

Abstract
We have previously shown that familial Alzheimer’s disease mutants of presenilin-2 (PS2) and, to a lesser extent, of presenilin-1 (PS1) lower the Ca2+ concentration of intracellular stores. We here examined the mechanism by which wild-type and mutant PS2 affect store Ca2+ handling. By using HeLa, SH-SY5Y and MEFs as model cells, and recombinant aequorins as Ca2+ probes, we show evidence that transient expression of either wild-type or mutant PS2 increases the passive Ca2+ leakage: both ryanodine- and IP3-receptors contribute to Ca2+ exit out of the ER, whereas the ribosome translocon complex is not involved. In SH-SY5Y cells and MEFs, wild-type and mutant PS2 potently reduce the uptake of Ca2+ inside the stores, an effect that can be counteracted by over-expression of SERCA-2B. On this line, in wild-type MEFs, lowering the endogenous level of PS2 by RNA interference, increases the Ca2+-loading capability of intracellular stores. Furthermore, we show that in PS double knockout MEFs, reduction of Ca2+ stores is mimicked by the expression of PS2-D366A, a loss-of-function mutant, uncleaved because also devoid of presenilinase activity but not by co-expression of the two catalytic active fragments of PS2. In summary, both physiological and increased levels of wild-type and mutant PS2 reduce the Ca2+ uptake by intracellular stores. To exert this newly described function, PS2 needs to be in its full-length form, even if it can subsequently be cleaved.
Keywords: presenilin, calcium stores, Alzheimer’s disease, SERCA, aequorin
Introduction
Alzheimer’s disease (AD) is responsible for the most part of the dementias in developed countries. Although the majority of AD cases are sporadic, a significant fraction of AD is inherited in a dominant pattern. The familial forms of AD (FAD) have been traced to mutations in genes for three proteins: the amyloid precursor protein (APP), presenilin-1 and -2 (PS1 and 2). PSs are essential components of the γ-secretase complex which, in turn, by cleaving APP in concert with β-secretase, produces the neurotoxic β-amyloid peptide (Aβ).
Much attention has recently been devoted to the fact that some FAD-linked PS mutants cause a dysregulation of cellular Ca2+ homeostasis. Ca2+ is a key parameter in neuronal physiopathology, as it controls many cellular functions, whereas alterations in Ca2+ levels are responsible for neuronal cell death in a number of genetic and sporadic diseases. It has been suggested that an imbalance of Ca2+ homeostasis may represent an early event in the pathogenesis of FAD, but the mechanisms through which FAD-linked PS mutants affect the control of cellular Ca2+ are controversial. In particular, it was observed that mutations in PS1 cause larger Ca2+ release from intracellular stores and increase excitotoxicity in neurons from transgenic mice [1–5]. The idea that FAD-linked PS mutations are somehow correlated to altered Ca2+ signalling was further supported by the fact that these mutations could alter the expression, or sensitivity, of endoplasmic reticulum (ER) Ca2+ release channels, ryanodine receptors (RyRs) and inositol (1,4,5)-trisphosphate receptors (IP3Rs) in different cell models [2, 6–11] and in neurons from AD mice [5, 12, 13]. Interestingly, similar observations were also made in non-neuronal cells, such as fibroblasts, lymphocytes and oocytes, indicating that PSs play a general role in Ca2+ homeostasis [11, 14]. These data lead to the ‘Ca2+ overload’ hypothesis [15, 16], stating that AD neuronal degeneration depends on exaggerated ER Ca2+ release because of ER Ca2+ overload. However, an increased ER Ca2+ content has not always been observed: different studies have described either no alteration or reduced ER Ca2+ stores in cells expressing wild-type (wt) or FAD-mutant PSs [11, 17–19]. In particular, we demonstrated that the FAD-linked PS2 mutations M239I and T122R reduce rather than increase Ca2+ release in fibroblasts from FAD patients and in cell lines stably or transiently expressing the PS2 mutants [18, 20, 21]. In addition, an extended investigation of other FAD-linked PS mutants (PS2−N141I, PS1-A246E, PS1-L286V, PS1-M146L, PS1-P117L), by monitoring directly the ER Ca2+ levels in different cell lines, confirmed that these FAD-linked PS2 mutations caused a reduction in ER Ca2+ levels and none of the PS1 mutations caused an increase [18].
Recently, a physiological role for wt PSs in ER Ca2+ handling has also been proposed, although divergent and contrasting data were reported [11, 18, 22–25]. In agreement, different mechanisms of PSs action have been proposed to explain their effect on Ca2+ homeostasis. PSs were demonstrated to form Ca2+ permeable channels in planar lipid bilayers [23], with FAD-linked PS mutants forming channels with reduced ionic conductance and thus being responsible for ER Ca2+ overload [23, 26]. An increased Ca2+ content has been also explained by an increased sarco-endoplasmic reticulum calcium-ATPase (SERCA)-2B activity, as deduced by accelerated cytosolic Ca2+ clearance following expression of wt PS1/2 or PS1−M146V in Xenopus laevis oocytes [25]. In contrast, Cheung et al. demonstrated that different FAD PS mutants increase IP3Rs’ sensitivity, leading to exaggerated Ca2+ responses, yet in the presence of reduced ER Ca2+ levels [11].
By employing different cell models, we here show that wt and mutant PS2 act primarily by reducing SERCA pumps’ activity and secondly by increasing the leak across ER Ca2+ channels (RyRs and IP3Rs).
Materials and methods
Cell lines and transfection
HeLa and SH-SY5Y cells were grown in DMEM supplemented with 10% FCS containing penicillin (100 U/ml) and streptomycin (100 μg/ml). Before transfection, cells were seeded on cover slips (13-mm-diameter) and allowed to grow to 50% confluence. At this stage, transfections of HeLa cells were carried out using the Ca2+-phosphate technique in the presence of 4 μg of DNA (3 μg PS2-cDNA or void vector plus 1 μg aequorin [Aeq] cDNA]. SH−SY5Y cells were transfected by means of Lipofectamine™ 2000 using 1.5 μg of DNA (1 μg PS2-cDNA or void vector plus 0.5 μg Aeq cDNA). Intracellular Ca2+ measurements were carried out 48 or 24 hrs after transfection by means of the Aeq technique as previously described [27] and summarized below. PS1/PS2-null (PS1−/−, PS2−/−) and wild-type mouse embryonic fibroblasts (MEFs), obtained as previously described [28, 29], were kindly provided by Dr. Bart De Strooper (Center for Human Genetics, KUL, VIB, Leuven, Belgium). Cells, grown in DMEM-F12 supplemented with 10% FCS and 100 U/ml penicillin/streptomycin, were transfected by Lipofectamine™ 2000 employing 2 μg of DNA (1.5 μg PS2-cDNA or void vector plus 0.5 μg Aeq cDNA). For γ-secretase activity assay [30] cells were transfected with 1.5 μg of PS2-cDNA and 0.5 μg of C99-GFP instead of Aeq. For RNA interference experiments, the growth medium was substituted 1 hr before transfection with antibiotics-free medium; siRNAs (murine IP3R-1 [GenBank accession no. NM-010585; nucleotides 505–523, 2254–2272, 3680–3698 and 5122–5140] and IP3R-3 [GenBank accession no. NM-080553; nucleotides 1114–1132, 1125–1143, 1219–1237 and 1459–1477]; mouse PS2, target sequence: GAUAUACUCAUCUGCCAUG; siGENOME RISC-Free Control siRNA; Dharmacon Research, Lafayette, CO) were added to the transfection mixes to a final concentration of 20–40 nM.
