

Abstract
Type 2 melastatin-related transient receptor potential channel (TRPM2), a member of the melastatin-related TRP (transient receptor potential) subfamily is a Ca2+-permeable channel activated by hydrogen peroxide (H2O2). We have investigated the role of TRPM2 channels in mediating the H2O2-induced increase in the cytoplasmic free Ca2+ concentration ([Ca2+]i) in insulin-secreting cells. In fura-2 loaded INS-1E cells, a widely used model of β-cells, and in human β-cells, H2O2 increased [Ca2+]i, in the presence of 3 mM glucose, by inducing Ca2+ influx across the plasma membrane. H2O2-induced Ca2+ influx was not blocked by nimodipine, a blocker of the L-type voltage-gated Ca2+ channels nor by 2-aminoethoxydiphenyl borate, a blocker of several TRP channels and store-operated channels, but it was completely blocked by N-(p-amylcinnamoyl)anthranilic acid (ACA), a potent inhibitor of TRPM2. Adenosine diphosphate phosphate ribose, a specific activator of TRPM2 channel and H2O2, induced inward cation currents that were blocked by ACA. Western blot using antibodies directed to the epitopes on the N-terminal and on the C-terminal parts of TRPM2 identified the full length TRPM2 (TRPM2-L), and the C-terminally truncated TRPM2 (TRPM2-S) in human islets. We conclude that functional TRPM2 channels mediate H2O2-induced Ca2+ entry in β-cells, a process potently inhibited by ACA.
Keywords: calcium influx, TRPM2, TRP channels, insulin-secreting cells, microfluorometry, N-(p-amylcinnamoyl)anthranilic acid, calcium signalling
Introduction
The type 2 melastatin-related transient receptor potential channel (TRPM2; formerly called TRPC7 and LTRPC2) is a non-specific cation channel permeable to Na+, K+ and albeit weakly to Ca2+[1]. In addition to the full-length TRPM2 (called TRPM2-L), a short form of TRPM2 (called TRPM2-S), where the four C-terminal trans-membrane domains and the putative pore-forming domain are deleted, have been described. TRPM2 is activated by hydrogen peroxide (H2O2), a model substance used as a paradigm of oxidative stress. TRPM2 channels are, thus, thought to be sensors for oxidative stress. Reactive oxygen species and oxidative stress have been implicated in the pathogenesis of diabetes. A number of studies have demonstrated that H2O2 induces β-cell death [2, 3]. There is evidence suggesting that TRPM2 mRNA is expressed in human islets [4]. TRPM2 current has been studied mostly in rat insulinoma RIN-5F, and Cambridge rat insulinoma G1 (CRI-G1) cells [5, 6]. In these cells, it has been demonstrated that TRPM2 channels are involved in insulin secretion [5]. Moreover, it has been shown that H2O2-induced death of insulinoma cells is prevented by antisense TRPM2 [7]. However, insulinoma cells of type RIN-5F and CRI-G1 are highly undifferentiated, poorly glucose responsive and thus, show limitations as models of β-cells. A better model of β-cells is INS-1E cells. These are highly differentiated rat insulinoma cells that are currently widely used in experimental diabetes research [8]. It is not known whether INS-1E cells express functional TRPM2 channels. Moreover, TRPM2 proteins have not yet been demonstrated in human β-cells. The main aims of this study were to: (i) investigate the role of the TRPM2 channels in H2O2-induced [Ca2+]i increase in INS-1E cells; (ii) to test N-(p-amylcinnamoyl)anthranilic acid (ACA) as an inhibitor of TRPM2 channels in these cells and (iii) to identify the TRPM2 proteins in human islets.
Materials and methods
Materials
Fura-2 AM was from Invitrogen (Stockholm, Sweden). H2O2 (30%[W/W]), adenosie diphosphate ribose, nimodipine, 2-aminoethoxydiphenyl borate (2-APB) were from Sigma. ACA was from Calbiochem (Stockholm, Sweden). INS-1E cells were from C. B. Wollheim, Geneva, Switzerland.
Cell culture
We used a highly differentiated rat insulinoma cell line INS-1E cells (S5 clone) [8, 9]. Glucose-stimulated insulin secretion in these cells is similar to that reported previously [8, 9]. The cells were cultured in RPMI-1640 medium, supplemented with 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 500 μM 2-mercaptoethanol (2-ME), 2.5% foetal bovine serum, 50 i.u./ml penicillin and 50 μg/ml streptomycin.
Isolation and culture of human islets
Human islets were provided by the Cell Isolation and Transplantation Centre at the University of Geneva School of Medicine. Use of islets for in vitro experiments was approved by the local ethical committee. Islets from three donors were used for experiments. Islets were purified by an automated procedure using a continuous digestion-filtration device [10, 11]. Islets were dispersed into single cells by repeated pipetting after digestion in 0.025% trypsin in Ca2+- and Mg2+-free HBSS. Dispersed cells were cultured on glass cover slips for measurement of [Ca2+]i.
