

Abstract
Chondrocytes’ hypertrophy includes metabolic changes, matrix remodelling, proliferation and apoptosis, characteristics associated with the progression of osteoarthritis. We investigated a possible association among Runt-related transcription factor 2 (RUNX-2), SOX-9 and fibroblast growth factor (FGF)-23 mRNA expressions in articular chondrocytes in order to elucidate their contribution in the osteoarthritic hypertrophic cartilage. SOX-9, FGF-23, RUNX-2 and matrix metalloproteinase (MMP)-13 mRNA expressions were evaluated in osteoarthritic and normal chondrocytes by real-time PCR whereas MMP-13 protein expression by immunofluorescense. RUNX-2, FGF-23 and SOX-9 were down-regulated using small interfering RNA technology and transfection with liposomes. The effect of human recombinant FGF-23 (hrFGF-23) on SOX-9 and RUNX-2 expression was tested in normal chondrocytes. We found higher expression of RUNX-2 and FGF-23 and a decreased expression of SOX-9 mRNA in osteoarthritic chondrocytes compared to normal (P < 0.0001). RUNX-2 down-regulation resulted in reduced MMP-13 expression in osteoarthritic chondrocytes and inhibition of SOX-9 in increased RUNX-2 and MMP-13 mRNA expression in normal chondrocytes, whereas inhibition of FGF-23 resulted in reduced RUNX-2 mRNA expression in osteoarthritic chondrocytes (all P < 0.0001). Silencing of RUNX-2 or FGF-23 did not affect SOX-9 mRNA levels in osteoarthritic chondrocytes. Moreover simultaneous down-regulation of SOX-9 and up-regulation of FGF-23 mRNA expressions in normal chondrocytes resulted in additive up-regulation of RUNX-2 mRNA expression. Treatment of normal chondrocytes with hrFGF-23 resulted in increased RUNX-2 mRNA expression, whereas it had no effect on SOX-9 mRNA expression. We demonstrated convincing associations among RUNX-2, SOX-9 and FGF-23 in relation to MMP-13 expression in osteoarthritic chondrocytes, contributing to a better understanding of the abnormal gene expression and cartilage degeneration processes associated with osteoarthritis.
Keywords: RUNX-2, SOX-9, FGF-23, osteoarthritis, chondrocytes
Introduction
Chondrogenesis is a multi-step cell differentiation pathway genetically characterized by a complex cascade of specific genes, including bone morphogenetic proteins, fibroblast growth factors (FGFs), their signalling molecules and transcription factors, such as homebox factors [1, 2]. Chondrocytes within articular cartilage are responsible for the maintenance of the specialized extracellular matrix and for its biomechanical properties. Extracellular matrix and chondrocytes integrity are based on the expression of several matrix genes, such as Col2a1, Col9a1, Col11a2, aggrecan and cartilage link protein (CRTL1), which are under the control of anabolic and catabolic transcription factors [3]. SOX-9 is an anabolic transcription factor, which belongs to the SOX (Sry-related high mobility group box) family and plays an important role during cartilage development [4]. It is expressed in chondrocytes and other tissues including central nervous system and urogenital systems [5–8]. SOX-9 has been shown to contribute to chondrogenesis by activating cartilage-specific genes such as Col2a1[3, 9], Col11a2[10] and aggrecan[11]. Moreover, SOX-9 has been found down-regulated in osteoarthritic cartilage compared to normal adult human articular chondrocytes, suggesting that SOX-9 might be involved in maintaining the chondrocytic phenotype in normal and osteoarthritic cartilage [12, 13]. In view of the importance of SOX-9 in the development and maintenance of chondrocytes phenotype, its down-regulation in OA is clearly likely to contribute to cartilage pathology.
Changes in chondrocyte behaviour, including matrix remodelling, proliferation and apoptosis, are associated with OA progression and these behaviours are characteristic of chondrocyte maturation and hypertrophy in developing growth plates [14–16]. When chondrocytes acquire a hypertrophic osteoarthritic phenotype, they start to express Col10a1 and not Col2a1[3]. Runt-related transcription factor 2 (RUNX-2), which was originally isolated for its ability to activate transcription of the osteoblast-specific osteocalcin gene [17–18], is known to induce chondrocyte hypertrophy [19–21]. Additionally several other transcription factors, such as Shox/Shox2, Dlx5, MEF2C, have been identified as positive or negative regulators of chondrocyte hypertrophy in mouse models [22–25]. Overexpression of RUNX-2 in chondrocytes promotes collagen type X and alkaline phosphatase expression, which are both up-regulated in OA cartilage [26–29]. As RUNX-2 activity is a key determinant of chondrocyte maturation in the growth plate and experimentally in permanent cartilage, it might as well be a focal point of the abnormal signalling mechanisms that promote the initiation and progression of OA [30]. Recent studies have shown that SOX-9 acts as a transcriptional repressor for osteoblasts differentiation and chondrocyte hypertrophy partly via inhibition of RUNX-2 transactivation [31].