Ca2+ measurements
Cells seeded on 13-mm-diameter cover slips and transfected with Aeq constructs were incubated at 37°C with coelenterazine (5 μM) for 1–2 hrs in a modified Krebs–Ringer buffer (mKRB, in mM:140 NaCl, 2.8 KCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 11 glucose, pH 7.4) and then transferred to the perfusion chamber. For reconstitution of ER-Aeq, luminal [Ca2+] was reduced before coelenterazine addition by exposing the cells to CPA (20 μM) in mKRB without CaCl2 (Ca2+-free mKRB) and containing EGTA (600 μM). Upon 1-hr incubation at 4°C in the same medium, the cells were extensively washed with Ca2+-free mKRB supplemented with EGTA (1 mM) and bovine serum albumin (BSA, 2%). All the luminescence measurements were carried out in mKRB at 37°C. For SH-SY5Y and MEF cells, a high potassium medium (in mM: KCl 100, NaCl 43, MgCl2 1, HEPES 10, pH 7.4) was used. The experiments were terminated by cell permeabilization with digitonin (100 μM) in a hypotonic Ca2+-rich solution (10 mM CaCl2 in H2O) to discharge the remaining unused Aeq pool. The light signal was collected as previously described [27].
For permeabilization, cells were exposed for 1–2 min. to digitonin (20–100 μM) in an intracellular medium containing (in mM): KCl 130, NaCl 10, KH2PO4 1, succinic acid 2, MgSO4 1, HEPES 20, EGTA 0.05 pH 7, at 37°C. The cells were then washed with the same intracellular medium containing EGTA 50 μM for 2–5 min. The Ca2+-buffer solution was prepared by adding to the intracellular medium: HEDTA, piruvic acid and MgCl2 (1 mM each), EGTA or BAPTA (2 mM) and CaCl2 at different concentrations (0.5−1.8 mM) and the pH was brought to 7 at 37°C. ATPNa2 (0.2 mM) was added to this Ca2+-buffered solution. The free [Ca2+] (0.1–2 μM) was estimated by MaxChelator2.5 and checked by fluorimetric measurements with fura-2.
Plasmids
pcDNA3 vectors, codifying different PS2 mutants (M239I, N141I, T122R, D366A) were created by site-directed mutagenesis of pcDNA3/PS2-wt (QuikChange Site-directed mutagenesis Kit, Stratagene, La Jolla, CA). The constructs were checked by sequence analysis (ABI Prism Genetic Analyzer 310, Applied Biosystems, Monza, Italy).
Protein extracts preparation and Western blot analysis
The different cell types were harvested and treated as previously reported [21]. Briefly, cells were washed twice with ice-cold phosphate-buffered saline (PBS) and harvested with RIPA buffer supplemented with proteases inhibitors cocktail (Complete Mini™, Roche, Basel, Schweiz). Samples were analysed in SDS−PAGE gel, and Western blotting immunodetection was carried out with the polyclonal antibody anti-PS2 (324–335; Ab-2, Calbiochem, Merck, Darmstadt, Germany) and with the monoclonal mouse antibody anti-PS2 (MMS-359S, Covance Research Products Inc., Princeton, NJ, USA). IP3R-1 and -3 were detected by a polyclonal antibody (PA3–901A, ABR-Affinity BioReagents, Inc., Golden, CO, USA) and a monoclonal mouse antibody (610312, BD Biosciences Pharmingen, San Jose, CA, USA), respectively. SERCA-2B detection was carried out with a polyclonal anti-SERCA-2 antibody (N-19, Santa Cruz Biotechnology, Inc.). AICD-GFP was detected by the polyclonal anti-GFP antibody ab290, purchased from abCAM (Cambridge Science Park, Cambridge, UK). Actin was detected by the monoclonal mouse antibody (A4700, Sigma-Aldrich, St. Louis, MO, USA) The proteins were visualized by the chemiluminescence reagent ECL (Amersham, GE Healthcare, UK Ltd., Amersham Place, Little Chalfont, Buckinghamshire, UK).
Chemicals and reagents
Antibiotics, sera, culture media, plasmids and Lipofectamine™ 2000 were purchased from Invitrogen (Carlsbad, CA); DAPT and MW167 (γ-secretase inhibitor II) were from Calbiochem (Merck KGaA; Darmstadt, Germany), whereas all other reagents were from Sigma Chemical Co. (St. Louis, MO), unless otherwise stated.
Statistical analysis
Data were analysed by Origin 7.5 SR5 (OriginLab Corporation, Northampton/Wellesley Hills, MA, USA). Averages are expressed as mean ± S.E.M. (n= number of independent experiments; *=P < 0.05, **=P < 0.01, ***=P < 0.001, unpaired Student’s t-test).
Results
Effect of PS2 variants on store calcium leak
Using cytosolic and organelle-targeted Aeq, we have previously demonstrated that expression of various PSs, especially the PS2 variants, reduces the steady-state free Ca2+ concentration of the endoplasmic reticulum ([Ca2+]ER) and Golgi apparatus ([Ca2+]GA), the major intracellular Ca2+ stores of mammalian cells [18]. For those experiments, we used primarily HeLa cells, a convenient and extensively used cell model. In the work here presented, we carried out the same type of measurements in two other cell types, the SH-SY5Y cell line, derived from a human neuroblastoma, and mouse embryonic fibroblasts (MEFs). The latter cell model offers the possibility that a PS double knockout (DKO) clone is also available [31].
SH-SY5Y cells were cotransfected with the cDNAs coding for a recombinant Aeq targeted to the endoplasmic reticulum (ER-Aeq) and for PS2-T122R, a FAD-linked mutant PS2 whose effect at the Ca2+ store level was originally described in human FAD fibroblasts and HeLa cells [18, 21]; control cells were transfected with ER-Aeq and vector alone (pcDNA3). Twenty-four to 48 hrs after transfection, Ca2+ stores were depleted in a Ca2+-free, EGTA-containing medium to allow ER-Aeq reconstitution (see Materials and Methods) and subsequently the refilling process was continuously monitored upon addition of CaCl2 (1 mM) to the bathing medium. Under these conditions, the [Ca2+]ER increased in a couple of minutes up to a plateau that stabilized at a significant lower level in PS2−T122R–expressing cells, with respect to control cells (Fig. 1A). Table 1 reports the steady-state [Ca2+]ER obtained with this protocol in all the cell types here investigated: HeLa, SH-SY5Y, wt and DKO MEFs. Note that PS2-T122R was maximally effective in SH-SY5Y cells (–53 ± 3%, mean ± S.E.M., n= 29).
Figure 1.
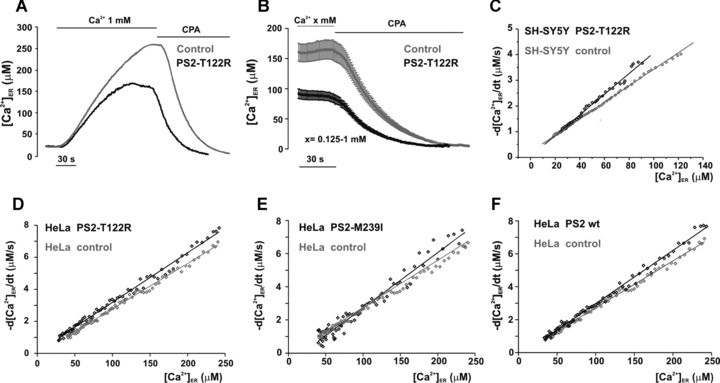
Effect of PS2 variants on passive ER Ca2+ leak. (A) Representative traces of [Ca2+]ER measurements in SH-SY5Y cells transiently cotransfected with the cDNAs coding for ER-Aeq and PS2-T122R (black) or with the void vector as control (grey). After Aeq reconstitution (see Materials and Methods), cells were washed and bathed in a Ca2+-free, EGTA (0.6 mM)-containing medium before exposure to CaCl2 (1 mM). The passive ER Ca2+ leak was estimated by addition, at the plateau, of CPA (20 μM) together with EGTA (1 mM). (B) For quantitative analysis of ER Ca2+ leak, different steady-states of [Ca2+]ER were obtained by the addition of CaCl2 ranging from 0.125 to 1 mM. The single traces were averaged and aligned to CPA addition (grey and black traces for control and PS2-T122R–expressing cells, respectively, mean ± S.E.M., n= 24). (C) The rate of ER Ca2+ loss (-d[Ca2+]/dt) was plotted as a function of the instantaneous [Ca2+]ER estimated from single traces as shown in (B) (black and grey symbols, for PS2-T122R-expressing and control cells, respectively). The S.E.M. was omitted for clarity. (D–F) Experiments with HeLa cells were carried out as described in (A) and analysed as shown in (B) and (C) (black and grey symbols for PS2-expressing and control cells, respectively, n= 6–9).
