

Abstract
Vasoactive intestinal peptide (VIP) was originally isolated as a vasodilator intestinal peptide, then as a neuropeptide. In the immune system, VIP is described as an endogenous macrophage-deactivating factor. VIP exerts its immunological actions in a paracrine and/or autocrine manner, through specific receptors. However, very little is known about the molecular regulation of VIP type 2 receptor (VPAC2) in the immune system. We now report that different toll-like receptor (TLR) ligands selectively regulate the VPAC2 receptor gene and show a gene repression system controlled by key protein kinase signalling cascades in macrophages. VPAC2 gene expression is regulated by gram-positive (TLR2 ligands) and gram-negative bacteria wall constituents (TLR4 ligands). Moreover, VPAC2 is tightly regulated: TLR2- or TLR2/6- but not TLR2/1-mediated mechanisms are responsible for the induction of VPAC2. TLR stimulation by viral or bacterial nucleic acids did not modify the VPAC2 mRNA levels. Remarkably, imiquimod – a synthetic TLR7 ligand – led to a potent up-regulation of VPAC2 gene expression. TLR5 stimulation by flagellin present in gram-positive and gram-negative bacteria did not affect VPAC2 mRNA. The p38 mitogen-activated protein kinase (MAPK) activity accounted for the TLR4-mediated induction of VPAC2 gene expression. Surprisingly, our data strongly suggest for the first time a tightly repressed control of VPAC2 mRNA induction by elements downstream of MAPK kinase 1/2, PI3K/Akt, and particularly Jun-NH2-terminal kinase signalling pathways.
Keywords: VIP, neuroimmunology, neuropeptides, inflammation, toll-like receptors
Introduction
Research on endogenous negative feedback or compensatory mechanisms that serve to limit deleterious immune responses is critical in the attempts to identify effective therapeutic targets [1, 2]. In recent years, vasoactive intestinal peptide (VIP) and VIP receptors have shown their relevance as endogenous factors that regulate inflammatory immune responses and immune tolerance [3, 4]. The mechanisms involved include the deviation towards Th2-driven inflammatory pathways, the specific recruitment and development of Th2 cells and the peripheral expansion of regulatory T cells [3]. Endogenously, VIP is produced and secreted by antigen-stimulated Th2 cells [5–7], resembling a cytokine-like acting molecule [3, 4]. Interestingly, VIP knockout mice expressed an asthmatic phenotype that is partially corrected after VIP treatment [8]. This is agreement with current views of bronchial asthma as a non-exclusive Th2 polarized immune response [9, 10]. VIP exerts its immunological actions in a paracrine and/or autocrine manner, through three different specific receptors: VIP type 1 receptor (VPAC1), VPAC2 and PAC1[11, 12]. Although seminal works in the immune system described VIP as an anti-inflammatory agent, acting as an endogenous macrophage-deactivating factor (see reviews [3, 11, 13]), very little is known about the molecular regulation of VPAC receptors in macrophages. Delgado et al. [14] have reported that in contrast to VPAC1 and PAC1 receptors – which seem to be constitutively expressed in mice macrophages – VPAC2 is selectively expressed only after lipopolysaccharide (LPS) stimulation. In lymphocytes, VPAC2 mRNA is an activation-induced gene in several experimental conditions [15–18]. Despite the inducible character of the VPAC2 gene, detailed data about the promoter and regulatory elements scarcely exist and are limited to the human genome [19, 20]. Very recently, Lutz and collaborators have identified the core promoter region of the mouse VPAC2 receptor gene [21].
The present work represents the first study of the regulation of VPAC2 mRNA by toll-like receptors (TLRs) in macrophages. TLRs are the best-characterized signal generating receptors among the pathogen-associated molecular patterns; they initiate key inflammatory responses and also shape the adaptive immunity [22, 23]. TLRs can be divided into two groups according to their cellular localization: TLRs 1, 2, 4, 5, 6 are located mainly on the cell surface and primarily recognize bacterial components, while TLRs 3, 7, 8 and 9 are mostly found in the endocytic compartments and mainly recognize viral products [22, 23]. TLR-mediated innate immune activation also induces several molecules shown to negatively regulate TLR signalling [24]. Although studies on VIP immunoregulatory activities are of notable therapeutic interest [3], the effects of TLRs on the induction of VPAC2 have not yet been examined. Thus, the VPAC2 regulation by TLRs may play an important role in positive and negative regulation of normal and aberrant immune responses.
Our findings indicate that the VPAC2 gene is differentially regulated by subsets of TLRs. Furthermore, VPAC2 mRNA is tightly repressed by key protein kinase pathways: c-Jun-NH2-terminal kinase (JNK), mitogen-activated protein kinase (MAPK) kinase 1/2 (MEK1/2), p38 MAPK and phosphatidylinositol-3 kinase (PI3K/Akt).
Materials and methods
Animals
Female C57BL6/JIco mice (4–8 weeks old) were maintained in the Animal Breeding Center of the University of Seville. Experiments were carried out under the supervision and guidelines of the Animal Welfare Committee.
Cell culture and reagents
Raw 264.7 cells (ECACC, Porton Down, Wiltshire, UK) were grown in RPMI 1640 medium supplemented with 10% heat-inactivated foetal bovine serum (BioWhitaker, Verviers, Belgium), 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma, St. Louis, MO, USA). Peritoneal macrophages were isolated and pooled after intraperitoneal injection of 1 ml of 3% thioglycolate at day 5 (Fluka, Milwaukee, WI, USA). TLR ligands were as follows: LPS (E. coli serotype 0127: B8, Sigma), oligodeoxynucleotide containing unmethylated CpG motifs (CpG+: CpG-DNA 1668: 5′-TCCATGACGTTCCTGATGCT-3′, TIB MolBiol, Berlin, Germany), polyinosinic: polycytidylic acid (poly I: C), flagellin, lipoteichoic acid (LTA), synthetic bacterial lipopeptide Pam3CSK4, synthetic lipoprotein derived from Mycoplasma (FSL-1), peptidoglycan (PGN), imiquimod and ssRNA40 were obtained from InvivoGen (San Diego, CA, USA). SB203580 (a p38 MAPK inhibitor), SP600125 (a JNK inhibitor), PD98059 (a MEK1/2 inhibitor), LY294002 (a PI3K/Akt inhibitor) were all from Sigma.
