

Abstract
The mdx mouse, a model of the human Duchenne muscular dystrophy, displays impaired contractile function in skeletal, cardiac and smooth muscles. We explored the possibility that ryanodine receptor (RYR) expression could be altered in vascular muscle. The three RYR sub-types were expressed in portal vein myocytes. As observed through mRNA and protein levels, RYR2 expression was strongly decreased in mdx myocytes, whereas RYR3 and RYR1 expression were unaltered. The use of antisense oligonucleotide directed against RYR sub-types indicated that caffeine-induced Ca2+ response and Ca2+ spark frequency depended on RYR2 and RYR1. In mdx mice, caffeine-induced Ca2+ responses were decreased in both amplitude and maximal rate of rise, and the frequency of Ca2+ sparks was also strongly decreased. The gentamycin treatment was able to increase both the expression of RYR2 and the caffeine-induced Ca2+ response to the same level as that observed in wild-type mice. Taken together, these results confirm that both RYR1 and RYR2 are required for vascular Ca2+ signalling and indicate that inhibition of RYR2 expression may account for the decreased Ca2+ release from the SR in mdx vascular myocytes. Finally, we suggest that gentamycin can restore the Ca2+ signalling in smooth muscle from mdx mice by increasing RYR2 and dystrophin expression. These results may help explain the reduced efficacy of contraction in vascular myocytes of mdx mice and Duchenne muscular dystrophy–afflicted patients. Gentamycin treatment could be a good therapeutic tool to restore the vascular function.
Keywords: ryanodine receptor, calcium signalling, muscular dystrophy, mdx, gentamycin, smooth muscle, vascular myocyte
Introduction
Dystrophin is a cytoskeletal structural protein normally expressed in skeletal, cardiac and smooth muscles [1]. The absence of dystrophin causes Duchenne muscular dystrophy (DMD), characterized by progressive muscle weakness. Much of our knowledge comes from experimental evidence obtained from the mdx mouse, the murine animal model of DMD that also lacks the expression of dystrophin. In mdx mice, elimination of dystrophin expression leads to the absence of the dystrophin-associated glycoprotein complex (DAG), which serves as a membrane anchor. Without DAG, the overall membrane integrity is compromised, leading to impaired Ca2+ homeostasis, for example, increased Ca2+ influx, activation of Ca2+-dependent proteases and muscle necrosis [2]. However, conflicting data have been reported for both the increase in resting Ca2+ concentration ([Ca2+]i) and the amplitude of Ca2+ responses upon stimulation [3–5], indicating that the molecular mechanisms of the disease are still unclear. The dysfunction of smooth muscle due to the absence of dystrophin in DMD was earlier suggested [6].
A key aspect of the Ca2+ signalling pathway is represented by its spatial and temporal complexity that regulates muscle excitation– contraction coupling. Two well-characterized Ca2+ channel complexes represent the central elements of this excitation–contraction coupling: the voltage-gated Ca2+ channel (CaV) and the ryanodine-sensitive Ca2+ release channel (RYR). In skeletal muscle, CaV and RYR channels interact directly at the triad junction, whereas in cardiac and smooth muscles, a Ca2+-induced Ca2+ release (CICR) mechanism appears to be the dominant process for the release of Ca2+ from the sarcoplasmic reticulum (SR). In addition, the three RYR sub-types are differentially expressed in muscles, as RYR1 and RYR2 are mainly found in skeletal and cardiac muscles, respectively [7, 8]. In vascular smooth muscle, both RYR1 and RYR2 are required for Ca2+ release from the SR, although the three RYR sub-types are expressed [9, 10]. The decrease of RYR2 expression in the duodenal muscles of mdx mice was reported [11], but it has been under debate in the case of cardiac myocytes [12, 13]. Finally, the contraction of portal vein in mdx mice is decreased [14].
In mdx mice and in patients with DMD, the absence of dystrophin is due to a point mutation generating a premature stop codon [15]. Aminoglycoside antibiotics, such as gentamycin, have been described to suppress the nonsense mutation in vivo[16]. This family of antibiotics can restore dystrophin function in mdx skeletal muscle [17] and the mechanotransduction and vasodilation in vascular muscle [18]. These results suggest that aminoglycoside treatment is a putative therapeutic approach for DMD [19].
In this study, we investigated the different expression of RYR sub-types in portal vein smooth muscle from mdx mice. We addressed this issue by using RT-PCR and Western blot to evaluate the expression of RYR sub-types in myocytes from wild-type and mdx mice changes in Ca2+ signals of mdx vascular myocytes by confocal microscopy with Fluo-4. We show for the first time that the RYR2 sub-type expression is selectively impaired in vascular myocytes of mdx mice, and that inhibition of RYR2 expression reduced Ca2+ release from the SR and spontaneous Ca2+ sparks, leading to impaired contractile function. Finally, we report that treatment of mdx mice with gentamycin restored both the expression level of RYR2 and the caffeine-induced Ca2+ response.
Materials and methods
Cell preparation
The investigation conforms to the European Community and French guiding principles in the care and use of animals (authorization to perform animal experiments: A-33-063-003). Wild-type control (C57Bl10) and mdx mice aged 5–8 months were killed by cervical dislocation. Isolated myocytes were obtained from the portal vein by enzymatic dispersion and maintained in culture [20]. Cells were seeded on glass slides in M199 culture medium containing 10% foetal calf serum, 20 units/ml penicillin and 20 μg/ml streptomycin. Cells were used for Ca2+ experiments within 24 hrs. To characterize the function of each RYR sub-type by antisense oligonucleotide strategy, the culture conditions were modified as previously described [21]. (In the results and figures, myocytes isolated from wild-type ‘control’ C57Bl10 and mdx mice are indicated as C57 and mdx, respectively.)