Furthermore, it has been shown that FGF signalling up-regulates RUNX-2 expression and activity in osteoblasts [32, 33]. FGF-23 is a novel member of the FGF family and is an important regulator of serum phosphorus, which is essential in chondrocyte differentiation and mineralization [34]. FGF-23 acts as a negative regulator of chondrogenesis and may induce premature exit of proliferating chondrocytes into hypertrophy leading to shorter bone growth [35]. Recently it was shown that FGF-23 is localized in the hypertrophic chondrocytes and may play a role in chondrocyte maturation [36]. Moreover, in phosphorus-loaded rats overexpression of FGF-23 resulted in smaller growth plate with significant reduction in chondrocyte proliferation, which was maintained by down-regulating the transcription factor RUNX-2 [37–39].
In order to advance our understanding regarding the pathways involved in the catabolic and degenerative processes in osteoarthritic articular cartilage, we investigated the possible associations among RUNX-2, FGF-23 and SOX-9 in osteoarthritic cartilage.
Materials and methods
Patients and cartilage tissue samples
Articular cartilage samples were obtained from femoral heads, femoral condyles and tibial plateaus of patients with primary osteoarthritis undergoing knee replacement surgery at the Orthopaedics Department of University Hospital of Larissa. A total of 33 patients were included in this study (28 females and 5 males; mean age 68.91 ± 6.97 years). Normal cartilage was obtained from 10 individuals (six females and four males; mean age 61.70 ± 18.17, range 27–78; mean BMI 23.80 ± 4.34). Radiographs were obtained before surgery and graded according to the Kellgren–Lawrence system. Twenty-six patients had score 4, six patients had score 3 and one patient had score 2. The radiographs were assessed by two independent observers who were blinded to all data of the individuals. Patients with rheumatoid arthritis and other autoimmune disease as well as chondrodysplasias, infection-induced OA and post-traumatic OA were excluded from the study. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the Local Ethical Committee of the University Hospital of Larissa.
Isolation of RNA from fresh human cartilage and cultured chondrocytes
Fresh tissue, within 1 hr from surgery, was dissected and total cellular RNA was extracted using Trizol reagent (Invitrogen, Life Technologies, Paisley, UK). Cultured chondrocytes were trypsinized and collected and total cellular RNA was extracted using Trizol reagent. Preservation of 28S and 18S ribosomal RNA species was used to assess RNA integrity and only these samples were included in the study.
Primary cultures of human articular chondrocytes, normal and osteoarthritic
Articular cartilage was transported from the surgical room in Hanks balanced salt solution medium, was immediately dissected and subjected to sequential digestion with 1 mg/ml pronase (Roche Applied Science, Roche Diagnostics, GmbH, Penzberg, Germany) for 90 min. and 1 mg/ml collagenase P (Roche Applied Science) for 3 hrs at 37°C. Chondrocytes were counted and checked for viability using trypan blue staining. More than 95% of the cells were viable after isolation. Chondrocytes were seeded in six-well plates with Dulbecco’s modified eagles medium/Ham’s F-12 (DMEM/F-12) (GIBCO BRL, UK) plus 5% foetal bovine serum and were incubated at 37°C.
RUNX-2, FGF-23, SOX-9 and matrix metalloproteinase (MMP)-13 quantitative mRNA expression by real-time PCR
Transcription of 0.1 μg RNA to cDNA was performed with the AMV Kit (Roche, Indianapolis, IN, USA). Retinoic acid receptor-α cDNA sequences were amplified in separate reactions as positive cDNA controls. Quantification of RUNX-2, FGF-23, SOX-9 and MMP-13 mRNA expressions were performed by real-time PCR. The cDNA synthesized was used as a template in 20 μl PCR reactions containing 10 μl SYBR Green Master mix (FastStart Universal SYBR Green Master, Roche), 1 μl primers, 2 μl cDNA synthesis reaction and 7 μl molecular grade water. Reactions were run and analysed on a BioRad PCR detection system (MJ Mini Opticon Thermal Cycler, BioRad, Hercules, CA, USA). PCR primers were designed for each cDNA target using Primer3 software and cycling parameters were optimized for each set of primers used. Data were analysed using the comparative Ct method as means of relative quantitation, normalized to an endogenous reference (GAPDH cDNA) and relative to a calibrator (normalized Ct value obtained from vehicle-treated cells) and expressed as 2−ΔΔCt.
Protein expression on chondrocytes
Osteoarthritic and normal cells were trypsinized and collected. The cell pellet was washed with PBS and lysed using Nonidet P-40 lysis buffer containing 30 mM Tris (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Nonidet P-40 and a cocktail of protease inhibitors. Protein concentration was quantified using the BioRad Bradford protein assay (BioRad Protein Assay, BioRad) with bovine serum albumen as standard.
Western blot analysis
Cell lysates from normal and OA chondrocytes were electrophoresed and separated on a 4–20% Tris-HCl gel (BioRad) and transferred to a Hybond-ECL nitrocellulose membrane (Amersan Biosciences, Piscataway, NJ, USA). The membrane was probed with anti-FGF-23 (1:1000 dilution, Novus Biologicals, Inc., Littleton, CO, USA) and signals were detected using anti-goat immunoglobulin IgG conjugated with horseradish peroxidase (1:5000 dilution). The results were normalized using anti-actin monoclonal antibody.