Cytokine assays
Supernatants from Raw 264.7 cells and thioglycolate-elicited peritoneal macrophages were harvested 24 hrs after TLR stimulation. Interleukin 6 (IL-6) ELISA determinations were carried out according to the manufacturer’s instructions (OptEIA Mouse IL-6 set, BD Pharmingen, San Diego, CA, USA).
Quantitative real-time PCR (qPCR)
Raw 264.7 cells were seeded into 12-well plates in a final volume of 2 ml. At 70–80% confluence, cells were stimulated with LPS (1 μg/ml), CpG+ (1 μg/ml, CpG-DNA 1668), Poly (I: C) acid (50 μg/ml,), LTA (10 μg/ml), Pam3 CSK4 (300 ng/ml), FSL-1 (1 μg/ml), PGN (10 μg/ml), imiquimod (10 μg/ml) or ssRNA40 (0.25 μg/ml). Peritoneal macrophages were adherent-isolated by overnight culture into 12-well plates in 1.5 ml RPMI 1640. Then, non-adherent cells were removed and peritoneal macrophages were treated as indicated above. Twenty-four hours later, RNA from cell cultures was extracted using Tripure™ isolation reagent (Roche, Basel, Switzerland) and qPCRs of VPAC2, IL-6 or the housekeeping gene HPRT were carried out. For qPCR determinations (MiniOpticon detector, BioRad, Hercules, CA, USA), 1 μg of RNA was reversely transcribed and genomic DNA removed using QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA, USA). One hundred nanograms of cDNA were amplified using 12.5 μl of 2× FastStart SYBR Green Master Mix (Roche), 200 nM of each primer and H2O up to 25 μl. The PCR amplification scheme was: 10 min. at 95°C; followed by 50 cycles at 95°C 15 sec., 65°C 30 sec. and 30 sec. at 72°C. HPRT (ID genebank accession number 15452) was used as housekeeping gene (forward 5′-GTAATGATCAGTCAACGGGGGAC-3′, reverse 5′-CCAGCAAGCTTGCAACCTTAACCA-3′ (174 bp in length spanning from nucleotide 464 to 638). The VPAC2 sequence (ID genebank accession number 22355) was used to design the VPAC2 primers. The oligonucleotides were as follow: forward 5′-GGACAGCAACTCGCCTCTCT-3′ (nt 825–844), reverse 5′-CCCTGGAAGGAACCAACACATAAC-3′ (nt 1129–1152) (328 bp in length). IL-6 primers (ID genebank accession number 16193) were: forward 5′-TTCCATCCAGTTGCCTTCTT-3′, reverse 5′-ATTTCCACGATTTCCCAGAG-3′ (170 bp in length spanning from nucleotide 55 to 225). SYBR-Green I detection was followed by generation of melting curves and visualization of the products to confirm specificity. Quantitative PCR results were obtained using the ΔΔCt method [25]. The induction of mRNA was calculated as 2−ΔΔCt.
Statistical significance
For statistical evaluation, Mann-Whitney rank sum tests were performed to differentiate between experimental groups.
Results and discussion
The VPAC2 gene is constitutively expressed in both Raw 264.7 cells and peritoneal macrophages (Fig. 1). VPAC2 mRNA levels were up-regulated upon LPS treatment, as previously reported by Delgado and collaborators [14]. In our experimental setting, we were able to detect a constitutive expression of VPAC2 mRNA in resting macrophages while Delgado and collaborators [14] did not detect it by conventional RT-PCR in resting Raw 264.7 cells. This discrepancy could be due to differences in the sensitivity of the systems used (conventional versus real time PCR) or the efficiency of the primers used in each study. Besides, what is constitutive or not relies on the experimental technique or reagents used in each case. In this sense, we reproduced the responsive character of the VPAC2 gene pioneered reported by Delgado and collaborators [14] and this information about the inducible nature of the VPAC2 gene is the relevant message in terms of biological interpretation.
Figure 1.
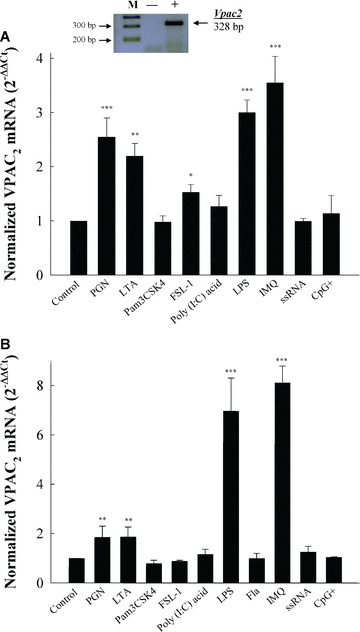
Differential TLRs regulation of VPAC2 mRNA in macrophages. (A) Raw 264.7 cells were seeded into 12-well plates in a final volume of 2 ml. At 70–80% confluence (750,000 cells/well), cells were stimulated with LPS (1 μg/ml), CpG+ (1 μg/ml, CpG-DNA 1668: 5′-TCCATGACGTTCCTGATGCT-3′), Poly (I: C) acid (50 μg/ml), LTA (10 μg/ml), Pam3 CSK4 (300 ng/ml), FSL-1 (1 μg/ml), PGN (10 μg/ml), imiquimod (10 μg/ml) or ssRNA40 (0.25 μg/ml). Twenty-four hours, RNA was extracted and qPCR of VPAC2 was carried out. Results are the mean ± S.D. of five independent experiments performed in triplicate. (Inset) Samples from qPCR samples on 1.7% agarose gel electrophoresis. Lanes showed the DNA molecular size markers (M), reaction performed in the absence of RT (–) and presence (+) of RT from resting Raw 264.7 RNA. (B) Female C57BL6/JIco (n= 20) peritoneal macrophages were isolated and pooled after i.p. injection of thioglycolate medium at day 5 as described in the ‘Materials and methods’ section. For qPCR determinations of VPAC2 mRNA levels, cDNA (100 ng) was amplified as described in the ‘Materials and methods’ section. Quantitative PCR results were obtained using the ΔΔCt method [25]. The induction of mRNA was calculated as 2−ΔΔCt (normalized for HPRT as housekeeping gene). For statistical evaluation, Mann-Whitney rank sum tests were performed. Asterisks indicate statistical significance (*, P < 0.05 versus control; **, P < 0.01 versus control; ***, P < 0.001 versus control).
