

Abstract
As the most common monogenic form of human obesity, about 130 naturally occurring melanocortin-4 receptor (MC4R) gene mutations have been identified. In this study, we reported detailed functional characterization of 10 novel human MC4R (hMC4R) mutants including R7C, C84R, S127L, S136F, W174C, A219V, P230L, F261S, I317V and L325F. Flow cytometry experiments showed that six mutants, including R7C, C84R, S127L, W174C, P230L and F261S, have decreased cell surface expression. The other four mutants are expressed at similar levels as the wild-type hMC4R. Binding assays showed that the mutants have similar binding affinities for the agonist and endogenous antagonist agouti-related protein. Signalling assays showed that S136F is defective in signalling. Multiple mutagenesis showed that S136 of hMC4R is required for the normal function of the receptor. To identify potential therapeutic approaches for patients with intracellularly retained MC4R mutants, we tested the effect of an MC4R inverse agonist, ML00253764, on C84R and W174C. We showed that ML00253764 could function as a pharmacological chaperone rescuing the mutant MC4Rs to the cell surface. The rescued mutants are functional with increased cAMP production in response to agonist stimulation. In conclusion, of 10 mutants we studied, 6 had decreased cell surface expression. Pharmacological chaperone is a potential approach for treating obesity caused by MC4R mutations that result in intracellular retention.
Keywords: melanocortin-4 receptor, obesity, trafficking, pharmacological chaperone, functional rescue, G protein-coupled receptor
Introduction
As one of the most significant epidemics in the world, human obesity is strongly associated with several diseases such as hypertension, type 2 diabetes mellitus, stroke, coronary artery diseases and certain types of cancer, thus representing a significant risk of morbidity and mortality [1]. During the past decade or so, mouse and human genetic studies together with detailed pharmacological investigations revealed that the leptin-regulated pathway plays a critical role in regulating food intake and energy expenditure. Mutations in seven genes including leptin (LEP) [2], leptin receptor (LEPR) [3], prohormone convertase 1 (PC1) [4], pro-opiomelanocortin (POMC) [5], melanocortin-3 receptor (MC3R) [6, 7], melanocortin-4 receptor (MC4R) [8, 9] and single-minded homolog 1 (SIM1) [10], cause monogenic obesity in human. Of these genes, the MC4R has been identified as a key component in the leptin-regulated pathway [11–14]. Mutations in the MC4R represent the most common monogenic form of human obesity [15–17], with up to 6% of the early-onset severe obesity caused by MC4R mutations in some populations [15].
As a member of the G protein-coupled receptors (GPCRs) superfamily, the MC4R shares the common structure of GPCRs including three extracellular loops, seven transmembrane domains (TMs) and three intracellular loops with its N- and C-termini facing the outside and inside of the cell, respectively. The MC4R couples primarily to the stimulatory G protein; therefore, hormone stimulation activates the adenylyl cyclase to increase the intracellular cAMP level [18]. The endogenous hormones that activate the MC4R are α-melanocyte stimulating hormone (α-MSH) and β-MSH. These hormones together with γ-MSH and ACTH are all derived from POMC through a tissue-specific posttranslational processing [19]. Agouti-related protein (AgRP) serves as the endogenous antagonist to inhibit the MC4R activity.
So far, about 130 mutations in the coding region of the MC4R have been identified from populations of various ethnic backgrounds. The mutations include frameshift, inframe deletions, nonsense and missense mutations. Many mutant MC4Rs has been functionally characterized in detail (reviewed in [17, 20]). Previously, several groups reported the identification of 10 novel MC4R mutants in obese children or adults of diverse ethnic groups. They are R7C [21] in the N-terminus, C84R [22] in TM2, S127L [23] and S136F [24] in TM3, W174C [25] in TM4, A219V [26] and P230L [23] in IL3, F261S [27] in TM6, I317V [25] and L325F [26] in the C terminal tail (Fig. 1). The clinical characteristics of the patients carrying these MC4R mutations are listed in Table 1. The functional properties of these mutants were either not investigated or only partially determined. In this study, we set to quantitatively measure both the cell surface and total expression levels of these mutants, and to analyse their functional properties including ligand binding and signalling properties.
Figure 1.
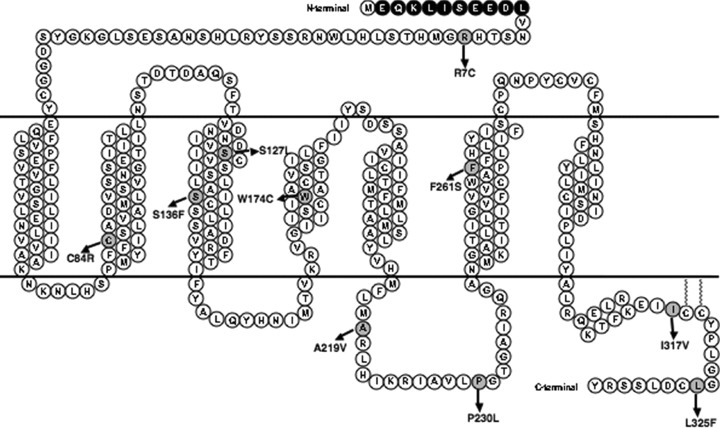
Schematic representation of the 10 hMC4R mutations characterized in this study. The c-myc epitope at the N-terminus is highlighted with dark spheres with white letters. The mutated residues are indicated with gray spheres.
Table 1.
Phenotypes of patients carrying the 10 MC4R mutations studied in the current report
| Mutation | Prevalence (%) | BMI | Age of obesity onset | References |
|---|---|---|---|---|
| R7C | 0.02* | 28.5 | Unknown | [21] |
| C84R | 0.35† | 26.2 | 13 | [22] |
| S127L | 0.25† | Unknown | Unknown | [23] |
| S136F | 0.98† | 33.2 | 2.3 | [24] |
| W174C | 0.51‡ | 59.0 | 8.0 | [25] |
| A219V | 0.13‡ | Unknown | Unknown | [26] |
| P230L | 0.12† | Unknown | Unknown | [23] |
| F261S | 0.39‡ | 36.8 | 1.0 | [27] |
| I317V | 0.51‡ | 39.2 | 10.0 | [25] |
| L325F | 0.13‡ | Unknown | Unknown | [26] |
BMI, body mass index.
From population-based samples.
From obese children and adolescents.
From obese adults.
Bouvier and colleagues pioneered the field of using small molecule antagonists as pharmacological chaperones for GPCRs [28]. These molecules act as folding templates, assisting the mutants to fold into conformations that allow their exit from the endoplasmic reticulum. Since then, pharmacological chaperones have been identified for naturally occurring mutations in rhodopsin and gonadotropin-releasing hormone receptor (GnRHR) [29–32] and wild-type (WT) or laboratory-generated mutants in several other GPCRs [33–39]. The rescuing effect does not depend on specific chemical structure. For example, three different classes of chemicals, including indoles, quinolines and erythromycin macrolides, can rescue almost all the naturally occurring GnRHR mutations that cause hypogonadotropic hypogonadism [32]. Recently, it has been shown that treatment of patients with nephrogenic diabetes insipidus harbouring transport-defective V2 vasopressin receptor mutations with the non-peptide antagonist SR49059 decreased urine volume and water intake [40], proving the clinical utility of pharmacological chaperones. Because the majority of the naturally occurring MC4R mutants are retained intracellularly, we tested the potential chaperone activity of a small molecule MC4R antagonist, ML00253764. This compound, [2-{2-[2-(5-bromo-2-methoxyphenyl)-ethyl]-3-fluorophenyl}-4,5-dihydro-1H-imidazolium hydrochloride], originally discovered by Vos and colleagues [41], was shown to be a MC4R inverse agonist [42].