Protein levels of RUNX-2 (1:1000 dilution) and SOX-9 (1:500 dilution) (Abcam, Cambrige, UK) were also evaluated 48 hrs after siRNA transfection against RUNX-2 and SOX-9, respectively. Moreover, SOX-9 protein levels were evaluated after human recombinant FGF-23 (hrFGF-23) treatment in normal chondrocytes. The results were normalized using anti-GAPDH monoclonal antibody. The nitrocellulose membranes were exposed to photographic film, which was scanned and the intensities of the protein bands were expressed as arbitrary units and determined by computerized densitometry.
Oligonucleotide transfections
Osteoarthritic and normal chondrocytes were seeded in six-well plates and were transfected with 50 nM siRNA against RUNX-2 or FGF-23 and SOX-9, respectively (Ambion, Applied Biosystems Inc., Austin, TX, USA) using siPORT NeoFX transfection agent. We used two different siRNAs against RUNX-2 and SOX-9 in order to exclude off-target effects. Transfection with 50 nM scrambled siRNA was used as a control. No cell toxicity was detected due to the transfection agent. Chondrocytes were treated with varying concentrations (20, 50 and 100 ng/ml) of hrFGF-23 (CYT-374, Biosupply, Bradford, UK). RNA was extracted 24 and 48 hrs after siRNA transfection and real-time PCR analysis was performed as described above. GAPDH levels were used as loading control. In each experiment, the results from two wells of six-well plates were averaged and considered as n= 1. There was no significant variance among the individual wells in each averaged group.
Immunofluorescence
Cells were seeded on fibronectin-coated cell culture slides (Becton Dickinson, New Jersey, NJ, USA), fixed for 10 min. in 3.7% PBS-buffered formaldehyde, blocked with 3% bovine serum albumin (BSA) and incubated with primary antisera for MMP-13 (1:500 dilution) (H230, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), for 2 hrs in 1% BSA in PBS, and with secondary antisera under the same conditions. Secondary sera used were anti-rabbit FITC-conjugated anti-IgG (Santa Cruz Biotech, Inc.).
Statistical analysis
Statistical significance was determined using Student’s t-test using a confidence level of 95% (P < 0.05).
Results
RUNX-2 mRNA expression in freshly isolated and cultured osteoarthritic and normal chondrocytes
We evaluated RUNX-2 mRNA expression in freshly isolated ex vivo osteoarthritic and normal chondrocytes using quantitative real-time PCR. We found that RUNX-2 mRNA expression was significantly higher in both freshly isolated (Fig. 1A) and cultured osteoarthritic chondrocytes compared to normal (Fig. 1B) (P < 0.0001).
Figure 1.
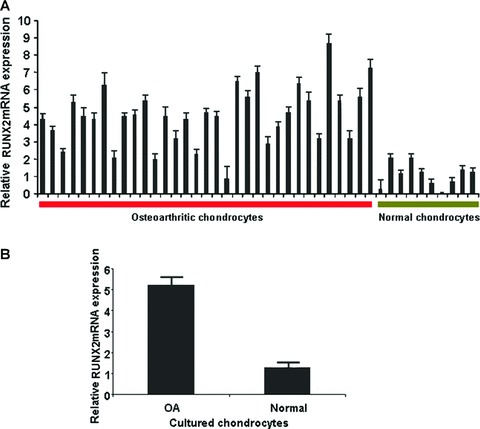
Quantitative RUNX-2 mRNA expression (A) freshly isolated (P < 0.0001) and (B) cultured normal and osteoarthritic chondrocytes (P < 0.0001).
Association between RUNX-2 mRNA expression and MMP-13 expression
RUNX-2 mRNA expression was blocked in osteoarthritic chondrocytes by transfection with two different siRNAs against RUNX-2 in order to exclude off-target effects. The effectiveness of this method was assessed by real-time PCR analysis of RUNX-2 mRNA expression levels normalized to GAPDH, a housekeeping reference gene. We first examined the percentage of the chondrocytes that expressed RUNX-2 after transfection with two different siRNAs in different time periods. We found that siRNAs against RUNX-2 were able to inhibit successfully its expression in similar levels and appear to have the same kinetics. Specifically, RUNX-2 was 60% and 74% down-regulated after 24 hrs with siRNA1 and siRNA2 transfection, respectively, and 78% and 80% down-regulated after 48 hrs with siRNA1 and siRNA2 transfection, respectively (Fig. 2A). Moreover, both siRNAs inhibited RUNX-2 protein expression in similar levels (Fig. 2B).
Figure 2.
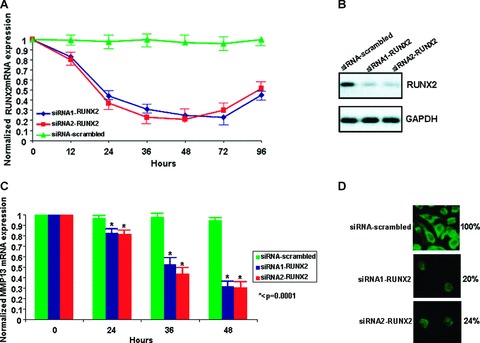
(A) RUNX-2 inhibition with siRNA1 and siRNA2 (B) RUNX-2 protein expression after 48 hrs siRNA1 and siRNA2 transfection (C) MMP-13 mRNA expression 0, 24, 36 and 48 hrs after RUNX-2 inhibition (P < 0.0001) (D) Assessment of MMP-13 expression by immunofluorescence after 48 hrs of RUNX-2 inhibition with two different siRNAs. All experiments were performed in OA chondrocytes. (A) and (B) were normalized to GAPDH. siRNA-scrambled was used as a control.