Table 1.
ER Ca2+ levels (μM)
| Cell type | Control | n | PS2-T122R | n | % change |
|---|---|---|---|---|---|
| HeLa | 414.4 ± 12.8 | 54 | 336.1 ± 11.5* | 30 | −19 |
| SH-SY5Y | 298.2 ± 9.6 | 28 | 140.5 ± 8.0*** | 29 | −53 |
| wt MEFs | 310.7 ± 27.0 | 27 | 180.0 ± 21.8*** | 9 | −42 |
| DKO MEFs | 306.5 ± 27.4 | 11 | 211.6 ± 16.1*** | 10 | −31 |
P < 0.05
P < 0.01
P < 0.001.
The question then arises as to the molecular mechanisms leading to this reduced steady-state [Ca2+]ER. A first possibility is that it is due to an increased Ca2+ leak out of the stores. The passive ER Ca2+ leak rate was thus measured directly using a previously described procedure [32]. Briefly, cells, cotransfected with the cDNAs coding for ER-Aeq and PS2-T122R (or the void vector), were first allowed to refill their emptied stores until they reached a steady-state. In the typical experiment with SH−SY5Y cells, shown in Fig. 1B, a wide range of external Ca2+ concentrations (0.125–0.25–0.5–1 mM) was used in order to obtain different levels of the steady-state [Ca2+]ER before leak measurement; typically, three cover slips, for both control and mutant PS2–expressing cells, were used at each Ca2+ concentration. At the plateau, the SERCA inhibitor cyclopiazonic acid, CPA (20 μM), was added in a Ca2+-free medium containing EGTA (1 mM) to remove extracellular Ca2+ and the rate of [Ca2+]ER decay was continuously monitored (Fig. 1A and B). The single traces were averaged and aligned to CPA addition (grey and black traces for control and PS2-T122R–expressing cells, respectively). Figure 1C shows the average rate of Ca2+ leakage (-d[Ca2+]ER/dt) as a function of the instantaneous [Ca2+]ER, estimated from the traces shown in Fig. 1B (grey and black symbols, for control and PS2-T122R–expressing cells, respectively). In cells expressing the mutant PS2, at [Ca2+]ER above 40 μM, the decay rates are significantly higher than those of controls. The linear fit of the experimental data shown in Fig. 1C thus shows a significantly greater slope for PS2-T122R–expressing cells compared with control ones, being respectively (s−1) 0.040 ± 0.003 and 0.032 ± 0.002 (n= 24, P < 0.001). The percentage increase in slope, averaged among different cell batches, was 17.1 ± 6.3% (n= 7). When similar experiments were carried out in HeLa cells (Fig. 1D–F), the estimated slopes were (s−1) 0.031 ± 0.3, 0.035 ± 0.001, 0.031 ± 0.004 and 0.028 ± 0.003 for cells transfected, respectively, with the cDNA coding for PS2-T122R, PS2-M239I, wt PS2 and the void vector. Compared with SH−SY5Y cells, expression of PS2-T122R in HeLa cells resulted in a smaller increase in slope (9.1 ± 2.3%, n= 3) (see also Table 1 and [18]).
It has to be mentioned that in control, void-vector transfected SH-SY5Y and HeLa cells (Figs 1C–F), the absolute values of the decay rates were rather similar for the same ER Ca2+ level, that is, around 2.8−3 μM/sec. at 100 μM [Ca2+]ER for the two cell types. Indeed, in control cells, ER leak rates covered higher ranges in HeLa (0−12 μM/sec.) than in SH-SY5Y (0−6 μM/sec.) being the latter cell type characterized by a lower maximal steady-state ER Ca2+ level (see Table 1).
The rate of [Ca2+]ER decay in intact cells can be influenced by factors other than the intrinsic ER Ca2+ leak, for example, the rate of Ca2+ extrusion across the plasma membrane or the rate of Ca2+ sequestration by mitochondria or other organelles. We thus investigated whether the reduction of the steady-state ER Ca2+ level, induced by transient expression of mutant PS2, could also be observed in digitonin permeabilized cells with the free [Ca2+] of the bathing solution ([Ca2+]o) buffered at a constant level by an EGTA-based buffer (see Materials and Methods). Under these conditions, SH−SY5Y cells cotransfected with the cDNAs coding for ER-Aeq and PS2-T122R showed a reduction of the [Ca2+]ER (compared with controls) similar to that found in intact cells: 53 and 47%, respectively, in intact and permeabilized SH−SY5Y cells (Fig. 2A). Similar results were also obtained in DKO MEFs (Fig. 2B). The capability of PS2-T122R to reduce the [Ca2+]ER level upon cell treatment with digitonin (20−100 μM, 1–2 min.) was confirmed also in an SH-SY5Y clone stably expressing PS2−T122R. The estimated [Ca2+]ER reductions, compared with the void-vector transfect clone, were 25.8 ± 2.9% (n= 20, P < 0.01) and 28.1 ± 5.4% (n= 22, P < 0.01), respectively, in intact and permeabilized cells.
Figure 2.
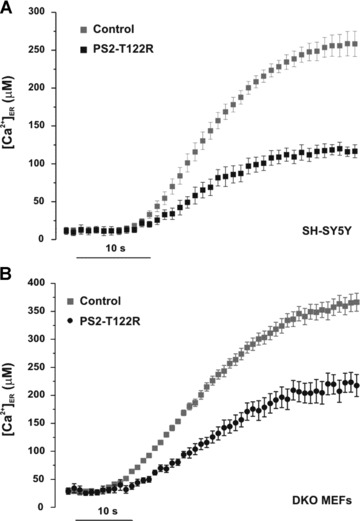
Effect of PS2-T122R on ER Ca2+ uptake. SH-SY5Y cells (A) and DKO MEFs (B) were transiently cotransfected with the cDNAs coding for ER-Aeq and PS2-T122R (black) or with the void vector as control (grey). Upon Aeq reconstitution (see Materials and Methods), the cells were washed and bathed in a Ca2+-free, EGTA (0.6 mM)-containing medium. The cells were then permeabilized for 2 min. with digitonin (20 μM) in an EGTA-containing intracellular solution. Upon washing, ER Ca2+ uptake was followed in the same solution containing an EGTA-based buffer (free [Ca2+]= 0.3 μM) and ATP (0.2 mM) (see Materials and Methods). Traces were aligned to Ca2+ addition, black and grey symbols for PS2-T122R expressing and control cells, respectively (mean ± S.E.M., n= 12 for SH-SY5Y and n= 16 for DKO MEFs).
