi: 10.1046/j.1365-2958.2003.03635.x. [DOI] [PubMed] [Google Scholar]
- 59.McIntyre J, Podlaska A, Sconeczna A, et al. Analysis of the spontaneous mutator phenotype associated with 20S proteasome deficiency in S. cerevisiae. Mut Res. 2006;593:153–63. doi: 10.1016/j.mrfmmm.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 60.Sconeczna A, McIntyre J, Sconeczny M, et al. Polymerase eta is a short-lived, proteasomally degraded protein that is temporarily stabilized following UV irradiation in Saccharomyces cerevisiae. J Mol Biol. 2007;366:1074–86. doi: 10.1016/j.jmb.2006.11.093. [DOI] [PubMed] [Google Scholar]
- 61.Miyase S, Tateishi S, Watanabe K, et al. Differential regulation of Rad18 through Rad6-dependent mono- and poly-ubiquitination. J Biol Chem. 2005;280:515–24. doi: 10.1074/jbc.M409219200. [DOI] [PubMed] [Google Scholar]
- 62.Takezawa J, Ishimi Y, Yamada K. Proteasome inhibitors remarkably prevent translesion replication in cancer cells but not normal cells. Cancer Sci. 2008;99:863–71. doi: 10.1111/j.1349-7006.2008.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang S, Zhou Y, Trusa S, et al. A novel DNA damage response: rapid degradation of the p12 subunit of DNA polymerase δ. J Biol Chem. 2007;282:15330–40. doi: 10.1074/jbc.M610356200. [DOI] [PubMed] [Google Scholar]
- 64.Heinrich MC, Silvey KV, Stone S, et al. Posttranscriptional cell cycle-dependent regulation of human FANCC expression. Blood. 2000;95:3970–7. [PubMed] [Google Scholar]
- 65.Nijman SM, Huang TT, Dirac AM, et al. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–9. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 66.Smogorzewska A, Matsuoka S, Vinciguerra P, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grompe M, van de Vrugt H. The Fanconi family adds a fraternal twin. Dev Cell. 2007;12:661–2. doi: 10.1016/j.devcel.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 68.Hardeland U, Steinacher R, Jiricny J, et al. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002;21:1456–64. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moschos SJ, Mo YY. Role of SUMO/Ubc9 in DNA damage repair and tumorigenesis. J Mol Hist. 2006;37:309–19. doi: 10.1007/s10735-006-9030-0. [DOI] [PubMed] [Google Scholar]
- 70.Masson M, de Murcia JM, Matei MG, et al. Poly(ADP-ribose) polymerase interacts with a novel human ubiquitin conjugating enzyme: hUbc9. Gene. 1997;190:287–96. doi: 10.1016/s0378-1119(97)00015-2. [DOI] [PubMed] [Google Scholar]
- 71.Gocke CB, Yu H, Kang J. Systematic identification and analysis of mammalian small ubiquitin-like modifier substrates. J Biol Chem. 2005;280:5004–12. doi: 10.1074/jbc.M411718200. [DOI] [PubMed] [Google Scholar]
- 72.Gudmundsdottir K, Lord CJ, Witt E, et al. DSS1 is required for Rad51 focus formation and genomic stability in mammalian cells. EMBO Rep. 2004;5:989–93. doi: 10.1038/sj.embor.7400255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sone T, Saeki Y, Toh-e A, et al. Sem1p is a novel subunit of the 26S proteasome from Saccharomyces cerevisiae. J Biol Chem. 2004;279:28807–16. doi: 10.1074/jbc.M403165200. [DOI] [PubMed] [Google Scholar]
- 74.Collavoli A, Comelli A, Rainaldi G, et al. A yeast-based genetic screening to identify human proteins that increase homologous recombination. FEMS Yeast Res. 2008;8:351–61. doi: 10.1111/j.1567-1364.2007.00343.x. [DOI] [PubMed] [Google Scholar]
- 75.Kovalenko OV, Plug AW, Haaf T, et al. Mammalian ubiquitin-conjugating enzyme Ubc9 interacts with Rad51 recombination protein and localizes in synaptonemal complexes. Proc Natl Acad Sci USA. 1996;93:2958–63. doi: 10.1073/pnas.93.7.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen Z, Pardington-Purtymun PE, Comeaux JC, et al. Associations of UBE2I with RAD52, UBL1, p53, and RAD51 proteins in a yeast two-hybrid system. Genomics. 1996a;37:183–6. doi: 10.1006/geno.1996.0540. [DOI] [PubMed] [Google Scholar]
- 77.Shen Z, Pardington-Purtymun PE, Comeaux JC, et al. UBL1, a human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics. 1996b;36:271–9. doi: 10.1006/geno.1996.0462. [DOI] [PubMed] [Google Scholar]