We next assessed MMP-13 mRNA expression levels after RUNX-2 down-regulation in two independent experiments (siRNA1-RUNX-2 and siRNA2-RUNX-2) and the results were evaluated 0, 24, 36 and 48 hrs siRNA after transfection. Our findings suggest that inhibition of RUNX-2 is associated with reduced MMP-13 mRNA expression more than 40% with the first siRNA, more than 55% with the second siRNA at 36 hrs after transfection, and more than 70% at 48 hrs after transfection with both siRNAs (Fig. 2C). We then localized MMP-13 in OA chondrocytes using immunofluorescence. Cultured osteoarthritic chondrocytes were stained for MMP-13 protein expression before and after RUNX-2 inhibition. MMP-13 expression was reduced (reduced number of green cells/field) after RUNX-2 inhibition with both siRNAs 48 hrs after transfection in OA chondrocytes. Specifically, we found that only 20% and 24% of chondrocytes expressed MMP-13 after RUNX-2 inhibition with siRNA1 and siRNA2, respectively (Fig. 2D).
Association between SOX-9 mRNA expression, RUNX-2 and MMP-13 expression
SOX-9 mRNA expression levels were evaluated in osteoarthritic and normal chondrocytes from freshly isolated ex vivo cartilage by real-time PCR. SOX-9 was highly down-regulated in osteoarthritic chondrocytes and was highly expressed in normal chondrocytes (P < 0.0001) (Fig. 3A).
Figure 3.
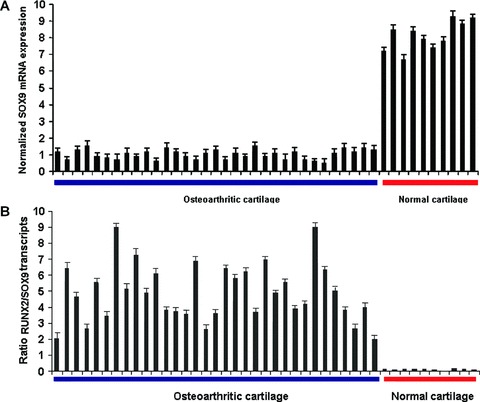
(A) Quantitative SOX-9 mRNA levels in normal and osteoarthritic primary chondrocytes normalized to GAPDH (P < 0.0001) (B) Ratio of RUNX-2 to SOX-9 transcripts in OA and normal cartilage (P < 0.0001).
Using real-time PCR we determined the relative expression levels of both SOX-9 and RUNX-2, which were normalized to the same reference gene, GAPDH and calculated the ratio of RUNX-2 to SOX-9 transcripts (RUNX-2/SOX-9). Our results demonstrate that the ratio RUNX-2/SOX-9 increases in osteoarthritic and decreases in normal cartilage (P < 0.0001) (Fig. 3B).
Subsequently, SOX-9 mRNA expression levels were blocked with two different siRNAs in normal chondrocytes and we confirmed its silencing by evaluating its protein levels (Fig. 4A).
Figure 4.
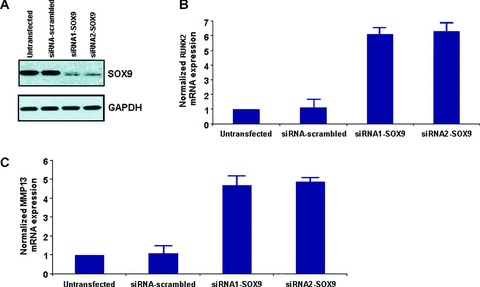
(A) SOX-9 protein expression after siRNA1 and siRNA2 transfection (B) RUNX-2 mRNA expression after SOX-9 inhibition by siRNA1 and siRNA2 (C) MMP-13 mRNA expression after SOX-9 inhibition by siRNA1 and siRNA2. All experiments were performed in normal chondrocytes. All data were normalized to GAPDH. siRNA-scrambled was used as a control.
We then tested RUNX-2 mRNA expression after silencing of SOX-9 by siRNA in normal chondrocytes. Our results showed that inhibition of SOX-9 mRNA expression by transfection with siRNA1 and siRNA2 against SOX-9 affects RUNX-2 mRNA expression leading to its up-regulation compared to the untransfected and the siRNA-scrambled 48 hrs after siRNA1 and siRNA2 transfection. Subsequently, it seems that SOX-9 mRNA expression is associated with reduced or absence of RUNX-2 mRNA expression in normal chondrocytes (Fig. 4B).
Considering that RUNX-2 expression is associated with increased MMP-13 expression, we proceeded to test, MMP-13 expression levels after inhibiting SOX-9 mRNA expression. Our results revealed that inhibition of SOX-9 expression with two different siRNAs, resulted in up-regulation of MMP-13 mRNA expression in normal chondrocytes (Fig. 4C).