LPS treatment caused about three-fold induction of VPAC2 mRNA levels in Raw 264.7 cells. Interestingly, PGN, a ligand for TLR2, or ligands for TLR2/6 heterodimers such as LTA or the lipopeptide FSL-1 – a synthetic analogue of mycoplasmal lipoprotein – were able to up-regulate VPAC2 gene expression in Raw 264.7 cells (Fig. 1A). In contrast, Raw 264.7 cells treated with Pam3 CSK4 – a TLR2/1 ligand – did not modify the mRNA levels of VPAC2 receptors (Fig. 1A). Therefore, VPAC2 gene expression is regulated by components of gram-positive (TLR2 ligands) and gram-negative bacteria (TLR4 ligands) cell walls. These data also imply that different TLR-mediated signalling pathways may result in the selective regulation of the VPAC2 gene.
VPAC2 is tightly regulated by TLR2- or TLR2/6-mediated mechanisms. TLR2/1 heterodimers recognize triacyl lipopeptides such as Pam3 CSK4, and TLR2/6 heterodimers recognize diacyl lipopeptides. Findings of differential responses mediated by TLR1 are scarcely known. Recently, defects in TLR1 recognition have been related to ineffective responses to lipoprotein antigens in mice and human beings, providing a link between TLR1 and acquired antibody responses [26]. The reason why VPAC2 up-regulation is unresponsive to TLR2/1 stimulation is uncertain. Synthetic bacterial lipopeptide Pam3CSK4, LTA and PGN have been shown to exert a beneficial decrease in the Th2 phenotype in allergic contexts [27–30], although there is no such a comprehensive comparative analysis of the Th1 responses elicited by these ligands. Although future studies should clarify the differences observed with Pam3CSK4 regarding VPAC2 expression, they might be related with specific features of the Th1-mediated response by Pam3CSK4.
This is the first report of VPAC2 regulation by TLRs sensing gram-positive bacteria. Recent studies support the concept that chronic or recurrent microbial infections may contribute to atherosclerotic disease acting through TLR2 [31, 32]. The putative role of VPAC2 in the modulation of innate responses affecting atherosclerosis is presently unknown and deserves further attention.
Optimal stimulation with CpG DNA motifs for TLR9, single stranded RNA for TLR7, or poly (I: C) acid as a dsRNA synthetic analogue for TLR3 did not modify VPAC2 mRNA levels (Fig. 1). Therefore, VPAC2 gene is not affected by TLR-stimulation detecting nucleic acids derived from viruses and bacteria. Imiquimod – a specific TLR7 ligand – is an imidazoquinoline-like molecule. Interestingly, imiquimod treatment of Raw 264.7 cells led to a potent up-regulation of VPAC2 mRNA levels, causing 3.5 ± 0.5 fold induction of VPAC2 mRNA levels in Raw 264.7 cells (Fig. 1A). There is still a long way to go regarding the imiquimod molecular mechanism of action and many questions remain unanswered. This is the reason why ssRNA and imiquimod does not always show similar effects. It has been shown that imiquimod has mechanisms of action that are TLR7-independent, suggesting a role for adenosine receptors [33]. Topical imiquimod cream is an FDA-approved treatment for superficial basal cell carcinomas [34]. Predominant among its actions is the induction, in transcutaneous immunization protocols, of efficient Th1-antitumoral cellular immunity [35, 36]. Fabricius and collaborators have reported an inhibitory role for VIP in the production of interferon (IFN)-α stimulated by TLR9 ligands in human plasmacytoid cells [37]. Recently, a strong inhibitory effect after TLR7 stimulation on the IFN-α production induced by TLR9 ligands has been shown [38]. Even though the regulation of specific TLR7-mediated responses by VIP has not been addressed, our results might provide grounds for a possible counter regulatory mechanism mediated by VPAC2 receptors.
We extended our observations to peritoneal macrophages, a more physiological context. Moreover, peritoneal macrophages, contrary to Raw 264.7 cells, express TLR5 [39, 40]. TLR5 stimulation is mediated through the interaction with gram-positive or gram-negative bacterial flagellin [22, 24]. VPAC2 mRNA levels were not affected by treatment with this bacterial constituent (Fig. 1B). Using peritoneal macrophages, we confirmed the pattern of VPAC2 gene regulation by TLR ligands (Fig. 1B). VPAC2 is differentially up-regulated by ligands of TLR2, TLR2/6 and TLR4 receptors. The synthetic ligand for TLR7, imiquimod, also increased the mRNA levels of the VPAC2 gene. Interestingly, the increase in VPAC2 gene expression by LPS or imiquimod was two-fold compared with that observed in Raw 264.7 cells, while the induction mediated by TLR2 and TLR2/6 remained at the same levels (Fig. 1B). To be certain that Raw 264.7 cells or peritoneal macrophages have responded appropriately to the different TLR ligands used, we determined IL-6 secretion or expression in both Raw 264.7 cells and peritoneal macrophages as a well-recognized marker of TLR-mediated activation [24] (Fig. 2).