PS2-T122R effect on ER Ca2+ channels
It has been suggested that, in PC12 cells and in cortical neurons, PS2-N141I (as well as PS1 mutations) alters Ca2+ homeostasis by interfering with ryanodine receptors (RyRs) [2, 9, 10]. Furthermore, it has been shown that, in triple transgenic mice, the knock-in of PS1-M146V up-regulates the expression of RyRs in the brain [5, 13] and in single-channel activity experiments, the cytosolic N-terminus of PS1 or PS2 potentiates mouse RyR open probability [33, 34]. We employed a pharmacological approach to investigate whether RyRs could be possibly involved in the effect of PS2-T122R on ER Ca2+ handling. In intact SH-SY5Y cells, an overnight treatment with the classical RyR inhibitor dantrolene (20 μM) caused only a minor reduction of the PS2-T122R effect. In fact, when the Ca2+ store content was estimated by cyt-Aeq following the addition of CPA (20 μM) plus bradykinin (Bk, 100 nM) (Fig. 3A), a small but significant recovery of the reduction caused by PS2-T122R was observed (+12%, P < 0.05) (Fig. 3B). A similar trend to a recovery, albeit not statistically significant (+5%), was found while monitoring the steady-state ER Ca2+ level in dantrolene-treated cells (data not shown). Consistently with the modest effect found upon RyR inhibition, the process of Ca2+-induced Ca2+ release seemed not to play a dominant role, since in permeabilized SH-SY5Y cells perfused with an intracellular solution containing BAPTA (a Ca2+ chelator acting faster than EGTA) the effect of PS2-T122R on the [Ca2+]ER was similar, being the estimated reductions of ER Ca2+ plateaus 39 ± 3% and 34 ± 3% (mean ± S.E.M., n= 3), respectively, with BAPTA- and EGTA-based buffers (see Materials and Methods).
Figure 3.
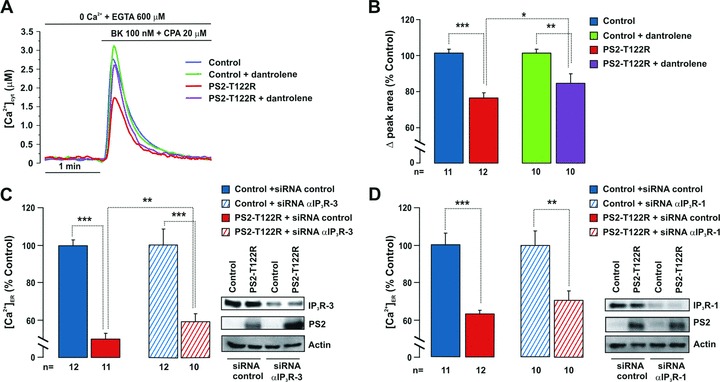
Role of RyRs and IP3Rs. (A) SH-SY5Y cells, transiently cotransfected with the cDNAs coding for cyt-Aeq and PS2-T122R (red trace), or with the void vector, control (blue trace), were overnight treated with dantrolene (20 μM) and continuously maintained in the presence of the drug (purple and green traces, respectively). Upon Aeq reconstitution (see Materials and Methods), the cells were washed in mKRB and then exposed to CPA (20 μM) plus bradykinin (Bk, 100 nM) in a Ca2+-free, EGTA (0.6 mM)-containing medium to fully discharge the intracellular Ca2+ stores. (B) Bars represent the average peak area measured above the baseline and expressed as percentage of control, void-vector transfected cells (mean ± S.E.M.). (C, D) wt MEFs were cotransfected with the cDNAs coding for ER-Aeq and PS2-T122R (or with the void vector as control) and siRNA specific for mouse IP3R-3 (40 nM) (C), IP3R-1 (20 nM) (D) or equivalent amounts of control siRNA (C, D). After 48 hrs, cells were harvested and probed for expression levels of PS2 and IP3Rs by Western blotting (right) or were employed to measure steady-state ER Ca2+ levels (left), according to the protocol described in Fig. 1A. Bars represent the average [Ca2+]ER expressed as percentage of control cells transfected with the void-vector and control siRNA (mean ± S.E.M.). Note interruption in the Y axes in (B–D).
IP3Rs are the other class of Ca2+ release channels that have also been involved in the modulation of Ca2+ handling by PSs [4, 7, 11, 12, 22]. When the steady-state [Ca2+]ER was monitored in permeabilized SH-SY5Y cells, transfected with ER-Aeq and PS2-T122R, addition of the IP3R antagonist heparin (100–200 μg/ml) had no effect or caused only a marginal recovery. On average, the reduction in the steady-state was maintained (40 ± 1.4% and 41 ± 6%, mean ± S.E.M., n= 5, respectively, with and without heparin). The other IP3R inhibitor 2−aminoethoxydiphenyl borate (2−APB) could not be used because it interfered with the ER-Aeq–based detection system already at 20 μM, a concentration smaller than that usually employed to block the receptor (unpublished data). In MEFs that, at variance with SH-SY5Y, express high levels of IP3R-3 in addition to IP3R-1 [22, 35], knocking down IP3R-3 by a specific siRNA, partially rescued (+10%, P < 0.01) the ER Ca2+ defect caused by expression of PS2-T122R in the same interfered cells (Fig. 3C); a siRNA specific for the type 1 IP3R was ineffective (Fig. 3D). Although siRNAs to both IP3Rs significantly and potently reduced the specific receptor level, no additional rescue was found (data not shown).
We also investigated another possible ER exit pathway for Ca2+ ions. It has recently been suggested that the protein import machinery across the ER membrane could be a relevant Ca2+o leak pathway. In particular, it has been shown that the protein import machinery, the so-called ribosomal–translocon complex (RTC), once freed of the newly synthesized protein, can allow the flux of Ca2+ ions and small sugars [36–38].
When SH-SY5Y cells were maintained in the presence of puromycin (200 μM), following a 10-min. pre-incubation at 37°C, a protocol sufficient to release nascent proteins and leave the RTC in an open configuration [36], the steady-state [Ca2+]ER was unaffected both in control and PS2-T122R–expressing cells (Fig. 4A). When a similar treatment was also employed in control SH-SY5Y cells expressing cyt-Aeq, the rate of store Ca2+ efflux (d[Ca2+]cyt/dt) induced by CPA addition was of the same magnitude with or without puromycin treatment (9.6 ± 0.5 nM/sec. and 8.9 ± 1.0 nM/sec., mean ± S.E.M., n= 3). The involvement of the RTC as a possible target of PS action was also investigated by employing anisomycin. This peptidyl-transferase inhibitor locks nascent chains in the ribosome, leaving the RTC in a closed configuration. Anisomycin was reported to prevent opening of the RTC if added simultaneously or before puromycin [36]. As shown in Fig. 4B, incubation for 1 hr at 4°C with anisomycin (200 μM) during the ER-Aeq reconstitution protocol, and continuous exposure to the drug during the experiment, did not prevent the reduction of the ER Ca2+ level, induced by PS2-T122R expression in SH-SY5Y cells. Interestingly, treatment with this drug, that should close a passive ER Ca2+ leak pathway, decreased the steady-state ER Ca2+ level in the control cells (–22 ± 6.8%, n= 6, P < 0.05).
Figure 4.