Measurement of [Ca2+]i
Cells cultured on cover slips were incubated in RPMI 1640 containing 0.1% bovine serum albumin (BSA) and 1 μM fura-2 AM for 35 min. The cover slips were then left for 10 min. at room temperature (RT) in a solution containing (in mM) 140 NaCl, 3.6 KCl, 0.5 NaH2PO4, 0.5 MgSO4, 1.5 CaCl2, 10 HEPES, 3 glucose and 0.1% BSA (pH 7.4). Nominally Ca2+-free medium was prepared by omitting Ca2+ from the solution and adding EGTA (0.5 mM). Cover slips were mounted at the bottom of an open perfusion chamber on the stage of an inverted microscope (Olympus CK40, Olympus, Solna, Sweden). The temperature within the chamber was maintained at 37°C. Single cells were studied by using a 40×, 1.3 NA oil immersion objective (40×, UV APO). Cells were excited alternately by 340 and 380 nm wavelengths selected by a monochromator (Photon Technology International, Seefeld, Germany). The emitted light selected by a 510 nm filter was monitored by a photomultiplier tube. The emissions at the excitation wavelengths of 340 nm (F340) and 380 nm (F380) were used to calculate the fluorescence ratio (R340/380). The excitation wavelengths were alternated to obtain one ratio data point per second. The [Ca2+]i was calculated from R340/380 as described before [12]. Rmax and Rmin were determined by using external standard solutions containing fura-2 free acid and sucrose (2M). The Kd for Ca2+-fura-2 was taken as 225 nM.
Recording of TRPM2 current
Cells were analysed with the patch-clamp technique in the whole-cell mode, using an EPC 9 patch-clamp amplifier equipped with a personal computer with Pulse and X chart software (HEKA, Lamprecht, Germany). The extracellular solution contained (in mM) 140 NaCl, 1.2 MgCl2, 1.2 CaCl2, 5 KCl, 10 HEPES, pH adjusted by NaOH to 7.4. For Na+-free solutions, Na+ was replaced by 150 mM N-methyl-D-glucamine (NMDG+) and pH was titrated with HCl. The pipette solution contained (in mM) 145 Cs-glutamate, 8 NaCl, 2 MgCl2, 1 Cs-EGTA, 0.88 CaCl2, 10 HEPES, pH adjusted by CsOH to 7.2. This solution contained 1 μM free Ca2+. If not otherwise stated cells were held at a potential of –60 mV at 22°C. The current–voltage (I–V) relations were obtained during voltage ramps from –90 to +60 mV applied for 400 ms.
Western blot analysis of human islets
Human islets were homogenized in an ice-cold buffer consisting of 150 mM NaCl, 20 mM Tris, pH 7.5, 1% NP40, 1 mM ethylenediaminetetraacetic acid and protease inhibitors. The homogenate was centrifuged at 20,000 rpm for 30 min. at 4°C. The supernatant containing the membrane proteins was collected, and protein concentration was measured by Bio-Rad protein assay kit (Biorad, Sundbyberg, Sweden). A total of 90 μg of protein was fractionated by 10% SDS-polyacrylamide gel electrophoresis and transferred to PVDF membrane. The membranes were blocked by 5% non-fat milk overnight at 4°C, in Tris-buffered saline with Tween-20 (TBS-T). This step was followed by overnight incubation at 4°C with the primary TRPM2 antibodies (dilution 1:300). The primary antibodies were: (i) an affinity-purified rabbit polyclonal IgG directed against an epitope on the N-terminal part of TRPM2 (anti-TRPM2-N) (BL 970. Cat. no. A300–414A, Bethyl Laboratories, Inc., Montgomery, TX, USA). (ii) Affinity-purified rabbit polyclonal IgG directed against the C-terminal part of the TRPM2 (anti-TRPM2-C) (BL969, Cat. no.A300–413A, Bethyl Laboratories, Inc.). The PVDF membranes were then washed with TBS-T buffer, and incubated with goat anti-rabbit IgG conjugated to horseradish-peroxidase (1:10,000) for 1 hr at RT. Membranes were washed and the immunoreactive bands were detected by enhanced chemiluminescence method and exposure to x-rays film. Antibody specificity was tested by using a blocking peptide (TRPM2 blocking peptide BP969, catalogue no. BP300–413, Bethyl Laboratories, Inc.).
Statistical analysis
Data are displayed as mean ± S.E.M., n indicating the number of independent experiments. Student’s t-test (unpaired) was used for testing statistical significance and P < 0.05 was accepted as statistically significant.
