

Abstract
The tumour necrosis factor (TNF) family member TWEAK activates the Fn14 receptor and has pro-apoptotic, proliferative and pro-inflammatory actions that depend on the cell type and the microenvironment. We explored the proliferative actions of TWEAK on cultured tubular cells and in vivo on renal tubules. Additionally, we studied the role of TWEAK in compensatory proliferation following unilateral nephrectomy and in an inflammatory model of acute kidney injury (AKI) induced by a folic acid overdose. TWEAK increased the proliferation, cell number and cyclin D1 expression of cultured tubular cells, in vitro. Exposure to serum increased TWEAK and Fn14 expression and the proliferative response to TWEAK. TWEAK activated the mitogen-activated protein kinases ERK and p38, the phosphatidyl-inositol 3-kinase (PI3K)/Akt pathway and NF-κB. TWEAK-induced proliferation was prevented by inhibitors of these protein kinases and by the NF-κB inhibitor parthenolide. TWEAK-induced tubular cell proliferation as assessed by PCNA and cyclin D1 expression in the kidneys of adult healthy mice in vivo. By contrast, TWEAK knock-out mice displayed lower tubular cell proliferation in the remnant kidney following unilateral nephrectomy, a non-inflammatory model. This is consistent with TWEAK-induced proliferation on cultured tubular cells in the absence of inflammatory cytokines. Consistent with our previously published data, in the presence of inflammatory cytokines TWEAK promoted apoptosis, not proliferation, of cultured tubular cells. In this regard, TWEAK knock-out mice with AKI displayed less tubular apoptosis and proliferation, as well as improved renal function. In conclusion, TWEAK actions in tubular cells are context dependent. In a non-inflammatory milieu TWEAK induces proliferation of tubular epithelium. This may be relevant for compensatory renal hyperplasia following nephrectomy.
Keywords: cyclin, Fn14, kidney, mitosis, tubular cell, TWEAK, uninephrectomy
Introduction
Kidney tissue cell number is carefully regulated through the balance between mitosis and cell death. An imbalance between these processes can result in disorders of cell number characterized by an excessive or insufficient cell number [1]. Compensatory renal hypertrophy and hyperplasia are observed following unilateral nephrectomy [2]. This occurs clinically following nephrectomy for benign or malignant conditions, in living kidney donors and in transplant recipients. Growth factors and cytokines, such as growth hormone and insulin-like growth factors, have been implicated in compensatory renal growth after uninephrectomy [3]. In addition, tubular cell proliferation is required to recover from tubular cell death in acute kidney injury (AKI). Tumour necrosis factor (TNF) superfamily cytokines participate both in parenchymal cell injury as well as in proliferation following loss of tissue mass. In the liver, TNF-α promotes hepatocyte apoptosis, but type 1 TNF receptor is also required for compensatory proliferation following partial hepatectomy [4, 5]. TNF-like weak inducer of apoptosis (TWEAK, TNFSF12) is a recently characterized member of the TNF superfamily of structurally related cytokines [6]. TWEAK regulates cell death, proliferation and inflammation in a cell- and microenvironment-dependent fashion [7–12]. Fibroblast growth factor-inducible 14 (Fn14) is the TWEAK receptor [13]. Fn14 and TWEAK are present in healthy adult kidney, and their expression is up-regulated in AKI, where TWEAK contributes to amplification of inflammation [6, 14–16]. TWEAK induces apoptosis in tubular cells cultured in an inflammatory milieu that includes interferon-γ (IFN-γ) and TNF-α, similar to that found during AKI [17]. However, there is no information on the role of TWEAK in tubular cell proliferation.
The cell division cycle is regulated by an intricate network of regulatory pathways that ensures the correct sequence and timing of each cycle phase [18]. Proliferative stimuli activate signal transduction pathways such as kinases and transcription factors that promote the progression through the cell cycle. This is controlled by the sequential formation, activation and subsequent inactivation of a series of specific cyclin–cyclin dependent kinase complexes. Cyclin levels fluctuate during the cell cycle. Cyclin D1 is a G1 cyclin and its up-regulation is required early in cell cycle progression [19].
We now report that TWEAK promotes NF-κB, mitogen-activated protein kinase (MAPK) and Akt activation in renal tubular cells, leading to tubular proliferation, when the microenvironment is not inflammatory. TWEAK-induced tubular cell proliferation was observed in healthy kidneys in vivo. By contrast, TWEAK KO mice displayed an impaired hyperplastic response to unilateral nephrectomy, suggesting that this pathway plays a role in compensatory renal cell proliferation in this model. We have previously shown that in an inflammatory environment characterized by the presence of IFN-γ and TNF-α, TWEAK has apoptotic effects [17] and it does not promote cultured tubular cell proliferation. In an AKI model characterized by increased local IFN-γ and TNF-α, absence of TWEAK limited both tubular apoptosis and proliferation and the overall effect was beneficial.
Methods
Cells and reagents
MCT cells are a cultured line of proximal tubular epithelial cells harvested originally from the renal cortex of SJL mice and have been extensively characterized [20]. MCT cells were cultured in RPMI 1640 (GIBCO, Grand Island, NY, USA), 10% decomplemented foetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, in 5% CO2 at 37°C [20].
Human proximal tubular epithelial (HK-2) cells (ATCC, Rockville, MD, USA) were grown on the same media as the MCT cells plus 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml sodium selenite, and 5 ng/ml hydrocortisone [15]. Results for these cells are only shown in Fig. 1E.
Figure 1.
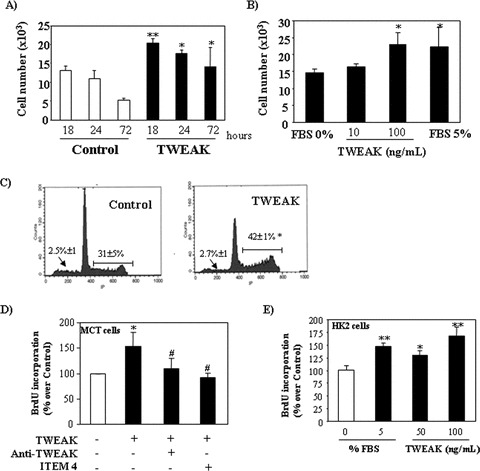
TWEAK increases cell proliferation in cultured murine (A–D) and human (E) renal tubular cells. (A) and (B) Quantification by flow cytometry of tubular cell number following treatment with TWEAK: (A) Time–response to 100 ng/ml TWEAK in serum-deprived cells, **P < 0.005 versus its corresponding control; *P < 0.05 versus control. Mean (±S.E.M.) of four independent experiments. (B) Dose–response at 18 hrs, *P < 0.02 versus 0% FBS. Mean (±S.E.M.) of four independent experiments. Cells cultured in 5% FBS medium were used as positive controls. (C) Treatment with 100 ng/ml TWEAK for 18 hrs induced cell proliferation (flow cytometry of DNA content). Note the increased proportion of cells in the S and M phase of the cell cycle (|––––|), without changes in the apoptotic rate (arrow). Mean (±S.E.M.) of four experiments, *P < 0.002 versus control. (D) ITEM-4, a neutralizing anti-Fn14 antibody and an anti-TWEAK antibody prevented proliferation induced by 100 ng/ml TWEAK at 18 hrs, as assessed by BrdU incorporation. Mean (±S.E.M.) of four independent experiments. *P < 0.005 versus control; #P < 0.003 versus TWEAK alone. (E) TWEAK-induced proliferation in human HK2 tubular cells as assessed by BrdU incorporation dose–response at 18 hrs, *P < 0.02 versus 0% FBS. **P < 0.001 versus 0% FBS. Mean (±S.E.M.) of four independent experiments. Cells cultured in 5% FBS medium were used as positive controls.