Microinjection of oligonucleotides
Sequences of phosphorothioate antisense oligonucleotides (asRYR1-3) used in the present study as well as the method of intranuclear oligonucleotide injection were previously described [9]. The vascular myocytes located in a marked area of glass slides were injected, whereas non-injected cells outside this marked area were used as controls. To control the quality of intranuclear microinjection we used Cy5-tagged antisense oligonucleotides, and to verify the efficiency of RYR sub-type deletion the immunostaining of RYR sub-type was performed on injected cells (data not shown).
Gentamycin treatment
The mdx mice were intraperitoneally injected daily with gentamycin (MP Biomedicals Illkirch, France; ref 194530; lot R16139) or saline solution (34 mg/kg per injection in 0.4 ml saline solution [18]). Mice were killed after 14 days of treatment.
RT-PCR
Total RNA was extracted from freshly dissociated portal vein myocytes using the RNA preparation kit from Epicentre (Madison, WI, USA) following the instructions of the supplier. The RNA concentration was determined by OD260 with Eppendorf biophotometer (Hamburg, Germany). The RT reaction was performed on 50 ng RNA using Sensiscript RT kit (Qiagen, Courtaboeuf, France). Total RNA was incubated with oligodT(15) primers at 65°C for 5 min. After a 5-min. cooling time at 4°C, the RT mix was added and the mixture was incubated for 60 min. at 37°C. PCR conditions and primer design were previously detailed [9]. Briefly, PCR was performed with 0.25 μg cDNA, 1.25 units of HotStartTaq DNA polymerase (Qiagen), 1 mmol/l of each primer and 200 μmol of each deoxynucleotide triphosphate, in a final volume of 25 μl. Amplicons were separated by electrophoresis (2% agarose gel) and visualized by Syber green staining. Gels were photographed with EDAS 120 and analyzed with KDS1D 2.0 software (Kodak Digital Science, Paris, France). To determine the relative effect of treatments on RYR expression at the mRNA level, the GAPH amplicon was used as a reference to normalized RYR amplicons. As described in our previous study, PCRs were carried out at different numbers of steps to determine the best number of cycles to quantify amplicons.
Western blot
Smooth muscles from the portal vein of C57 and mdx mice treated or not with gentamycin were homogenized in an appropriate volume of SDS 10% supplemented with protease inhibitor cocktail (Sigma, Lyon, France). After centrifugation (10 min., 10,000 ×g), supernatants were collected and the protein content was measured with a DC protein assay kit (Biorad, Marne la Coquette, France). Equal amounts of protein (50 or 100 μg) from C57, mdx and gentamycin-treated mdx tissues were heated at 95°C for 3 min. in Laemmli-loaded buffer, separated by 4–12% SDS-PAGE (Geba Gels, Interchim, Montlucon, France) and electrically transferred to PVDF membranes (70 min., 100 V, 4°C). Non-specific binding was blocked by incubating membrane in phosphate buffer saline/Tween (0.1%) containing 5% non-fat dry milk for 1 hr and blots were incubated (overnight, 4°C) with anti-RyR2 (1:1000), anti-RYR1 (1:1000), anti-RYR3 (1:500), anti-InsP3R3 (1:1000) and anti-dystrophin (1:1000) antibodies. Primary antibodies were detected with a horseradish peroxidase–coupled secondary antibody (Santa Cruz Biotechnology, Heidelberg, Germany) using an enhanced chemoluminescence kit (Amersham Biosciences, Orsay, France). Photographic films were analyzed with the KDS1D 2.0 software. Each experiment was performed three times.
Ca2+ measurements
In experiments using laser scanning confocal microscopy, cells were loaded with a physiological solution containing 4 μmol/l fluo-4-AM (20 min. at 35°C). These cells were washed and allowed to cleave the dye to the active fluo-4 compound for at least 10 min. Images were acquired in the line-scan mode (2 msec per scan) of a Bio-Rad MRC 1024ES with Lasersharp2000 software (Biorad). Briefly, Fluo-4 was excited at 488 nm, and the emitted fluorescence was filtered and measured at 540 ± 30 nm. Images were analyzed with IDL software (RSI) as previously detailed [21]. The fluorescence value of each pixel in the line (F) was divided by the fluorescence value of the same pixel at rest levels (F0); fluorescence signals are thus expressed as pixel-per-pixel fluorescence ratios (F/F0). Amplitudes of the responses are expressed as F/F0, which represents the difference between maximal ratio and ratio at the rest level. An algorithm was developed to characterize Ca2+ sparks in the line-scan image by their spatial diffusion (2 μm), amplitude, time to half-decay and upstroke velocity. All variables were measured and compared in control and mdx mice.
To verify that the Ca2+ probe has similar fluorescence properties in both control and mdx mice, we performed ratiometric measurements of [Ca2+]i with an indo-1 set-up, as described elsewhere [11]. No difference was measured in both probe loading and resting [Ca2+]i level between myocytes from control and mdx mice (not shown).
Caffeine was applied by pressure ejection from a glass pipette for the period indicated in the figures. All experiments were carried out at 26 ± 1°C.
Solutions
The physiological solution contained (in mM) NaCl 130, KCl 5.6, MgCl2 1, CaCl2 2, glucose 11 and HEPES 10, pH 7.4, with NaOH. Caffeine was applied to the recorded cell by pressure ejection for the period indicated in the records.