Results
H2O2 increased [Ca2+]i by inducing Ca2+ entry through TRPM2 channels
INS-1E cells were cultured in the presence of 2-ME. During the measurement of [Ca2+]i, 2-ME was omitted from the medium. To limit damage to the cells during the experiments, the duration of the experiments was kept as short as possible. To be able to detect relatively small changes in [Ca2+]i, we loaded cells with low concentration of fura-2, because the chelating action of high concentrations of fura-2 makes detection of small [Ca2+]i changes difficult [13]. We first, established the concentrations of H2O2 that could increase [Ca2+]i reproducibly, without causing major damage to the cells. As shown in Fig. 1A, after the application of H2O2 (200 μM), [Ca2+]i increased to a plateau in ∼2–3 min., the magnitude of maximal increase being 29 ± 3 nM (n= 16). All [Ca2+]i changes were confirmed to be true [Ca2+]i changes, by examining the respective F340 and F380 traces, which moved in opposite directions with change of [Ca2+]i. No [Ca2+]i change was observed for a period of up to 10 min. in cells where H2O2 was not applied (Fig. 1C). After the washout of H2O2, [Ca2+]i returned to the baseline indicating that there was no major drift in the baseline (Fig. 1A). A return of [Ca2+]i to the basal level also suggested that the cells were not severely damaged by exposure to 200 μM H2O2. 50 μM H2O2 increased [Ca2+]i, the magnitude of which was comparable to that obtained with 200 μM H2O2 (Fig. 1E). However, [Ca2+]i increase by 50 μM H2O2 was more variable compared to that obtained by 200 μM H2O2 (Fig. 1E). [Ca2+]i increases by higher concentrations of H2O2 (e.g. 500 μM and 1 mM) were not completely reversible on washout which suggested persistent activation of the Ca2+-entry pathways (Fig. 1B and D). [Ca2+]i response to a given concentration of H2O2 (i.e. 200 μM or 500 μM) varied, but on the average, the magnitude of [Ca2+]i increase obtained by 200 μM H2O2 was similar to that obtained by 500 μM H2O2 (Fig. 1E). For these reasons, we used either 200 μM or 500 μM of H2O2 in subsequent experiments. As shown in Fig. 1F and G, H2O2 (200 μM and 500 μM) increased [Ca2+]i also in human β-cells.
Figure 1.
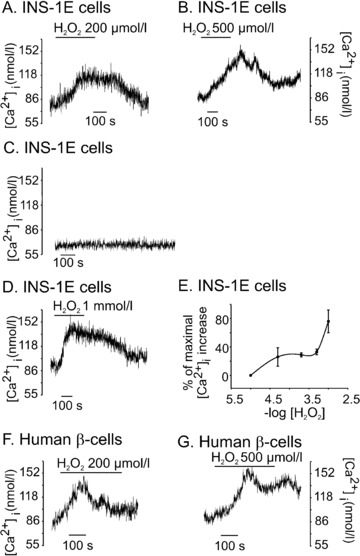
H2O2 increased [Ca2+]i in INS-1E cells and human β-cells. Cells were perfused with physiological solution containing 3 mM glucose and [Ca2+]i was measured in fura-2 loaded cells by microfluorometry. (A) H2O2 (200 μM) increased [Ca2+]i in a reversible manner (n= 16). (B) and (D) [Ca2+]i increase by 500 μM, (n= 24) and 1 mM H2O2, (n= 9) did not completely reverse after washout of H2O2. (C) baseline [Ca2+]i remained stable when no H2O2 was added. (E) the concentration-response curve for H2O2-induced [Ca2+]i increase. Data are expressed as percentage of maximal [Ca2+]i increase obtained by 1 mM H2O2. Each point represents a mean of 5–24 experiments. (F) and (G) show the effects of H2O2 on [Ca2+]i in single human β-cells. H2O2 increased [Ca2+]i in 10 out of 12 cells for 200 μM and 4 out of 6 for 500 μM.
When Ca2+ was omitted from the extracellular medium, [Ca2+]i response to 200 μM H2O2 was abolished (Fig. 2B and C). In these experiments, cells were exposed to nominally Ca2+-free medium for 1 min. before addition of H2O2. In separate experiments, we established that exposure of cells to nominally Ca2+-free medium for such short period, did not deplete the ER Ca2+ store, because carbachol increased [Ca2+]i by releasing Ca2+ from the ER under such conditions (data not shown). The maximal [Ca2+]i changes in the Ca2+-containing and in the Ca2+-free medium were 39 ± 2 nM and –5 ± 10 nM, respectively (P= 0.01, n= 6). In Fig. 2D, [Ca2+]i was first raised by 500 μM H2O2 in the presence of Ca2+-containing extracellular medium. When [Ca2+]i increased to a plateau, the medium was switched to the nominally Ca2+-free medium. This resulted in the return of [Ca2+]i to the basal level, indicating that the [Ca2+]i increase by H2O2 was due to the entry of Ca2+ across the plasma membrane (Fig. 2D, c.f.Fig. 1B).
Figure 2.
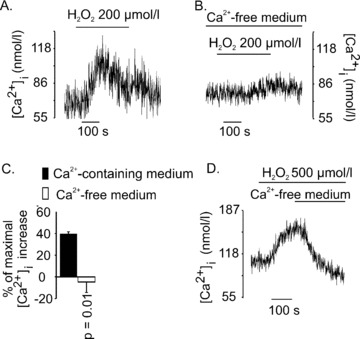
H2O2-induced [Ca2+]i increase was due to Ca2+ entry across the plasma membrane. H2O2 (200 μM) was applied to INS-1E cells in the presence of 1.5 mM extracellular Ca2+ (A) or in nominally Ca2+-free medium (B). Maximal [Ca2+]i change in Ca2+ containing medium was 39±2 nM (n= 3) and that in nominally Ca2+-free medium was –5±10 nM (n= 3) (C). In (D), cells were first exposed to H2O2 (500 μM) in the presence of 1.5 mM extracellular Ca2+, and the solution was switched to a nominally Ca2+-free medium at the time indicated by the horizontal bar(n= 5) (c.f.Fig. 1B).