Recombinant human TWEAK (Alexis, Läufelfingen, Switzerland) and ITEM-4 neutralizing anti-Fn14 antibody (eBioscience, San Diego, CA) were dissolved in water. Unless otherwise specified the concentration of TWEAK was 100 ng/ml. Blocking anti-TWEAK mAb (clone P2D10, BiogenIdec, Inc., Cambridge, MA, USA) was used at 10 μg/ml [15]. Murine TNF-α (PrePotech, London, UK) 30 ng/ml and interferon-γ (INF-γ) (PrePotech) 30 U/ml were used in some experiments. The kinase inhibitors SB203580, PD98059 (Stressgen Bioreagent, Brussels, Belgium), Wortmannin (Calbiochem, Gibbstown, NJ, USA) and LY294002 (Sigma, St. Louis, MO, USA) were dissolved in DMSO and used at 5 μM, 20 μM, 50 nM and 50 μM, respectively, 1 hr prior to TWEAK. These concentrations inhibit phosphorylation of their targets in MCT cells. Final concentration of DMSO, 0.05%, did not modulate cell death or proliferation [21]. Parthenolide (Sigma) was used at 10 μM.
Cell proliferation
Cells seeded in 12-well plates (Costar, Cambridge, MA, USA) in 10% FBS RPMI overnight, were rested in serum-free medium for 24 hrs and 100 ng/ml TWEAK was added. For assessment of cell cycle, apoptosis and quantification of cell number, adherent cells were pooled with spontaneously detached cells, and stained in 100 μg/ml propidium iodide, 0.05% NP-40, 10 μg/ml RNAse A in PBS and incubated at 4°C for >1 hr. This assay permeabilizes the cells and propidium iodide stains DNA. The total cell number, the percentage of apoptotic cells with decreased DNA staining and proliferating cells with increased DNA content (S+M) was counted by flow cytometry using BD CellQuest Software (BD Biosciences, San Jose, CA, USA) [17].
Cell proliferation was also quantified by a colorimetric immunoassay of BrdU incorporation into DNA (Roche, Penzberg, Germany). Cells were seeded in 96-well plates in 10% FBS RPMI overnight, and then synchronized for 24 hrs with RPMI without FBS and stimulated with TWEAK for 18 hrs. Every condition was tested in triplicate. BrdU (100 μM) was added 14 hrs before the end of the assay. Absorbance was quantified using a spectrophotometer plate reader (Microplate Absorbance Reader Anthos 2020, Anthos, Wals, Austria).
Western blot
Cell samples were homogenized in lysis buffer (50 mM TrisHCl, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0,2% Triton X-100, 0,3% NP-40, 0,1 mM PMSF and 1 μg/ml pepstatin A) then separated by 12% SDS-PAGE under reducing conditions. After electrophoresis, samples were transferred to PVDF membranes (Millipore, Bedford, MA), blocked with 5% skimmed milk in PBS/0.5% v/v Tween 20 for 1 hr, washed with PBS/Tween and incubated with rabbit polyclonal anti-TWEAK (1:500), rabbit polyclonal anti-p-Akt (1:1000), rabbit polyclonal anti-Akt (1:2000), mouse monoclonal anti-p-ERK (1:500), goat polyclonal anti-ERK (1:2000), mouse monoclonal anti-p-p38 (1:500), goat polyclonal anti-p38 (1:2500), or rabbit polyclonal anti-PCNA (1:1000) all from Santa Cruz Biotechnology (Santa Cruz, CA, USA) or mouse monoclonal anti-cyclin D1 (1:2000), rabbit polyclonal anti-Fn14 (1:1000) or rabbit polyclonal anti-cleaved caspase 3 active (1:500) from Cell Signaling (Danvers, MA, USA). Antibodies were diluted in 5% milk PBS/Tween. Blots were washed with PBS/Tween and incubated with appropriate horseradish peroxidase-conjugated secondary antibody (1:2000, Amersham, Aylesbury, UK). After washing with PBS/Tween the blots were developed with the chemiluminescence method (ECL) (Amersham). Blots were then probed with mouse monoclonal anti-α-tubulin antibody (1:2000, Sigma) and levels of expression were corrected for minor differences in loading [21].
RNA extraction and real-time polymerase chain reaction
Total RNA was extracted from MCT cells or kidneys by the TRI Reagent method (Sigma) and 1 μg of RNA was reverse transcribed with High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). Pre-developed primer and probe assays for Fn14, TWEAK, TNF-α, INF-γ, and GAPDH (murine) were from Applied (Applied Biosystems). Quantitative PCR was performed by 7500 Real Time PCR System with the Prism 7000 System SDS Software (Applied Biosystems) and RNA expression of different genes was corrected for GAPDH [15].
NF-κ B luciferase reporter gene assay
MCT cells were plated at a density of 8 × 104 cells in six-well plates 24 hrs before transfection with FuGENE™ 6 (Roche, Indianapolis, IN, USA). pNF-κB-Luc (Stratagene, CA, USA) and pRLTK vectors, that contain the luciferase gene Renilla (Promega, WI, USA) were used in a ratio of 10:1. The medium was replaced with RPMI without serum 4 hrs after transfection and cells were pre-treated for 1 hr with the NF-κB inhibitor parthenolide or the kinase inhibitors SB203580, PD98059 or Wortmannin and then stimulated with 100 ng/ml TWEAK. Luciferase activity was determined by a luciferase assay system (Promega) and a luminometer (Berthold, Nashua, NH, USA) and normalized to Renilla activity to control for differences in transfection efficiency [15].
In vivo administration of TWEAK to adult healthy mice
All animal studies were conducted in accord with the NIH Guide for the Care and Use of Laboratory Animals. Balb/c 5- to 7-week-old mice were obtained from IFFA-CREDO (Barcelona, Spain). Mice were killed at 4 or 24 hrs after a single intraperitoneal injection of either: (i) 0.5 μg/mouse TWEAK (n= 5) or (ii) vehicle (200 μl of 0.9% NaCl) (n= 5). The dose of TWEAK in vivo (0.5 mg/mouse) was calculated based on in vitro experiments for an extracellular volume of 5 ml/mouse and on in vivo experience of renal effects of the systemic administration of this dose of TWEAK (15).
Experimental model of acute kidney injury
Folic acid nephropathy is a classical model of AKI, characterized by tubular cell death and proliferation and inflammation, all of them features of human AKI [15]. The time course of tubular cell death and proliferation was characterized in C57/BL6 mice (12- to 14-week old) that received a single i.p. injection of folic acid (Sigma) 250 mg/kg in 0.3 mol/l sodium bicarbonate or vehicle and mice were killed 24 or 72 hrs. In a second set of experiments, C57/BL6 TWEAK KO mice and WT littermates mice [21] (BiogenIdec, Inc.) were injected with of folic acid and killed 24 hrs, 72 hrs or 7 days later (n= 6 per group). In a third set of experiments, anti-TWEAK antibodies were administered to wild-type animals with folic acid induced AKI and the data have been described elsewhere [15].