Association between FGF-23 mRNA expression and RUNX-2 expression
We determined FGF-23 mRNA and protein expression in normal and osteoarthritic cartilage by real-time PCR and Western blotting, respectively. Our results indicated that FGF-23 is highly up-regulated in osteoarthritic cartilage and has low expression levels in normal cartilage (Fig. 5AP < 0.0001, Fig. 5B).
Figure 5.
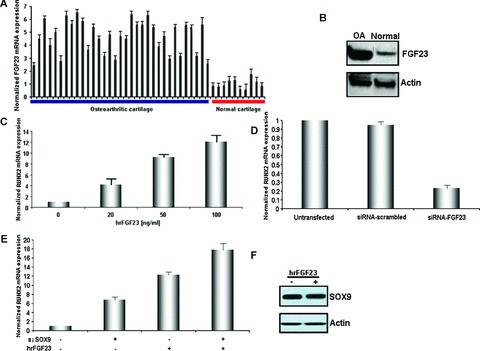
(A) Quantitative FGF-23 mRNA levels in normal and OA cartilage (P < 0.0001) (B) FGF-23 protein expression in osteoarthritic and normal chondrocytes normalized to actin (C) RUNX-2 mRNA expression after treatment of normal chondrocytes with 0, 20, 50 and 100 ng/ml with hrFGF-23. (D) RUNX-2 mRNA expression in OA chondrocytes after FGF-23 down-regulation. (E) Effect of SOX-9 and FGF-23 modulation on RUNX-2 expression in normal chondrocytes. (F) Effect of (100 ng/ml) hrFGF-23 on SOX-9 protein expression in normal chondrocytes.
We next investigated whether the addition or inhibition of FGF-23 could modulate RUNX-2 expression. Normal chondrocytes were treated with different concentrations of hrFGF-23 for 2 days and subsequently RUNX-2 mRNA expression was determined.
We found that hrFGF-23 increases RUNX-2 mRNA levels in a dose-dependent manner (20, 50, 100 ng/ml) in normal chondrocytes and the maximum increase of RUNX-2 mRNA expression was observed after 100 ng/ml treatment with hrFGF-23 (Fig. 5C).
Moreover, we checked RUNX-2 expression after FGF-23 inhibition with siRNA against FGF-23 in OA chondrocytes (48 hrs after transfection). We found that inhibition of FGF-23 blocked more than 80% of RUNX-2 mRNA expression in OA chondrocytes compared to a negative control (Fig. 5D).
FGF-23 and SOX-9 activate RUNX-2 expression in normal chondrocytes
Normal chondrocytes express high levels of SOX-9 and low levels of FGF-23. In order to investigate whether SOX-9 and FGF-23 act in the same pathway to regulate RUNX-2 mRNA expression, we inhibited SOX-9 expression with siRNA against SOX-9 and activated FGF-23 expression by treating normal chondrocytes with hrFGF-23. We then determined RUNX-2 mRNA expression levels, normalized to GAPDH reference gene. Detection of the same or very similar RUNX-2 fold induction by hrFGF-23 or siRNA- SOX-9 would suggest that these two proteins exist in the same pathway. However, in our case we detected a 6.5-fold increase in RUNX-2 expression levels when treated with siRNA against SOX-9 and a 12-fold increase in RUNX-2 expression levels when treated with hrFGF-23 (Fig. 5E). The 5.5-fold difference in RUNX-2 induction between hrFGF-23 and siRNA-SOX-9 stimuli does not suggest that these two proteins act in the same pathway.
In order to identify if FGF-23 regulates SOX-9, we treated normal chondrocytes with hrFGF-23 (100 ng/ml) and tested SOX-9 protein expression (Fig. 5F). We did not find any changes in SOX-9 levels suggesting that FGF-23 does not regulate SOX-9 expression. Moreover inhibition of FGF-23 or RUNX-2 in osteoarthritic chondrocytes by siRNA did not affect SOX-9 mRNA levels (data not shown). These findings suggest that SOX-9 is a suppressor, whereas FGF-23 is an activator of RUNX-2 expression in chondrocytes (Fig. 6).
Figure 6.
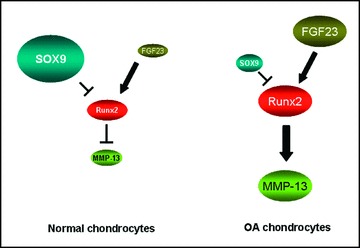
Model of associations among RUNX-2, SOX-9 and FGF-23 in human chondrocytes. SOX-9 is up-regulated in normal chondrocytes and acts as a repressor of RUNX-2, which in turn suppresses the expression of MMP-13. On the other hand, FGF-23 is highly expressed in OA chondrocytes and activates RUNX-2 expression which in turn up-regulates MMP-13 expression.
Discussion
The onset and progression of osteoarthritis is associated with changes in gene expression in chondrocytes resulting in an imbalance between matrix production and matrix degradation. In the degrading cartilage, chondrocytes acquire a hypertrophic phenotype, producing proteinases, which cause further cartilage degradation at the centre of the joint and osteophyte formation at the periphery [40]. RUNX-2 is implicated in this imbalance inducing chondrocytes’ hypertrophy and apoptosis under the influence of certain transcription factors. In the present study we tested the possible associations among SOX-9, FGF-23 and RUNX-2 in normal and osteoarthritic chondrocytes.