[Ca2+]i increase by H2O2 (500 μM) was not inhibited by nimodipine (5 μM), a blocker of the L-type voltage-gated Ca2+ channels (Fig. 3A and B). The maximal [Ca2+]i increases by H2O2 in the control group and in the nimodipine group were 27 ± 5 and 23 ± 6 nM, respectively (P= 0.63 n= 15) (Fig. 3C). 2-APB (50 μM), a blocker of several TRP channels and some store-operated Ca2+ channels, also did not inhibit H2O2-induced Ca2+ entry (Fig. 3D and E). The magnitudes of [Ca2+]i increases in the control group and in the 2-APB group were 13 ± 2 and 14 ± 1 nM, respectively (P= 0.75, n= 10) (Fig. 3F). Flufenamic acid and econazole are two inhibitors of TRPM2 [14, 15]. We found that both flufenamic acid and econazole increased [Ca2+]i by themselves and were thus, not suitable for use in further experiments (data not shown). Instead, we tested the effect of ACA, a potent blocker of TRPM2 [16]. In separate experiments, we demonstrated that ACA (20 μM) itself, did not increase [Ca2+]i in INS-1E cells (Fig. 3G). As shown in Fig. 3H (trace a), H2O2 (200 μM) induced a typical increase of [Ca2+]i in the control cells. When H2O2 was applied in the presence of ACA (20 μM), there was no increase of [Ca2+]i (trace b). Instead, on the average, [Ca2+]i decreased in the ACA-treated cells, despite continued presence of H2O2. The maximal changes of [Ca2+]i by H2O2 in the absence of, and in the presence of ACA were 23 ± 1 and –9 ± 6 nM, respectively (P≤; 0.01, n= 6) (Fig 3I). These observations indicated that the H2O2-induced Ca2+ entry was due to the activation of the TRPM2 channels.
Figure 3.
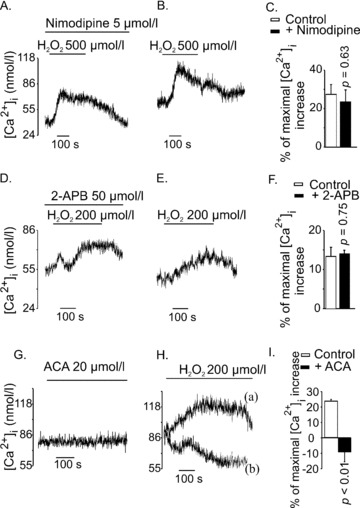
Effects of different channel blockers on H2O2-induced [Ca2+]i increase in INS-1E cells. (A) and (B) Nimodipine did not inhibit H2O2-induced [Ca2+]i response. Cells were pre-incubated with Nimodipine (5 μM) for 10 min. H2O2 (500 μM) was applied in the continued presence of nimodipine (the trace is representative of seven independent experiments). (B) Shows control experiments for (A) (the trace is representative of eight independent experiments). (D) and (E) 2-APB (50 μM) did not alter the [Ca2+]i response to H2O2 (200 μM). Cells were treated with 2-APB (50 μM) for 10 min. during pre-incubation and H2O2 was applied in the continued presence of 2-APB. (E) The control for (A) showing [Ca2+]i response to H2O2 in the absence of 2-APB (traces D and E are representatives of three to five independent experiments). (G) ACA (20 μM) did not increase [Ca2+]i in INS-1E cells. (H) ACA (20 μM) completely inhibited H2O2-induced [Ca2+]i response. Trace (a) shows [Ca2+]i response to H2O2 (200 μM) in the absence of ACA (the trace is representative of three independent experiments). In trace (b), ACA (20 μM) was applied 1 min. before application of H2O2 (200 μM) and was continuously present in the perfusion (the trace is representative of three independent experiments). (I) Maximal Ca2+ changes by H2O2 in controls and in the ACA-treated cells were 23 ± 1 and –9 ± 6 nM, respectively (P < 0.01, n= 6). In (C), (F) and (I), the bars represent mean [Ca2+]i increase obtained by H2O2, expressed as the percentage of maximal [Ca2+]i increase obtained by 25 mM KCl in respective experiments.