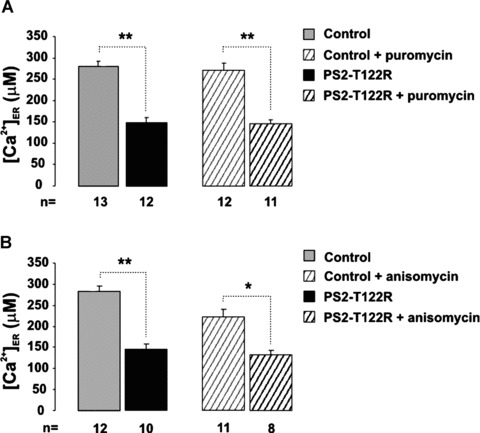
Role of the ribosomal–translocon complex. (A) SH-SY5Y cells were transiently cotransfected with the cDNAs coding for ER-Aeq and PS2-T122R (or with the void vector as control). Upon ER-Aeq reconstitution, cells were treated for 10 min. with puromycin (0.2 mM) at 37°C and continuously maintained in the presence of the drug. ER Ca2+ levels were measured as described in Fig. 1A. (B) SH-SY5Y cells were transfected as described in (A). Anisomycin (0.2 mM) was present during ER-Aeq reconstitution and throughout the experiments. Bars represent the average [Ca2+]ER (μM) (mean ± S.E.M.).
Based on indirect evidence, that is, cytosolic Ca2+ sequestration, LaFerla and colleagues recently suggested that wt PS1 and PS2, as well as an FAD mutant PS1 (M146V), increase SERCA activity [25]. To directly address whether and how PS2 affects the activity of SERCA pumps, we carried out a detailed analysis in permeabilized SH−SY5Y cells by means of ER-Aeq. Ca2+ uptake rates were measured in different ranges of free [Ca2+]o, set by distinct Ca2+-EGTA–based buffers (see Materials and Methods). The maximal values of the first derivative of the instantaneous [Ca2+]ER (d[Ca2+]ER/dt) were plotted as a function of the imposed free [Ca2+]o (Fig. 5A). Expression of PS2-T122R significantly and consistently reduced the maximal rate of Ca2+ uptake, at all the [Ca2+]o tested. Similar reductions in ER Ca2+ uptake were observed in DKO MEFs (Fig. 5B) and wt MEFs (data not shown) expressing PS2-T122R, upon cell permeabilization with digitonin (20 μM). Table 2 reports the estimated Km and Vmax values of Ca2+ uptake obtained by double-reciprocal plots; in all cell types, the trend was similar: expression of PS2-T122R strongly reduced the Vmax (22–35%) with modest increases in the Km (7–16%). Noteworthy, in DKO MEFs, the ER Ca2+ uptake rates were also similarly reduced upon expression of wt PS2 (12.2 ± 0.9 μM/sec. and 9.4 ± 0.9 μM/sec., for control and wt PS2–expressing cells with the free [Ca2+] buffered at 300 nM; mean ± S.E.M., n= 7, P < 0.05; Fig. 5B, triangles). It should be stressed that these values represent the initial rates of the Ca2+ refilling process, that is, when the PS2 effect on leak rates should be negligible, being the ER lumen practically empty of Ca2+. Differences in maximal uptake rates were maintained even in the presence of heparin (200 μg/ml; data not shown).
Figure 5.
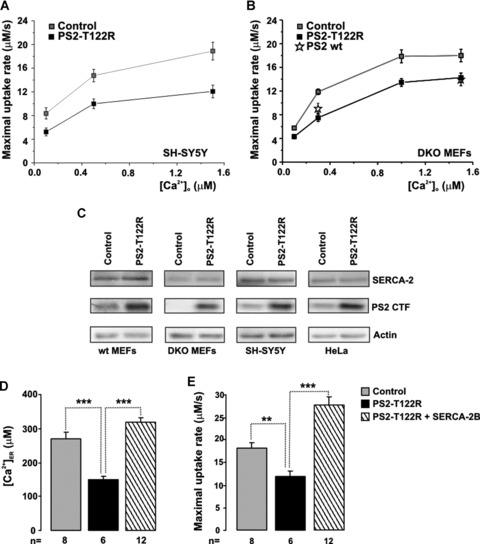
Effect of PS2-T122R on ER Ca2+ uptake. (A, B) ER Ca2+ uptake was followed in permeabilized SH-SY5Y cells and DKO MEFs at different free [Ca2+]o as described in Fig. 2. The maximal values of the first derivative of the instantaneous [Ca2+]ER (d[Ca2+]ER/dt) were plotted as a function of the free external [Ca2+]o (mean ± S.E.M., n ranging from 6 to 20). (B) also shows the values obtained with PS2 wt (empty stars): 9.4 ± 0.9 μM/sec. (n= 7) and 14.4 ± 0.8 μM/sec. (n= 8), respectively, at 0.3 and 1.5 μM [Ca2+]o. (C) Western blots showing SERCA-2 and PS2 levels in control and PS2-T122R–expressing cell lines. (D, E) SH-SY5Y cells were transiently cotransfected with the cDNAs coding for ER-Aeq and PS2-T122R in the absence (black bars) or presence of SERCA-2B cDNA (hatched bars); control cells were cotransfected with ER-Aeq cDNA and with the void vector (grey). Bars represent the average [Ca2+]ER (μM) (D) and maximal uptake rates (μM/sec.) (E) (mean ± S.E.M.).
Table 2.
Kinetic parameters of ER Ca2+ uptake
| Cell type | Km (nM) | % change | Vmax (μM/sec.) | % change | ||
|---|---|---|---|---|---|---|
| Control | PS2-T122R | Control | PS2-T122R | |||
| SH-SY5Y | 150 | 167 | 11 | 19.3 | 12.8 | −34 |
| wt MEFs | 164 | 175 | 7 | 14.5 | 10.1 | −30 |
| DKO MEFs | 277 | 321 | 16 | 21 | 16.4 | −22 |
Because expression of PS2-T122R reduces the ER Ca2+ uptake without reducing the SERCA-2 protein level (Fig. 5C), the simplest interpretation of these results is that PS2 variants affect directly or indirectly the activity of the pump. Experiments were carried out to determine whether the ER Ca2+ depletion induced by PS2-T122R could be compensated by increasing the number of SERCA-2B. Figure 5D and E shows the results obtained by monitoring ER Ca2+ levels in digitonin permeabilized SH-SY5Y cells: over-expression of SERCA-2B together with PS2-T122R allowed to fully recover both the maximal uptake rate and the ER Ca2+ plateau found in control cells.
Conformation of PS2 and store Ca2+ handling
It has been reported that only the full-length (FL), immature forms of wt PSs (but not FAD-linked mutants) can form ER Ca2+ leak channels [23, 26] (but see also [11]). We asked which form of the protein, the FL or the dimeric complex formed by the N- and C-terminal fragments (NTF, CTF), is responsible for the reduced ER Ca2+ level here reported. In DKO MEFs, transient expression of the loss-of-function mutant PS2-D366A, which is devoid of γ-secretase as well as presenilinase activity [39] (Fig. 6A), reduced the ER Ca2+ level by an amount similar to that observed upon expression of wt or mutant PS2 (Fig. 6B).
Figure 6.
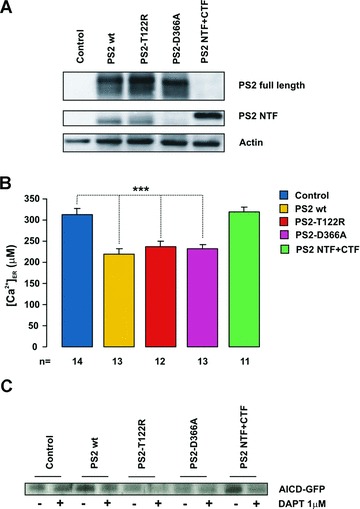
Effect of PS2 conformation on ER Ca2+ levels. (A) DKO MEFs were cotransfected with the cDNA coding for ER-Aeq and a PS2 variant (wt, T122R or D366A) or with a bicistronic vector coding for the NTF and CTF of wt PS2. The Western blot shows the expression level of PS2 (FL and NTF). The PS2 NTF of cells transfected with the bicistronic vector migrates at higher MW because of a myc-tag. (B) Bars represent the average [Ca2+]ER (μM) for DKO MEFs described in (A) (mean ± S.E.M.). (C) The γ-secretase activity was checked in DKO-MEFs transfected with the cDNA coding for C99-GFP as suitable substrate. Co-expression of wt PS2, PS2-T122R or PS2 NTF+CTF, but not PS2-D366A, led to AICD-GFP generation, as detected by Western blot with an anti-GFP antibody. For each condition, cells were also overnight treated with DAPT (1 μM).