Materials and methods
Cells, plasmids and peptides
Human embryonic kidney (HEK) 293 and 293T cells (American Type Culture Collection, Manassas, VA, USA) were incubated at 5% CO2 in Dulbecco’s modified Eagles medium supplemented with 10% newborn calf serum, 10 mM HEPES and 100 units/ml of penicillin and 100 μg/ml streptomycin (all from Invitrogen, Carlsbad, CA, USA). The N-terminal myc-tagged human MC4R (hMC4R) was described previously [43]. The superpotent agonist of α-MSH, [Nle4, D-Phe7]-α-MSH (NDP-MSH) and the MC4R antagonist AgRP(86–132) were purchased from Peptides International (Louisville, KY, USA). ML00253764 was synthesized by BioMol (Plymouth Meeting, PA, USA). [125I]-NDP-MSH was obtained from the Peptide Radioiodination Service Center at the University of Mississippi (Dr. R. C. Speth, Director; University, MS, USA).
Site-directed mutagenesis of hMC4R
Mutant hMC4Rs were generated by site-directed mutagenesis using N-terminal myc-tagged hMC4R as template and the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) as previously described [43]. The mutants generated were verified by nucleotide sequencing (performed at The University of Chicago Cancer Research Center DNA Sequencing Facility, Chicago, IL, USA). Plasmids prepared with an IsoPure Maxi Prep kit (Denville Scientific, Metuchen, NJ, USA) were used for transfection.
Plasmid transfection and establishment of non-clonal stable cell lines
For transient expression of the hMC4Rs, HEK293T cells were plated on gelatin-coated 35-mm six-well plates and transfected with 4 μg of plasmid per well using the calcium precipitation method [44]. The cells were then incubated for 48 hrs before used for radioligand binding and signalling assays. For establishment of non-clonal stable cells, HEK293 cells were plated on gelatin-coated 100-mm dishes and transfected with 10 μg of plasmid per dish as described above. After 24 hrs incubation, the cells were trypsinized and then transferred to T75 flasks in media containing 250 μg/ml geneticin (Research Products International, Mt. Prospect, IL, USA). Selection was continued for 3 weeks. The geneticin-resistant cells were maintained in media containing 250 μg/ml geneticin.
Radioligand binding assay
Cells were washed twice with warm Waymouth’s MB752/1 media (Sigma-Aldrich, St. Louis, MO, USA) modified to contain 1 mg/ml bovine serum albumin (BSA) (referred to as Waymouth/BSA). Fresh Waymouth/BSA was added to each well, together with 50 μl of 125I-NDP-MSH (∼100,000 cpm) with or without different concentrations of unlabeled NDP-MSH or AgRP(86–132). The total volume in each well was 1 ml. The final concentration of unlabeled ligand ranged from 10−11 to 10−6 M. After 1 hr incubation, cells were washed twice with cold Hank’s balanced salt solution (Sigma-Aldrich) modified to contain 1 mg/ml BSA. Then, 100 μl of 0.5 N NaOH was added to each well. Cells were collected from each well using cotton swabs, and ligand binding was counted in a gamma counter. Maximal binding capacity (Bmax) and IC50 values were calculated using Prism software version 4 (GraphPad Software, San Diego, CA, USA).
NDP-MSH stimulation of intracellular cAMP generation
Transfected cells were washed twice with warm Waymouth/BSA. Then 1 ml of fresh Waymouth/BSA containing 0.5 mM isobutylmethylxanthine (Sigma-Aldrich) was added to each well. After 15 min. incubation, either buffer alone or different concentrations of NDP-MSH were added. The final concentration of the unlabeled ligand ranged from 10−12 to 10−6 M. After 1 hr incubation, cells were then placed on ice, media aspirated and intracellular cAMP were extracted with 0.5 N percholoric acid containing 180 μg/ml theophylline and measured using radioimmunoassay [45]. All determinations were performed in triplicate. Maximal responses (Rmax) and EC50 values were calculated using Prism 4.
Flow cytometry assay of HEK293 cells stably expressing WT or mutant hMC4Rs
This assay was performed as described before [46]. Briefly, cells were plated into six-well plates the day before the experiment. On the day of the experiment, cells were washed once with filtered PBS-IH (in mM, consisting of 137 NaCl, 2.7 KCl, 1.4 KH2PO4 and 4.3 Na2HPO4[pH 7.4]), fixed with 4% paraformaldehyde for 30 min., permeabilized (only for determination of total expression) with 1% Triton-X-100 in PBS-IH for 4 min. and then incubated with blocking solution (5% BSA in PBS-IH) for 1 hr. Cells were then incubated for 1 hr with fluorescein-conjugated 9E10 monoclonal antibody (Affinity Bioreagents, Golden, CO, USA) diluted 1:20 in PBS-IH containing 1 mg/ml BSA. Cells were then washed three times with PBS-IH and assayed with a MoFlo7-color flow cytometer and high-performance sorter (Dakocytomation, Fort Collins, CO, USA). The expression level of the mutant receptors was calculated as a percentage of WT expression using the formula: (mutant-pcDNA3)/(WT-pcDNA3) × 100%.
ML00253764 treatment of the HEK293 cells
One million HEK293 cells stably expressing WT or mutant hMC4Rs were plated on gelatin-coated six-well plates and allowed to attach for 24 hrs. Cells were then rinsed three times with Waymouth/BSA before 900 μl of Waymouth/BSA were added to each well together with 100 μl of ML00253764 (10−4 M, dissolved first in dimethyl sulfoxide, then diluted in saline containing BSA) or buffer was added to each well to reach a final concentration of 10−5 M of the drug. The cells then were cultured for another 24 hrs before assayed for expression and signalling.
Confocal microscopy
The day before the experiment, 80,000 HEK293 cells stably expressing WT or mutant hMC4Rs were plated into each well of an eight-well chamber slide (Biocoat cellware purchased from Falcon, Bedford, MA, USA) and cultured for 24 hrs. On the day of the experiment, cells were washed three times with Waymouth/BSA and 450 μl of Waymouth/BSA was added into each well thereafter. Either 50 μl of buffer alone or 50 μl of ML00235764 (10−4 M) were added into each well and the cells were then cultured for another 24 hrs. The cells were then washed three times with filtered PBS-IH, fixed with 4% paraformaldehyde for 30 min., and then incubated with blocking solution (5% BSA in PBS-IH) for 1 hr. Cells were then incubated for 1 hr with fluorescein-conjugated 9E10 monoclonal antibody diluted 1:200 in PBS-IH containing 1 mg/ml BSA. After cells were washed twice with PBS-IH, the cover slips were mounted to the slides using Vetashield HardSet(tm) mounting medium (Alexis, San Diego, CA, USA) and the slides were viewed with a Bio-Rad (Bio-Rad, Hercules, CA, USA) confocal microscope. All the steps were performed at room temperature.
Statistical analyses
Data were presented as mean ± S.E.M. Statistical analyses were performed with Prism 4. For comparisons on cell surface and total expression of the WT and mutant hMC4Rs and cell surface maximal binding and maximal signalling, one-sample unpaired t-test was used. For comparisons of IC50 and EC50, a two-sample unpaired t-test was used. Differences with a P < 0.05 were considered statistically significant.