We observed that osteoarthritic chondrocytes had significantly higher RUNX-2 expression levels compared to normal chondrocytes in accordance with previous studies [30, 40, 41]. It has been suggested that overexpression of RUNX-2 in cartilage of transgenic mice promotes chondrocyte hypertrophic maturation and endochondral ossification, while inhibiting or eliminating RUNX-2 activity delays or blocks chondrocyte hypertrophic maturation [21, 30, 41]. Because this transcription factor functions as a rate-limiting regulator of chondrocyte hypertrophic maturation, the increased levels of RUNX-2 expression in osteoarthritic chondrocytes may contribute to similar changes leading to abnormal gene expression and cartilage degeneration seen in osteoarthritis. Additionally, it has been shown that RUNX-2 may contribute to increased expression of its downstream catabolic target MMP-13 [30]. It is known that the proximal promoter of MMP-13 gene contains one conserved osteoblasts specific site 2 site and binding of RUNX-2 in this increases gene transcription [42, 43]. In accordance with this observation, we found that RUNX-2 down-regulation by transfection with siRNA against RUNX-2 was associated with reduced MMP-13 mRNA and protein expression, suggesting that chondrocytes’ hypertrophy and MMP-13 induction during osteoarthritis progression might be partly mediated by RUNX-2.
As a key transcription activator for osteoblasts differentiation and chondrocyte maturation, it is likely that RUNX-2 function is regulated by diverse cofactors during different developmental stages for proper bone and cartilage formation. It has been previously reported that SOX-9 can act as a transcriptional repressor regulating negatively RUNX-2 expression in osteoblasts [31]. Both SOX-9 and RUNX-2 act during chondrogenic cell fate commitment and later during chondrogenesis. Due to this co-expression during mesenchymal condensation, the previous findings suggest the dominance of SOX-9 function over RUNX-2 during this early first step in the progenitor cell fate decision between osteoblastic versus chondrogenic lineages [31]. In accordance with previous reports, we found that SOX-9 mRNA expression was reduced in osteoarthritic cartilage and was highly expressed in normal cartilage. Moreover, the increased RUNX-2 mRNA expression after silencing of SOX-9 shows an inverse relationship between SOX-9 and RUNX-2 expressions. In addition, silencing of SOX-9 resulted in increased MMP-13 mRNA expression. These findings suggest a negative correlation between RUNX-2 and MMP-13 and SOX-9 mRNA expressions.
Furthermore, FGF-23 acts as a negative regulator of chondrogenesis because its up-regulation may induce premature exit of proliferating chondrocytes into hypertrophy leading to shorter bone growth [36]. We found that FGF-23 was highly expressed in osteoarthritic chondrocytes and had low expression levels in normal chondrocytes. We also showed that FGF-23 and RUNX-2 had similar expression levels, and more importantly that FGF-23 is associated with increased RUNX-2 expression in normal chondrocytes. It is known that RUNX-2 expression and activity can be up-regulated through FGF signalling in osteoblasts [32]. De Crombrugghe et al. have shown that FGF2 activates SOX-9 expression through MAPK signalling pathway [44]. However, different FGF family members have diverse biological functions. FGF-23 belongs in the FGF19 subfamily, whose members have been correlated with SOX-9 activity [45]. In addition Bobick et al. demonstrated that treatment with members of FGF family (FGF-8, -4, -2) decreased SOX-9 mRNA expression levels in chondrocyte cultures, while it elevated MAPK activity through ERK phosphorylation [46]. All above results suggest that each member of the FGF family exerts different functions, thus positive, negative or no effects on SOX-9 activity. Additionally, it was shown recently that FGF-23 transgenic mice had increased RUNX-2 expression levels, whereas SOX-9 levels were not reported to be affected [47]. These results coincide with our hypothesis that FGF-23 does not affect SOX-9 expression, whereas it is a strong inducer of RUNX-2 expression.
In conclusion in the present study, we demonstrated convincing associations among RUNX-2, and SOX-9 and FGF-23 in relation to MMP-13 expression in osteoarthritic chondrocytes, suggesting that simultaneous FGF-23 up-regulation and SOX-9 down-regulation could have a positive additive effect on RUNX-2 expression, contributing thus to a better understanding of osteoarthritis pathophysiology.