Figure 2.
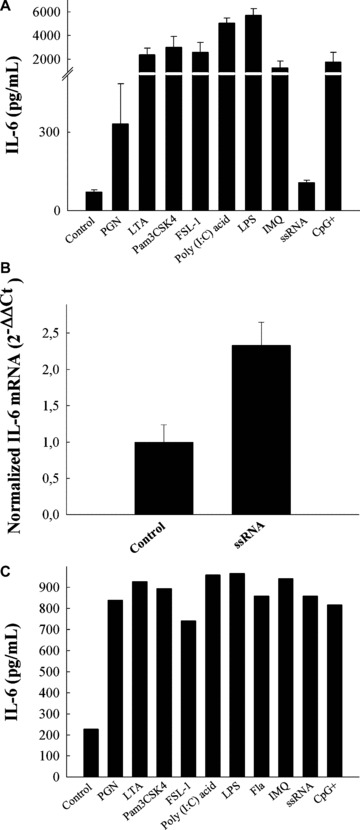
IL-6 production after TLRs stimulation in macrophages. (A) Raw 264.7 cells were seeded into 12-well plates in a final volume of 2 ml. At 70–80% confluence, cells (750,000 cells/well) were stimulated with LPS (1 μg/ml), CpG+ (1 μg/ml, CpG-DNA 1668: 5′-TCCATGACGTTCCTGATGCT-3′), Poly (I: C) acid (50 μg/ml), LTA (10 μg/ml), Pam3 CSK4 (300 ng/ml), FSL-1 (1 μg/ml), PGN (10 μg/ml), imiquimod (10 μg/ml) or ssRNA40 (0.25 μg/ml). Twenty-four hours later, supernatants were harvested for IL-6 ELISA determination. (B) mRNA IL-6 stimulation after ssRNA treatment in Raw 264.7 cells. RNA was extracted for IL-6 gene expression study after ssRNA treatment as described in the ‘Materials and methods’ section. For qPCR determinations of IL-6 mRNA levels, cDNA (100 ng) was amplified as described in the ‘Materials and methods’ section. Quantitative PCR results were obtained using the ΔΔCt method [25]. The induction of mRNA was calculated as 2−ΔΔCt. Results are the mean ± S.D. of five independent experiments performed in triplicate. (C) Female C57BL6/JIco (n= 20) peritoneal macrophages were isolated and pooled after intraperitoneal injection of 1 ml of 3% thioglycolate at day 5 as described above. Afterwards, non-adherent cells were removed and peritoneal macrophages were treated as indicated above and 24 hrs later, supernatants were harvested for IL-6 ELISA determination.
TLRs have been shown to activate four major intracellular kinase signalling pathways; p38 MAPK, JNK, MEK1/2 and PI3K/Akt [24, 41, 42]. We studied the signalling pathways involved in the induction of VPAC2 mRNA by TLR4, TLR2, TLR2/6 and TLR7 in Raw 264.7 macrophages. Previously, we checked whether the specific inhibitors of the different kinase signalling pathways or the TLR ligands used in the present study affected cell viability at the concentration used herein (Fig. S1). No statistically significant differences were observed in terms of cell viability compared to the control experimental situation. The proteasome inhibitor MG132 was used as a positive control for cytotoxicity.
The induction of VPAC2 expression triggered by LPS was completely abrogated by the blockade of the p38 MAPK-dependent signalling pathway with the specific inhibitor SB203580 (Fig. 3) [43, 44]. However, the up-regulation of VPAC2 mRNA levels observed after treatment with PGN (TLR2), lipoteichoic acid (TLR2/6) or imiquimod (TLR7) were not dependent on p38 MAPK activity (Fig. 3). This is the first report that ascribes a signal transduction cascade to the LPS induction of VPAC2 gene expression, although we cannot exclude a partial indirect effect mediated by pro-inflammatory cytokines triggered by LPS treatment as it has been reported in the epidermal keratinocyte cell line DJM-1 for VPAC1[45]. Strikingly, SP600125 – a highly selective inhibitor of JNK activity [46]– increased the mRNA levels of VPAC2 up to 12.5 ± 1.4 fold compared to control values (Fig. 3). The co-treatment of the different TLR ligands with the specific JNK inhibitor always led to a dramatic increase in VPAC2 gene expression despite TLR2-, TLR2/6- or TLR7-mediated stimulation without a synergistic effect (Fig. 3). Whether LPS triggers additional counter mechanisms is presently unknown, but only the LPS co-treatment with SP600125 abolished the effect that the JNK inhibitor produced by itself in Raw 264.7 cells (Fig. 3). Addition of the MEK1/2 signalling blocker [44, 47] PD98059 led to an increase in VPAC2 mRNA levels which did not synergize with the TLR stimulation (Fig. 3). However, LPS-induced VPAC2 expression was not reversed by blocking MEK1/2 activity, as was the case when p38 MAPK activity was inhibited (Fig. 3), nor did LPS affect the up-regulation of VPAC2 mRNA levels by MEK/ inhibition as in the case of the inhibition of JNK (Fig. 3).
Figure 3.