Results
Cell surface and total expression levels of the mutant hMC4Rs
Previous functional studies of the naturally occurring human MC4R mutations identified from various patient populations showed that reduced cell surface expression is the most prevalent defect. Therefore, we performed a quantitative analysis of cell surface expression of the mutant hMC4Rs. Flow cytometry assay was used to measure the cell surface expression levels in HEK293 cells stably expressing hMC4Rs tagged at the N terminus with a myc-tag. The cell surface expression of the WT hMC4R was defined as 100%. Six mutants, including R7C, C84R, S127L, W174C, P230L and F261S, had significantly reduced cell surface expression levels compared to the WT hMC4R (Fig. 2A). The other four mutants, including S136F, A219V, I317V and L325F, had similar cell surface expression level as that of the WT hMC4R (Fig. 2A).
Figure 2.
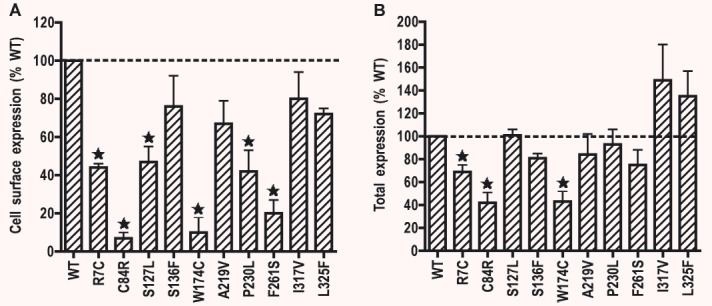
Cell surface (A) and total (B) expressions of WT and mutant hMC4Rs stably expressed in HEK293 cells. Establishment of the stable cell lines was performed as described in ‘Materials and methods’. Cell surface and total expression levels of the WT and mutant hMC4Rs were quantitatively determined by flow cytometry. The results are expressed as percentage of total or cell surface expression levels of the WT hMC4R. Shown are mean ± S.E.M. of three experiments. Star (*) indicates significant difference from WT hMC4R (P < 0.05).
We also measured the total expression levels of the mutant hMC4Rs by flow cytometry because decreased cell surface expression of the mutant hMC4Rs might be caused by either decreased total expression or intracellular retention of the mutant receptors or both. Cells were permeabilized with Triton X-100 and then stained with anti-myc antibody. As shown in Fig. 2B, three mutants, including R7C, C84R and W174C, had significantly decreased total expression levels compared with the WT hMC4Rs. The other seven mutants had similar total expression levels as the WT hMC4R. Taken together, these data indicated that the decreased cell surface expression of S127L, P230L and F261S hMC4Rs were caused by intracellular retention of the mutant receptors, not decreased total expression. The decreased cell surface expression of R7C, C84R and W174C is partly due to decreased total expression levels of these mutants. From the relative decrease in total and cell surface expression, we conclude that C84R and W174C are also defective in intracellular trafficking.
Ligand binding properties of the mutant hMC4Rs
We further investigated the ligand binding properties of the WT and mutant hMC4Rs using whole-cell binding assays in HEK293T cells transiently transfected with WT or mutant hMC4Rs. Both NDP-MSH and AgRP(86–132) were used to compete with [125I]-NDP-MSH for binding to the cell surface binding sites. As shown in Fig. 3A and summarized in Table 2, four mutants, including S127L, A219V, P230L and F261S, had decreased maximal binding compared to the WT hMC4R. No specific binding could be measured for C84R and W174C. Four mutants, including R7C, S136F, I317V and L325F, had similar Bmax as the WT hMC4R. S136F, F261S and L325F had significantly lower IC50s. There was no significant difference in the binding affinities between WT and the other five mutant hMC4Rs. When AgRP(86–132) was used as the competitor, the WT hMC4R had an IC50 of 4.16 nM. S127L and A219V had increased IC50s for AgRP compared with WT hMC4R whereas S136F and F261S had decreased IC50. The other four mutants had similar IC50 as the WT hMC4R (Fig. 3B and Table 2).
Figure 3.
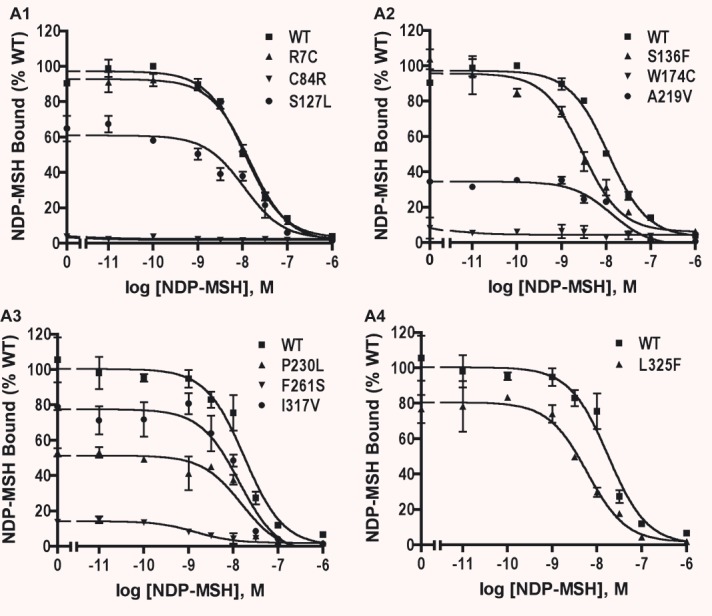
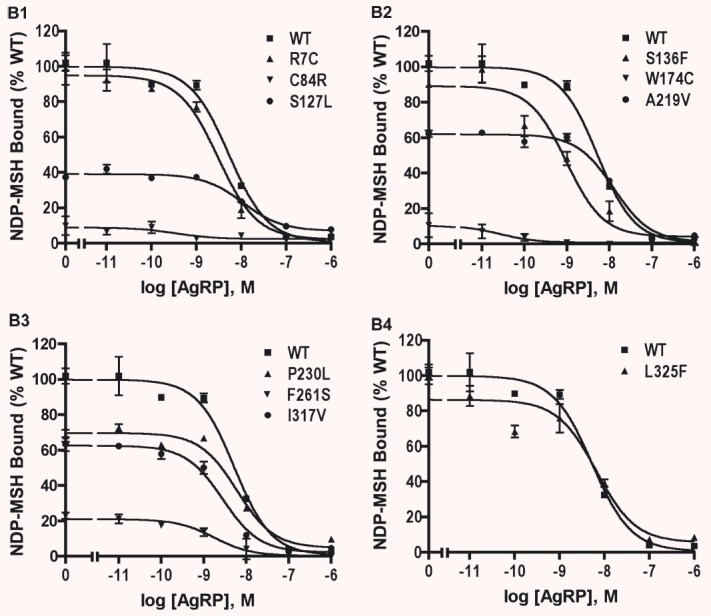
NDP-MSH binding assay of the WT and mutant hMC4Rs transiently expressed in HEK293T cells. HEK293T cells were transiently transfected with the indicated hMC4R constructs and binding assays were performed as described in ‘Materials and methods’. Different concentrations of unlabeled NDP-MSH (A) or AgRP(86–132) (B) were used to displace the binding of [125I]-NDP-MSH to hMC4Rs on intact cells. Results are expressed as percentage of the maximal binding for WT hMC4R from duplicate determinations within one experiment. Shown are mean ± S.E.M. All experiments were performed at least three times.
Table 2.