TRPM2 current in INS-1E cells
In patch-clamp experiments 0.6 mM ADP ribose was dialysed into the cells through the patch pipette. The pipette solution also contained 1 μM Ca2+ to facilitate development of ADP ribose-dependent current [17]. Figure 4A shows rapid development of inward currents of ∼1 nA after establishment of the whole-cell configuration. The inward currents were minimized when extracellular Na+ was substituted by NMDG+, which is impermeable through TRPM2. In the absence of ADP ribose, no such current developed, even if cells were infused with pipette solution containing 1 μM Ca2+ (see first 150 sec. in the recording of Fig. 4B). After extracellular application of H2O2 (∼10 mM), inward currents developed gradually and the currents were immediately suppressed when the bath solution contained NMDG+ (Fig. 4B). The currents could be repeatedly restored by reperfusion with standard bath solution, even when the solution did not contain H2O2 (Fig. 4B). After external application of ACA (50 μM), the current gradually declined to almost basal levels. After washout of ACA, currents were partially restored demonstrating that the ACA effect was reversible (Fig. 4B). The corresponding I–V relation is shown in Fig. 4C. The H2O2-evoked currents showed a reversal potential close to 0 mV and inward currents were minimized in the presence of NMDG+, which is characteristic for a non-selective cation current (NSCC) like TRPM2.
Figure 4.

Whole-cell currents induced by ADP ribose and H2O2 in INS-1E cells. The whole-cell configuration was attained at the point indicated with ‘w.c.’. Recordings were performed at RT and the holding potential was –60 mV. Bars indicate times where the standard bath solution was changed to a solution containing either NMDG+ or the TRPM2 channel inhibitor ACA. (A) Whole-cell current recorded in the presence of intracellular ADP ribose. The pipette solution contained 0.6 mM ADP ribose and 1 μM Ca2+. (B) Whole cell currents recorded without ADP ribose and after application of 1–2 μl 30% H2O2 directly into the recording chamber. The estimated final concentration of H2O2 in the chamber was ∼10 mM. The pipette solution contained 1 μM free Ca2+. (C) Current–voltage relationship of H2O2-induced currents as derived from (B), recorded during voltage ramps from –90 to +60 mV of 400 ms duration.
TRPM2 proteins in human islets
We performed Western blotting with membrane preparations from human islets. The blot was probed with anti-TRPM2-N and anti-TRPM2-C antibodies. The immunogen for anti-TRPM2-N was the peptide ILKELSKEEEDTDSSEEMLA, which represents the amino acids 658–677 of human TRPM2 encoded within exon 13. The immunogen for anti-TRPM2-C was the peptide KAAEEPDAEPGGRKKTEEPGDS, which represents amino acids 1216–1237 of human TRPM2 encoded within exon 25. In Western blotting of human islets ∼171 kD bands representing TRPM2-L were detected by both anti-TRPM2-N (Fig. 5A) and anti-TRPM2-C antibodies (Fig. 5A1). As expected, a ∼95 kD band representing the TRPM2-S was detected by the anti-TRPM2-N antibody (Fig. 5A) but not by the anti-TRPM2-C antibody (Fig. 5A1). In control experiments we found that the ∼75 kD bands were non-specific ones, because they were detected even when the membranes were treated with the corresponding blocking peptides.
Figure 5.
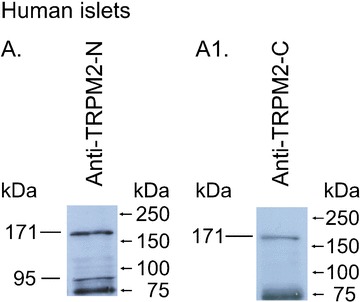
Western blot analysis of TRPM2 proteins in human islets. A total of 90 μg of membrane proteins from human islets were separated in two lanes (A, A1) by 10% SDS-PAGE electrophoresis. In A, the blots were probed with an anti-TRPM2-N antibody (1:300). In A1, the blots were probed with an anti-TRPM2-C antibody (1:300). Standard proteins are shown on the right side. The experiments have been repeated at least three times with similar results.
Discussion
Effects of H2O2 on [Ca2+]i changes in INS-1E cells have been reported before [2]. However, it remained unknown whether H2O2 can trigger Ca2+ influx in these cells, and in that case, what could be the identity of the Ca2+ influx pathways. The emergence of TRPM2 as a H2O2-sensitive channel prompted us to examine if TRPM2 could be a link between H2O2 and [Ca2+]i increase in INS-1E cells. We demonstrate that a short (∼5 min.) exposure of H2O2 (50–500 μM) to INS-1E cells, increased [Ca2+]i solely by inducing Ca2+ entry across the plasma membrane. This was evident from the observation that no [Ca2+]i increase by H2O2 was observed when Ca2+ was omitted from the extracellular medium. H2O2 activates TRPM2 by acting on the cytoplasmic side of the channel [18]. Externally applied H2O2 does not freely pass through the plasma membrane [19]. For this reason, and because of the presence of catalase in the cytoplasm, the effective cytoplasmic concentration of H2O2 that increased [Ca2+]i in our experiments, is likely to be lower. The high [Ca2+]i increase caused by 1 mM H2O2 is due to multiple non-specific mechanisms including the release of Ca2+ from the mitochondria and the ER, as has been described before [20, 21]. In our study, H2O2-activated Ca2+ entry was not blocked by nimodipine, indicating that the L-type voltage-gated Ca2+ channels did not mediate the Ca2+ entry. Moreover, 2-APB did not block the Ca2+ entry suggesting a lack of involvement of some of the store-operated Ca2+ channels or the inositol 1,4,5 trisphosphate receptor [22]. As an inhibitor of TRPM2-channel, the role of 2-APB remains controversial [23, 24]. On the other hand, H2O2-induced Ca2+ entry was completely blocked by ACA, a potent blocker of TRPM2 channel [16]. In fact, in ACA-treated cells there was, on the average, a small decrease in [Ca2+]i suggesting some basal activity of TRPM2 channels at 37°C. ACA, however, is not entirely specific for TRPM2, because it also blocks TRPM8 and TRPC6 [16]. Nevertheless, Ca2+ entry that is induced by H2O2 and ADP ribose, and is blocked by ACA is most likely to be mediated through the TRPM2 channels.