Chemicals and drugs
Collagenase was obtained from Worthington (Freehold, NJ, USA). M199 medium, streptomycin, penicillin, glutamine and pyruvate were from Invitrogen (Cergy-Pontoise, France). Foetal calf serum was from Biowest (Nuaillé, France). Caffeine was from Merck (Darmstadt, Germany). The anti-RYR1, anti-RYR2, anti-RYR3 and anti-dystrophin–specific antibodies were from Millipore (Molsheim, France). The mouse anti-InsP3R3 antibody was from Transduction Laboratory (BD Bioscience, Le Pont de Claix, France). Fluo 4-acetoxymethylester (fluo-4-AM) was from Interchim. All other products were from Sigma (St. Louis, MO).
Data analysis
Data are expressed as means ± S.E.M.; n represents the number of cells or experiments as indicated in the legend of the figure. Significance was tested by means of paired Student’s t-test when cells were their own control; otherwise, unpaired test was used. P values <0.05 were considered as significant. To measure the effect of gentamycin treatment, a one-way ANOVA was used in association with Dunett’s test. P values <0.05 were considered as significant.
Results
RYR sub-types expression in myocytes from mdx mice
Expression of RYR sub-types was first detected with RT-PCR in vascular myocytes. To compare expression levels obtained from independent experiments, we normalized the data by using GAPDH as an internal standard. RYR1, RYR2 and RYR3 mRNAs were detected in isolated portal vein myocytes from control and mdx mice. For 30 and 35 cycles, similar levels of RYR1 and RYR3 mRNAs were observed in control and mdx mice, whereas the expression of RYR2 mRNA was strongly inhibited in mdx portal vein myocytes (Fig. 1). In the same manner, we investigated the expression of InsP3-activated Ca2+ release channels (InsP3R) and voltage-dependent Ca2+ channels (CaV). Neither expressions of both InsP3R and CaV appeared modified (not shown).
Figure 1.
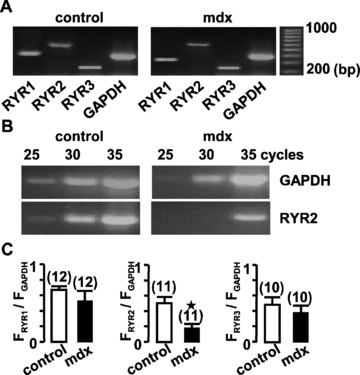
Expression of RYRs in portal vein muscle from control and mdx mice. (A) RT-PCR analysis of RYR1, RYR2, RYR3 and GAPDH mRNAs in freshly dissociated portal vein myocytes for 35 cycles. (B) RT-PCR analysis of RYR2 and GAPDH mRNAs for different cycle protocols. (C) Relative mRNA expression levels of the three RYR sub-types were compared with GAPDH expression in control (open bars) and mdx portal vein myocytes (filled bars). Molecular sizes standards are indicated in bp. Data are means ± S.E., with the number of experiments indicated in parentheses. ⋆, values significantly different from those obtained in control mice (P < 0.05).
These results were confirmed by Western blots in which 100 μg of total proteins were loaded. The loading control protein has been chosen by its molecular weight in a similar range of RYR. We investigated the expression of InsP3R as revealed by RT-PCR, which indicated that InsP3R3 was the most potentially expressed and it was not modified in mdx mice (not shown). Then, the expression of InsP3R3 was measured in the three experiments pooling five different control and mdx mice, and it did not differ between control and mdx mice. Therefore, InsP3R3 was used as a loading control to normalize the expression of RYR sub-types. In portal vein muscle, the RYR2 protein level was significantly inhibited in mdx mice, whereas the expression of RYR1 and RYR3 was not affected (Fig. 2).
Figure 2.
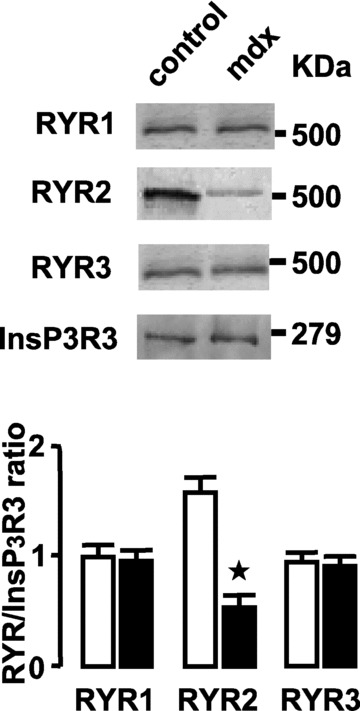
Western blotting of RYR sub-types expressed in portal vein muscles. RYR sub-types were revealed in proteins extracted from control and mdx mice. InsP3R3 expression was used as control loading (upper panel). Results from three mdx (filled bars) and control (open bars) preparations were normalized to the loading control (lower panel). Data are means ± S.E. with the number of experiments in parentheses. ⋆, values significantly different from those obtained in control mice (P < 0.05).
Only the expression of RYR2 was affected in mdx mice, but to verify that the decrease in RYR2 expression may affect the vascular responses of mdx mice, we studied the Ca2+ responses in isolated portal vein myocytes.
Caffeine-activated Ca2+ signal in mouse portal vein myocytes
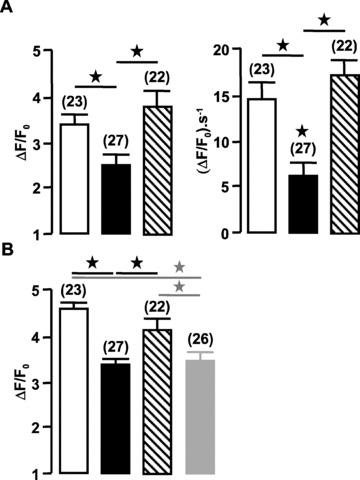
Applications of caffeine (10 mmol/l) activated propagation of Ca2+ waves in control and mdx myocytes (Fig. 3A). These caffeine-induced Ca2+ waves were characterized by their mean amplitude, time to peak and upstroke velocity. In mdx portal vein myocytes, caffeine-activated Ca2+ waves were significantly decreased compared with control myocytes, that is, the peak amplitude and maximal rate of rise were decreased, whereas the time to peak was increased (Fig. 3B). The inhibition of caffeine-activated Ca2+ responses by application of 1 μmol/l thapsigargin (n= 9) or 10 μmol/l ryanodine for 30 min. (n= 11) indicates that they depend on Ca2+ release from the SR by RYR.