- 78.Sacher M, Pfander B, Hoege C, et al. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat Cell Biol. 2006;8:1284–90. doi: 10.1038/ncb1488. [DOI] [PubMed] [Google Scholar]
- 79.Ohuchi T, Seki M, Branzei D, et al. Rad52 sumoylation and its involvement in the efficient induction of homologous recombination. DNA Repair. 2008;7:879–89. doi: 10.1016/j.dnarep.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 80.Foster RE, Nnakwe C, Woo L, et al. Monoubiquitination of the nonhomologous end joining protein XRCC4. Biochem Biophys Res Commun. 2006;341:175–83. doi: 10.1016/j.bbrc.2005.12.166. [DOI] [PubMed] [Google Scholar]
- 81.Yurchenko V, Xue Z, Sadofsky MJ. SUMO modification of human XRCC4 regulates its localization and function in DNA double-strand break repair. Mol Cell Biol. 2006;26:1786–94. doi: 10.1128/MCB.26.5.1786-1794.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee KY, Myung K. PCNA modifications for regulation of post-replication repair pathways. Mol Cells. 2008;26:5–11. [PMC free article] [PubMed] [Google Scholar]
- 83.Plosky BS, Vidal AE, Fernández de Henestrosa AR, et al. Controlling the subcellular localization of DNA polymerases ι and η via interactions with ubiquitin. EMBO J. 2006;25:2847–55. doi: 10.1038/sj.emboj.7601178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang QE, Zhu Q, Wani G, et al. DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res. 2005;33:4023–34. doi: 10.1093/nar/gki684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Russell SJ, Reed SH, Huang W, et al. The 19S regulatory complex of the proteasome functions independently of proteolysis in nucleotide excision repair. Mol Cell. 1999;3:687–95. doi: 10.1016/s1097-2765(01)80001-0. [DOI] [PubMed] [Google Scholar]
- 86.Gillette TG, Huang W, Russell SJ, et al. The 19S complex of the proteasome regulates nucleotide excision repair in yeast. Genes Dev. 2001;15:1528–39. doi: 10.1101/gad.869601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reed SH, Gillette TG. Nucleotide excision repair and the ubiquitin proteasome pathway-Do all roads lead to Rome. DNA Repair. 2007;6:149–56. doi: 10.1016/j.dnarep.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 88.Gillette TG, Yu S, Zhou Z, et al. Distinct functions of the ubiquitin-proteasome pathway influence nucleotide excision repair. EMBO J. 2006;25:2529–38. doi: 10.1038/sj.emboj.7601120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu G, Warbrick E. The p66 and p12 subunits of DNA polymerase delta are modified by ubiquitin and ubiquitin-like proteins. Biochem Biophys Res Commun. 2006;349:360–6. doi: 10.1016/j.bbrc.2006.08.049. [DOI] [PubMed] [Google Scholar]
- 90.Jones J, Wu K, Yang Y, et al. A targeted proteomic analysis of the ubiquitin-like modifier Nedd8 and associated proteins. J Proteome Res. 2008;7:1274–87. doi: 10.1021/pr700749v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Norman JA, Shiekhattar R. Analysis of Nedd8-associated polypeptides: a model for deciphering the pathway for ubiquitin-like modifications. Biochemistry. 2006;45:3014–9. doi: 10.1021/bi052435a. [DOI] [PubMed] [Google Scholar]
- 92.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 93.Brooks CL, Gu W. Dynamics in the p53-Mdm2 ubiquitination pathway. Cell Cycle. 2004;3:895–9. [PubMed] [Google Scholar]
- 94.Schoenfeld AR, Apgar S, Dolios G, et al. BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Mol Cell Biol. 24:7444–55. doi: 10.1128/MCB.24.17.7444-7455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Adimoolam S, Ford JM. p53 and regulation of DNA damage recognition during nucleotide excision repair. DNA Repair. 2003;2:947–54. doi: 10.1016/s1568-7864(03)00087-9. [DOI] [PubMed] [Google Scholar]
- 96.Gatz SA, Wiesmüller L. p53 in recombination and repair. Cell Death Differ. 2006;13:1003–16. doi: 10.1038/sj.cdd.4401903. [DOI] [PubMed] [Google Scholar]
- 97.Hartman AR, Ford JM. BRCA1 and p53: compensatory roles in DNA repair. J Mol Med. 2003;81:700–7. doi: 10.1007/s00109-003-0477-0. [DOI] [PubMed] [Google Scholar]
- 98.Wang Y, Cortez D, Yazdi P, et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–39. [PMC free article] [PubMed] [Google Scholar]