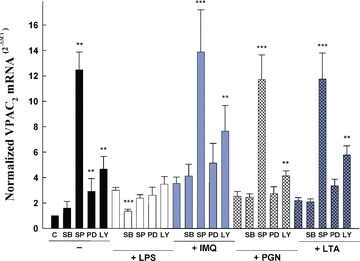
Effects of protein kinase inhibitors on TLR-mediated VPAC2 regulation in Raw 264.7 macrophages. Raw 264.7 cells were grown and treated as above. Briefly, at 70–80% confluence, cells (750,000 cells/well) were stimulated for 24 hrs with LPS (1 μg/ml), LTA (10 μg/ml), PGN (10 μg/ml) or imiquimod (10 μg/ml) after 30 min. pre-treatment in the presence or absence of 10 μM SB203580 (a p38 MAPK inhibitor), 25 μM SP600125 (a JNK inhibitor), 50 μM PD98059 (a MEK1/2 inhibitor) and 10 μM LY294002 (a PI3K/Akt inhibitor). Twenty-four hours later, RNA was extracted and qPCR of VPAC2 was carried out according to the ‘Materials and methods’ section. Results are the mean ± S.D. of three independent experiments performed in triplicate. For statistical evaluation, Mann-Whitney rank sum tests were performed. Asterisks indicate statistical significance (*, P < 0.05; **, P < 0.01; ***, P < 0.0001 versus the experimental condition in the absence of inhibitor).
We blocked PI3K/Akt activity with LY294002 [48, 49]. PI3K/Akt inhibition produced a 4.7 ± 1.0 fold induction of VPAC2 mRNA in Raw 264.7 macrophages (Fig. 3). PI3K/Akt inhibition did not reverse the TLR-mediated up-regulation of VPAC2 mRNA. Moreover, PI3K/Akt inhibition synergized with TLR ligands for TLR7, but not for TLR4, TLR2 or TLR2/6 (Fig. 3). These data strongly suggest a tightly repressed control of VPAC2 mRNA induction by elements downstream of MEK1/2, PI3K/Akt and especially JNK signalling pathways.
Recent studies have been highlighting that VIP decreases the up-regulation of TLR2 and TLR4 in human monocytes [50], as well as in lymphocytes, macrophages and dendritic cells after the trinitrobenzene sulfonic acid induced colitis in mice [13, 51, 52]. Interestingly, VIP has also been reported to inhibit the LPS-induced RNA expression of downstream adapters molecules involved in TLR4 signalling pathways in fibroblast-like synoviocytes from patients with rheumatoid arthritis or osteoarthritis [53]. We should notice the importance of understanding how the TLR, associated kinases pathways and VIP receptors serve to maintain the homeostasis and the integrity of the immune response. In this sense, it has been shown very recently that VIP suppresses TLR4 expression in macrophages via PI3K/Akt [54]. This is in agreement with increasing experimental evidence showing that PI3K/Akt signalling pathway has a negative regulatory role in inflammatory processes [55–57]. The VIP-mediated down-regulation of the pro-inflammatory response is partly explained by activation of PKA and subsequent inhibition of p38 MAPK activity and JNK activity in macrophages [1, 3]. Thus, it is conceivable to propose that up-regulation of VPAC2 expression might be part of a counter balance mechanism that allows a VIP-mediated anti-inflammatory effect in situations that produce extensive inhibition of the PI3K/Akt signalling pathway. In addition, the up-regulation of VPAC2 expression could be part of the inflammatory deactivating cascade produced by VIP, partly due to its inhibitory effects on p38 MAPK and JNK activities. Further experiments should address protein expression levels of VPAC2. Unfortunately, although there are no reliable/specific antibodies against VPAC2 receptors available so far, our data point out to new fine-tuned mechanisms of control of VPAC2 receptors. Very recently, two findings highlight the importance of the expression levels of VIP receptors in the onset and evolution of autoimmune/inflammatory human diseases. Delgado and collaborators have shown a genetic association involving VPAC1 receptor genetic polymorphisms and susceptibility of human rheumatoid arthritis, suggesting that an impaired expression of VPAC1 could be related to the pathogenesis of rheumatoid arthritis [58]. Juarranz and collaborators have shown that the inhibitory effects of VIP on fibroblast-like synoviocytes production of pro-inflammatory mediators were mimicked by VPAC1 and VPAC2 specific agonists in these cells from patients affected by osteoarthritis and rheumatoid arthritis, respectively, with a predominant role for VPAC2 receptor in the latter [59]. Taken together, despite difficulties of homogenizing cell cultures from synovial tissues from patients with different degrees of rheumatoid arthritis and osteoarthritis, it is evident that VPAC receptors regulate the threshold of autoreactivity in these diseases where TLRs and their signalling pathways might have great potential as additional targets for new treatments. Last, we should not rule out the possibility that the observed changes in the VPAC2 gene expression were based on post-transcriptional regulation of its mRNA stability due to the inhibition of p38 MAPK, JNK or PI3K/Akt signalling pathways as growing experimental evidence has been showing [60–62].
Figure 4A summarizes our findings. Based on these results, we postulate that up-regulation of VPAC2 by TLRs represents an additional mechanism to ensure the modulator capabilities of VIP, while at the same time a down-regulation of TLRs occurs. In this sense, we used the Jaspar database (http://jaspar.genereg.net) to search for transcription factors possibly involved in TLR-mediated signalling (Fig. 4B). The 3000 nucleotide long upstream region of the Mus musculus VPAC2 gene (ENSMUSG00000011171) was obtained from the Ensembl database (http://www.ensembl.org). We found putative binding sites with a relative score threshold of 0.8 in the positive strand for CREB1, Fos, GATA2, GATA 3 and IRF/ transcription factors. It must be noted that most of the predictions will correspond to sites bound in vitro, and therefore their functional significance must be further analysed.
Figure 4.