Figure 3.
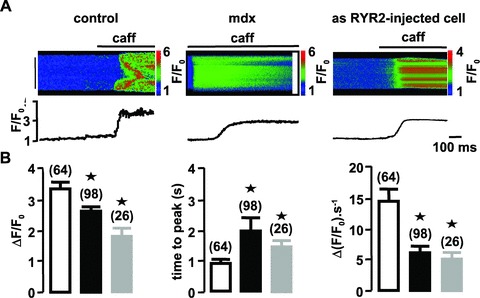
Caffeine-induced Ca2+ responses in control, mdx mice and asRYR2-injected portal vein myocytes. (A) Line-scan images and averaged fluorescence from the entire images evoked by external applications of 10 mmol/l caffeine in C57 control and asRYR2-injected and mdx portal vein myocytes. (B) Parameters of caffeine-induced Ca2+ responses (F/F0, time to peak and upstroke velocity) in vascular myocytes from control (open bars), mdx mice (filled bars) and asRYR2-injected C57 myocytes (grey bars), with the number of cells tested in parentheses. Cells were obtained from six different batches. ⋆, values significantly different from those obtained in control mice (P < 0.05). Myocytes were loaded with fluo 4-AM.
To investigate if the decreased expression of RYR2 may be responsible for the decrease of caffeine-induced Ca2+ response, we inhibited RYR2 expression in control myocytes by injecting antisense oligonucleotide directed against the RYR2 sub-type in C57 portal vein myocytes. As previously shown on rat and mouse myocytes [9, 22], (i) the inhibition of one RYR sub-type did not modify the expression of the others and (ii) the maximum inhibitory effect of asRYR on RYR sub-type expression was observed 2–3 days after nuclear injection. After each experiment, the decrease in RYR2 expression induced by asRYR2 treatment was verified by RYR2 immunostaining (not shown). Based on this culture time scale, the caffeine-induced Ca2+ responses were measured in myocytes injected with 10 μmol/l asRYR2. Firstly, the culture conditions cannot modify the caffeine-induced Ca2+ release (amplitude: 3.30 ± 0.4 ratio units; time to peak: 0.78 ± 0.11 sec; upstroke velocity 15 ± 2 ΔF/F0·s−1). Amplitude, time to peak and upstroke velocity of caffeine-induced Ca2+ waves were significantly reduced in asRYR2-injected cells compared with non-injected cells (Fig. 3A and B).
As described previously in rat, caffeine-induced Ca2+ responses were totally inhibited by the injection of the cocktail asRYR1 + asRYR2, and similar inhibitions as those obtained in asRYR2-injected cells were obtained in asRYR1-injected cells. In contrast, no inhibitory effect on the Ca2+ responses was observed in cells injected with asRYR3. It is noteworthy that injection of scrambled sequences of asRYR1 or asRYR2 had no effect on the caffeine-induced Ca2+ responses (Supplementary Information). These results indicate that as described in rat, both RYR1 and RYR2 are required for triggering Ca2+ responses in mouse portal vein myocytes [9], and the inhibition of RYR2 was sufficient to significantly inhibit partially caffeine-induced Ca2+ response.
The SERCA function was not modified in mdx mice; we applied thapsigargin (1 μM) to follow the Ca2+ leak induced by SERCA inhibition. The responses induced by thapsigargin in portal vein myocytes from control and mdx mice were similar.
Spontaneous Ca2+ sparks in mouse portal vein myocytes
RYR2 in association with RYR1 is also needed to encode spontaneous localized Ca2+ events (i.e., Ca2+ sparks) [9]. In control vascular myocytes, analysis of line-scan fluorescence images revealed spontaneous, spatially localized, Ca2+ sparks in about 28% of the cells tested (Fig. 4A). The means of amplitude (ΔF/F0), time at half decay (s) and upstroke velocity (ΔF/F0·s−1) of Ca2+ sparks were measured (Fig. 4B). The mean full width at half-maximum amplitude was estimated to 1.05 ± 0.09 μm (n= 18; four dissociations were tested). In portal vein myocytes from mdx mice, spontaneous Ca2+ sparks were revealed only in 6% of the cells tested (four dissociations were tested). The kinetic variables of these Ca2+ sparks were not significantly different from those measured in control myocytes (Fig. 4B). As illustrated in Fig. 4, it has been possible to visualize two different Ca2+ spark sites in a control cell, whereas it never happened in mdx myocytes. In asRYR2-treated cells, Ca2+ sparks were not observed in the 26 tested cells from three different cell cultures, which indicates that RYR2 was implicated in Ca2+ sparks in vascular myocytes.
Figure 4.