ACA has generally been used as an inhibitor of phospholipase A2 (PLA2), which plays an important role in mediating insulin secretion and [Ca2+]i oscillations in β-cell [25–29]. However, in our experiments, activation of TRPM2 by H2O2 did not involve activation of PLA2. This is evident from the observation that H2O2 activated TRPM2 current, even when cells were internally dialysed and the recordings were performed at 22°C (Fig. 4B). In fact, it is known that H2O2 does not activate PLA2, rather it inhibits the enzyme [30]. Given that ACA is now established as a potent blocker of TRPM2, caution is needed in interpreting previous reports where ACA was used as the sole inhibitor of PLA2. Another inhibitor of PLA2, namely AACOCF3 does not inhibit TRPM2 [16]. However, AACOCF3 was not suitable for use in our experiments, because it is oxidized by H2O2.
Consistent with H2O2-induced [Ca2+]i increase, we recorded NSCC induced by H2O2 and ADP ribose in INS-1E cells. The NSCC was carried mainly by Na+ in the inward direction. Such Na+ currents were minimized by NMDG+, in spite of the presence of 1.2 mM Ca2+ in the external solution. In a previous study, we demonstrated that very small Ca2+ currents could be detected when external Na+ was replaced by NMDG+, even when the external Ca2+ concentration was raised to 10 mM [18]. The contribution of Ca2+ to the overall currents was not resolved in the experiments shown in Fig. 4. It is known from previous studies that Ca2+ is only weakly permeable through TRPM2 channel, the permeability ratio pCa:pNa being ∼0.6–0.7 [31]. However, the permeability of Ca2+ measured in the presence of high extracellular Ca2+ can lead to an underestimation of Ca2+ fluxes under physiological conditions [32]. Functionally, the importance of the Ca2+ fluxes through the TRPM2 channels is evident from the increase in [Ca2+]i detected by fura-2 after activation of TRPM2 by H2O2. In patch-clamp experiments, we also used 0.6 mM ADP ribose together with 1 μM free Ca2+. This protocol allowed rapid development of a sizable current after attaining the whole-cell configuration (Fig. 4A). In contrast, the development of currents after extracellular application of H2O2, even in the presence of intracellular Ca2+, was delayed. Such delay may be due to the fact that H2O2 does not gate TRPM2 directly, rather it may act by increasing the concentration of intracellular ADP ribose [33, 34]. The activation of TRPM2 currents by H2O2 was long lasting and persisted even after wash out of H2O2, an observation consistent with previous reports [18, 31]. After substitution of the NMDG+-containing bath solution (which suppressed inward currents) with normal bath solution (which supported inward currents) the inward currents increased immediately to the maximal level. This indicated that the channel activation persisted after the initial exposure to H2O2. In contrast, the release from ACA-block showed much slower kinetics, an observation consistent with a decrease in the open probability of the channel by ACA [16]. Thus, we have demonstrated an inward current that is activated by ADP-ribose (Fig. 4A) and by H2O2 (Fig. 4B) and is inhibited by ACA. This pharmacological profile establishes that the current is mediated through the TRPM2 channels.
Furthermore, by Western blotting, we have demonstrated for the first time, the presence of TRPM2 proteins in human islets. By using anti-TRPM2-N and anti-TRPM2-C antibodies, we identified two isoforms of TRPM2 in these cells [35]. The anti-TRPM2-N antibody detected not only the full length TRPM2-L, but also the C-terminally truncated TRPM2-S, in human islets. TRPM2-S isoform appeared as a ∼95 kD band detected by the anti-TRPM2-N, and not by the anti-TRPM2-C. TRPM2-S itself does not form a channel but instead it acts as a dominant negative of TRPM2-L [35]. Thus, human β-cells have not only the H2O2-sensitive isoform but also the protective isoform of TRPM2. The relative abundance of these isoforms may determine the extent of H2O2-induced TRPM2-mediated Ca2+ influx [36].