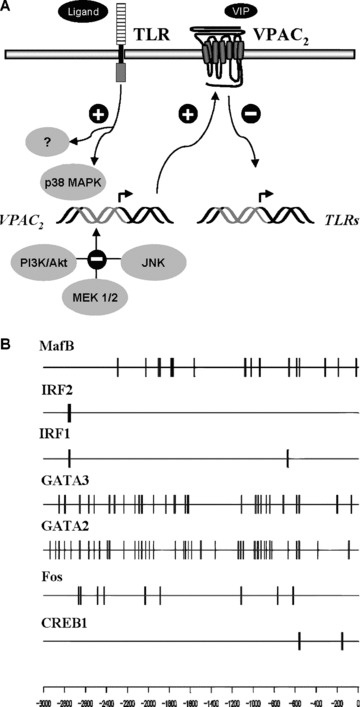
TLR-mediated control of VPAC2. (A) VPAC2 up-regulation by TLRs represents an additional mechanism to ensure the modulator capabilities of VIP while at the same time a down-regulation of TLRs occurs. (B) Distribution of putative binding sites for transcription factors involved in TLR signalling across the 3000 long upstream region of mouse VPAC2 gene. Thicker lines correspond to more densely packed putative binding sites.
The present data shed light on how TLRs differentially decode innate stimuli, not only to achieve protection but also to avoid damage by uncontrolled inflammatory responses, in this case, by means of VPAC2 receptor.
Acknowledgments
This work was supported by grants from the Spanish Ministry of Health (PI052056; PI061641 to D.P.) and PAIDI programme from the Regional Government (BIO323 to D.P.). The authors declare no conflict of interests.
Appendix S1 Lactate dehydrogenase (LDH) release
LDH release from cells was measured to quantify the cytotoxicity by using the Cytotoxicity Detection kit (Roche). Raw 264.7 cells were previously seeded into 96-well plates in a final volume of 100 μL. After treatment, 100 μL of the supernatants and 100 μL of 1% Triton X100-lysed cells were assayed for LDH content under each experimental condition. LDH determinations were carried out according to the manufacturer’s guidelines. LDH release was calculated according to the following formula: LDH release =[LDH supernatant/(LDH supernatant + LDH cells)]*100. The proteasome inhibitor MG-132 (Sigma) was used as a positive control for cytotoxicity.
Supporting Information
Fig. S1 Effects of TLR ligands and protein kinaseinhibitors on cytotoxicity. Raw 264.7 cells were previously seededinto 96-well plates to a final volume of 100 μl. After 24-hrstreatment with protein kinase inhibitors, LDH release wascalculated according to the following equation: LDH release ∇[LDH supernatant/(LDH supernatant + LDHcells)]*100. The proteasome inhibitor MG-132 (6 μM) wasused as a positive control for cytotoxicity. Results are the mean± S.D. of two independent experiments performed in duplicate.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
References
- 1.Anderson P, Delgado M. Endogenous anti-inflammatory neuropeptides and proresolving lipid mediators: a new therapeutic approach for immune disorders. J Cell Mol Med. 2008;12:1830–47. doi: 10.1111/j.1582-4934.2008.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pozo D. Immune-based disorders: the challenges for translational immunology. J Cell Mol Med. 2008;12:1085–6. doi: 10.1111/j.1582-4934.2008.00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez-Rey E, Chorny A, Delgado M. Regulation of immune tolerance by anti-inflammatory neuropeptides. Nat Rev Immunol. 2007;7:52–63. doi: 10.1038/nri1984. [DOI] [PubMed] [Google Scholar]
- 4.Pozo D, Delgado M. The many faces of VIP in neuroimmunology: a cytokine rather a neuropeptide. FASEB J. 2004;18:1325–34. doi: 10.1096/fj.03-1440hyp. [DOI] [PubMed] [Google Scholar]
- 5.Martinez C, Delgado M, Abad C, et al. Regulation of VIP production and secretion by murine lymphocytes. J Neuroimmunol. 1999;93:126–38. doi: 10.1016/s0165-5728(98)00216-1. [DOI] [PubMed] [Google Scholar]
- 6.Delgado M, Ganea D. Cutting edge: is vasoactive intestinal peptide a type 2 cytokine. J Immunol. 2001;166:2907–12. doi: 10.4049/jimmunol.166.5.2907. [DOI] [PubMed] [Google Scholar]
- 7.Huang MC, Miller AL, Wang W, et al. Differential signaling of T cell generation of IL-4 by wild-type and short-deletion variant of type 2 G protein-coupled receptor for vasoactive intestinal peptide (VPAC2) J Immunol. 2006;176:6640–6. doi: 10.4049/jimmunol.176.11.6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szema AM, Hamidi SA, Lyubsky S, et al. Mice lacking the VIP gene show airway hyperresponsiveness and airway inflammation, partially reversible by VIP. Am J Physiol Lung Cell Mol Physiol. 2006;291:L880–6. doi: 10.1152/ajplung.00499.2005. [DOI] [PubMed] [Google Scholar]
- 9.Traves SL, Donnelly LE. Th17 cells in airway diseases. Curr Mol Med. 2008;8:416–26. doi: 10.2174/156652408785160998. [DOI] [PubMed] [Google Scholar]
- 10.Ford JG, Rennick D, Donaldson DD, et al. Il-13 and IFN-gamma: interactions in lung inflammation. J Immunol. 2001;167:1769–77. doi: 10.4049/jimmunol.167.3.1769. [DOI] [PubMed] [Google Scholar]
- 11.Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004;56:249–90. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- 12.Harmar A, Arimura A, Gozes I, et al. International union of pharmacology. XVIII. Nomenclature of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Pharmacol Rev. 1998;50:265–70. [PMC free article] [PubMed] [Google Scholar]
- 13.Arranz A, Juarranz Y, Leceta J, et al. VIP balances innate and adaptive immune responses induced by specific stimulation of TLR2 and TLR4. Peptides. 2008;29:948–56. doi: 10.1016/j.peptides.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 14.Delgado M, Munoz-Elias E, Kan Y, et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumor necrosis factor α transcriptional activation by regulating factor-κb and cAMP response element-binding protein/c-jun. J Biol Chem. 1998;273:31427–36. doi: 10.1074/jbc.273.47.31427. [DOI] [PubMed] [Google Scholar]