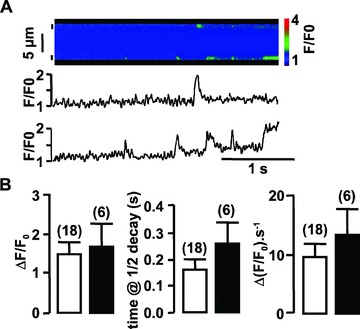
Typical Ca2+ sparks obtained spontaneously in portal vein myocytes from control mice. (A) Traces show (from top to bottom) the line-scan fluorescence image and averaged fluorescence from two 2-μm regions (indicated by small vertical bars) of the line-scan image. (B) Amplitude, time at half decay and upstroke velocity of spontaneous Ca2+ sparks observed in control (open bars) and mdx mice (filled bars). Data are means ± S.E. with the number of cells presenting Ca2+ sparks in parentheses. Cells were obtained from six different batches. Myocytes were loaded with fluo 4-AM.
Taken together, these results confirm that full expression of RYR2 is required for triggering Ca2+ sparks and waves in mouse vascular myocytes because inhibition of RYR2 in mdx myocytes inhibited the frequency of Ca2+ sparks and reduced caffeine-induced Ca2+ responses.
Gentamycin treatment has been shown to increase the expression of full-length dystrophin in mdx muscles. Therefore, we investigated the effect of gentamycin on RYR-dependent Ca2+ signalling in portal vein myocytes.
Effect of gentamycin on RYR expression
Gentamycin is an aminoglycoside compound. A property of this molecule family is its ability to shunt stop codon. Here it has been used to ‘improve’ the aberrant stop codon produced by the mutation observed in mdx mice. The mdx mice were treated by intraperitoneal injection of gentamycin sulphate (gentamycin-treated mdx mice) or injected with saline solution (untreated mdx mice) during 14 days. The duration of the treatment was described to be sufficient to reverse the decrease of vessel contractility observed in mdx mice [18]. Firstly, the efficiency of gentamycin treatment was verified by Western blot analysis of dystrophin expression. Dystrophin was revealed in control mice as well as in gentamycin-treated mdx mice, but dystrophin was not expressed in untreated mdx mice (Fig. 5). Secondly, the effect of gentamycin treatment on the expression of RYR sub-types was measured. From 25 to 35 cycles, the expression of RYR2 was significantly increased in myocytes from gentamycin-treated mdx mice in comparison with untreated mdx mice. The level of RYR2 expression was restored at the same level as was observed in myocytes from control mice (Fig. 6A). The expression of RYR1 was not significantly modified by the gentamycin treatment. The RYR2 sub-type could be negatively regulated by a dominant negative splice variant of RYR3 [23, 24]. The investigation of expression of both dominant negative and full-length RYR3 isoforms was performed by RT-PCR, which indicated that the level of each isoform was not modified by the gentamycin treatment (Fig. 6A). The expression levels of RYR1 and RYR3 were similar in control, gentamycin-treated and untreated mdx mice. The restoration by gentamycin treatment of RYR2 protein level was confirmed by Western blots, in which 100 and 50 μg of total proteins were loaded (Fig. 6B).
Figure 5.
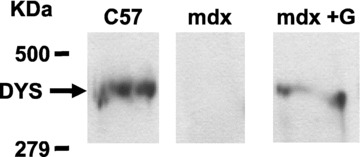
Western blotting of dystrophin in skeletal muscle. Dystrophin was revealed in proteins extracted from control, mdx and gentamycin-treated mdx mice. Each group of gentamycin-treated mdx mice was tested to verify the efficiency of gentamycin treatment.
Figure 6.
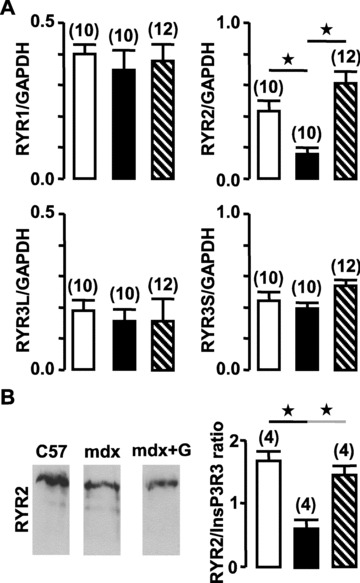
Effect of gentamycin on expression of RYRs in portal vein muscle. (A) Relative mRNA expression levels of the three RYR sub-types were revealed by RT-PCR (35 cycles) and compared with GAPDH expression in control (open bars), mdx (filled bars) and gentamycin-treated mdx mice (hatched bars). (B) RYR sub-types were revealed in proteins extracted (100 μg) from control, mdx and gentamycin-treated mdx mice. Results from four control (open bar), mdx (filled bar) and gentamycin-treated mdx mice (hatched bars) preparations were normalized to the loading control. Data are means ± S.E. with the number of experiments in parentheses. ⋆, values significantly different from those obtained in control mice (P < 0.05).
Effect of gentamycin on Ca2+ signalling
The amplitude and upstroke velocity of caffeine-activated Ca2+ release were significantly increased in myocytes from gentamycin-treated mdx mice in comparison with untreated mdx mice. It is noticeable that the caffeine-activated Ca2+ responses were identical in control C57 mice and in gentamycin-treated mdx mice (Fig. 7A). This last result indicated that the RYR-dependent Ca2+ release was restored by gentamycin treatment. The most important role of RYR in smooth muscle is the amplification of InsP3-dependent Ca2+ response activated by neurotransmitters; therefore, we investigated the effect of gentamycin on ACh-induced Ca2+ response. As illustrated in Fig.7B, the amplitude of ACh-evoked Ca2+ response was significantly reduced in mdx mice in comparison with those observed in control mice. This effect was similar to those observed in cells injected with asRYR2, indicating that the decrease of RYR2 expression was sufficient to decrease the CICR mechanism. The treatment with gentamycin increased the amplitude of the ACh-induced Ca2+ response in mdx mouse myocytes to a similar level as that observed in control mice.
Figure 7.