Lendahl and colleagues [40] have previously demonstrated that, in DB8 cells KO for both PSs, co-expression of the NTF and CTF of wt PS2, by means of a bicistronic vector, allows the recovery of γ-secretase activity. We confirmed this result in DKO MEFs by employing a recently developed γ-secretase cell assay [30] (Fig. 6C); NTF and CTF co-expression, however, failed to mimic the effect of wt or mutant PS2 on Ca2+ handling (Fig. 6B).
Role of endogenous PS2 on ER Ca2+ uptake
All the above results were obtained in cells over-expressing PS2. To test whether physiological PS2 levels play a role in ER Ca2+ handling, wt MEFs were cotransfected with siRNA specific for mouse PS2 and the cDNA coding for ER-Aeq. As shown in Fig. 7, the protein level was reduced by 50–80%, 24–48 hrs after transfection (see Materials and Methods). When ER Ca2+ plateaus and uptake rates were evaluated, upon digitonin permeabilization, a small but significant increase in both parameters was detected in cells knocked down for PS2, compared with cells treated with control siRNA (Fig. 7A and B). On the other hand, when cells were transfected with the cDNA coding for ubiquilin1 [41] (Fig. 7C) or were treated overnight with the presenilinase inhibitor MW167 (15 μM) [42] (Fig. 7D), both treatments known to increase FL PS levels by interfering with PS processing, significant decreases of ER Ca2+ plateaus were observed in wt but not in DKO MEFs.
Figure 7.
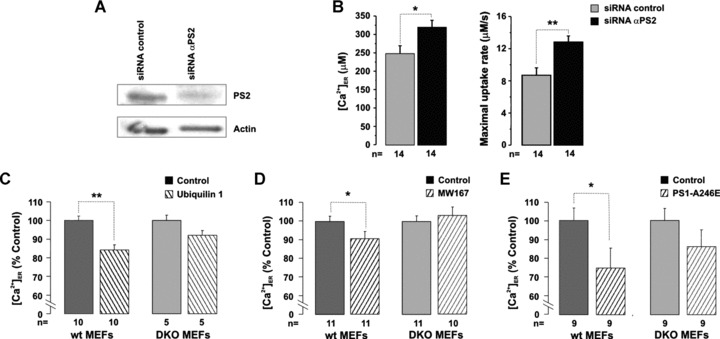
Effect of endogenous PS2 on ER Ca2+ uptake. (A) wt MEFs were transfected with the cDNA coding for ER-Aeq and siRNA specific for mouse PS2 or control siRNA (20 nM). After 48 hrs, part of the cells was harvested to check the expression level of PS2 by Western blotting. (B) The same cells were used to estimate ER Ca2+ uptake upon cell permeabilization with the protocol described in Fig. 2. Bars represent the average [Ca2+]ER (μM) (left) and maximal uptake rates (μM/sec.) (right) (mean ± S.E.M.). (C) wt and DKO MEFs were transfected with the cDNA coding for ER-Aeq and ubiquilin1. (D) wt and DKO MEFs were transfected with the cDNA coding for ER-Aeq and overnight treated with MW167 (15 μM) or vehicle (DMSO). (E) wt and DKO MEFs were transfected with the cDNA coding for ER-Aeq and PS1-A246E or its void vector (control). Bars represent the average [Ca2+]ER (mean ± S.E.M.) expressed as percentage of control cells transfected with the void vector (C, E) or treated with vehicle (D); note interruption in the Y axes. (C–E) Upon Aeq reconstitution, steady-state ER levels were measured as described in Fig. 1A.
We had previously shown that, in HeLa and SH-SY5Y cells as well as in human FAD fibroblasts, PS1 mutants partially mimicked the ER Ca2+–depleting effect of PS2 mutants [18]. We here evaluated the hypothesis that the former mutants can exert an ER-depleting effect only in the presence of PS, that is, in an endogenous PS background. In fact, the transient expression of PS1-A246E significantly decreased the ER Ca2+ plateau by 25% in wt MEFs (243 ± 34 μM and 325 ± 21 μM, mean ± S.E.M., P < 0.05, n= 9) but not in DKO MEFs (270 ± 28 μM and 314 ± 20 μM, mean ± S.E.M., n= 9; Fig. 7E).
Discussion
We have previously reported that different FAD-linked PS1 and PS2 mutants instead of causing a Ca2+ overload, reduce the ER Ca2+ content in different model cells, including fibroblasts from FAD patients and rat primary neurons. The effect is consistent and more dramatic with PS2 variants, and it is also mimicked by over-expression of wt PS2 [18, 20, 21]. Similar conclusions have also been reached in neuroblastoma cells by over-expression of the wt forms of PS2 and PS1 [19]. Mild reductions of store Ca2+ levels rather than Ca2+ overloads were also recently reported in DT40 cells expressing PS1−M146L [11]. Taken together, these data, while questioning the ‘Ca2+ overload’ hypothesis [15, 43], strongly suggest that PSs might be key determinants in setting the ER Ca2+ level. We here investigated the molecular mechanism by which wt and mutant PS2 reduce the ER Ca2+ content of intracellular stores. To this aim, in addition to HeLa cells, we also used neuroblastoma SH-SY5Y cells and two MEFs clones, with and without endogenous PSs (wt and DKO MEFs, respectively).
When the effect of the transient expression of PS2-T122R was taken as a reference to compare the different cell types, we invariably observed a reduction in the steady-state [Ca2+]ER, ranging from about 50% to 20%, with the following efficacy order: SH-SY5Y > wt MEFs ≥ DKO MEFs > HeLa cells. All these models were thus employed to untangle the likely common mechanisms that underlie the PS2 effect.
By means of ER-Aeq, we initially verified whether the amount of passive ER Ca2+ leak was increased by expression of wt and mutant PS2. In the absence of Ca2+ and in the presence of a SERCA inhibitor, the decay rate of the [Ca2+]ER was modestly but significantly accelerated by expression of PS2-T122R. The effect was more pronounced in SH-SY5Y (+17%) than HeLa cells (+9%). Of note, an increased leakage was also found upon expression of PS2-M239I or wt PS2.
Passive ER leak may be accounted for by classical Ca2+ release channels. Indeed, PS2 has been suggested to increase number and/or sensitivity of both RyRs [9, 10, 34, 44] and IP3Rs [7, 11, 12, 22]. In SH-SY5Y and wt MEF cells, where the PS2 effect was more pronounced, pharmacological (dantrolene and heparin) and genetic (siRNA) approaches, allowed us to estimate an increase in leak, due to both RyRs and IP3Rs (type 3), that corresponds to about 15%, a value not far from that found in HeLa and SH-SY5Y cells by measuring passive ER Ca2+ leakage in the presence of CPA (ranging from 9% to 17%).
We also verified that PS2 did not exert its effect by acting on the protein import machinery, the so-called RTC, which was recently suggested to represent a relevant ER Ca2+ efflux pathway [36–38]. Puromycin, a known RTC opener [36], was not able to mimic the PS2 effect on both the cytosolic and ER Ca2+ levels of SH-SY5Y cells. Notably, puromycin by itself did not increase the rate of Ca2+ exit from the stores. In contrast, anisomycin, a drug that should keep the RTC closed, did not rescue the ER Ca2+ loss caused by mutant PS2. Taken together, these data indicate that, at least in SH-SY5Y cells, the RTC does not significantly contribute to the resting ER Ca2+ leakage or to that induced by expressed PS2.