- 15.Delgado M, Martinez C, Johnson M, et al. Differential expression of vasoactive intestinal peptide receptors 1 and 2 (VIP-R1 and VIP-R2) mRNA in murine lymphocytes. J Neuroimmunol. 1996;68:27–38. doi: 10.1016/0165-5728(96)00063-x. [DOI] [PubMed] [Google Scholar]
- 16.Lara-Marquez ML, O’Dorisio MS, O’Dorisio TM, et al. Selective gene expression and activation-dependent regulation of vasoactive intestinal peptide receptor type 1 and type 2 in human T cells. J Immunol. 2001;166:2522–30. doi: 10.4049/jimmunol.166.4.2522. [DOI] [PubMed] [Google Scholar]
- 17.Voice JK, Dorsam G, Lee H, et al. Allergic diathesis in transgenic mice with constitutive T cell expression of inducible vasoactive intestinal peptide receptor. FASEB J. 2001;15:2489–96. doi: 10.1096/fj.01-0671com. [DOI] [PubMed] [Google Scholar]
- 18.Sun W, Hong J, Zang YC, et al. Altered expression of vasoactive intestinal peptide receptors in T lymphocytes and aberrant Th1 immunity in multiple sclerosis. Int Immunol. 2006;18:1691–700. doi: 10.1093/intimm/dxl103. [DOI] [PubMed] [Google Scholar]
- 19.Lutz EM, Shen S, Mackay M, et al. Structure of the human VIPR2 gene for vasoactive intestinal peptide receptor type 2. FEBS Lett. 1999;458:197–203. doi: 10.1016/s0014-5793(99)01135-7. [DOI] [PubMed] [Google Scholar]
- 20.Asano E, Kuivaniemi H, Huq AH, et al. A study of novel polymorphisms in the upstream region of vasoactive intestinal peptide receptor type 2 gene in autism. J Child Neurol. 2001;16:357–63. doi: 10.1177/088307380101600509. [DOI] [PubMed] [Google Scholar]
- 21.Steel G, Lutz EM. Characterisation of the mouse vasoactive intestinal peptide receptor type 2 gene, VIPR2, and identification of a polymorphic line-1-like sequence that confers altered promoter activity. J Neuroendocrinol. 2007;19:14–25. doi: 10.1111/j.1365-2826.2006.01498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 23.Trinchieri G, Sher A. Cooperation of toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–90. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 24.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔc(t)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Alexopoulou L, Thomas V, Schnare M, et al. Hyporesponsiveness to vaccination with borrelia burgdorferi ospa in humans and in TLR1- and TLR2-deficient mice. Nat Med. 2002;8:878–84. doi: 10.1038/nm732. [DOI] [PubMed] [Google Scholar]
- 27.Velasco G, Campo M, Manrique OJ, et al. Toll-like receptor 4 or 2 agonists decrease allergic inflammation. Am J Respir Cell Mol Biol. 2005;32:218–24. doi: 10.1165/rcmb.2003-0435OC. [DOI] [PubMed] [Google Scholar]
- 28.Revets H, Pynaert G, Grooten J, et al. Lipoprotein I, a TLR2/4 ligand modulates Th2-driven allergic immune responses. J Immunol. 2005;174:1097–103. doi: 10.4049/jimmunol.174.2.1097. [DOI] [PubMed] [Google Scholar]
- 29.Horiuchi Y, Bae S, Katayama I, et al. Lipoteichoic acid-related molecule derived from the streptococcal preparation, OK-432, which suppresses atopic dermatitis-like lesions in NC/NGA mice. Arch Dermatol Res. 2006;298:163–73. doi: 10.1007/s00403-006-0674-0. [DOI] [PubMed] [Google Scholar]
- 30.Horiuchi Y, Bae S, Katayama I, et al. Therapeutic effects of streptococcal preparation OK-432 on atopic dermatitis-like lesions in NC/NGA mice: possible shift from a Th2- to Th1-predominance. J Dermatol Sci. 2004;35:187–97. doi: 10.1016/j.jdermsci.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by toll-like receptor 2. J Clin Invest. 2005;115:3149–56. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansson GK, Libby P, Schonbeck U, et al. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–91. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 33.Schon MP, Schon M. Imiquimod: mode of action. Br J Dermatol. 2007;157:8–13. doi: 10.1111/j.1365-2133.2007.08265.x. [DOI] [PubMed] [Google Scholar]
- 34.Vidal D, Matias-Guiu X, Alomar A. Fifty-five basal cell carcinomas treated with topical imiquimod: outcome at 5-year follow-up. Arch Dermatol. 2007;143:266–8. doi: 10.1001/archderm.143.2.266. [DOI] [PubMed] [Google Scholar]
- 35.Redondo P, Del Olmo J, Lopez-Diaz de Cerio A, et al. Imiquimod enhances the systemic immunity attained by local cryosurgery destruction of melanoma lesions. J Invest Dermatol. 2007;127:1673–80. doi: 10.1038/sj.jid.5700777. [DOI] [PubMed] [Google Scholar]
- 36.Schon M, Schon MP. The antitumoral mode of action of imiquimod and other imidazoquinolines. Curr Med Chem. 2007;14:681–7. doi: 10.2174/092986707780059625. [DOI] [PubMed] [Google Scholar]
- 37.Fabricius D, O’Dorisio MS, Blackwell S, et al. Human plasmacytoid dendritic cell function: inhibition of IFN-alpha secretion and modulation of immune phenotype by vasoactive intestinal peptide. J Immunol. 2006;177:5920–7. doi: 10.4049/jimmunol.177.9.5920. [DOI] [PubMed] [Google Scholar]
- 38.Berghofer B, Haley G, Frommer T, et al. Natural and synthetic TLR7 ligands inhibit CpG-A- and CpG-C-oligodeoxynucleotide-induced IFN-alpha production. J Immunol. 2007;178:4072–9. doi: 10.4049/jimmunol.178.7.4072. [DOI] [PubMed] [Google Scholar]