Kidneys were cold saline perfused in situ before removal. One kidney from each mouse was fixed in buffered formalin, embedded in paraffin and used for immunohistochemistry. The other kidney was snap-frozen in liquid nitrogen for RNA and protein studies.
Experimental model of compensatory renal growth following uninephrectomy
Compensatory renal growth takes place following uninephrectomy for malignant or benign lesions or in living kidney donors. Both hypertrophy and hyperplasia contribute to kidney growth [2]. Female, 12- to 14- week-old TWEAK KO mice and WT littermate mice were used (n= 7/group) [22]. Nephrectomy of the left kidney was performed following ligation of the renal vessels and ureter. The remaining kidneys were removed 48 hrs following nephrectomy, the time of peak tubular cell mitosis [2]. Kidneys were perfused with cold saline in situ before removal. Half of each kidney was fixed in buffered formalin, embedded in paraffin and used for immunohistochemistry. The other half kidney was snap-frozen in liquid nitrogen for RNA and protein (Western) studies.
In a second set of experiments wild-type mice were subjected to left nephrectomy, and 0.5 μg TWEAK (n= 5) or vehicle (0.9% NaCl) were administered 24 hrs later. Mice were killed 48 hrs after nephrectomy.
Immunohistochemistry
Immunohistochemistry was carried out in paraffin-embedded tissue sections 5-μm thick. The slides were deparaffinized with xylene and graded concentrations of ethanol and then rehydrated. Endogenous peroxidase was blocked by incubation in 3% H2O2/methanol (1:1) at 25°C for 30 min. The slides were incubated in PBS with 4% bovine serum albumin (BSA) and 6% horse serum, for 1 hr at 37°C to reduce non-specific background staining, and then incubated overnight at 4°C with anti-cyclin D1 (1:100, Cell Signaling), anti-active caspase 3 (1:100, Promega) or anti-Fn14 (1:20, Cell Signaling) antibodies in PBS, 4% BSA, 1% serum. For PCNA staining, slides were incubated in PBS with 1.5% horse serum for 1 hr at 37°C to reduce non-specific background staining, and then incubated overnight at 4°C with anti-PCNA antibody (1:150, Santa Cruz Biotechnology) in PBS containing 1.5% serum. After washing with PBS, sections were incubated with a secondary biotin-labelled antibody, washed and developed with AB streptavidin complex to cyclin D1, to caspase 3 active and to Fn14 and with an anti-IgG HRP-conjugated antibody to PCNA. Finally, the sections were washed and stained with DAB (Dako Diagnostics, Barcelona, Spain). Sections were counterstained with Carazzi’s haematoxylin. Negative controls included incubation with isotype IgG.
Apoptotic cell death was assessed by enzymatic in situ labelling of DNA strand breaks using the terminal deoxynucleotidyl-transferasa-mediated dUTP nick-end labelling (TUNEL) (In Situ Cell Death Detection Kit; Promega).
The total number of PCNA and TUNEL+ nuclei was quantitated in 20 randomly chosen fields (40×) per kidney using Image Pro Plus Software (Media cybernetics, Bethesda, MD, USA). Cyclin D1 and caspase 3 active staining was evaluated by a quantitative scoring system with the same software in 10 randomly chosen fields (20×) per kidney [15].
Statistics
Statistical analysis was performed with SPSS 11.0 statistical software. Results are expressed as mean ± S.E.M. Significance at the P < 0.05 level was assessed by Student’s t-test for two groups of data and anova for three of more groups.
Results
TWEAK induces tubular cell proliferation
TWEAK increased the number of cultured tubular cells in a time- and dose-dependent manner (Fig. 1A and B). TWEAK, also increased the percentage of cells in the S and M phase of the cell cycle without changes in the apoptotic rate (Fig. 1C). These findings suggest that the changes in cell number are the consequence of TWEAK-induced proliferation. Indeed, TWEAK increased BrdU incorporation during DNA synthesis in a dose-dependent manner (Figs 1D and 3D).
Figure 3.
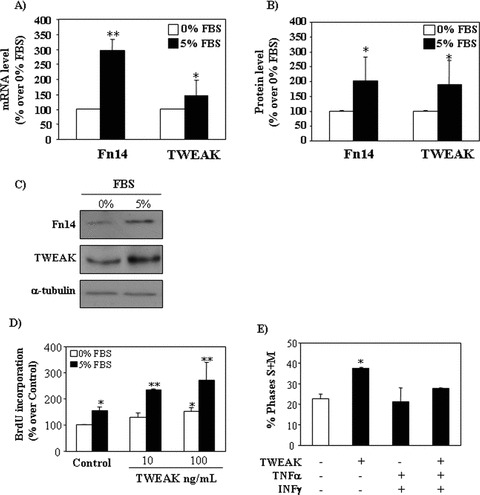
Modulation of the proliferative effect of TWEAK by the microenvironment. (A) TWEAK and Fn14 mRNA expression (real time RT-PCR) in tubular cells cultured with 0% or 5% FBS for 24 hrs. Mean (±S.E.M.) of three independent experiments. *P < 0.05 versus 0% FBS; **P < 0.001 versus 0% FBS. (B) Quantification of Fn14 and TWEAK protein in tubular cells cultured with 0% or 5% FBS for 24 hrs. Mean (±S.E.M.) of three independent experiments. *P < 0.05 versus 0% FBS. (C) Representative Western blot of TWEAK and Fn14 in tubular cells cultured with 0% or 5% FBS for 24 hrs. (D) TWEAK for 18 hrs in serum-free medium or in 5% FBS increased BrdU incorporation during DNA synthesis. Mean (±S.E.M.) of four independent experiments. *P < 0.005 versus 0% FBS control, **P < 0.0005 versus 5% FBS control. (E) In the presence of 30 ng/ml TNF-α/30 U/ml INF-γ the proliferative effect of TWEAK was abolished (cells in the S+M phase of the cycle assessed by flow cytometry of DNA content). Mean (±S.E.M.) of three independent experiments. *P < 0.005 versus control.
Fn14 is the characterized signalling TWEAK receptor. However, the existence of additional TWEAK receptors has been suggested [7, 23] and TWEAK may translocate to the nucleus [24]. Anti-Fn14 neutralizing antibody, ITEM-4, inhibited TWEAK-induced cell proliferation (Fig. 1D), suggesting that TWEAK induces proliferation in tubular epithelium through Fn14 activation. TWEAK blockade with anti-TWEAK antibody also prevented TWEAK-induced proliferation, confirming the specificity of the observation (Fig. 1D). TWEAK also induced proliferation in human proximal tubular HK2 cells (Fig. 1E). Cyclin D1 expression regulates the transit through the G1 phase of the cell cycle and is a key step in cell proliferation. TWEAK increases cyclin D1 protein expression in a time- and dose-dependent manner (Fig. 2A and B).
Figure 2.