Previous studies have examined the effects of H2O2 on insulin secretion, [Ca2+]i changes, membrane potential changes, glucose metabolism, mitochondrial metabolism and β-cell death [2, 20, 21, 37]. It is evident from these reports that exposure of β-cells to relatively high concentration of H2O2 for prolonged period inhibits metabolism, leading to opening of the KATP channels, hyperpolarization of membrane potential and inhibition of insulin secretion [2, 20]. More recently, Pi et al. demonstrated that 1–4 μM H2O2 induces insulin release from INS-1 cells suggesting that H2O2 may act as a signal for insulin secretion [3]. These investigators used a clone of INS-1 cells which is different from ours. The authors did not report whether 1–4 μM H2O2 increased [Ca2+]i in their clone of INS-1 cells. Maechler et al. demonstrated that the threshold concentration of H2O2 for insulin secretion from INS-1E cells was 200 μM, a concentration that invariably increased [Ca2+]i in our experiments [2]. However, in our patch-clamp experiments, we used 10 mM H2O2 to obtain a large current that displays a fingerprint of current properties of TRPM2 channel. Even when TRPM2 current was activated by 10 mM H2O2, the current could be completely blocked by ACA. TRPM2 is a temperature sensitive channel and its regulation by temperature has been studied by Togashi et al.[5]. Whereas, our microfluorometry experiments were performed at 37°C, the patch-clamp experiments were done at 22°C for technical reasons. It may be mentioned that patch-clamp experiments are performed under conditions that are not strictly physiological, and it is often necessary to use 10 mM H2O2 for inducing TRPM2 currents in native cells when experiments are performed at 22°C [18, 37]. However, in transfected cells where TRPM2 is overexpressed, a sizable current can often be detected by micromolar H2O2[7].
In summary, we have demonstrated that in INS-1E cells, H2O2 (50–500 μM) applied for a short period (5–10 min.) increased [Ca2+]i by triggering Ca2+ entry through the TRPM2 channels. Consistent with this, H2O2- and ADP ribose-activated TRPM2 current was detected in INS-1E cells. By Western blotting, two major isoforms of TRPM2 were identified in human islets. It has been demonstrated that glucose increases production of H2O2 in INS-1(832/13) cells [3]. However, we demonstrate that concentrations of H2O2 that could be relevant from signalling point of view (e.g. 1–4 μM) fail to activate TRPM2 channel. Nevertheless, given the known roles of TRPM2 channels in redox- and cytokine-mediated cell death, these channels may be of relevance in the pathogenesis of type 1 and type 2 diabetes, where β-cell damage contributes to the disease process. Inhibitors of TRPM2 channels are thus, of potential interest as therapeutic agents.
Acknowledgments
This work was supported by the Swedish Research Council grant K2006–72X-20159–01-3, funds from Karolinska Institutet, Swedish Medical Society, Stiftelsen Irma och Arvid Larsson-Rösts minne, Stiftelsen Göljes Minne, Svenska Diabetesstiftelsen. Human islets were provided by the Cell Isolation and Transplantation Centre at the University of Geneva, School of Medicine, thanks to the ECIT ‘islets for research’ distribution program sponsored by the Juvenile Diabetes Research Foundation. A.J.G. is funded by Karolinska Institutet’s M.D. Ph.D. program and Stockholm County Council. This paper is dedicated to the memory of Rolf Luft.
References
- 1.Perraud AL, Fleig A, Dunn CA, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–9. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 2.Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem. 1999;274:27905–13. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- 3.Pi J, Bai Y, Zhang Q, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–91. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 4.Qian F, Huang P, Ma L, et al. TRP genes: candidates for nonselective cation channels and store-operated channels in insulin-secreting cells. Diabetes. 2002;51:S183–9. doi: 10.2337/diabetes.51.2007.s183. [DOI] [PubMed] [Google Scholar]
- 5.Togashi K, Hara Y, Tominaga T, et al. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006;25:1804–15. doi: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inamura K, Sano Y, Mochizuki S, et al. Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J Membr Biol. 2003;191:201–7. doi: 10.1007/s00232-002-1057-x. [DOI] [PubMed] [Google Scholar]
- 7.Hara Y, Wakamori M, Ishii M, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–73. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 8.Merglen A, Theander S, Rubi B, et al. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145:667–78. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- 9.Bruton JD, Lemmens R, Shi CL, et al. Ryanodine receptors of pancreatic beta-cells mediate a distinct context-dependent signal for insulin secretion. FASEB J. 2003;17:301–3. doi: 10.1096/fj.02-0481fje. [DOI] [PubMed] [Google Scholar]
- 10.Ricordi C, Lacy PE, Scharp DW. Automated islet isolation from human pancreas. Diabetes. 1989;38:140–2. doi: 10.2337/diab.38.1.s140. [DOI] [PubMed] [Google Scholar]
- 11.Brandhorst H, Brandhorst D, Brendel MD, et al. Assessment of intracellular insulin content during all steps of human islet isolation procedure. Cell Transplant. 1998;7:489–95. doi: 10.1177/096368979800700508. [DOI] [PubMed] [Google Scholar]