Ligand binding and agonist-stimulated cAMP response of the WT and naturally occurring mutant hMC4Rs
| hMC4R | n | NDP-MSH binding | NDP-MSH-stimulated cAMP | n | AgRP binding IC50 (nM) | ||
|---|---|---|---|---|---|---|---|
| IC50 (nM) | Bmax (% WT) | EC50 (nM) | Rmax (% WT) | ||||
| WT | 9 | 13.16 ± 1.91 | 100 | 1.22 ± 0.19 | 100 | 4 | 4.16 ± 0.70 |
| R7C | 3 | 12.32 ± 0.99 | 99 ± 3 | 3.06 ± 0.06‡ | 102 ± 4 | 3 | 3.60 ± 0.36 |
| C84R | 3 | N/A | N/A | 8.73 ± 0.43‡ | 20 ± 1‡ | 3 | N/A |
| S127L | 3 | 14.99 ± 2.57 | 32 ± 2‡ | 1.78 ± 1.07 | 57 ± 3† | 3 | 18.25 ± 4.85‡ |
| S136F | 3 | 3.72 ± 0.77† | 101 ± 9 | 7.61 ± 0.23‡ | 11 ± 1‡ | 3 | 0.41 ± 0.05‡ |
| W174C | 3 | N/A | N/A | 14.80 ± 0.35‡ | 31 ± 1‡ | 3 | N/A |
| A219V | 3 | 20.11 ± 3.55 | 40 ± 3† | 24.19 ± 1.27‡ | 56 ± 3† | 3 | 12.83 ± 0.33† |
| P230L | 3 | 12.82 ± 1.31 | 62 ± 6* | 12.31 ± 0.67‡ | 122 ± 4* | 3 | 4.01 ± 1.09 |
| F261S | 3 | 2.49 ± 0.94† | 15 ± 1‡ | 1.48 ± 0.12 | 49 ± 1‡ | 3 | 1.33 ± 0.59* |
| I317V | 3 | 18.44 ± 2.53 | 94 ± 8 | 3.60 ± 0.14‡ | 100 ± 2 | 3 | 3.56 ± 0.19 |
| L325F | 3 | 5.12 ± 0.42† | 92 ± 6 | 2.62 ± 0.06† | 94 ± 4 | 3 | 6.17 ± 0.12 |
Significantly different from WT hMC4R, P < 0.05.
Significantly different from WT hMC4R, P < 0.01.
Significantly different from WT hMC4R, P < 0.001.
Data shown are the mean ± S.E.M. of the number of experiments indicated. The Rmax of WT hMC4R was 948 ± 86 pmol cAMP/106 cells (mean ± S.E.M. of nine experiments). IC50 is the concentration of NDP-MSH that is needed to cause 50% inhibition in the binding assay. EC50 is the concentration of NDP-MSH that results in 50% stimulation of the maximal response.
Signalling properties of the mutant hMC4Rs upon NDP-MSH stimulation
To investigate the signalling properties of the hMC4Rs, HEK293T cells transiently transfected with WT or mutant hMC4Rs were stimulated with different concentrations of NDP-MSH, and accumulation of intracellular cAMP was measured. As shown in Fig. 4 and Table 2, R7C, I317V and L325F had similar Rmax as the WT hMC4R. P230L had a small but significant increase in Rmax compared with the WT hMC4R. The other six mutants had significantly decreased Rmax compared to the WT hMC4R. S127L and F261S had similar EC50s as the WT hMC4R. The other eight mutants had increased EC50s (Fig. 4 and Table 2).
Figure 4.
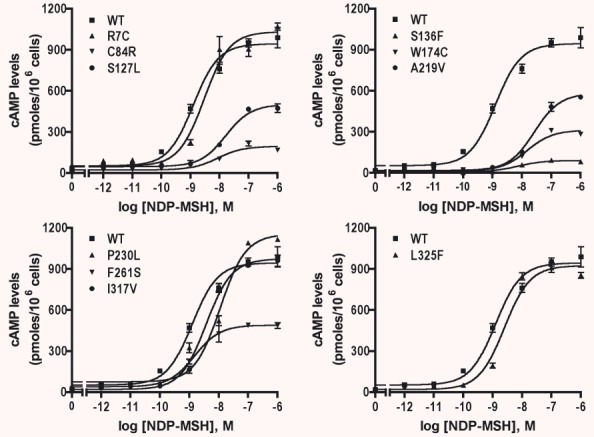
Signalling of the WT and mutant hMC4Rs transiently expressed in HEK293T cells. HEK293T cells were transiently transfected with indicated hMC4R constructs and stimulated with various concentrations of NDP-MSH and intracellular cAMP levels were measured as described in ‘Materials and methods’. Results are expressed as the mean ± S.E.M. of triplicate determinations within one experiment and all experiments were performed at least three times.
Previous studies suggested that loss of basal signalling might be one defect of the receptor in MC4R mutants identified from obese patients [47]. Therefore, we also determined the constitutive activities of the mutants in this study. As shown in Fig. 5, when compared with the WT hMC4R, S127L and P230L had significantly increased constitutive activities, whereas W174C and A219V had significantly decreased constitutive activities. The other six mutants had similar constitutive activities as the WT hMC4R (Fig. 5).
Figure 5.
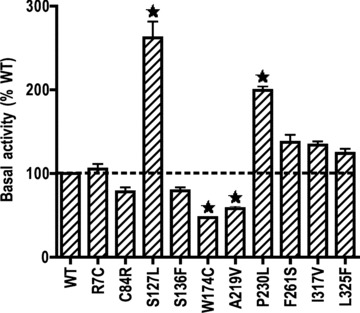
Basal activities of the WT and mutant hMC4Rs. The basal activities of WT and mutant hMC4Rs were assessed by measuring intracellular cAMP levels in HEK293T cells transiently expressing WT or mutant hMC4Rs without hormone stimulation. The results are expressed as percentage of basal cAMP level of WT hMC4R-expressing HEK293T cells. Shown are mean ± S.E.M. of three or more experiments. The basal cAMP level in the WT hMC4R was 181.9 ± 76.9 pmol/106 cells (mean ± S.E.M. of six experiments). Star (⋆) indicates significant difference from WT hMC4R (P < 0.05).
Amino acid requirement at codon 136 of the hMC4R for receptor function
Data presented above showed that S136F had normal total and cell surface expression and ligand binding. However, it does not exhibit any signalling. To gain a better understanding of the side-chain requirement at codon S136 of hMC4R for NDP-MSH signalling, we generated six additional mutants, including replacing the serine with hydrophobic amino acids such as alanine and leucine, polar amino acids such as threonine, tyrosine and cysteine, and charged amino acids such as aspartate and arginine. These mutants were expressed in HEK293T cells and ligand binding and signalling properties of the mutants were measured. As shown in Fig. 6A and Table 3, all seven mutants had normal Bmax compared to the WT hMC4R (S136D even had a small but significant increase in Bmax). Except for S136R and S136T that had significantly higher IC50s, the other five mutants had similar IC50s as the WT hMC4R. All mutants can signal in response to NDP-MSH stimulation. S136L had a small but significant increase in Rmax. However, the maximal responses were decreased in S136A, S136C and S136R mutants (Fig. 6B and Table 3). Four mutants, including S136A, S136L, S136R and S136Y, had significantly increased EC50s compared to the WT MC4R, whereas S136C, S136D and S136T had normal EC50s (Table 3).
Figure 6.