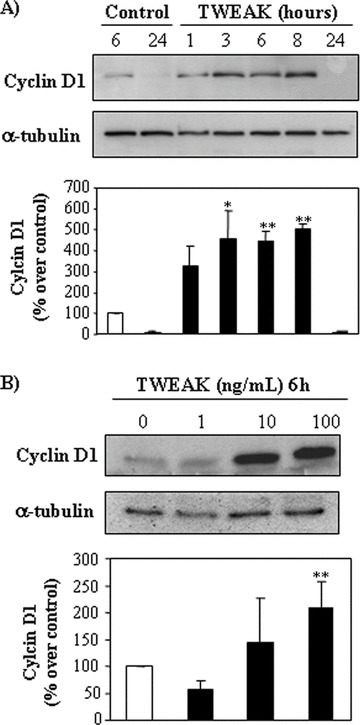
TWEAK induces expression of cyclin D1 in a dose- and time-dependent manner. Representative Western blots and quantification showing the effect of TWEAK on cyclin D1 levels in tubular cell lysates. (A) Time-course. tubular cells were stimulated with 100 ng/ml TWEAK, or (B) dose–response at 6 hrs. Mean (±S.E.M.) of four experiments. *P < 0.04 versus control; **P < 0.02 versus control.
Modulation of the proliferative effect of TWEAK by the microenvironment
Renal tubular cells constitutively express TWEAK and Fn14 [17]. The presence of serum increased TWEAK expression at the protein, as measured by Western blot analysis and mRNA levels (Fig. 3A–C) Constitutive Fn14 expression was increased by serum at the protein and mRNA levels (Fig. 3A–C). In this regard, the presence of serum increased the proliferative effect of TWEAK (Fig. 3D).
The inflammatory cytokines TNF-α and IFN-γ also increase Fn14 expression in tubular cells [15]. Interestingly, the proliferative effect of TWEAK was lost in the presence of IFN-γ and TNF-α (Fig. 3E). Tubular cell apoptosis induction by the combination of TWEAK, IFN-γ and TNF-α was previously reported and characterized by our group [17] and we now confirmed a four-fold increase in hypodiploid cells in the presence of these cytokines (data not shown).
TWEAK-induced tubular cell proliferation is mediated by p38 kinase, ERK1/2 and AKT
Next, we studied the effect of TWEAK on signalling pathways that control cell proliferation. TWEAK induced phosphorylation of ERK1/2 (Fig. 4A), p38 kinase (Fig. 4B) and Akt (Fig. 4C). “to” TWEAK promoted phosphorylation of ERK1/2 (Fig. 4A), p38 kinase (Fig. 4B) and Akt (Fig. 4C).
Figure 4.
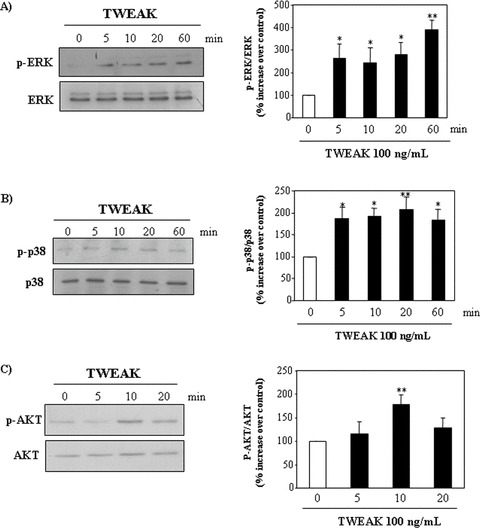
TWEAK activates the MAP kinases, p38 and ERK1/2, and promotes Akt phosphorylation in tubular cells. Tubular cells were stimulated with 100 ng/ml TWEAK. Cell lysates were analyzed by Western blot for phospho-ERK1/2 MAP kinase (A), phospho-p38 MAP kinase (B) and phospho-Akt kinase (C). Western blots representative of five independent experiments. Each blot was stripped and reprobed with anti-ERK, anti-p38 MAPK and anti-Akt antibody respectively. The band intensities for each phospho-protein were normalized to the corresponding band intensities for each total protein. Mean (±S.E.M.) of five independent experiments. **P < 0.005 versus 0 min., *P < 0.04 versus 0 min.
To explore the role of MAP kinases and PI3kinase/Akt signalling in TWEAK-induced proliferation, we used the specific kinase inhibitors Wortmannin (PI3kinase), PD-98059 (ERK1/2) and SB-203580 (p38). In the presence of these inhibitors TWEAK failed to increase the number of cultured tubular cells (Fig. 5A) as well as BrdU incorporation into DNA (Fig. 5B). Flow cytometry of DNA content confirmed a decreased percentage of inhibitor-treated cells in the S or M phases of the cycle as well as absence of apoptosis induction (not shown). The inhibitors of ERK1/2 (PD-98059) or Akt (Wortmannin and LY294002) prevented the increased expression of cyclin D1 in response to TWEAK, whereas the effect of the p38 inhibitor SB203580 was partial (Fig. 5C).
Figure 5.
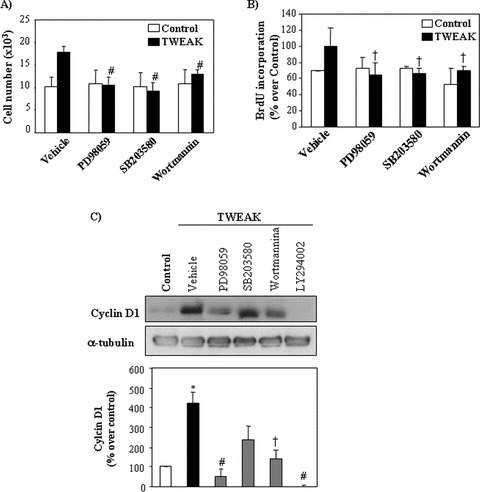
Inhibition of TWEAK-induced tubular cell proliferation with specific inhibitors of MAPKs and PI3K. (A), (B) Cells were pre-treated with inhibitors of ERK1/2 (20 μM PD98059), p38 MAPK (5 μM SB203580) or PI3K (50 nM Wortmannin) for 1 hr and then stimulated with 100 ng/ml TWEAK for 18 hrs. (A) Quantification of cell number by flow cytometry. Mean (±S.E.M.) of four independent experiments. #P < 0.005 versus TWEAK alone. (B) Cell proliferation assessed by BrdU incorporation. Mean (±S.E.M.) of four independent experiments. †P < 0.05 versus TWEAK alone. (C) Effect of kinase inhibitors on TWEAK-induced up-regulation of cyclin D1 at 8 hrs. Inhibition of ERK1/2 or Akt prevented the up-regulation of cyclin D1 induced by TWEAK. Representative Western blot and quantification. Mean (±S.E.M.) of four experiments, *P < 0.005 versus control; †P < 0.05 versus TWEAK alone; #P < 0.005 versus TWEAK alone. LY294002 (10 μM) is a PI3K inhibitor.
NF-κB controls TWEAK-induced proliferation in a different pathway than MAP-kinases and PI3 kinase/Akt
NF-κB plays a role in cell proliferation [25]. In tubular cells TWEAK induces NF-κB activation and this is prevented by parthenolide [15]. Inhibition of NF-κB activation by parthenolide prevented TWEAK-induced increase in cell number (Fig. 6A). At this concentration parthenolide does not induce apoptosis in cultured tubular cells as assessed by flow cytometry analysis of DNA content (Fig. 6B). By contrast, parthenolide decreased TWEAK-induced proliferation (Fig. 6C).