- 39.Applequist SE, Wallin RPA, Ljunggren H-G. Variable expression of toll-like receptor in murine innate and adaptive immune cell lines. Int Immunol. 2002;14:1065–74. doi: 10.1093/intimm/dxf069. [DOI] [PubMed] [Google Scholar]
- 40.Mizel SB, Honko AN, Moors MA, et al. Induction of macrophage nitric oxide production by gram-negative flagellin involves signaling via heteromeric toll-like receptor 5/toll-like receptor 4 complexes. J Immunol. 2003;170:6217–23. doi: 10.4049/jimmunol.170.12.6217. [DOI] [PubMed] [Google Scholar]
- 41.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 42.Miggin SM, O’Neill LA. New insights into the regulation of TLR signaling. J Leukoc Biol. 2006;80:220–6. doi: 10.1189/jlb.1105672. [DOI] [PubMed] [Google Scholar]
- 43.Cuenda A, Rouse J, Doza YN, et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–33. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 44.Ranganathan P, Agrawal A, Bhushan R, et al. Expression profiling of genes regulated by TGF-beta: Differential regulation in normal and tumour cells. BMC Genomics. 2007;8:98. doi: 10.1186/1471-2164-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kakurai M, Fujita N, Murata S, et al. Vasoactive intestinal peptide regulates its receptor expression and functions of human keratinocytes via type I vasoactive intestinal peptide receptors. J Invest Dermatol. 2001;116:743–9. doi: 10.1046/j.1523-1747.2001.01306.x. [DOI] [PubMed] [Google Scholar]
- 46.Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee PJ, Zhang X, Shan P, et al. Erk1/2 mitogen-activated protein kinase selectively mediates IL-13-induced lung inflammation and remodeling in vivo. J Clin Invest. 2006;116:163–73. doi: 10.1172/JCI25711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Kholy W, Macdonald PE, Lin JH, et al. The phosphatidylinositol 3-kinase inhibitor LY294002 potently blocks k(v) currents via a direct mechanism. FASEB J. 2003;17:720–2. doi: 10.1096/fj.02-0802fje. [DOI] [PubMed] [Google Scholar]
- 49.Gharbi SI, Zvelebil MJ, Shuttleworth SJ, et al. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J. 2007;404:15–21. doi: 10.1042/BJ20061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foster N, Lea SR, Preshaw PM, et al. Pivotal advance: Vasoactive intestinal peptide inhibits up-regulation of human monocyte TLR2 and TLR4 by LPS and differentiation of monocytes to macrophages. J Leukoc Biol. 2007;81:893–903. doi: 10.1189/jlb.0206086. [DOI] [PubMed] [Google Scholar]
- 51.Gomariz RP, Arranz A, Abad C, et al. Time-course expression of toll-like receptors 2 and 4 in inflammatory bowel disease and homeostatic effect of VIP. J Leukoc Biol. 2005;78:491–502. doi: 10.1189/jlb.1004564. [DOI] [PubMed] [Google Scholar]
- 52.Arranz A, Abad C, Juarranz Y, et al. Vasoactive intestinal peptide as a healing mediator in Crohn’s disease. Neuroimmunomodulation. 2008;15:46–53. doi: 10.1159/000135623. [DOI] [PubMed] [Google Scholar]
- 53.Arranz A, Gutierrez-Canas I, Carrion M, et al. VIP reverses the expression profiling of TLR4-stimulated signaling pathway in rheumatoid arthritis synovial fibroblasts. Mol Immunol. 2008;45:3065–73. doi: 10.1016/j.molimm.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 54.Arranz A, Androulidaki A, Zacharioudaki V, et al. Vasoactive intestinal peptide suppresses toll-like receptor 4 expression in macrophages via Akt1 reducing their responsiveness to lipopolysaccharide. Mol Immunol. 2008;45:2970–80. doi: 10.1016/j.molimm.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 55.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 56.Fukao T, Yamada T, Tanabe M, et al. Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat Immunol. 2002;3:295–304. doi: 10.1038/ni768. [DOI] [PubMed] [Google Scholar]
- 57.Williams DL, Ozment-Skelton T, Li C. Modulation of the phosphoinositide 3-kinase signaling pathway alters host response to sepsis, inflammation, and ischemia/reperfusion injury. Shock. 2006;25:432–9. doi: 10.1097/01.shk.0000209542.76305.55. [DOI] [PubMed] [Google Scholar]
- 58.Delgado M, Robledo G, Rueda B, et al. Genetic association of vasoactive intestinal peptide receptor with rheumatoid arthritis: Altered expression and signal in immune cells. Arthritis Rheum. 2008;58:1010–9. doi: 10.1002/art.23482. [DOI] [PubMed] [Google Scholar]
- 59.Juarranz Y, Gutierrez-Canas I, Santiago B, et al. Differential expression of vasoactive intestinal peptide and its functional receptors in human osteoarthritic and rheumatoid synovial fibroblasts. Arthritis Rheum. 2008;58:1086–95. doi: 10.1002/art.23403. [DOI] [PubMed] [Google Scholar]
- 60.Pei Y, Zhu P, Dang Y, et al. Nuclear export of NF90 to stabilize IL-2 mRNA is mediated by Akt-dependent phosphorylation at ser647 in response to CD28costimulation. J Immunol. 2008;180:222–9. doi: 10.4049/jimmunol.180.1.222. [DOI] [PubMed] [Google Scholar]
- 61.Khabar KS. Rapid transit in the immune cells: the role of mRNA turnover regulation. J Leukoc Biol. 2007;81:1335–44. doi: 10.1189/jlb.0207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Valledor AF, Sanchez-Tillo E, Arpa L, et al. Selective roles of MAPKs during the macrophage response to IFN-gamma. J Immunol. 2008;180:4523–9. doi: 10.4049/jimmunol.180.7.4523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item