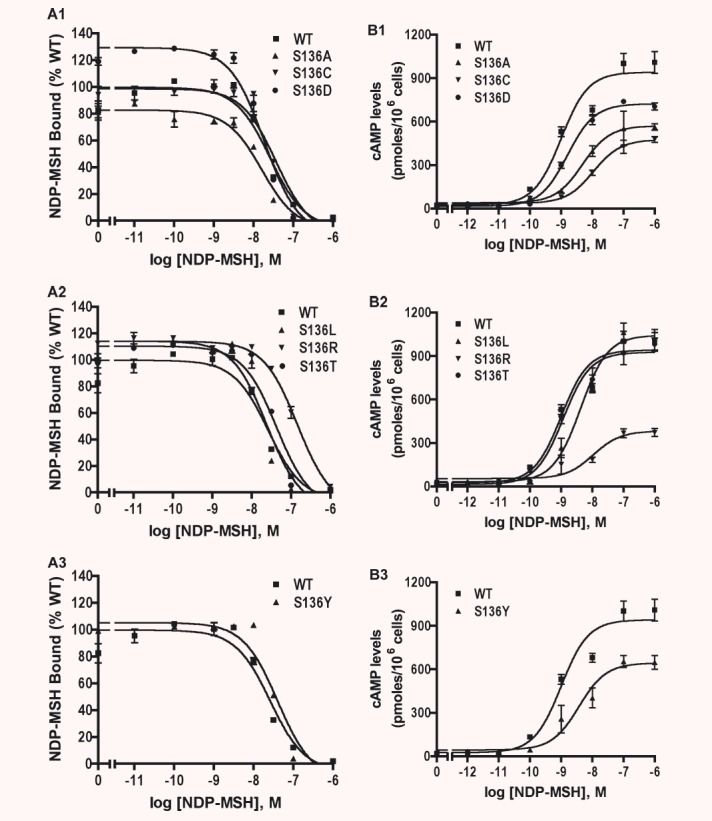
Ligand binding (A) and signalling (B) properties of multiple mutants at S136. HEK293T cells were transiently transfected with indicated hMC4R constructs and binding assays and cAMP response were performed as described in ‘Materials and methods’. Results are expressed as the mean ± S.E.M. of duplicate (for binding assay) or triplicate determinations (for cAMP response) within one experiment and all experiments were performed at least three times.
Table 3.
Ligand binding and agonist-stimulated cAMP response of the WT hMC4R and S136 mutants
| hMC4R | n | NDP-MSH binding | NDP-MSH-stimulated cAMP | ||
|---|---|---|---|---|---|
| IC50 (nM) | Bmax (% WT) | EC50 (nM) | Rmax (% WT) | ||
| WT | 4 | 25.21 ± 0.35 | 100 | 1.50 ± 0.60 | 100 |
| S136A | 4 | 24.84 ± 3.60 | 108 ± 10 | 8.06 ± 2.11* | 77 ± 8 * |
| S136C | 4 | 37.37 ± 6.26 | 116 ± 8 | 1.07 ± 0.40 | 37 ± 7† |
| S136D | 4 | 26.74 ± 8.65 | 132 ± 6† | 2.85 ± 0.54 | 87 ± 5 |
| S136L | 4 | 21.91 ± 3.79 | 103 ± 5 | 10.38 ± 2.30† | 129 ± 10* |
| S136R | 3 | 151.80 ± 7.01‡ | 97 ± 4 | 10.73 ± 0.71† | 40 ± 5† |
| S136T | 4 | 43.77 ± 5.64* | 116 ± 7 | 2.45 ± 0.91 | 116 ± 7 |
| S136Y | 4 | 43.64 ± 9.26 | 104 ± 6 | 7.98 ± 1.46† | 85 ± 11 |
Significantly different from WT hMC4R, P < 0.05.
Significantly different from WT hMC4R, P < 0.01.
Significantly different from WT hMC4R, P < 0.001.
Data shown are the mean ± S.E.M. of the number of experiments indicated. The Rmax (maximal response) was 1225 ± 88 pmol cAMP/106 cells (mean ± S.E.M. of four experiments). IC50 is the concentration of NDP-MSH that is needed to cause 50% inhibition in the binding assay. EC50 is the concentration of NDP-MSH that results in 50% stimulation of the maximal response.
Functional rescue of C84R and W174C hMC4Rs
Our data presented above showed that C84R and W174C had minimal cell surface expression. Their ligand binding was undetectable although both mutants retained some residual signalling. To investigate whether small molecule MC4R ligands can act as pharmacological chaperones, we studied the effect of ML00253764 on the cell surface expression of these two mutant receptors. Cells stably transfected with the empty vector pcDNA3.1 were used as the negative control. HEK293 cells stably expressing WT, C84R or W174C hMC4Rs were treated with 10−5 M ML00253764 for 24 hrs. Confocal microscopy and flow cytometry were then performed. As shown in Fig. 7A, the cell surface expression levels of both mutants were increased after ML00253764 treatment. Quantitative measurement by flow cytometry revealed that after treatment with ML00253764, the cell surface expression levels of C84R and W174C were increased to ∼35% of that of the untreated WT hMC4R (Fig. 7B). The cell surface expression of the WT hMC4R was also increased to ∼152% after ML00253764 treatment.
Figure 7.
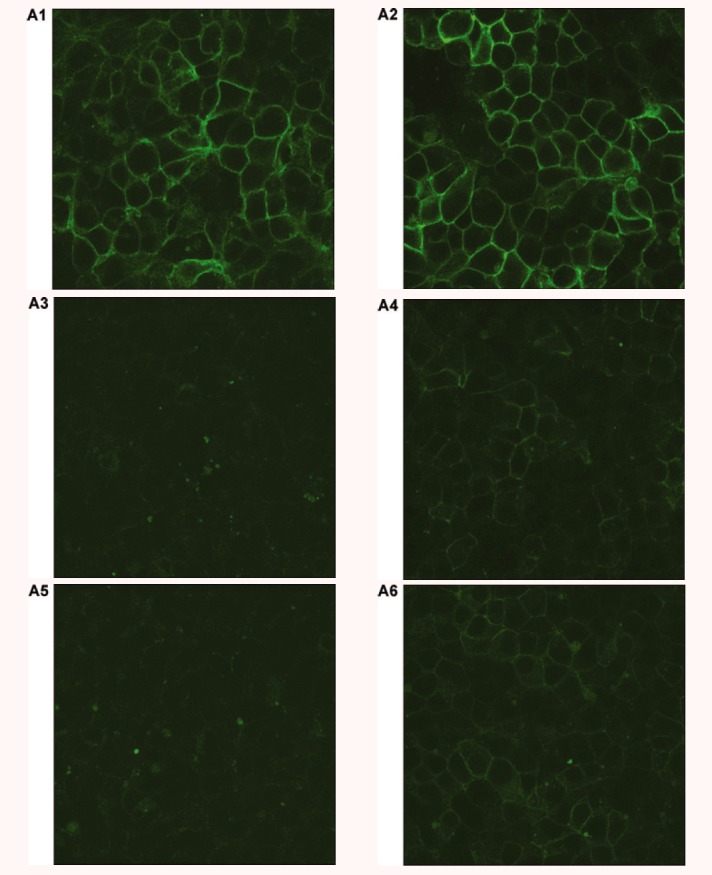
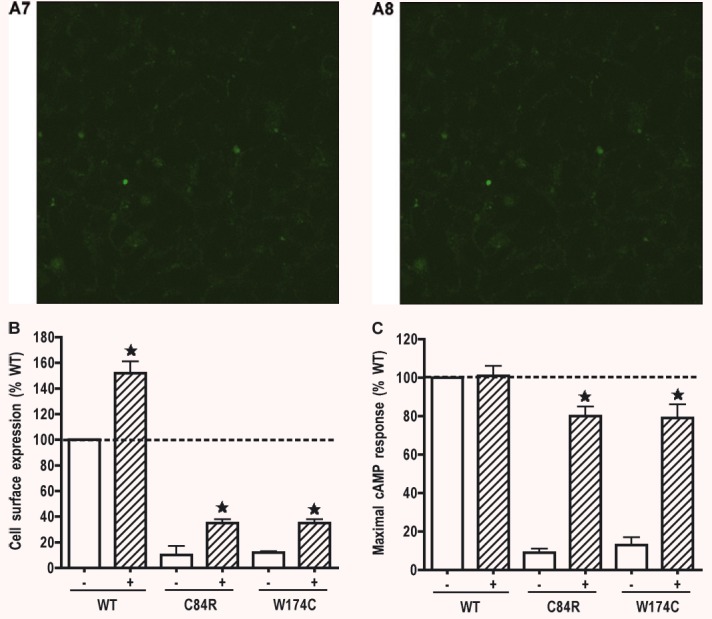
Pharmacological rescue of C84R and W174C by ML00253764. HEK293 cells stably expressing WT, C84R or W174C hMC4Rs or empty vector pcDNA3 were incubated with 10−5 M ML00253764 for 24 hrs. Cells were then stained with anti-myc antibody and visualized with a confocal microscope (A), or sorted with flow cytometry (B) to quantitatively measure cell surface expression. Cell surface expression of WT hMC4R treated with vehicle was set as 100%. In (A), the constructs transfected were WT (A1 and A2), C84R (A3 and A4), W174C (A5 and A6), and pcDNA3 (A7 and A8). A1, A3, A5, and A7 were treated with vehicle; A2, A4, A6 and A8 were treated with ML00253764. In (C), cells were stimulated with 10−6 M NDP-MSH and intracellular cAMP accumulation measured by RIA. Cyclic AMP generated by WT hMC4R treated with vehicle and stimulated with NDP-MSH was set as 100%. Star (⋆) indicates significant difference from corresponding vehicle treated controls (P < 0.05).