Figure 6.
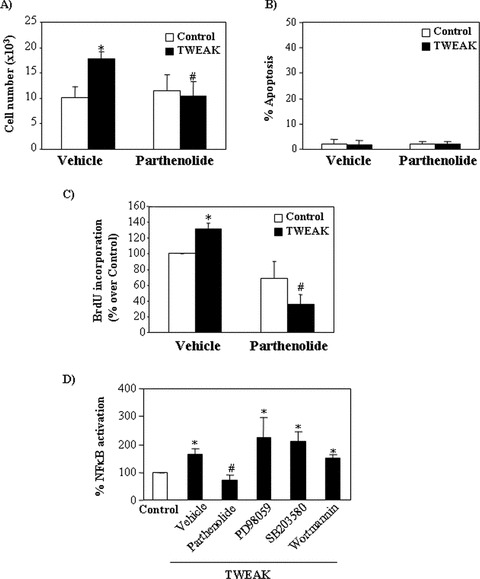
TWEAK-induced proliferation is NF-κB dependent. Tubular cells were pre-treated with the NF-κB inhibitor parthenolide for 1 hr and then stimulated with 100 ng/ml TWEAK for 18 hrs. Parthenolide prevents TWEAK-induced increase in cell number (A), without changing the rate of apoptosis (B). Parthenolide prevents TWEAK-induced proliferation as assessed by BrdU incorporation (C). Mean (±S.E.M.) of four experiments. *P < 0.005 versus control, #P < 0.005 versus TWEAK alone. (D) Parthenolide, but not ERK1/2 (PD98059), p38 MAPK (SB203580) or Akt (Wortmannin) inhibitors prevented TWEAK-induced NF-κB activation as assessed by a luciferase gene reporter assay at 24 hrs. Mean (±S.E.M.) of three experiments. *P < 0.05 versus control, #P < 0.03 versus TWEAK alone.
Because NF-κB can be activated by MAP kinases and Akt [26, 27], the role of kinases in TWEAK-induced NF-κB activation was studied by a reporter NF-κB luciferase gene assay. Inhibition of ERK and p38 MAP kinase or phosphatidyl-inositol 3-kinase (PI3K)/AKT did not decrease NF-κB activation (Fig. 6D).
In vivo actions of TWEAK on tubular cell proliferation
Tubular epithelium is the main site of Fn14 expression in the normal kidney [17]. TWEAK administration to healthy adult wild-type mice resulted in an increased number of tubular cell nuclei positive for PCNA and increased total kidney PCNA levels (Fig. 7A and B). This was associated with up-regulation of cyclin D1 in tubular epithelium (Fig. 7C).
Figure 7.
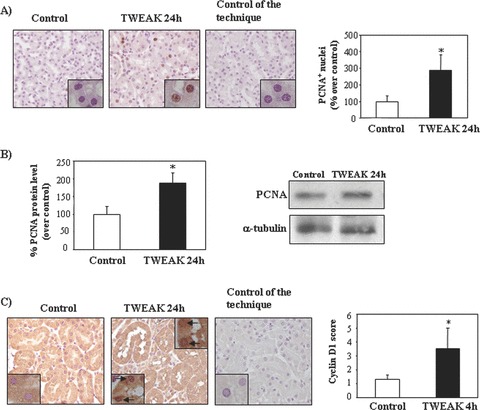
TWEAK induces tubular cell proliferation in healthy kidneys from wild-type-mice in vivo. (A) Increased number of PCNA+ cells in kidney tubular epithelium 24 hrs after injection of recombinant TWEAK as compared to vehicle. Original magnification ×400. Inset, original magnification ×1000. Mean (±S.E.M.) n= 5. *P < 0.005 versus control. (B) Whole kidney PCNA expression assessed by Western blot. Representative Western blot and quantification. Mean (±S.E.M.) n= 5. *P < 0.05 versus control. (C) Increased expression of cyclin D1 in kidney tubular epithelium 4 hrs following TWEAK administration. Original magnification ×400. Inset: note nuclear localization of Cyclin D1 (arrow), original magnification ×1000. Mean (±S.E.M.) of 5 animals per group. *P < 0.05 versus control.
Because our data indicated that TWEAK has proliferative activities on renal tubular epithelium, we explored whether the TWEAK pathway may be involved in renal cell proliferation following unilateral nephrectomy. The expression of Fn14 was increased in remnant kidneys, both at mRNA (Fig. 8A) and protein levels (Fig. 8B and C), while TWEAK expression did not change (Fig. 8A–C). Immuohistochemistry localized the increased Fn14 expression to tubular cells in remnant kidneys following uninephrectomy (Fig. 8E). Renal expression of the inflammatory cytokines TNF-α and IFN-γ was not increased in this model, in accordance with previous reports [28] (Fig. 8D). Compensatory kidney growth takes place following uninephrectomy both through hyperplasia and hypertrophy. Cell proliferation peaks at 48 hrs [2]. There was a trend towards a lower increase in kidney weight in TWEAK KO mice versus wild-type (17 ± 4 versus 31 ± 7 mg, P= 0.08). TWEAK KO mice displayed lower tubular cell proliferation in the remnant kidney than wild-type mice (Fig. 9A and B). Administration of exogenous TWEAK to uninephrectomized wild-type mice led to a further increase in renal cell proliferation, thus supporting the involvement of TWEAK in renal tubular proliferation (Fig. S1).
Figure 8.
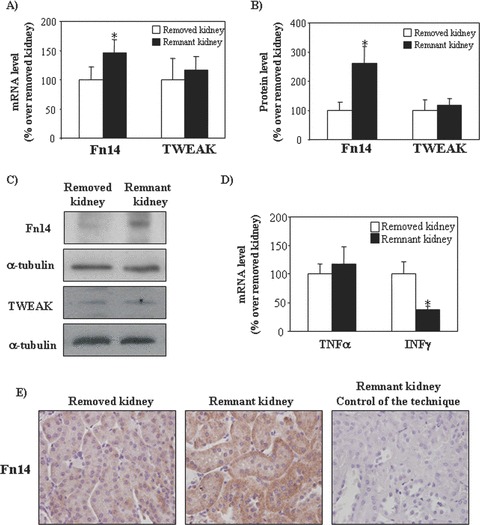
Expression of Fn14 and TWEAK in compensatory renal growth following uninephrectomy. (A) Total kidney Fn14 and TWEAK mRNA expression in TWEAK WT mice as measured by real time PCR. Mean (±S.E.M.) of seven animals. *P < 0.05 versus removed kidney. (B) Total kidney Fn14 and TWEAK protein expression in TWEAK WT mice as measured by Western blot. Mean (±S.E.M.) of 7 animals. *P < 0.05 versus removed kidney. (C) Representative Western blot showing Fn14 and TWEAK protein expression in removed and remnant kidneys following uninephrectomy. (D) No increase in renal TNF-α or IFN-γ mRNA expression in remnant kidneys, as assessed by quantitative PCR. In fact, IFN-γ mRNA was decreased. Mean (±S.E.M.) of seven animals. *P < 0.05. (E) Tubular cell localization of increased Fn14 expression in remnant kidney, when compared with removed kidney. Immunohistochemistry. Original magnification ×400.