We also verified whether there was an effect of PS on ER Ca2+ uptake. In intact cells, uptake rates do not simply reflect pump activity, as they are also affected by Ca2+ influx/extrusion processes across the plasma membrane and mitochondria Ca2+ buffering. To overcome these uncertainties, studies were thus carried out in digitonin permeabilized cells with the free [Ca2+] of the bathing medium buffered at different values (0.1–2 μM). At these Ca2+ concentrations, the contribution of mitochondria to Ca2+ uptake is negligible. Under these conditions, in cells expressing PS2-T122R, reductions of Ca2+ uptake rates were found at each Ca2+ concentration tested. Similar results were also obtained in DKO MEFs expressing wt PS2. In SH-SY5Y, over-expression of SERCA-2B together with PS2-T122R rescued both ER Ca2+ uptake rates and steady-state levels at, or above, the values observed in control, void-vector transfected cells. Recently, Green et al.[25] have shown that wt PS1 and PS2 as well as an FAD-linked mutant (PS1-M146V) increase SERCA-2 activity. Those data, however, have been obtained by monitoring cytosolic Ca2+ clearance in intact Xenopus oocytes, that is, under conditions where other factors such as the amplitude of capacitative Ca2+ influx, the activity of the plasma membrane Ca2+ ATPase and the contribution of mitochondrial Ca2+ uptake may complicate data interpretation. From our results obtained in permeabilized cells, we could conclude that PS2-T122R affects the maximal capacity of the pump rather than its Km, which appears to be set around 0.15–0.3 μM, a value not far from that reported for purified SERCA-2B (0.17 μM) [45].
With these results in mind, one would expect to find ER Ca2+ overloads in DKO MEFs, if compared with wt MEFs. On the contrary, we found similar ER Ca2+ steady-state levels and about a 20% increase in ER Ca2+ uptake rates in permeabilized DKO MEFs. It should be noted that previous data using these model cells found both ER Ca2+ overload [23] and ER Ca2+ reduction [22, 25], despite the fact that these clones all derive from the same laboratory (B. De Strooper’s lab; [31]). The different results do not depend on different approaches to test Ca2+ handling, since both the fura-2 [23, 25] and recombinant aequorin [22] were used, as we did. Moreover, we could not confirm in the DKO MEFs differences in the expression levels of IP3Rs [22] or SERCAs [25]. These discrepancies indicate that, despite originating from the same transgenic mice, cell clones can substantially differ one from the other (at least in terms of Ca2+ handling) and, more important, that a cause–effect relationship between alterations in Ca2+ homeostatic machinery and lack of PS expression cannot be unambiguously established using these cells. On the contrary, such a cause–effect relationship appears more easily and consistently found upon transient expression of wt or mutant PS1/2 in each clone. We followed this approach to test the hypothesis that endogenous PS2 works as a brake on ER Ca2+ uptake. Along the same line, in wt MEFs, knocking down the endogenous level of PS2 by siRNAs increased both ER Ca2+ pumping and steady-state levels, thus unmasking the inhibitory role played by PS2.
In DKO MEFs, the effect of mutant PS2 on ER Ca2+ handling does not qualitatively differ from that exerted by either wt PS2 or PS2-D366A, a loss-of-function, non-pathogenic mutant also devoid of presenilinase activity (see Fig. 6A and [39]). These findings suggest that PS2 and possibly SERCA-2 interact independently of the γ-secretase activity and the intra-molecular cut of PS2. To further address this issue, we have co-expressed the NTF and CTF of wt PS2 in DKO MEFs using a bicistronic vector [40]. Expression of the two separate fragments, while being able to rescue the γ-secretase activity, was unable to mimic the effect of PS2 on ER Ca2+ handling. Furthermore, the Ca2+-depleting effect of PS2 over-expression could be partially mimicked by ubiquilin1 over-expression [41] or inhibition of presenilinase activity by MW167 [42], two treatments that have been previously suggested to stabilize the FL form of endogenous PS2.
We previously found that transient or stable expression of some FAD-linked PS1 mutants also causes ER Ca2+ depletion. This effect, however, is quantitatively much smaller than that caused by PS2 expression and is dependent on the type of mutation (A246E, M146L, P117L but not L286V; [18]) and the cell system employed. For example, PS1-A246E was effective in HeLa cells and wt MEFs but not in SH-SY5Y cells and primary rat neurons (this work and [18]). These results might be explained taking into account recent findings that suggest the capability of PS1 mutants to shift the balance from PS1 to PS2 containing γ-secretase complexes [46]. Thus, given the inhibitory role played by endogenous PS2 on ER Ca2+ uptake, the store-depleting effect of some PS1 mutants could be indirect and reside in their capability to potentiate the effect of endogenous PS2, either as single molecule or as part of a complex. This is, however, only a working hypothesis, and additional experiments are required to address this specific point.
In conclusion, we here provide evidence showing that PS2 has a dual role on ER Ca2+ homeostasis: (i) it inhibits SERCAs and (ii) increases Ca2+ leak through ER Ca2+ channels (RyRs and IP3R-3). The effect on Ca2+ uptake was dominant in SH-SY5Y with respect to HeLa cells. This finding is consistent with the fact that cells of the former type, like neurons, have very low resting ER Ca2+ levels. Altogether, these results suggest that PS2 plays a direct, primary role on Ca2+ handling by intracellular stores while strengthening the idea of a complex interplay between PS1 and PS2.
Acknowledgments
We thank I. Moressa. and V. Rocchetto for performing some of the experiments; G. Ronconi and M. Santato for technical assistance. This work was supported by grants from the Italian Ministry of University and Scientific Research (MURST) to C.F., P.P. and T.P.; FIRB (grant no. RBIN042Z2Y) to P.P.; the Veneto Region (Biotech 2) and the CARIPARO Foundation to T.P. We thank G. Binetti for the cDNAs coding for wild-type and mutant presenilins (PSs); B. De Strooper for wild-type and PS double knockout mouse embryonic fibroblasts; U. Lendhal for the bicistronic vector coding for PS2 N-and C-terminal fragments; M. J. Monteiro for the cDNA coding for ubiquilin1. We are grateful to P. Magalhães for useful suggestions. L. Brunello’s Ph.D. fellowship is granted by the CARIPARO Foundation.