To investigate whether the rescued mutants were functional, the cells were stimulated with 10−6 M NDP-MSH. Intracellular cAMP accumulation was measured. As shown in Fig. 7C, after treatment with ML00253764, the rescued mutants were functional. Cyclic AMP generation was increased to up to 80% of that of the maximal signalling generated by the WT hMC4R treated with vehicle.
Discussion
Mutations in the MC4R gene were identified as a cause for obesity in 1998 [8, 9]. Since then, studies from numerous groups have identified about 130 distinct mutations in the coding region of the MC4R gene from both obese and normal weight persons. Based on the life cycle of GPCRs and modelled after low-density lipoprotein receptor and cystic fibrosis transmembrane conductance regulator [48, 49], we proposed that mutations in the MC4R could be divided into five classes [43]. The class I mutants are defective in receptor biosynthesis. The class II mutants are synthesized but are defective in trafficking onto the cell surface. The class III mutants can be synthesized and transported onto the cell surface normally. However, they are defective in ligand binding. The class IV mutants are defective in signalling but with normal cell surface expression and ligand binding. Mutants that are normal in cell surface expression, ligand binding and signalling are class V mutants.
In this study, we functionally characterized 10 hMC4R mutants. According to our classification scheme described above, R7C is a class I mutant, C84R, S127L, W174C, P230L and F261S are class II mutants, A219V is a class III mutant, S136F belongs to class IV, I317V and L325F are class V mutants.
R7C was previously shown to have normal cell surface expression and slightly decreased cAMP response upon stimulation by NDP-MSH [21]. We observed a similar but statistically significant decrease in maximal response (Fig. 4 and Table 2). Its total expression was decreased; therefore, a class I mutant. S127L, was identified independently in several studies [23, 50, 51]. Previous reports showed that S127L is normal in cell surface expression [23, 50] whereas its cAMP response is impaired with decreased Rmax and increased EC50 upon stimulation by either α-MSH [23, 50, 51], β-MSH [51] or γ1-MSH [51]. In our experiments, S127L had a significantly decreased Rmax with normal EC50. Importantly, although it had normal total expression, S127L had decreased cell surface expression compared to the WT hMC4R. Thus, S127L is a class II mutant. W174C and I317V hMC4Rs were identified from severely obese Italian adults and were not functionally characterized [25]. We showed that W174C had a significantly decreased total expression compared to the WT hMC4R and undetectable cell surface expression (Fig. 2). No ligand binding and cAMP response could be detected (Figs 3 and 4). In contrast, the cell surface expression, ligand binding and cAMP response of I317V are similar to the WT hMC4R. Its total expression is even higher than that of the WT hMC4R. Therefore, I317V might represent a rare polymorphism with no overt functional defect.
Functional studies of Hinney and colleagues showed that P230L is a constitutively active mutant with increased Rmax and decreased EC50 upon stimulation by α-MSH [23]. We confirmed that P230L is constitutively active. We further showed that P230L had significantly decreased cell surface expression, with a corresponding decrease in Bmax. Its total expression was normal. Therefore, P230L is defective in intracellular trafficking and a class II mutant. F261S was identified from obese Chinese and preliminary characterization showed that it has decreased Rmax and increased IC50[27]. Intracellular retention of the mutant receptor was recently reported [52]. Our data are consistent with these observations. Furthermore, we showed that the cell surface expression of F261S was significantly decreased to 35% of that of the WT hMC4R, although the total expression is similar to the WT hMC4R. Corresponding decreases in Bmax and Rmax were observed whereas the IC50 and EC50 were not significantly altered. Thus, F261S is also defective in intracellular trafficking, hence a class II mutant. Mutants A219V and L325F were identified from German patients [26]. Using NDP-MSH as the ligand, we showed that A219V had significantly decreased maximal binding, although its cell surface and total expression levels were similar to the WT hMC4R (Fig. 2). The decreased Bmax resulted in significantly decreased Rmax as well as a 10-fold increase in EC50 compared to the WT hMC4R (Figs 3 and 4, Table 2). Therefore, A219V is a class III mutant. L325F were relatively normal in both binding and signalling, a class V mutant, different from the results obtained by Larsen and colleagues [26].
Mutant S136F was reported as a loss-of-function mutant with normal cell surface expression [24]. Our data are consistent with these observations. We further showed that total expression and ligand binding properties of S136F were also normal compared to the WT hMC4R. Therefore, S136F is a class IV mutant. A recent study reported similar data [53]. Multiple mutagenesis experiments showed that S136 is important for signalling. Mutations into alanine, cysteine and arginine, resulted in decreased maximal response, whereas mutations into cysteine, leucine, arginine and tyrosine, resulted in increased EC50s (Fig. 6 and Table 3). Tan and colleagues showed that mutation of S136 into proline resulted in decreased cell surface expression, with corresponding decreases in Bmax and Rmax [53].
Vaisse and colleagues suggested that loss of constitutive activity might be one cause of obesity in MC4R mutation carriers [47]. In the current study, we measured the constitutive activities of the 10 mutants. W174C and A219V had decreased constitutive activities, whereas S127L and P230L had significantly increased constitutive activities (Fig. 5). The reason for the association of increased constitutive activity with obesity is unclear (reviewed in [54]). It might be argued that S127L and P230L cause obesity due to decreased cell surface expression and maximal binding; S127L also had decreased maximal response. We also noticed a disconnect between constitutive activity and maximal response. For example, C84R and F261S had significantly decreased Rmax but normal basal activities. S127L had increased basal activity but decreased Rmax. Similar observations were observed before. For example, in the follitropin receptor, we showed that L460R mutation causes constitutive activation but no response to follitropin stimulation [55]. These results support the currently accepted dogma that GPCRs exist in a myriad of conformations, including constitutively active and ligand-induced/stabililized active conformations [56].