Figure 9.
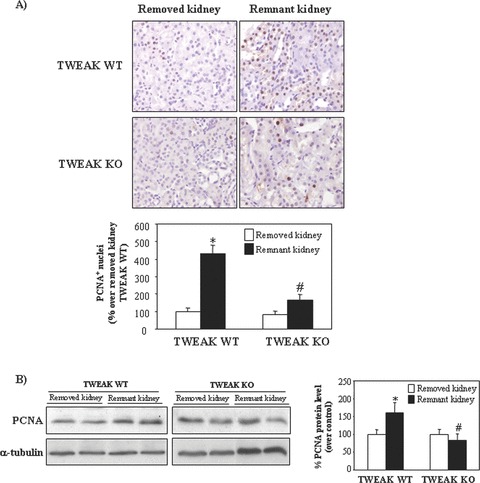
TWEAK deficiency decreases tubular proliferation in compensatory renal growth following uninephrectomy. (A) The increase in the number of PCNA+ nuclei is blunted in remnant kidneys from TWEAK KO as compared to TWEAK WT mice. Remnant kidneys studied 48 hrs following uninephrectomy. Original magnification ×400. Mean (±S.E.M.) of seven animals per group. *P < 0.0001 versus removed kidney of TWEAK WT mice; #P < 0.005 versus remnant kidney of TWEAK WT mice. (B) Whole kidney PCNA expression assessed by Western blot. Representative Western blot and quantification. Mean (±S.E.M.) n= 7. *P < 0.03 versus removed kidney of TWEAK WT mice; #P < 0.01 versus remnant kidney of TWEAK WT mice.
We next assessed the role of TWEAK on AKI. During AKI an initial wave of cell death is followed by tubular cell proliferation that leads to recovery (Fig. 10A). Renal inflammation is a feature of AKI. In particular, renal expression of mRNA for the inflammatory cytokines TNF-α (11-fold over healthy control, P < 0.05) and IFN-γ (2.5-fold over healthy control, P < 0.05) was increased in AKI induced by a folic acid overdose [15]. TWEAK KO mice had both a decreased early apoptosis peak (Fig. 10B–D), as well as a decreased late cell proliferation peak (Fig. 11A and B). Overall, renal function was better in TWEAK KO mice and there was no impact over functional recovery (Fig. 11C). These data are consistent with the pro-apoptotic action of TWEAK in an inflammatory milieu in cultured tubular cells [15]. Prophylactic treatment with anti-TWEAK antibodies [15] also decreased the rates of tubular cell apoptosis as assessed by TUNEL staining (by 55%, P < 0.05 versus control) and tubular cell proliferation, as assessed by PCNA staining (by 61%, P < 0.05 versus control) (data not shown).
Figure 10.
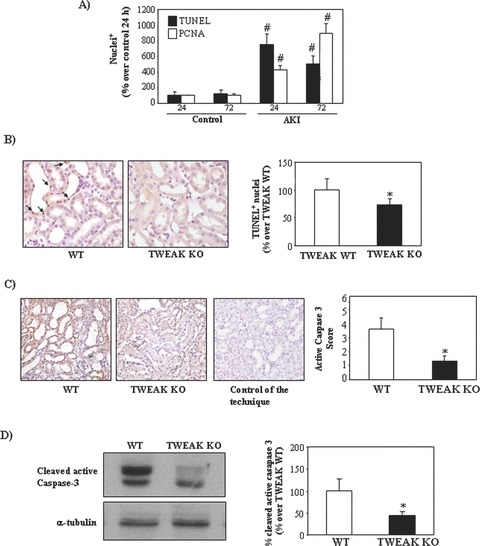
TWEAK neutralization decreases apoptosis in tubular epithelium during acute kidney injury (AKI). AKI is characterized by a local inflammatory milieu (15). (A) Time course of apoptosis, assessed by TUNEL and proliferation assessed as PCNA staining. Peak apoptosis precedes peak proliferation. Mean (±S.E.M.) of six animals per group at each time-point. *P < 0.005 versus control. (B) Apoptotic nuclei were stained by TUNEL. TWEAK deficiency decreases TUNEL+ nuclei 24 hrs following induction of AKI. Original magnification ×400. Mean of six animals per group, *P < 0.05 versus TWEAK WT. (C) Active caspase 3 was localized to tubular epithelium 24 hrs following induction of AKI and caspase 3 activation was decreased in TWEAK KO mice. Original magnification ×200. Mean (±S.E.M.) of 4 animals per group. *P < 0.05 versus TWEAK WT. (D) Whole kidney caspase 3 activation 24 hrs following induction of AKI assessed by Western blot of active caspase 3. Representative Western blot and quantification. *P < 0.05 versus TWEAK WT.
Figure 11.
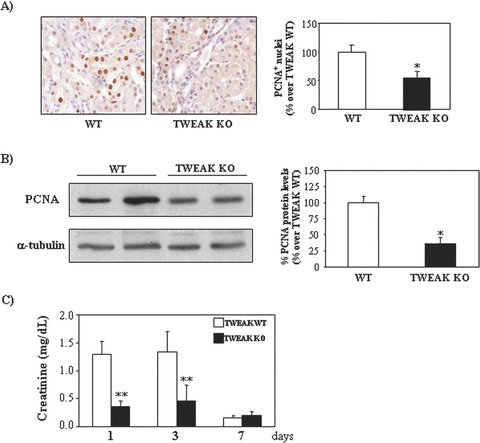
Lack of TWEAK decreases proliferation in tubular epithelium during acute kidney injury (AKI). (A) The number of PCNA+ nuclei is reduced in TWEAK KO mice at 72 hrs following induction of AKI. Original magnification ×400. Mean (±S.E.M.) of 6 animals per group.*P < 0.05 versus TWEAK WT. (B) Whole kidney PCNA expression assessed by Western blot. Representative Western blot and quantification. Mean (±S.E.M.) n= 6. *P < 0.009 versus control. (C) Time-course of serum creatinine in TWEAK KO or WT mice with AKI. Mean (±S.E.M.) of six animals per group at each time. **P < 0.001 versus TWEAK WT.
Discussion
TWEAK is a member of the TNF superfamily of cytokines that regulates apoptosis and inflammation in tubular cells [15, 17]. We now report that TWEAK also induces tubular cell proliferation, identify intracellular signalling cascades implicated in this action and show that the proliferative effect is increased in the presence of serum and lost in the presence of inflammation. Moreover, TWEAK-induced proliferation contributes to tubular hyperplasia following unilateral nephrectomy. By contrast, in the inflammatory milieu of AKI TWEAK targeting has anti-apoptotic actions that are associated with preservation of renal function without impairing recovery.
Although TWEAK was originally identified as an apoptosis-inducing factor, in quiescent tubular cells it stimulates cell proliferation. Other TNF family members such as FAS ligand or TNF can either induce apoptosis or proliferation depending upon experi mental conditions [29–31]. TWEAK actions on renal cells are modulated by the cell microenvironment [17]. The proliferative effect is enhanced in the presence of the mitotic factors from FBS. Serum increased both TWEAK and Fn14 expression and this may facilitate an increased proliferative response. Inflammatory cytokines also increase Fn14 expression [17] However, in their presence TWEAK potentiates cell death [17]. Because our studies identify the Fn14 receptor as the mediator of both the proliferative and the lethal potential of TWEAK [17] we have to hypothesize that serum and inflammatory cytokines may have opposite effects on other cell cycle regulatory proteins. In this regard, they have different effects on cell survival proteins. Thus, TNF increases Bax and SMAC/Diablo expression and down-regulates BclxL, while serum has opposite effects [21, 32].