Effect of gentamycin on caffeine- and ACh-induced Ca2+ responses in mouse portal vein myocytes. (A) Amplitude and upstroke velocity of caffeine-induced Ca2+ responses in vascular myocytes from control (open bars), mdx (filled bars) and gentamycin-treated mdx mice (hatched bars). (B) Amplitude of ACh-induced Ca2+ response in portal vein from control (open bar), mdx (filled bar), gentamycin-treated mdx mice (hatched bar) and asRYR2-injected myocytes (grey bar). The number of cells tested is indicated in parentheses, and cells were obtained from three different batches (⋆ and ⋆, P < 0.05). Myocytes were loaded with fluo 4-AM.
Discussion
In patients with Duchenne muscular dystrophy and mdx mice, it is generally accepted that the missing link between the absence of dystrophin and muscle hypotonicity is due to alterations in Ca2+ homeostasis, but the molecular mechanisms are still poorly understood. We show here that RYR1 and RYR2 are essential for Ca2+ signals in mice vascular myocytes and that expression of the RYR2 sub-type is selectively decreased in vascular smooth muscles of mdx mice. In contrast, RYR3 and RYR1 expressions were not affected. In mdx vascular myocytes where RYR2 expression was strongly decreased, the caffeine-induced Ca2+ responses and the frequency of Ca2+ sparks were reduced. The injection of gentamycin in mdx mice led to restoration of both dystrophin and RYR2 expression to the same levels as those observed in control mice. Caffeine-induced Ca2+ response was also rescued by gentamycin treatment.
As previously reported in rat portal vein myocytes [9], both RYR1 and RYR2 are required in mouse portal vein myocytes for triggering caffeine-induced Ca2+ waves and Ca2+ sparks. In the urinary bladder from RYR1-KO mouse, RYR2 and RYR3 are sufficient for encoding spontaneous Ca2+ sparks and caffeine-activated Ca2+ waves, whereas the frequency of depolarization-activated Ca2+ sparks was decreased but the KO of RYR1 was not compensated by RYR2 over-expression [25]. From these data and the present ones, we may postulate that vascular RYR clusters are formed by both RYR1 and RYR2 (as homotetramers), and therefore suppression of either RYR1 or RYR2 decreases the caffeine-induced Ca2+ responses and the frequency of Ca2+ sparks. Interestingly, in mdx vascular myocytes, the reduction of RYR2 expression was also associated with the decrease of frequency of Ca2+ sparks. This observation strengthens the idea that both RYR1 and RYR2 are required for triggering Ca2+ sparks in mouse portal vein myocytes. The potential consequences could be the decrease of vascular tone because Ca2+ sparks activate transient outward K+ currents, which exert a tonic hyperpolarizing and inhibitory influence on the contraction of vascular myocytes [26, 27]. In the absence of Ca2+ sparks, myocytes should be depolarized and incompletely relaxed, thus limiting contractility. In patients with DMD and mdx mice, the decrease of relaxation of α-adrenergic-induced contraction was attributed to the decrease of NO muscle production [28, 29]. Our data suggest that the vasoregulation by relaxation could also be blunted by the decrease of Ca2+ sparks frequency.
The decrease of RYR2 expression observed in mdx mice induced the same decrease of caffeine-induced Ca2+ response as that observed in asRYR2-injected cells from control mice, indicating that the CICR mechanism was affected in the portal vein, as previously described in duodenum myocytes [11]. In fact, the portal vein displays a spontaneous contractile activity regulated by action potentials. The contraction phase is activated by depolarization owing to CaV channel activation that induces the CICR mechanism. The relaxation is due to the repolarization phase as a result of Ca2+-dependent K+ currents. In mdx mice, the CICR mechanism and activation of Ca2+-dependent K+ currents are both affected, and our data suggest that a Ca2+ signalling defect could explain the decrease of spontaneous contraction of portal vein from the mdx mice [14] or the deregulation of vascular function observed in arteries [18, 30]. In theory, the spontaneous Ca2+ sparks could be responsible for the relaxation side of the basal vascular tone, but a change in the basal vascular tone was not clearly identified [14]. The contraction could be also activated by agonists. The following of ACh-induced Ca2+ response indicates that the amplification of Ca2+ signals by the CICR mechanism was decreased in mdx mice. This result also potentially explains the decrease of vascular reactivity observed in portal vein from mdx mice [14] or also vessels [30]. In mdx skeletal muscle, the absence of dystrophin disturbs the plasma membrane architecture that modifies the action potential–induced Ca2+ signal [31, 32]. The resulting increase of resting [Ca2+] induced cell proteolysis and cell death [33]. In smooth muscle from mdx mice, the modification of Ca2+ homeostasis induced by the decrease of RYR2 expression could be interpreted as a vascular tissue adaptation to the fragility because of the absence of dystrophin, as proposed in carotid arteries [30]. The regulation of RYR expression appears as the subtle mechanism to regulate Ca2+ signalling in smooth muscle not only in physiological conditions such as during pregnancy [23] but also in specific environmental stimulation such as microgravity [34]. Our results also confirm that the adaptation of mdx vessels as observed in mesenteric arteries appears insensitive to vasodilator agents. Moreover, this study indicates that the contractility regulation does not have the same amplitude as those observed in control mice [35]. Finally, our results suggest that a functional linkage might exist between dystrophin and RYR in smooth muscle, and the regulation of RYR expression could be an adaptation of smooth muscle physiology. We can speculate that the decrease of CICR via RYR expression is able to limit Ca2+-induced cell death in mdx smooth muscle.