Previous studies from several groups, including ours, revealed that a significant proportion of the MC4R mutants identified from obese patients result in defective cell surface expression (class II mutants) (reviewed in [17, 20]). To investigate whether small molecule MC4R ligands could act as pharmacological chaperones rescuing the MC4R mutants to the cell surface, we tested the effect of treatment with ML00253764. Our results demonstrated clearly that treatment of cells stably expressing C84R and W174C with ML00253764 increased the cell surface expression of the mutant receptors (Fig. 7A and B). We then tested the functions of the rescued mutants. Both mutants had robust signalling, amounting to 80% of the signalling elicited by WT hMC4R treated with vehicle (Fig. 7C). Importantly, cells stably expressing WT hMC4R had increased cell surface expression after ML00253764 treatment. The maximal signalling was not increased in these cells, perhaps due to the fact that some ML00253764 molecules were still in the cells, partially antagonizing the binding and hence stimulation elicited by NDP-MSH. This is supported by the fact that we could not perform ligand binding studies in these cells using NDP-MSH as the radioligand (data not shown). If indeed pharmacological chaperones such as ML00253764 can increase the expression and signalling of the WT hMC4R in neurons, they could be used as a therapeutic strategy for treating obese patients without MC4R mutations. Because ML00253764 treatment increased cell surface expression of WT hMC4R, it can be concluded that WT hMC4R maturation is not optimal even in stably transfected cells (which is more favourable than in transiently transfected condition [57]). This is not uncommon. It is well known that the maturation of GPCR is rate limiting in many receptors (reviewed in [58]). A caveat is that these studies were done in HEK293 cells. Similar studies need to be done in neuronal cells (in vitro) and in transgenic animals expressing the mutant receptors (in vivo). These are the future directions of our studies.
In summary, we performed detailed functional characterization of 10 MC4R mutations recently identified from obese patients. Furthermore, we showed that a small molecule MC4R antagonist could act as a pharmacological chaperone, rescuing mutant MC4Rs to the cell surface. The rescued mutants were functional. We speculate that pharmacological chaperone might be a novel therapeutic approach for treating obesity caused by MC4R mutations resulting in intracellularly retained receptors. Because it can also increase the cell surface expression of the WT hMC4R, it might also be used for treating common obesity by increasing the cell surface expression of the WT hMC4R.
Acknowledgments
We thank Dr. Ira Gantz (formally at University of Michigan, Ann Arbor, MI) for kindly providing the hMC4R cDNA. We thank the Flow Cytometry Core Facility at Auburn University College of Veterinary Medicine (Dr. R. C. Bird, Director) and Advanced Microscopy & Imaging Laboratory of Auburn University (Dr. M. Miller, Director) for their assistances in the flow cytometry and confocal microscopy experiments, respectively. This study was supported by Diabetes Action Research and Education Foundation and Animal Health and Disease Research Program of Auburn University College of Veterinary Medicine (to Y.-X. T.).
References
- 1.Comuzzie AG, Allison DB. The search for human obesity genes. Science. 1998;280:1374–7. doi: 10.1126/science.280.5368.1374. [DOI] [PubMed] [Google Scholar]
- 2.Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 3.Clement K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 4.Jackson RS, Creemers JW, Ohagi S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–6. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 5.Krude H, Biebermann H, Luck W, et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–7. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 6.Lee YS, Poh LK, Loke KY. A novel melanocortin 3 receptor gene (MC3R) mutation associated with severe obesity. J Clin Endocrinol Metab. 2002;87:1423–6. doi: 10.1210/jcem.87.3.8461. [DOI] [PubMed] [Google Scholar]
- 7.Tao YX, Segaloff DL. Functional characterization of melanocortin-3 receptor variants identify a loss-of-function mutation involving an amino acid critical for G protein-coupled receptor activation. J Clin Endocrinol Metab. 2004;89:3936–42. doi: 10.1210/jc.2004-0367. [DOI] [PubMed] [Google Scholar]
- 8.Vaisse C, Clement K, Guy-Grand B, et al. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–4. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 9.Yeo GS, Farooqi IS, Aminian S, et al. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–2. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 10.Holder JL, Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–8. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 11.Huszar D, Lynch CA, Fairchild-Huntress V, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–41. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 12.Ollmann MM, Wilson BD, Yang YK, et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–8. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 13.Graham M, Shutter JR, Sarmiento U, et al. Overexpression of Agrt leads to obesity in transgenic mice. Nat Genet. 1997;17:273–4. doi: 10.1038/ng1197-273. [DOI] [PubMed] [Google Scholar]
- 14.Fan W, Boston BA, Kesterson RA, et al. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–8. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 15.Farooqi IS, Keogh JM, Yeo GS, et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 16.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–8. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 17.Tao YX. Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity. Mol Cell Endocrinol. 2005;239:1–14. doi: 10.1016/j.mce.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Gantz I, Fong TM. The melanocortin system. Am J Physiol. 2003;284:E468–74. doi: 10.1152/ajpendo.00434.2002. [DOI] [PubMed] [Google Scholar]
- 19.Smith R, Healy E, Siddiqui S, et al. Melanocortin 1 receptor variants in an Irish population. J Invest Dermatol. 1998;111:119–22. doi: 10.1046/j.1523-1747.1998.00252.x. [DOI] [PubMed] [Google Scholar]
- 20.Tao YX. Inactivating mutations of G protein-coupled receptors and diseases: Structure-function insights and therapeutic implications. Pharmacol Ther. 2006;111:949–73. doi: 10.1016/j.pharmthera.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Hinney A, Bettecken T, Tarnow P, et al. Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population-based sample and obese adults from Germany. J Clin Endocrinol Metab. 2006;91:1761–9. doi: 10.1210/jc.2005-2056. [DOI] [PubMed] [Google Scholar]
- 22.Hainerova I, Larsen LH, Holst B, et al. Melanocortin 4 receptor mutations in obese Czech children: studies of prevalence, phenotype development, weight reduction response, and functional analysis. J Clin Endocrinol Metab. 2007;92:3689–96. doi: 10.1210/jc.2007-0352. [DOI] [PubMed] [Google Scholar]
- 23.Hinney A, Hohmann S, Geller F, et al. Melanocortin-4 receptor gene: case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J Clin Endocrinol Metab. 2003;88:4258–67. doi: 10.1210/jc.2003-030233. [DOI] [PubMed] [Google Scholar]
- 24.Rettenbacher E, Tarnow P, Brumm H, et al. A novel non-synonymous mutation in the melanocortin-4 receptor gene (MC4R) in a 2-year-old Austrian girl with extreme obesity. Exp Clin Endocrinol Diabetes. 2007;115:7–12. doi: 10.1055/s-2007-949150. [DOI] [PubMed] [Google Scholar]