TWEAK-induced proliferation is cell type-specific. TWEAK induces proliferation in endothelial cells, aortic smooth muscle cells, astrocytes, progenitor cells of the mesenchymal lineage, liver oval cells and human hepatocellular carcinomas but not in mature hepatocytes or dermal fibroblasts [11, 12, 33–35]. However, there is little information on the molecular pathways leading to TWEAK-mediated proliferation. As is the case for tubular cells, in endothelial cells TWEAK potentiates the mitogenic activity of other mitogens that increase Fn14 receptor expression, such as FGF-2 and VEGF-A. In endothelial cells TWEAK activated NF-κB, ERK1/2 and JNK1/2, but not p38 MAPK [35]. In addition TWEAK activated PI3K/Akt in osteoblastic cells [36] but inhibited insulin-induced Akt activation in hepatocytes [37]. The role of these pathways in TWEAK-induced cell proliferation was not explored. TWEAK induction of NF-κB activation in tubular cells has been extensively characterized in relation to its pro-inflammatory role [15]. In addition, in tubular cells ERK, p38 MAPK, PI3K/Akt and NF-κB cooperate in TWEAK-induced proliferation in a manner that disruption of one of them prevents proliferation.
The proliferative action of TWEAK was also observed in vivo in healthy kidneys from wild-type mice injected with exogenous TWEAK. In addition, TWEAK contributed to renal cell proliferation in compensatory renal growth following nephrectomy. This is a situation characterized by tubular cell proliferation in the absence of tubular injury or increased expression of inflammatory cytokines [2]. No increased expressions of TNF-α or IFN-γ, factors that in association with TWEAK promote tubular cell death [17] were noted in this model. This is in accordance with previous reports [28]. A decreased IFN-γ, expression by T cells had previously been observed following unilateral nephrectomy [38]. Increased renal Fn14 expression may be the facilitator event for tubular cell proliferation in compensatory renal growth following nephrectomy, sensitizing the cells to TWEAK and cooperating with other mitogens as observed in culture.
By contrast to the proliferative properties of TWEAK/Fn14 in cultured cells and kidneys in the absence of inflammation, TWEAK promotes death of cultured tubular cells when the microenvironment includes additional pro-inflammatory cytokines, such as TNF-α and IFN-γ, as characterized in detail by our group [17]. Consistent with the cell culture data, in an AKI model characterized by renal inflammation and increased local levels of TWEAK, Fn14, TNF-α and IFN-γ[15, 17], the lack of TWEAK decreased tubular cell apoptosis. In this model, as in other models of AKI, an initial wave of cell death is followed by tubular cell proliferation (Fig. 10A) that repopulates the tubules and promotes recovery. In this regard, TWEAK KO mice displayed a decreased peak of tubular cell apoptosis followed a decreased peak of proliferation. Based on: (i) the cell culture data that indicate that TWEAK promotes cell death in an inflammatory environment, (ii) the temporal relationship between cell death and proliferation in the AKI model, where cell death precedes proliferation and (iii) the beneficial effect of the absence of TWEAK for renal function during AKI, without compromise of functional recovery, we speculate that the absence of TWEAK decreased the early cell death peak, thus leading to a lesser need of repair proliferation during recovery. We cannot exclude some effect of TWEAK on tubular cell proliferation in this context. However, the initial effect of the lack of TWEAK on apoptosis appears to overwhelm any hypothetical deleterious effect of the lack of TWEAK on tubular cell proliferation that might impair recovery.
In conclusion, in tubular cells the actions of TWEAK depend on the cellular context. TWEAK has a proliferative effect on tubular epithelial cells cultured in a non-inflammatory environment, in healthy kidneys and in compensatory renal growth following nephrectomy, a model where the expression of inflammatory cytokines such as TNF-α and IFN-γ is low. By contrast, TWEAK promotes cell death in cultured tubular cells exposed to TNF-α and IFN-γ[17], as well as in AKI, an inflammatory model where there is a high local expression of TWEAK, TNF-α and IFN-γ[15]. This information is useful to design therapeutic strategies targeting TWEAK in renal injury.
Acknowledgments
We acknowledge the following grant support: FIS CP04/00060, 06/0046, SAF03/884, SAF2005–03378, EU QLG1-CT-2002–01215, Sociedad Española de Nefrologia, ISCIII-RETIC REDinREN/RD06/0016, Comunidad de Madrid/FRACM/S-BIO0283/2006, SAF 2007–60896. We also acknowledge the following salary support: FIS to P.J. and FIS and CAM to A.B.S., MEC to M.D.SN. and Programa Intensificación Actividad Investigadora (ISCIII/Agencia Laín-Entralgo/CM) to A.O. We thank Susana Carrasco for technical help.