- 12.Gustafsson AJ, Ingelman-Sundberg H, Dzabic M, et al. Ryanodine receptor-operated activation of TRP-like channels can trigger critical Ca2+ signaling events in pancreatic beta-cells. FASEB J. 2005;19:301–3. doi: 10.1096/fj.04-2621fje. [DOI] [PubMed] [Google Scholar]
- 13.Neher E. The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–42. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- 14.Hill K, McNulty S, Randall AD. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:227–37. doi: 10.1007/s00210-004-0981-y. [DOI] [PubMed] [Google Scholar]
- 15.Hill K, Benham CD, McNulty S, et al. Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology. 2004;47:450–60. doi: 10.1016/j.neuropharm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 16.Kraft R, Grimm C, Frenzel H, et al. Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br J Pharmacol. 2006;148:264–73. doi: 10.1038/sj.bjp.0706739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heiner I, Eisfeld J, Warnstedt M, et al. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem J. 2006;398:225–32. doi: 10.1042/BJ20060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wehage E, Eisfeld J, Heiner I, et al. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem. 2002;277:23150–6. doi: 10.1074/jbc.M112096200. [DOI] [PubMed] [Google Scholar]
- 19.Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Krippeit-Drews P, Kramer C, Welker S, et al. Interference of H2O2 with stimulus-secretion coupling in mouse pancreatic beta-cells. J Physiol. 1999;514:471–81. doi: 10.1111/j.1469-7793.1999.471ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakazaki M, Kakei M, Yaekura K, et al. Diverse effects of hydrogen peroxide on cytosolic Ca2+ homeostasis in rat pancreatic beta-cells. Cell Struct Funct. 2000;25:187–93. doi: 10.1247/csf.25.187. [DOI] [PubMed] [Google Scholar]
- 22.Bootman MD, Collins TJ, MacKenzie L, et al. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002;16:1145–50. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- 23.Xu SZ, Zeng F, Boulay G, et al. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: a differential, extracellular and voltage-dependent effect. Br J Pharmacol. 2005;145:405–14. doi: 10.1038/sj.bjp.0706197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Togashi K, Inada H, Tominaga M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB) Br J Pharmacol. 2008;153:1324–30. doi: 10.1038/sj.bjp.0707675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harteneck C, Frenzel H, Kraft R. N-(p-amylcinnamoyl)anthranilic acid (ACA): a phospholipase A(2) inhibitor and TRP channel blocker. Cardiovasc Drug Rev. 2007;25:61–75. doi: 10.1111/j.1527-3466.2007.00005.x. [DOI] [PubMed] [Google Scholar]
- 26.Larsson-Nyren G, Grapengiesser E, Hellman B. Phospholipase A2 is important for glucose induction of rhythmic Ca2+ signals in pancreatic beta cells. Pancreas. 2007;35:173–9. doi: 10.1097/MPA.0b013e318053e022. [DOI] [PubMed] [Google Scholar]
- 27.Olsen HL, Norby PL, Hoy M, et al. Imidazoline NNC77–0074 stimulates Ca2+-evoked exocytosis in INS-1E cells by a phospholipase A2-dependent mechanism. Biochem Biophys Res Commun. 2003;303:1148–51. doi: 10.1016/s0006-291x(03)00505-9. [DOI] [PubMed] [Google Scholar]
- 28.Simonsson E, Karlsson S, Ahrén B. Ca2+-independent phospholipase A2 contributes to the insulinotropic action of cholecystokinin-8 in rat islets: dissociation from the mechanism of carbachol. Diabetes. 1998;47:1436–43. doi: 10.2337/diabetes.47.9.1436. [DOI] [PubMed] [Google Scholar]
- 29.Konrad RJ, Jolly YC, Major C, et al. Inhibition of phospholipase A2 and insulin secretion in pancreatic islets. Biochim Biophys Acta. 1992;1135:215–20. doi: 10.1016/0167-4889(92)90139-3. [DOI] [PubMed] [Google Scholar]
- 30.Song H, Bao S, Ramanadham S, et al. Effects of biological oxidants on the catalytic activity and structure of group VIA phospholipase A. Biochemistry. 2006;45:6392–406. doi: 10.1021/bi060502a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraft R, Grimm C, Grosse K, et al. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286:C129–37. doi: 10.1152/ajpcell.00331.2003. [DOI] [PubMed] [Google Scholar]
- 32.Frings S, Seifert R, Godde M, et al. Profoundly different calcium permeation and blockage determine the specific function of distinct cyclic nucleotide-gated channels. Neuron. 1995;15:169–79. doi: 10.1016/0896-6273(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 33.Fonfria E, Marshall IC, Benham CD, et al. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol. 2004;143:186–92. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perraud AL, Takanishi CL, Shen B, et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–48. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, Chu X, Tong Q, et al. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem. 2003;278:16222–9. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- 36.Miller BA. The role of TRP channels in oxidative stress-induced cell death. J Membr Biol. 2006;209:31–41. doi: 10.1007/s00232-005-0839-3. [DOI] [PubMed] [Google Scholar]
- 37.Herson PS, Lee K, Pinnock RD, et al. Hydrogen peroxide induces intracellular calcium overload by activation of a non-selective cation channel in an insulin-secreting cell line. J Biol Chem. 1999;274:833–41. doi: 10.1074/jbc.274.2.833. [DOI] [PubMed] [Google Scholar]