- 25.Buono P, Pasanisi F, Nardelli C, et al. Six novel mutations in the proopiomelanocortin and melanocortin receptor 4 genes in severely obese adults living in southern Italy. Clin Chem. 2005;51:1358–64. doi: 10.1373/clinchem.2005.047886. [DOI] [PubMed] [Google Scholar]
- 26.Larsen LH, Echwald SM, Sorensen TI, et al. Prevalence of mutations and functional analyses of melanocortin 4 receptor variants identified among 750 men with juvenile-onset obesity. J Clin Endocrinol Metab. 2005;90:219–24. doi: 10.1210/jc.2004-0497. [DOI] [PubMed] [Google Scholar]
- 27.Shao XY, Jia WP, Cai SB, et al. Cloning and functional analysis of melanocortin 4 receptor mutation gene F261S. Zhonghua Yi Xue Za Zhi. 2005;85:366–9. [PubMed] [Google Scholar]
- 28.Morello JP, Salahpour A, Laperriere A, et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–95. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noorwez SM, Kuksa V, Imanishi Y, et al. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J Biol Chem. 2003;278:14442–50. doi: 10.1074/jbc.M300087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noorwez SM, Malhotra R, McDowell JH, et al. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004;279:16278–84. doi: 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- 31.Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab. 2002;87:3255–62. doi: 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- 32.Janovick JA, Goulet M, Bush E, et al. Structure-activity relations of successful pharmacologic chaperones for rescue of naturally occurring and manufactured mutants of the gonadotropin-releasing hormone receptor. J Pharmacol Exp Ther. 2003;305:608–14. doi: 10.1124/jpet.102.048454. [DOI] [PubMed] [Google Scholar]
- 33.Chaipatikul V, Erickson-Herbrandson LJ, Loh HH, et al. Rescuing the traffic-deficient mutants of rat í-opioid receptors with hydrophobic ligands. Mol Pharmacol. 2003;64:32–41. doi: 10.1124/mol.64.1.32. [DOI] [PubMed] [Google Scholar]
- 34.Petaja-Repo UE, Hogue M, Bhalla S, et al. Ligands act as pharmacological chaperones and increase the efficiency of ™ opioid receptor maturation. EMBO J. 2002;21:1628–37. doi: 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan J, Perry SJ, Gao Y, et al. A point mutation in the human melanin concentrating hormone receptor 1 reveals an important domain for cellular trafficking. Mol Endocrinol. 2005;19:2579–90. doi: 10.1210/me.2004-0301. [DOI] [PubMed] [Google Scholar]
- 36.Van Craenenbroeck K, Clark SD, Cox MJ, et al. Folding efficiency is rate-limiting in dopamine D4 receptor biogenesis. J Biol Chem. 2005;280:19350–7. doi: 10.1074/jbc.M414043200. [DOI] [PubMed] [Google Scholar]
- 37.Robert J, Auzan C, Ventura MA, et al. Mechanisms of cell-surface rerouting of an endoplasmic reticulum-retained mutant of the vasopressin V1b/V3 receptor by a pharmacological chaperone. J Biol Chem. 2005;280:42198–206. doi: 10.1074/jbc.M510180200. [DOI] [PubMed] [Google Scholar]
- 38.Hawtin SR. Pharmacological chaperone activity of SR49059 to functionally recover misfolded mutations of the vasopressin V1a receptor. J Biol Chem. 2006;281:14604–14. doi: 10.1074/jbc.M511610200. [DOI] [PubMed] [Google Scholar]
- 39.Fortin JP, Dziadulewicz EK, Gera L, et al. A nonpeptide antagonist reveals a highly glycosylated state of the rabbit kinin B1 receptor. Mol Pharmacol. 2006;69:1146–57. doi: 10.1124/mol.105.019976. [DOI] [PubMed] [Google Scholar]
- 40.Bernier V, Morello JP, Zarruk A, et al. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17:232–43. doi: 10.1681/ASN.2005080854. [DOI] [PubMed] [Google Scholar]
- 41.Vos TJ, Caracoti A, Che JL, et al. Identification of 2-[2-[2-(5-bromo-2-methoxyphenyl)-ethyl]-3-fluorophenyl]-4,5-dihydro-1H-imidazole (ML00253764), a small molecule melanocortin 4 receptor antagonist that effectively reduces tumor-induced weight loss in a mouse model. J Med Chem. 2004;47:1602–4. doi: 10.1021/jm034244g. [DOI] [PubMed] [Google Scholar]
- 42.Nicholson JR, Kohler G, Schaerer F, et al. Peripheral administration of a melanocortin 4-receptor inverse agonist prevents loss of lean body mass in tumor-bearing mice. J Pharmacol Exp Ther. 2006;317:771–7. doi: 10.1124/jpet.105.097725. [DOI] [PubMed] [Google Scholar]
- 43.Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544–51. doi: 10.1210/en.2003-0524. [DOI] [PubMed] [Google Scholar]
- 44.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–52. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan ZC, Sartin JL, Tao YX. Molecular cloning and pharmacological characterization of porcine melanocortin-3 receptor. J Endocrinol. 2008;196:139–48. doi: 10.1677/JOE-07-0403. [DOI] [PubMed] [Google Scholar]
- 46.Wang SX, Fan ZC, Tao YX. Functions of acidic transmembrane residues in human melanocortin-3 receptor binding and activation. Biochem Pharmacol. 2008;76:520–30. doi: 10.1016/j.bcp.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srinivasan S, Lubrano-Berthelier C, Govaerts C, et al. Constitutive activity of the melanocortin-4 receptor is maintained by its N-terminal domain and plays a role in energy homeostasis in humans. J Clin Invest. 2004;114:1158–64. doi: 10.1172/JCI21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hobbs HH, Russell DW, Brown MS, et al. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24:133–70. doi: 10.1146/annurev.ge.24.120190.001025. [DOI] [PubMed] [Google Scholar]
- 49.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–4. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 50.Lubrano-Berthelier C, Durand E, Dubern B, et al. Intracellular retention is a common characteristic of childhood obesity-associated MC4R mutations. Hum Mol Genet. 2003;12:145–53. doi: 10.1093/hmg/ddg016. [DOI] [PubMed] [Google Scholar]
- 51.Valli-Jaakola K, Lipsanen-Nyman M, Oksanen L, et al. Identification and characterization of melanocortin-4 receptor gene mutations in morbidly obese Finnish children and adults. J Clin Endocrinol Metab. 2004;89:940–5. doi: 10.1210/jc.2003-031182. [DOI] [PubMed] [Google Scholar]
- 52.Fang QC, Jia WP, Cai SB, et al. Intracellular retention of human melanocortin-4 receptor: a molecular mechanism underlying early-onset obesity in F261S pedigree of Chinese. Biomed Environ Sci. 2008;21:280–5. doi: 10.1016/S0895-3988(08)60042-2. [DOI] [PubMed] [Google Scholar]
- 53.Tan K, Pogozheva ID, Yeo GS, et al. Functional characterization and structural modeling of obesity-associated mutations in the melanocortin 4 receptor. Endocrinology. 2008;53:114–25. doi: 10.1210/en.2008-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tao YX. Constitutive activation of G protein-coupled receptors and diseases: Insights into mechanism of activation and therapeutics. Pharmacol Ther. 2008;120:129–48. doi: 10.1016/j.pharmthera.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tao YX, Abell AN, Liu X, et al. Constitutive activation of G protein-coupled receptors as a result of selective substitution of a conserved leucine residue in transmembrane helix III. Mol Endocrinol. 2000;14:1272–82. doi: 10.1210/mend.14.8.0503. [DOI] [PubMed] [Google Scholar]
- 56.Feng X, Muller T, Mizrachi D, et al. An intracellular loop (IL2) residue confers different basal constitutive activities to the human lutropin receptor and human thyrotropin receptor through structural communication between IL2 and helix 6, via helix 3. Endocrinology. 2008;149:1705–17. doi: 10.1210/en.2007-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tao YX, Johnson NB, Segaloff DL. Constitutive and agonist-dependent self-association of the cell surface human lutropin receptor. J Biol Chem. 2004;279:5904–14. doi: 10.1074/jbc.M311162200. [DOI] [PubMed] [Google Scholar]
- 58.Conn PM, Ulloa-Aguirre A, Ito J, et al. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev. 2007;59:225–50. doi: 10.1124/pr.59.3.2. [DOI] [PubMed] [Google Scholar]