References
- 1.Guo Q, Fu W, Sopher BL, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 2.Chan SL, Mayne M, Holden CP, et al. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195–200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- 3.Schneider I, Reverse D, Dewachter I, et al. Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J Biol Chem. 2001;276:11539–44. doi: 10.1074/jbc.M010977200. [DOI] [PubMed] [Google Scholar]
- 4.Stutzmann GE, Caccamo A, LaFerla FM, et al. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–13. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stutzmann GE, Smith I, Caccamo A, et al. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–9. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo Q, Furukawa K, Sopher BL, et al. Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid β-peptide. Neuroreport. 1996;8:379–83. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- 7.Leissring MA, Paul BA, Parker I, et al. Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J Neurochem. 1999;72:1061–8. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- 8.Popescu BO, Cedazo-Minguez A, Benedikz E, et al. γ-secretase activity of presenilin 1 regulates acetylcholine muscarinic receptor-mediated signal transduction. J Biol Chem. 2004;279:6455–64. doi: 10.1074/jbc.M306041200. [DOI] [PubMed] [Google Scholar]
- 9.Lee SM, Lee JW, Song YS, et al. Ryanodine receptor-mediated interference of neuronal cell differentiation by presenilin 2 mutation. J Neurosci Res. 2005;82:542–50. doi: 10.1002/jnr.20655. [DOI] [PubMed] [Google Scholar]
- 10.Lee SY, Hwang DY, Kim YK, et al. PS2 mutation increases neuronal cell vulnerability to neurotoxicants through activation of caspase-3 by enhancing of ryanodine receptor-mediated calcium release. FASEB J. 2006;20:151–3. doi: 10.1096/fj.05-4017fje;1. [DOI] [PubMed] [Google Scholar]
- 11.Cheung KH, Shineman D, Muller M, et al. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–83. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leissring MA, Parker I, LaFerla FM. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1, 4,5-trisphosphate-mediated calcium signals. J Biol Chem. 1999;274:32535–8. doi: 10.1074/jbc.274.46.32535. [DOI] [PubMed] [Google Scholar]
- 13.Smith IF, Green KN, LaFerla FM. Calcium dysregulation in Alzheimer’s disease: recent advances gained from genetically modified animals. Cell Calcium. 2005;38:427–37. doi: 10.1016/j.ceca.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 14.Leissring MA, LaFerla FM, Callamaras N, et al. Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol Dis. 2001;8:469–78. doi: 10.1006/nbdi.2001.0382. [DOI] [PubMed] [Google Scholar]
- 15.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–72. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 16.Thinakaran G, Sisodia SS. Presenilins and Alzheimer disease: the calcium conspiracy. Nat Neurosci. 2006;9:1354–5. doi: 10.1038/nn1106-1354. [DOI] [PubMed] [Google Scholar]
- 17.Lessard CB, Lussier MP, Cayouette S, et al. The overexpression of presenilin2 and Alzheimer’s-disease-linked presenilin2 variants influences TRPC6-enhanced Ca2+ entry into HEK293 cells. Cell Signal. 2005;17:437–45. doi: 10.1016/j.cellsig.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Zatti G, Burgo A, Giacomello M, et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium. 2006;39:539–50. doi: 10.1016/j.ceca.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Fedrizzi L, Lim D, Carafoli E, et al. Interplay of the Ca2+-binding protein dream with presenilin in neuronal Ca2+ signaling. J Biol Chem. 2008;283:27494–503. doi: 10.1074/jbc.M804152200. [DOI] [PubMed] [Google Scholar]
- 20.Zatti G, Ghidoni R, Barbiero L, et al. The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol Dis. 2004;15:269–78. doi: 10.1016/j.nbd.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Giacomello M, Barbiero L, Zatti G, et al. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol Dis. 2005;18:638–48. doi: 10.1016/j.nbd.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 22.Kasri NN, Kocks SL, Verbert L, et al. Up-regulation of inositol 1,4,5-trisphosphate receptor type 1 is responsible for a decreased endoplasmic-reticulum Ca2+ content in presenilin double knock-out cells. Cell Calcium. 2006;40:41–51. doi: 10.1016/j.ceca.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 23.Tu H, Nelson O, Bezprozvanny A, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–93. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yagi T, Giallourakis C, Mohanty S, et al. Defective signal transduction in B lymphocytes lacking presenilin proteins. Proc Natl Acad Sci USA. 2008;105:979–84. doi: 10.1073/pnas.0707755105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Green KN, Demuro A, Akbari Y, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–16. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson O, Tu H, Lei T, et al. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007;117:1230–9. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brini M, Marsault R, Bastianutto C, et al. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c): a critical evaluation. J Biol Chem. 1995;270:9896–903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- 28.Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci USA. 1999;96:11872–7. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nyabi O, Pype S, Mercken M, et al. No endogenous Aβ production in presenilin-deficient fibroblasts. Nat Cell Biol. 2002;4 doi: 10.1038/ncb0702-e164a. : E164; author reply E5–6. [DOI] [PubMed] [Google Scholar]
- 30.Florean C, Zampese E, Zanese M, et al. High content analysis of gamma-secretase activity reveals variable dominance of presenilin mutations linked to familial Alzheimer’s disease. Biochim Biophys Acta. 2008;1783:1551–60. doi: 10.1016/j.bbamcr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Herreman A, Serneels L, Annaert W, et al. Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol. 2000;2:461–2. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- 32.Pinton P, Ferrari D, Magalhães P, et al. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J Cell Biol. 2000;148:857–62. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rybalchenko V, Hwang SY, Rybalchenko N, et al. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008;40:84–97. doi: 10.1016/j.biocel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 34.Hayrapetyan V, Rybalchenko V, Rybalchenko N, et al. The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein-protein interaction. Cell Calcium. 2008;44:507–18. doi: 10.1016/j.ceca.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Wojcikiewicz RJ. Type I, II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J Biol Chem. 1995;270:11678–83. doi: 10.1074/jbc.270.19.11678. [DOI] [PubMed] [Google Scholar]
- 36.Roy A, Wonderlin WF. The permeability of the endoplasmic reticulum is dynamically coupled to protein synthesis. J Biol Chem. 2003;278:4397–403. doi: 10.1074/jbc.M207295200. [DOI] [PubMed] [Google Scholar]
- 37.Flourakis M, Van Coppenolle F, Lehen’kyi V, et al. Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store operated channels activation. FASEB J. 2006;20:1215–7. doi: 10.1096/fj.05-5254fje. [DOI] [PubMed] [Google Scholar]
- 38.Ong HL, Liu X, Sharma A, et al. Intracellular Ca2+ release via the ER translocon activates store-operated calcium entry. Pflugers Arch. 2007;453:797–808. doi: 10.1007/s00424-006-0163-5. [DOI] [PubMed] [Google Scholar]
- 39.Walker ES, Martinez M, Brunkan AL, et al. Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Abeta 42/40 ratios. J Neurochem. 2005;92:294–301. doi: 10.1111/j.1471-4159.2004.02858.x. [DOI] [PubMed] [Google Scholar]
- 40.Stromberg K, Hansson EM, Laudon H, et al. gamma-Secretase complexes containing N- and C-terminal fragments of different presenilin origin retain normal gamma-secretase activity. J Neurochem. 2005;95:880–90. doi: 10.1111/j.1471-4159.2005.03415.x. [DOI] [PubMed] [Google Scholar]
- 41.Massey LK, Mah AL, Monteiro MJ. Ubiquilin regulates presenilin endoproteolysis and modulates gamma-secretase components, Pen-2 and nicastrin. Biochem J. 2005;391:513–25. doi: 10.1042/BJ20050491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campbell WA, Reed ML, Strahle J, et al. Presenilin endoproteolysis mediated by an aspartyl protease activity pharmacologically distinct from gamma-secretase. J Neurochem. 2003;85:1563–74. doi: 10.1046/j.1471-4159.2003.01799.x. [DOI] [PubMed] [Google Scholar]
- 43.Cowburn RF, Popescu BO, Ankarcrona M, et al. Presenilin-mediated signal transduction. Physiol Behav. 2007;92:93–7. doi: 10.1016/j.physbeh.2007.05.053. [DOI] [PubMed] [Google Scholar]
- 44.Pack-Chung E, Meyers MB, Pettingell WP, et al. Presenilin 2 interacts with sorcin, a modulator of the ryanodine receptor. J Biol Chem. 2000;275:14440–5. doi: 10.1074/jbc.m909882199. [DOI] [PubMed] [Google Scholar]
- 45.Lytton J, Westlin M, Burk SE, et al. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem. 1992;267:14483–9. [PubMed] [Google Scholar]
- 46.Placanica L, Tarassishin L, Yang G, et al. PEN2 and presenilin-1 modulate the dynamic equilibrium of presenilin-1 and presenilin-2 gamma -secretase complexes. J Biol Chem. 2008;284:2967–77. doi: 10.1074/jbc.M807269200. [DOI] [PMC free article] [PubMed] [Google Scholar]