As described in severe cystic fibrosis, the use of aminoglycoside restores CFTR function by overcoming the premature stop codon [36], and clinical assays were performed [37]. However, as reviewed recently [38], the restoration of dystrophin expression in mdx mice by gentamycin was effective in vitro and in vivo, but its use was limited by the nature of mutation and was not efficient for all patients with DMD. The exon-skipping property of gentamycin was proposed to restore dystrophin expression in patients with DMD and in mdx mice [17]. We propose here that in smooth muscle, gentamycin restores the RYR2 expression level and the associated Ca2+ signalling. Together with results showing the restoration of NOS expression associated with NO-dependent dilatation [18], our results reinforced that gentamycin treatment could be proposed to patients with DMD. Recently, a study indicated that the restoration of dystrophin, only in vascular smooth muscle, improves aberrant regulation observed in mdx mouse vessels [39]. Our results, obtained in mdx mouse portal vein, prove that the restoration of dystrophin is linked to the improvement of Ca2+ signalling by the restoration of RYR2 expression. They also suggest that the RYR2 sub-type could be implicated in the Ca2+-dependent mechanism of relaxation via spontaneous Ca2+ sparks, whereas RYR1, by its function in depolarization-activated Ca2+ sparks, was more strongly implicated in contraction.
In conclusion, our study suggests that the restoration of dystrophin and RYR2 expression by gentamycin in mdx mice induces the restoration of Ca2+ signals implicated in vascular tone.
Acknowledgments
We acknowledge N. Biendon for technical support and Dr. A. Prévot for proofreading. This work was supported by grants from Association Française contre les myopathies (AFM) and Centre National de la Recherche Scientifique (CNRS), France.
Supporting Information
Fig. S1. Effects of asRYRI, asRYR2, asRYR3 and asSCBantisense oligonucleotides on caffeine-induced Ca2+responses in control portal vein myocytes. Typical Ca2+waves obtained from the entire line-scan images induced by 10mmol/L caffeine in non-injected control cells (A); in cellsinjected with 10 μmol/L asRYR2 antisense oligonucleotide(B) and in cells injected with 10 μmol/L asRYRI antisenseoligonucleotide (C). Compiled data showing the effects of 10μmol/L asRYRI, asRYR2, asRYR3 and asSCB (antisenseoligonucleotide corresponding to the scramble sequence of asRYR2)on both peak amplitude (D) and upstroke velocity (E)of caffeine-induced Ca2+ waves. Data are means ± S.E. in non-injected cells (open bars) and antisense oligonucleotide-injected cells (filled bars) with the number of cells tested in parentheses. Cells were obtained from three different batches. *, values significantly different from those obtained in non-injected cells (p < 0.05). Myocytes were loaded with fluo 4-AM.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
References
- 1.Gillis JM. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J Muscle Res Cell Motil. 1999;20:605–25. doi: 10.1023/a:1005545325254. [DOI] [PubMed] [Google Scholar]
- 2.Turner PR, Westwood T, Regen CM, et al. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 1988;335:735–8. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- 3.Bakker AJ, Head SI, Williams DA, et al. Ca2+ levels in myotubes grown from the skeletal muscle of dystrophic (mdx) and normal mice. J Physiol. 1993;460:1–13. doi: 10.1113/jphysiol.1993.sp019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Head SI, Williams DA, Stephenson DG. Abnormalities in structure and function of limb skeletal muscle fibres of dystrophic mdx mice. Proc Biol Sci. 1992;248:163–9. doi: 10.1098/rspb.1992.0058. [DOI] [PubMed] [Google Scholar]
- 5.Rivet-Bastide M, Imbert N, Cognard C, et al. Changes in cytosolic resting ionized calcium level and in calcium transients during in vitro development of normal and Duchenne muscular dystrophy cultured skeletal muscle measured by laser cytofluorometry using indo-1. Cell Calcium. 1993;14:563–71. doi: 10.1016/0143-4160(93)90077-j. [DOI] [PubMed] [Google Scholar]
- 6.Miyatake M, Miike T, Yoshioka K, et al. Possible systemic smooth muscle layer dysfunction due to a deficiency of dystrophin in Duchenne muscular dystrophy. J Neurol Sci. 1989;93:11–7. doi: 10.1016/0022-510x(89)90157-3. [DOI] [PubMed] [Google Scholar]
- 7.Takeshima H, Iino M, Takekura H, et al. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature. 1994;369:556–9. doi: 10.1038/369556a0. [DOI] [PubMed] [Google Scholar]
- 8.Takeshima H, Komazaki S, Hirose K, et al. Embryonic lethality and abnormal cardiac myocytes in mice lacking ryanodine receptor type 2. EMBO J. 1998;17:3309–16. doi: 10.1093/emboj/17.12.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coussin F, Macrez N, Morel JL, et al. Requirement of ryanodine receptor subtypes 1 and 2 for Ca2+-induced Ca2+ release in vascular myocytes. J Biol Chem. 2000;275:9596–603. doi: 10.1074/jbc.275.13.9596. [DOI] [PubMed] [Google Scholar]
- 10.Mironneau J, Coussin F, Morel JL, et al. Calcium signalling through nucleotide receptor P2×1 in rat portal vein myocytes. J Physiol. 2001;536:339–50. doi: 10.1111/j.1469-7793.2001.0339c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morel JL, Rakotoarisoa L, Jeyakumar LH, et al. Decreased expression of ryanodine receptors alters calcium-induced calcium release mechanism in mdx duodenal myocytes. J Biol Chem. 2004;279:21287–93. doi: 10.1074/jbc.M311124200. [DOI] [PubMed] [Google Scholar]