Supporting Information
Fig. S1 TWEAK administration increases kidneyproliferation in remnant kidney following uninephrectomy.(A) Whole kidney PCNA expression assessed by Western blot.TWEAK was administered to wild-type mice following uninephrectomyand mice were killed 24 hrs later (48 hrs after nephrectomy).Representative Western blot and quantification. Mean(±S.E.M.) n = 6. * P < 0.005 versus removedkidney; # P < 0.005 versus remnant control. (B)haematoxylin and eosin staining. Note mitotic figures (hyperplasia)and increased cell size (hypertrophy) in remnant kidneys.Magnification ×400.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
References
- 1.Ortiz A, Justo P, Catalan MP, et al. Apoptotic cell death in renal injury: the rationale for intervention. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2:181–92. [PubMed] [Google Scholar]
- 2.Sun J, Langer WJ, Devish K, et al. Compensatory kidney growth in estrogen receptor-alpha null mice. Am J Physiol Renal Physiol. 2006;290:F319–23. doi: 10.1152/ajprenal.00271.2005. [DOI] [PubMed] [Google Scholar]
- 3.Flyvbjerg A, Bennett WF, Rasch R, et al. Compensatory renal growth in uninephrectomized adult mice is growth hormone dependent. Kidney Int. 1999;56:2048–54. doi: 10.1046/j.1523-1755.1999.00776.x. [DOI] [PubMed] [Google Scholar]
- 4.Webber EM, Bruix J, Pierce RH, et al. Tumor necrosis factor primes hepatocytes for DNA replication in the rat. Hepatology. 1998;28:1226–34. doi: 10.1002/hep.510280509. [DOI] [PubMed] [Google Scholar]
- 5.Yamada Y, Webber EM, Kirillova I, et al. Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: requirement for type 1 but not type 2 receptor. Hepatology. 1998;28:959–70. doi: 10.1002/hep.510280410. [DOI] [PubMed] [Google Scholar]
- 6.Chicheportiche Y, Bourdon PR, Xu H, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem. 1997;272:32401–10. doi: 10.1074/jbc.272.51.32401. [DOI] [PubMed] [Google Scholar]
- 7.Sanz AB, Moreno JA, Sanchez-Nino MD, et al. TWEAKing renal injury. Front Biosci. 2008;13:580–9. doi: 10.2741/2703. [DOI] [PubMed] [Google Scholar]
- 8.Campbell S, Michaelson J, Burkly L, et al. The role of TWEAK/Fn14 in the pathogenesis of inflammation and systemic autoimmunity. Front Biosci. 2004;9:2273–84. doi: 10.2741/1395. [DOI] [PubMed] [Google Scholar]
- 9.Harada N, Nakayama M, Nakano H, et al. Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2002;299:488–93. doi: 10.1016/s0006-291x(02)02670-0. [DOI] [PubMed] [Google Scholar]
- 10.Tran NL, McDonough WS, Savitch BA, et al. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFkappaB pathway activation and BCL-XL/BCL-W expression. J Biol Chem. 2005;280:3483–92. doi: 10.1074/jbc.M409906200. [DOI] [PubMed] [Google Scholar]
- 11.Jakubowski A, Ambrose C, Parr M, et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest. 2005;115:2330–40. doi: 10.1172/JCI23486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girgenrath M, Weng S, Kostek CA, et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J. 2006;25:5826–39. doi: 10.1038/sj.emboj.7601441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiley SR, Winkles JA. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 2003;14:241–9. doi: 10.1016/s1359-6101(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Z, Burkly LC, Campbell S, et al. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol. 2007;179:7949–58. doi: 10.4049/jimmunol.179.11.7949. [DOI] [PubMed] [Google Scholar]
- 15.Sanz AB, Justo P, Sanchez-Nino MD, et al. The cytokine TWEAK modulates renal tubulointerstitial inflammation. J Am Soc Nephrol. 2008;19:695–703. doi: 10.1681/ASN.2007050577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meighan-Mantha RL, Hsu DK, Guo Y, et al. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem. 1999;274:33166–76. doi: 10.1074/jbc.274.46.33166. [DOI] [PubMed] [Google Scholar]
- 17.Justo P, Sanz AB, Sanchez-Nino MD, et al. Cytokine cooperation in renal tubular cell injury: the role of TWEAK. Kidney Int. 2006;70:1750–8. doi: 10.1038/sj.ki.5001866. [DOI] [PubMed] [Google Scholar]
- 18.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–39. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 19.Mulloy R, Salinas S, Philips A, et al. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene. 2003;22:5387–98. doi: 10.1038/sj.onc.1206839. [DOI] [PubMed] [Google Scholar]
- 20.Haverty TP, Kelly CJ, Hines WH, et al. Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J Cell Biol. 1988;107:1359–68. doi: 10.1083/jcb.107.4.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ortiz A, Lorz C, Catalan MP, et al. Expression of apoptosis regulatory proteins in tubular epithelium stressed in culture or following acute renal failure. Kidney Int. 2000;57:969–81. doi: 10.1046/j.1523-1755.2000.00925.x. [DOI] [PubMed] [Google Scholar]
- 22.Campbell S, Burkly LC, Gao HX, et al. Proinflammatory effects of TWEAK/Fn14 interactions in glomerular mesangial cells. J Immunol. 2006;76:1889–98. doi: 10.4049/jimmunol.176.3.1889. [DOI] [PubMed] [Google Scholar]
- 23.Bover LC, Cardo-Vila M, Kuniyasu A, et al. A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol. 2007;178:8183–94. doi: 10.4049/jimmunol.178.12.8183. [DOI] [PubMed] [Google Scholar]
- 24.Baxter FO, Came PJ, Abell K, et al. IKKbeta/2 induces TWEAK and apoptosis in mammary epithelial cells. Development. 2006;133:3485–94. doi: 10.1242/dev.02502. [DOI] [PubMed] [Google Scholar]
- 25.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 26.Firestein GS, Manning AM. Signal transduction and transcription factors in rheumatic disease. Arthritis Rheum. 1999;42:609–21. doi: 10.1002/1529-0131(199904)42:4<609::AID-ANR3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 27.Reddy SA, Huang JH, Liao WS. Phosphatidylinositol 3-kinase in interleukin 1 signaling. Physical interaction with the interleukin 1 receptor and requirement in NFkappaB and AP-1 activation. J Biol Chem. 1997;272:29167–73. doi: 10.1074/jbc.272.46.29167. [DOI] [PubMed] [Google Scholar]
- 28.Higashitsuji H, Arii S, Furutani M, et al. Expression of cytokine genes during liver regeneration after partial hepatectomy in rats. J Surg Res. 1995;58:267–74. doi: 10.1006/jsre.1995.1042. [DOI] [PubMed] [Google Scholar]
- 29.Alderson MR, Armitage RJ, Maraskovsky E, et al. Fas transduces activation signals in normal human T lymphocytes. J Exp Med. 1993;178:2231–5. doi: 10.1084/jem.178.6.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearl-Yafe M, Stein J, Yolcu ES, et al. Fas transduces dual apoptotic and trophic signals in hematopoietic progenitors. Stem Cells. 2007;25:3194–203. doi: 10.1634/stemcells.2007-0402. [DOI] [PubMed] [Google Scholar]
- 31.Mitsiades CS, Poulaki V, Fanourakis G, et al. Fas signaling in thyroid carcinomas is diverted from apoptosis to proliferation. Clin Cancer Res. 2006;12:3705–12. doi: 10.1158/1078-0432.CCR-05-2493. [DOI] [PubMed] [Google Scholar]
- 32.Justo P, Sanz A, Lorz C, et al. Expression of Smac/Diablo in tubular epithelial cells and during acute renal failure. Kidney Int Suppl. 2003:S52–6. doi: 10.1046/j.1523-1755.64.s86.10.x. [DOI] [PubMed] [Google Scholar]
- 33.Desplat-Jego S, Varriale S, Creidy R, et al. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J Neuroimmunol. 2002;133:116–23. doi: 10.1016/s0165-5728(02)00368-5. [DOI] [PubMed] [Google Scholar]
- 34.Lynch CN, Wang YC, Lund JK, et al. TWEAK induces angiogenesis and proliferation of endothelial cells. J Biol Chem. 1999;274:8455–9. doi: 10.1074/jbc.274.13.8455. [DOI] [PubMed] [Google Scholar]
- 35.Donohue PJ, Richards CM, Brown SA, et al. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arterioscler Thromb Vasc Biol. 2003;23:594–600. doi: 10.1161/01.ATV.0000062883.93715.37. [DOI] [PubMed] [Google Scholar]
- 36.Ando T, Ichikawa J, Wako M, et al. TWEAK/Fn14 interaction regulates RANTES production, BMP-2-induced differentiation, and RANKL expression in mouse osteoblastic MC3T3-E1 cells. Arthritis Res Ther. 2006;8:R146. doi: 10.1186/ar2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng F, Wang L, Albanese N, et al. TWEAK attenuates the action of insulin in hepatocytes. Endocrinology. 2008;149:1505–13. doi: 10.1210/en.2007-1119. [DOI] [PubMed] [Google Scholar]
- 38.Stalder M, Birsan T, Hausen B, et al. Immunosuppressive effects of surgery assessed by flow cytometry in nonhuman primates after nephrectomy. Transpl Int. 2005;18:1158–65. doi: 10.1111/j.1432-2277.2005.00119.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item