References
- 1.Erlebacher A, Filvaroff EH, Gitelman SE, et al. Toward a molecular understanding of skeletal development. Cell. 1995;80:371–8. doi: 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- 2.Reddi AH. Role of morphogenetic proteins in skeletal tissue engineering and regeneration. Nat Biotechnol. 1998;16:247–52. doi: 10.1038/nbt0398-247. [DOI] [PubMed] [Google Scholar]
- 3.Zhao Q, Eberspaecher H, Lefebvre V, et al. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev Dyn. 1997;209:377–86. doi: 10.1002/(SICI)1097-0177(199708)209:4<377::AID-AJA5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 4.De Crombrugghe B, Lefebvre V, Nakashima K. Regulatory mechanisms in the pathways of cartilage and bone formation. Curr Opin Cell Biol. 2001;13:721–7. doi: 10.1016/s0955-0674(00)00276-3. [DOI] [PubMed] [Google Scholar]
- 5.Wright E, Hargrave MR, Christiansen J, et al. The Sry-related gene SOX9 is expressed during chrondrogenesis in mouse embryos. Nat Genet. 1995;9:15–20. doi: 10.1038/ng0195-15. [DOI] [PubMed] [Google Scholar]
- 6.Kent J, Wheatley SC, Andrews JE, et al. A male-specific role for SOX9 in vertebrate sex determination. Development. 1996;122:2813–22. doi: 10.1242/dev.122.9.2813. [DOI] [PubMed] [Google Scholar]
- 7.Morais da Silva S, Hacker A, Harley V, et al. SOX9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat Genet. 1996;14:62–8. doi: 10.1038/ng0996-62. [DOI] [PubMed] [Google Scholar]
- 8.Ng LJ, Wheatley S, Muscat GE, et al. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol. 1997;183:108–21. doi: 10.1006/dbio.1996.8487. [DOI] [PubMed] [Google Scholar]
- 9.Lefebvre V, Huang W, Harley VR, et al. SOX9 is a potent activator of the chondrocyte specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol. 1997;17:2336–46. doi: 10.1128/mcb.17.4.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bridgewater LC, Lefebvre V, De Crombrugghe B. Chondrocyte-specific enhancer elements in the Col11a2 gene resemble the Col2a1 tissue-specific enhancer. J Biol Chem. 1998;273:14998–5006. doi: 10.1074/jbc.273.24.14998. [DOI] [PubMed] [Google Scholar]
- 11.Sekiya I, Tsuji K, Koopman P, et al. SOX9 enhances aggrecan gene promoter/enhancer activity and is up- regulated by retinoic acid in a cartilage-derived cell line, TC6. J Biol Chem. 2000;275:10738–44. doi: 10.1074/jbc.275.15.10738. [DOI] [PubMed] [Google Scholar]
- 12.Aigner T, Gebhard PM, Schmid E, et al. SOX9 expression does not correlate with type II collagen expression in adult articular chondrocytes. Matrix Biol. 2003;22:363–72. doi: 10.1016/s0945-053x(03)00049-0. [DOI] [PubMed] [Google Scholar]
- 13.Brew CJ, Andrew JG, Boot-Handford R, et al. Late osteoarthritic cartilage shows down regulation of SOX9 and aggrecan expression but little evidence of chondrocyte hypertrophy. Trans Orthop Res Soc. 2004;50:938. [Google Scholar]
- 14.Shum L, Nuckolls G. The life cycle of chondrocytes in the developing skeleton. Arthritis Res. 2002;4:94–106. doi: 10.1186/ar396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aigner T, Kim HA. Apoptosis and cellular vitality: issues in osteoarthritic cartilage degeneration. Arthritis Rheum. 2002;46:1986–96. doi: 10.1002/art.10554. [DOI] [PubMed] [Google Scholar]
- 16.Fukui N, Purple CR, Sandell LJ. Cell biology of osteoarthritis: the chondrocytes’ response to injury. Curr Rheumatol Rep. 2001;3:496–505. doi: 10.1007/s11926-001-0064-8. [DOI] [PubMed] [Google Scholar]
- 17.Ducy P, Zhang R, Geoffroy V, et al. Osf2/ Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 18.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development [review] Dev Cell. 2002;2:389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 19.Kim IS, Otto F, Zabel B, et al. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 1999;80:159–70. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- 20.Enomoto H, Enomoto-Iwamoto M, Iwamoto M, et al. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J Biol Chem. 2000;275:8695–702. doi: 10.1074/jbc.275.12.8695. [DOI] [PubMed] [Google Scholar]
- 21.Ueta C, Iwamoto M, Kanatani N, et al. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol. 2001;153:87–100. doi: 10.1083/jcb.153.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu L, Liu H, Yan M, et al. Shox2 is required for chondrocyte proliferation and maturation in proximal limb skeleton. Dev Biol. 2007;306:549–59. doi: 10.1016/j.ydbio.2007.03.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chin HJ, Fisher MC, Li Y, et al. Studies on the role of Dlx5 in regulation of chondrocyte differentiation during endochondral ossification in the developing mouse limb. Dev Growth Differ. 2007;49:515–21. doi: 10.1111/j.1440-169X.2007.00940.x. [DOI] [PubMed] [Google Scholar]
- 24.Arnold MA, Kim Y, Czubryt MP, et al. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell. 2007;12:377–89. doi: 10.1016/j.devcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Solomon LA, Berube NG, Beier F. Transcriptional regulators of chondrocyte hypertrophy. Birth Defects Res C Embryo Today. 2008;84:123–30. doi: 10.1002/bdrc.20124. [DOI] [PubMed] [Google Scholar]