- 12.Rohman MS, Emoto N, Takeshima Y, et al. Decreased mAKAP, ryanodine receptor, and SERCA2a gene expression in mdx hearts. Biochem Biophys Res Commun. 2003;310:228–35. doi: 10.1016/j.bbrc.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol. 2007;292:H846–55. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- 14.Mancinelli R, Tonali P, Romani R, et al. Mechanical properties of smooth muscle portal vein in normal and dystrophin-deficient (mdx) mice. Exp Physiol. 1999;84:929–40. [PubMed] [Google Scholar]
- 15.Sicinski P, Rowiński J, Warchoł JB, et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–80. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 16.Burke JF, Mogg AE. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 1985;13:6265–72. doi: 10.1093/nar/13.17.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barton-Davis ER, Cordier L, Shoturma DI, et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–81. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loufrani L, Dubroca C, You D, et al. Absence of dystrophin in mice reduces NO-dependent vascular function and vascular density: total recovery after a treatment with the aminoglycoside gentamicin. Arterioscler Thromb Vasc Biol. 2004;24:671–6. doi: 10.1161/01.ATV.0000118683.99628.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura S, Ito K, Miyagi T, et al. A novel approach to identify Duchenne muscular dystrophy patients for aminoglycoside antibiotics therapy. Brain Dev. 2005;27:400–5. doi: 10.1016/j.braindev.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 20.Fritz N, Mironneau J, Macrez N, et al. Acetylcholine-induced Ca2+ oscillations are modulated by a Ca2+ regulation of InsP3R2 in rat portal vein myocytes. Pflugers Arch. 2008;456:277–83. doi: 10.1007/s00424-007-0379-z. [DOI] [PubMed] [Google Scholar]
- 21.Fritz N, Macrez N, Mironneau J, et al. Ryanodine receptor subtype 2 encodes Ca2+ oscillations activated by acetylcholine via the M2 muscarinic receptor/cADP-ribose signalling pathway in duodenum myocytes. J Cell Sci. 2005;118:2261–70. doi: 10.1242/jcs.02344. [DOI] [PubMed] [Google Scholar]
- 22.Dabertrand F, Morel JL, Sorrentino V, et al. Modulation of calcium signalling by dominant negative splice variant of ryanodine receptor subtype 3 in native smooth muscle cells. Cell Calcium. 2006;40:11–21. doi: 10.1016/j.ceca.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Dabertrand F, Fritz N, Mironneau J, et al. Role of RYR3 splice variants in calcium signaling in mouse non-pregnant and pregnant myometrium. Am J Physiol Cell Physiol. 2007;293:C848–54. doi: 10.1152/ajpcell.00069.2007. [DOI] [PubMed] [Google Scholar]
- 24.Jiang D, Xiao B, Li X, et al. Smooth muscle tissues express a major dominant negative splice variant of the type 3 Ca2+ release channel (ryanodine receptor) J Biol Chem. 2003;278:4763–9. doi: 10.1074/jbc.M210410200. [DOI] [PubMed] [Google Scholar]
- 25.Fritz N, Morel JL, Jeyakumar LH, et al. RyR1-specific requirement for depolarization-induced Ca2+ sparks in urinary bladder smooth muscle. J Cell Sci. 2007;120:3784–91. doi: 10.1242/jcs.009415. [DOI] [PubMed] [Google Scholar]
- 26.Mironneau J, Arnaudeau S, Macrez-Lepretre N, et al. Ca2+ sparks and Ca2+ waves activate different Ca2+-dependent ion channels in single myocytes from rat portal vein. Cell Calcium. 1996;20:153–60. doi: 10.1016/s0143-4160(96)90104-9. [DOI] [PubMed] [Google Scholar]
- 27.Nelson MT, Cheng H, Rubart M, et al. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–7. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 28.Thomas GD, Sander M, Lau KS, et al. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci USA. 1998;95:15090–5. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sander M, Chavoshan B, Harris SA, et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000;97:13818–23. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dye WW, Gleason RL, Wilson E, et al. Altered biomechanical properties of carotid arteries in two mouse models of muscular dystrophy. J Appl Physiol. 2007;103:664–72. doi: 10.1152/japplphysiol.00118.2007. [DOI] [PubMed] [Google Scholar]
- 31.Woods CE, Novo D, DiFranco M, et al. The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J Physiol. 2004;557:59–75. doi: 10.1113/jphysiol.2004.061291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Weisleder N, Collet C, et al. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat Cell Biol. 2005;7:525–30. doi: 10.1038/ncb1254. [DOI] [PubMed] [Google Scholar]
- 33.Deconinck N, Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 34.Morel JL, Boittin FX, Halet G, et al. Effect of a 14-day hindlimb suspension on cytosolic Ca2+ concentration in rat portal vein myocytes. Am J Physiol. 1997;273:H2867–75. doi: 10.1152/ajpheart.1997.273.6.H2867. [DOI] [PubMed] [Google Scholar]
- 35.Loufrani L, Henrion D. Vasodilator treatment with hydralazine increases blood flow in mdx mice resistance arteries without vascular wall remodelling or endothelium function improvement. J Hypertens. 2005;23:1855–60. doi: 10.1097/01.hjh.0000183944.25832.81. [DOI] [PubMed] [Google Scholar]
- 36.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–9. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 37.Sermet-Gaudelus I, Renouil M, Fajac A, et al. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med. 2007;5:5. doi: 10.1186/1741-7015-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bidou L, Hatin I, Perez N, et al. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther. 2004;11:619–27. doi: 10.1038/sj.gt.3302211. [DOI] [PubMed] [Google Scholar]
- 39.Ito K, Kimura S, Ozasa S, et al. Smooth muscle-specific dystrophin expression improves aberrant vasoregulation in mdx mice. Hum Mol Genet. 2006;15:2266–75. doi: 10.1093/hmg/ddl151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item