- 26.Hoyland JA, Thomas JT, Donn R, et al. Distribution of type X collagen mRNA in normal and osteoarthritic human cartilage. Bone Miner. 1991;15:51–63. doi: 10.1016/0169-6009(91)90005-k. [DOI] [PubMed] [Google Scholar]
- 27.Nah HD, Swoboda B, Birk DE, et al. Type IIA procollagen: expression in developing chicken limb cartilage and human osteoarthritic articular cartilage. Dev Dyn. 2001;220:307–22. doi: 10.1002/dvdy.1109. [DOI] [PubMed] [Google Scholar]
- 28.Pfander D, Swoboda B, Kirsch T. Expression of early and late differentiation markers (proliferating cell nuclear antigen, syndecan-3, annexin VI, and alkaline phosphatase) by human osteoarthritic chondrocytes. Am J Pathol. 2001;159:1777–83. doi: 10.1016/S0002-9440(10)63024-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gebhard PM, Gehrsitz A, Bau B, et al. Quantification of expression levels of cellular differentiation markers does not support a general shift in the cellular phenotype of osteoarthritic chondrocytes. J Orthop Res. 2003;21:96–101. doi: 10.1016/S0736-0266(02)00094-3. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Manner PA, Horner A, et al. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage. 2004;12:963–73. doi: 10.1016/j.joca.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Zhou G, Zheng Q, Engin F, et al. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci USA. 2006;103:19004–9. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao G, Jiang D, Gopalakrishnan R, et al. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J Biol Chem. 2002;277:36181–7. doi: 10.1074/jbc.M206057200. [DOI] [PubMed] [Google Scholar]
- 33.Kim HJ, Kim JH, Bae SC, et al. The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J Biol Chem. 2003;278:319–26. doi: 10.1074/jbc.M203750200. [DOI] [PubMed] [Google Scholar]
- 34.van Donkelaar CC, Janssen XJ, de Jong AM. Distinct developmental changes in the distribution of calcium, phosphorus and sulphur during fetal growth plate development. J Anat. 2007;210:186–94. doi: 10.1111/j.1469-7580.2006.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabbagh Y, Carpenter TO, Demay MB. Hypophosphatemia leads to rickets by impairing caspase – mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci USA. 2005;102:9637–42. doi: 10.1073/pnas.0502249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez CP, He YZ. Bone growth during daily or intermittent calcitriol treatment during renal failure with advanced secondary hyperparathyroidism. Kidney Int. 2007;72:582–91. doi: 10.1038/sj.ki.5002375. [DOI] [PubMed] [Google Scholar]
- 37.Sanchez CP, He YZ, Leiferman E, et al. Bone elongation in rats with renal failure and mild or advanced secondary hyperparathyroidism. Kidney Int. 2004;65:1740–8. doi: 10.1111/j.1523-1755.2004.00577.x. [DOI] [PubMed] [Google Scholar]
- 38.MacLean HE, Guo J, Knight MC, et al. The cyclin-dependent kinase inhibitor p57(Kip2) mediates proliferative actions of PTHrP in chondrocytes. J Clin Invest. 2004;113:1334–43. doi: 10.1172/JCI21252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo J, Chung UI, Yang D, et al. PTH/ PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev Biol. 2006;292:116–28. doi: 10.1016/j.ydbio.2005.12.044. [DOI] [PubMed] [Google Scholar]
- 40.Kawaguchi H. Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol Cells. 2008;25:1–6. [PubMed] [Google Scholar]
- 41.Kamekura S, Kawasaki Y, Hoshi K, et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;54:2462–70. doi: 10.1002/art.22041. [DOI] [PubMed] [Google Scholar]
- 42.D’Alonzo RC, Selvamurugan N, Karsenty G, et al. Physical interaction of the activator protein-1 factors c-Fos and c-Jun with Cbfa1 for collagenase-3 promoter activation. J Biol Chem. 2002;277:816–22. doi: 10.1074/jbc.M107082200. [DOI] [PubMed] [Google Scholar]
- 43.Jiménez MJ, Balbìn M, Alvarez J, et al. A regulatory cascade involving retinoic acid, Cbfa1, and matrix metalloproteinases is coupled to the development of a process of perichondrial invasion and osteogenic differentiation during bone formation. J Cell Biol. 2001;155:1333–44. doi: 10.1083/jcb.200106147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murakami S, Kan M, McKeehan WL, et al. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 2000;93:1113–8. doi: 10.1073/pnas.97.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukumoto S. Actions and mode of actions of FGF19 subfamily members. Endocr J. 2008;55:23–31. doi: 10.1507/endocrj.kr07e-002. [DOI] [PubMed] [Google Scholar]
- 46.Bobick BE, Thornhill TM, Kulyk WM. Fibroblast growth factors 2, 4, and 8 exert both negative and positive effects on limb, frontonasal, and mandibular chondrogenesis via MEK-ERK activation. J Cell Physiol. 2007;211:233–43. doi: 10.1002/jcp.20923. [DOI] [PubMed] [Google Scholar]
- 47.Hollberg K, Marsell R, Norgård M, et al. Osteoclast polarization is not required for degradation of bone matrix in rachitic FGF23 transgenic mice. Bone. 2008;42:1111–21. doi: 10.1016/j.bone.2008.01.019. [DOI] [PubMed] [Google Scholar]